

While much focus is placed on cholera epidemics, the greatest burden occurs in settings where cholera is endemic, including areas of South Asia, Africa, and now Haiti1,2. Dhaka, Bangladesh is a megacity that is hyper-endemic for cholera, and experiences two regular seasonal outbreaks of cholera each year3. Despite this, a detailed understanding of the diversity of Vibrio cholerae strains circulating in this setting, and their relationships to annual outbreaks, has not yet been obtained. In this study, we performed whole-genome sequencing of V. cholerae across several levels of focus and scale, at the maximum possible resolution. We analyzed bacterial isolates to define cholera dynamics at multiple levels, ranging from infection within individuals, to disease dynamics at the household level, to regional and intercontinental cholera transmission. Our analyses provide a genomic framework for understanding cholera diversity and transmission in an endemic setting.
Between 2002 and 2005, V. cholerae was isolated from index patients admitted to the International Centre for Diarrhoeal Disease Research, Bangladesh (icddr,b)4. Over a surveillance period of three weeks, follow-up samples were collected from household contacts of the index patient (i.e., those that shared a cooking pot with the index patient during the three days prior to the presentation of the index case at icddr,b). In total, 303 V. cholerae isolates were obtained from 224 individuals, representing 103 households. V. cholerae was isolated more than once from the stool of 45 of these individuals in the three-week follow-up period. In order to characterize the diversity of these isolates, we performed whole-genome sequencing and defined single nucleotide variants (SNVs) by mapping each sample to the V. cholerae O1 El Tor N16961 reference genome with putative recombinant sites removed from the alignments (see online Methods).
To investigate the diversity of circulating strains, we determined how the 303 V. cholerae sampled from Dhaka from 2002–2005 are related evolutionarily to one another. Our phylogenetic analysis revealed that six sublineages of seventh pandemic V. cholerae El Tor (7PET) were circulating concurrently in Dhaka during our study period (Fig. 1). These included two serotype O1 Ogawa sublineages (B5, B6), two O1 Inaba sublineages (B1, B2), and two O139 serogroup sublineages (B3, B4). Three non-toxigenic V. cholerae O1 were isolated from patients with diarrhea, and these were not phylogenetically related to the 7PET lineage (Supplementary Fig. 1). Each 7PET sublineage displayed a strong temporal signal over the study period, during which five sublineages waxed and waned, whereas only a single sublineage (B1) was present throughout the study (Fig. 2). Our data correspond well to the surveillance data collected by icddr,b over the time period (Supplementary Fig. 2) as well as to published literature describing cholera outbreaks within Bangladesh5–7. Interestingly, we did not observe geographical clustering of isolates by sublineage across Dhaka (Fig. 2).
Figure 1. Phylogeny and household network of Vibrio cholerae 7th pandemic strains.

Bayesian phylogenetic reconstruction of the 300 7PET V. cholerae strains circulating in Dhaka from 2002–2005 along with the reference isolate N16961. The scale bar denotes number of substitutions per variable site. Labeled nodes delineate sublineages. The phylogeny was rooted on the wave 1 clade containing N16961. V. cholerae serogroup O139 lineages are B3 (n= 9) and B4 (n= 41). All other lineages are V. cholerae serogroup O1; B1 (n= 109) and B2 (n= 1) are serotype Inaba, and B5 (n= 34) and B6 (n= 106) are serotype Ogawa. The contact network was constructed by anchoring all household contacts to their respective index case on the phylogenetic tree. Colors denote household relationships when anchored to the same index case. Due to the number of households (n= 102), colors are non-unique.
Figure 2. Spatiotemporal dynamics of 7th pandemic strains.
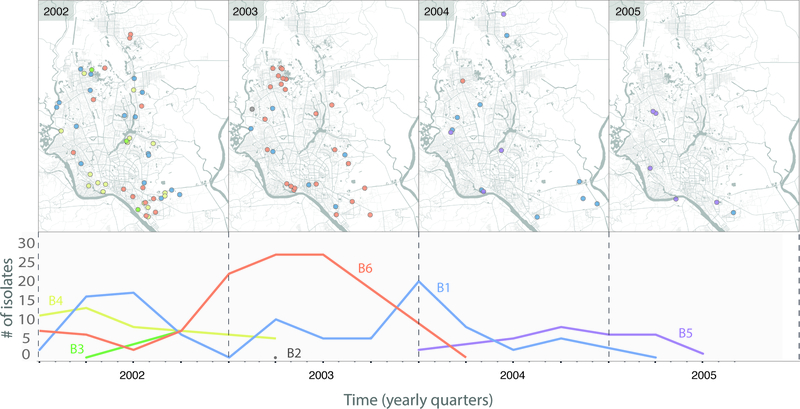
The top panel displays the spatial distribution of the 7PET sublineages (as defined in Figure 1) within Dhaka city for each year of the study. The number of isolates sampled per sublineage over time is shown on the bottom panel.
In endemic settings, it is common that multiple individuals contract cholera within a household4,8–10. To determine the diversity of V. cholerae seen within household members we linked the household contact information to the phylogeny (Fig. 1). Strikingly, this revealed that in eighteen (23%) of the 79 households from which more than one individual had been enrolled, cholera infections were caused by different V. cholerae sublineages, which were largely, but not exclusively, members of the 7PET lineage. These included infections with V. cholerae O1 and O139 serogroups, as well as mixed V. cholerae O1 serotypes Inaba (belonging to sublineages B1/B2) and Ogawa (sublineages B5/B6) within one household (Fig. 1). Two households were infected simultaneously with both 7PET and non-7PET V. cholerae. Comparison of pairwise genetic differences between 7PET isolates showed that isolates sampled from within the same household (median = 1 SNV) were significantly more similar to one another than to those sampled from different households (median = 55 SNVs; P= 4.25 × 10−47; Mood’s median test; Fig. 3, Supplementary Fig. 3A).
Figure 3. Pairwise comparisons of SNVs within and between households and longitudinal samples.
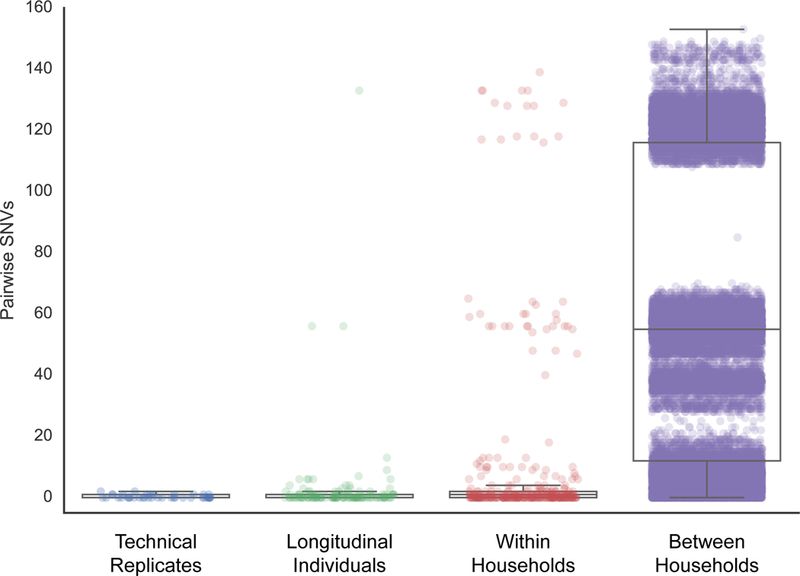
The technical replicates consist of five colonies that were isolated from a single glycerol stock, which was repeated using four different starting stocks from different individuals. A total of 420 data points represent the pairwise comparisons between the five replicates from each of the four stocks. The longitudinal dataset comprises the 45 individuals sampled at more than one time point. For each individual, we calculated all pairwise comparisons between samples, for a total of 129 data points across all longitudinal individuals. The within household dataset comprises the comparisons within each of the 79 households with more than one individual, for a total of 200 individuals and 339 data points. Only unique person-person samples were compared in this set. The between household dataset includes comparisons between all 102 households within the study, and comprises comparisons across 224 individuals for a total of 39,802 data points. Box plots depict the mean and quartiles for each dataset and the lengths of the whiskers correspond to 1.5 times the interquartile range. Each dot represents the number of SNVs between two isolates and their opacity scales with the number of observations. The apparent tri-modal distributions seen in the plots are due to the underlying population structure of the isolates (see Figure 1).
Ninety-four household contacts (42%) did not manifest clinical symptoms over the sampling period despite a positive stool culture for V. cholerae, compared to 127 contacts with both diarrhea and a positive culture (Supplementary Table 1). Although we did not observe an association between asymptomatic individuals and specific sublineages of V. cholerae, we did find a significant relationship between clinical presentation and immune response, as determined by a four-fold increase in vibriocidal antibody titer from day 4 to day 21 (P= 3.1 × 10−4; chi-squared test). Among individuals with low initial vibriocidal titers on day 2 (≤ 100), 70% of asymptomatic carriers (n = 40/57) had a four-fold increase compared to 95% among symptomatic individuals (n = 72/76). Importantly, these data show that if using vibriocidal antibody titer as a surrogate marker of infection, 9.5% of infections (n = 21/221) would not have been detected here.
Previous work has demonstrated that shedding of V. cholerae from cholera patients peaks two days post-presentation of symptoms, declining steadily for seven days thereafter11. We compared the pairwise genetic distances between isolates to the number of days between their isolation, within each household. We found that V. cholerae sampled within six days of each other are highly clonal (median SNVs for days 0 to 6 is <= 1) (Fig. 4). Seven of the eighteen mixed-infection households had isolates sampled between six and fourteen days of each other. Taken together, these data demonstrate the high degree of genetic relatedness between V. cholerae isolated within six days from members of the same household. This strongly implies that these infections are due to within-household transmission or exposure to a common source. The finding of isolates unrelated to the index case highlights the importance of repeat introductions of new isolates to the overall incidence of cholera within households in this setting.
Figure 4. Effect of time of sampling on genetic diversity within a household.
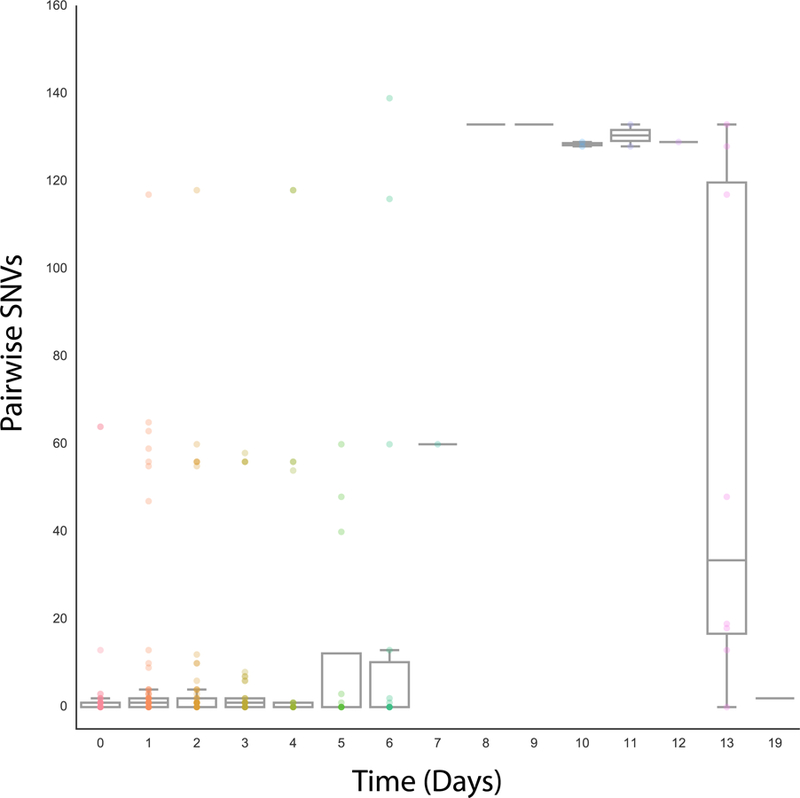
For each household with more than one isolate (n = 79), we calculated the pairwise number of SNVs and the number of days between sampling dates for each isolate. A total of 303 pairwise comparisons were performed. The opacity of the data points scales with the number of observations. Box plots for each time period display the mean and quartiles of the number of SNVs, with the whiskers extending to 1.5 times the interquartile range.
Informed by these household-level data, we investigated the V. cholerae diversity within the 45 individuals from whom V. cholerae was identified in stool more than once over the three-week follow-up period. Since the SNV differences between isolates taken from the same patient were predicted to be small12, we guarded against artifacts from sequencing and culturing procedures by sequencing five technical replicates from each of four patients (Supplementary Fig. 3C). Pairwise comparisons within these technical replicates revealed a maximum difference of two SNVs (mean = 0.35, median = 0). Pairwise comparisons of the strains isolated from contacts in whom V. cholerae was isolated more than once over 21 days (n = 43) revealed a median of 0 SNVs (range 0–133 SNVs) between time points over the infection (Fig 3; Supplementary Fig. 3B). Using the technical replicates to set a threshold of 2 SNVs, we identified eight individuals with longitudinal samples separated by this threshold (Supplementary Fig. 4). Two individuals appear to have mixed-sublineage infections, since the isolates are separated by more than 60 SNVs and fall within different 7PET sublineages (Fig. 3, Supplementary Fig. S4).
In addition to the SNV differences, we observed that V. cholerae isolated from one individual appeared to lose the CTX bacteriophage encoding the cholera toxin (ctxAB genes) during the course of an infection (Supplementary Fig. 5). Within this individual, CTXφ is detected on days two and five, but on days three and four, CTXφ was not present in the sequence data (Supplementary Fig. 5). Consistent with this, the phylogenetic relationship between these isolates suggests that two populations of V. cholerae existed within this patient.
To understand the spatiotemporal spread of cholera within and out of this hyper-endemic region, we placed the isolates from Dhaka (n = 300) into the context of a global collection of 513 additional 7PET genomes collected between 1957 and 2014. The phylogeny revealed a complex history of cholera in South Asia (Fig. 5), and suggests that a highly-connected network exists by which cholera is transmitted regionally. For instance, the 2005 Dhaka epidemic involved an O1 Ogawa strain resistant to streptomycin, tetracycline, and erythromycin13,14. This strain was first detected in our phylogeny in Kolkata as early as 2003, suggesting this strain was imported into Dhaka from elsewhere. This also suggests that Dhaka is part of a wider transmission network for cholera across this region. Consistent with this, several sublineages follow the same routes of dissemination from the area. Sublineages B1 and B5 both circulate regionally around the Ganges Delta and then proceed to seed outbreaks in other South Asian regions, such as in Nepal and Pakistan in 201015,16. However, sublineage B6 is only seen briefly in Dhaka and Kolkata between 2002 and 2004 (Fig. 5), and does not appear to have radiated out of the region. Sublineages in Dhaka are also the ancestors of strains that eventually seed epidemics outside of South Asia, such as the massive epidemic in Haiti in 201015,17 and outbreaks in Africa (Fig. 5).
Figure 5. Maximum likelihood phylogeny of global 7th pandemic strains.

The Dhaka isolates were put into the context of a global collection of 7th pandemic V. cholerae, for a total of 813 isolates. The maximum likelihood phylogeny was estimated under the GTR model on 6,241 variable sites with 500 bootstrap replicates. The phylogeny was rooted using the pre-seventh pandemic strain M66. Nodes are colored according to the origin of the isolates. Sublineages seen in Dhaka are indicated, as well as important transmission events.
The detection of a strong temporal signal within our molecular data allowed us to scale our phylogeny into time units (Supplementary Fig. 6). This dated phylogeny shows that the main sublineages circulating in South Asia all have a most recent common ancestor dated to 1989 [1988–1990] (Supplementary Fig. 7). Bangladesh suffered a massive diarrheal epidemic (> 1.3 million cases) in 1987–1988, brought on by severe flooding in many areas of the country18. V. cholerae was isolated in 45% and 38% of stools tested in 1987 and 1988, respectively, and was the predominant etiological agent detected for this epidemic18. Our phylogenetic data indicate that around this time, several successful sublineages emerged from the background V. cholerae diversity and have subsequently persisted in the human population until the present day. Thus, this regional circulation network is not only important for understanding endemic V. cholerae transmission within South Asia, but also shows that radiations of these sublineages seed global cholera epidemics.
Here, we demonstrate the repeated circulation of discrete V. cholerae sublineages within this hyper-endemic setting for cholera. We show that over a four year study period, although the 7PET lineage is a constant, sublineages within 7PET are highly dynamic and oscillate with different patterns. A high proportion of individuals had asymptomatic infections across these sublineages, despite low genetic diversity amongst V. cholerae isolates. This suggests that in this setting, host factors, including partial immunity and genetics, may play significant roles in the progression of disease. We also show that V. cholerae genomes sequenced after this study period fall within these sublineages. We conclude that these sublineages reflect the broad diversity of 7PET currently circulating within South Asia, and our data define the evolutionary space from which pandemic waves of 7PET cholera emerge and propagate worldwide. Our data strongly suggest extensive and longitudinal interaction between humans and multiple sublineages of 7PET V. cholerae in this highly endemic area.
Supplementary Material
Acknowledgements
This research was supported in part by NIAID grants R01 AI106878 to E.T.R., F.Q., S.B.C., F.C., A.I.K., Y.A.B., R.C.C., an R01 AI103055 to J.B.H., F.Q., R.C.L., a U01 AI058935 to S.B.C., F.Q., E.T.R., R.C.L., J.B.H., a U01 AI077883 to E.T.R., F.Q., the Fogarty International Center-NIH D43 TW005572 to M.I.U., T.R.B., as well as K43 TW010362 to T.R.B. This work was supported by the Wellcome Trust (grant 098051) to N.R.T. M.J.D. is supported by a Wellcome Trust Sanger Institute PhD Studentship. R.C.C was supported by the Robert Wood Johnson Foundation Harold Amos Medical Faculty Development Program (grant 72424). We thank A. J. Page, J. Keane and the sequencing teams at the Wellcome Trust Sanger Institute. This work was supported by the International Centre for Diarrhoeal Disease Research, Bangladesh (icddr,b) which is grateful to the Government of Bangladesh, Canada, Sweden and the UK for providing core/unrestricted support.
Footnotes
Competing Financial Interests
The authors declare no competing financial interests.
Data and code availability
All source code is available at https://figshare.com/s/48e1568811b79112ffce.
URL section
SMALT (http://www.sanger.ac.uk/science/tools/smalt-0); MapBox (http://mapbox.com/); QGIS (http://www.qgis.org/)
Accession Codes
Raw sequence data have been submitted to the European Nucleotide Archive (ENA) under accession PRJEB2215. All individual accession numbers for genomes used in this study can be found in Supplementary table 2.
References:
- 1.Ali M, Nelson AR, Lopez AL & Sack DA Updated Global Burden of Cholera in Endemic Countries. PLoS Negl. Trop. Dis 9, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clemens JD, Nair GB, Ahmed T, Qadri F & Holmgren J Cholera. The Lancet 0, (2017). [DOI] [PubMed] [Google Scholar]
- 3.Longini IM et al. Epidemic and Endemic Cholera Trends over a 33-Year Period in Bangladesh. J. Infect. Dis 186, 246–251 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Kendall EA et al. Relatedness of Vibrio cholerae O1/O139 Isolates from Patients and Their Household Contacts, Determined by Multilocus Variable-Number Tandem-Repeat Analysis. J. Bacteriol 192, 4367–4376 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Faruque SM et al. Reemergence of Epidemic Vibrio cholerae O139, Bangladesh. Emerg. Infect. Dis 9, 1116–1122 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schwartz BS et al. Diarrheal epidemics in Dhaka, Bangladesh, during three consecutive floods: 1988, 1998, and 2004. Am. J. Trop. Med. Hyg 74, 1067–1073 (2006). [PMC free article] [PubMed] [Google Scholar]
- 7.Chowdhury F et al. Vibrio cholerae Serogroup O139: Isolation from Cholera Patients and Asymptomatic Household Family Members in Bangladesh between 2013 and 2014. PLoS Negl Trop Dis 9, e0004183 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harris JB et al. Susceptibility to Vibrio cholerae Infection in a Cohort of Household Contacts of Patients with Cholera in Bangladesh. PLOS Negl Trop Dis 2, e221 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sugimoto JD et al. Household Transmission of Vibrio cholerae in Bangladesh. PLOS Negl Trop Dis 8, e3314 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.George CM et al. Genetic relatedness of Vibrio cholerae isolates within and between households during outbreaks in Dhaka, Bangladesh. BMC Genomics 18, 903 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weil AA et al. Clinical Outcomes in Household Contacts of Patients with Cholera in Bangladesh. Clin. Infect. Dis 49, 1473–1479 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levade I et al. Vibrio cholerae genomic diversity within and between patients. Microb. Genomics 3, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Faruque SM et al. An Improved Technique for Isolation of Environmental Vibrio cholerae with Epidemic Potential: Monitoring the Emergence of a Multiple-Antibiotic–Resistant Epidemic Strain in Bangladesh. J. Infect. Dis 193, 1029–1036 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Faruque ASG et al. Emergence of Multidrug-resistant Strain of Vibrio cholerae O1 in Bangladesh and Reversal of Their Susceptibility to Tetracycline after Two Years. J. Health Popul. Nutr 25, 241–243 (2007). [PMC free article] [PubMed] [Google Scholar]
- 15.Hendriksen RS et al. Population Genetics of Vibrio cholerae from Nepal in 2010: Evidence on the Origin of the Haitian Outbreak. mBio 2, e00157–11 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shah MA et al. Genomic Epidemiology of Vibrio cholerae O1 Associated with Floods, Pakistan, 2010. Emerg. Infect. Dis 20, 13–20 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eppinger M et al. Genomic Epidemiology of the Haitian Cholera Outbreak: a Single Introduction Followed by Rapid, Extensive, and Continued Spread Characterized the Onset of the Epidemic. mBio 5, e01721–14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Siddique AK et al. Cholera epidemics in Bangladesh: 1985–1991. J. Diarrhoeal Dis. Res 10, 79–86 (1992). [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.