

Abstract
Dasatinib is a multi-tyrosine kinase inhibitor approved for treatment of Ph+ acute lymphoblastic leukemia (ALL), but its efficacy is limited by resistance. Recent preclinical studies suggest that dasatinib may be a candidate therapy in additional ALL subtypes including pre-BCR+ ALL. Here we utilized shRNA library screening and global transcriptomic analysis to identify several novel genes and pathways that may enhance dasatinib efficacy or mitigate potential resistance in human pre-BCR+ ALL. Depletion of the transcriptional co-activator CBP increased dasatinib sensitivity by downregulating transcription of the pre-BCR signaling pathway previously associated with dasatinib sensitivity. Acquired resistance was due in part to upregulation of alternative pathways including WNT through a mechanism suggesting transcriptional plasticity. Small molecules that disrupt CBP interactions with the CREB KID domain or β-catenin showed promising preclinical efficacy in combination with dasatinib. These findings highlight novel modulators of sensitivity to targeted therapies in human pre-BCR+ ALL, which can be reversed by small molecules inhibitors. They also identify promising therapeutic approaches to ameliorate dasatinib sensitivity and prevent resistance in ALL.
Keywords: acute lymphoblastic leukemia; pre-BCR, E2A-PBX1; drug resistance; shRNA screen
Introduction
Although the treatment of acute lymphoblastic leukemia (ALL) has improved during the last decades (1), 25% of children and 65% of adults die within five years after diagnosis with relapse and chemotherapy-related complications being among the main causes of death (2,3). Several ALL subtypes have been described based on their karyotype, cell type, immunophenotype, and gene-expression profile. The pre-BCR+ ALL subtype is characterized by expression of the pre-B cell receptor (pre-BCR) (4) and in 50% of cases is associated with the chromosomal translocation t(1;19), coding for the chimeric fusion protein E2A-PBX1 (5), which is present in about 5% of pediatric and adult ALL (6). Using preclinical models, we and others have suggested that dasatinib, a small molecule multi-tyrosine kinase inhibitor, is a candidate therapy in pre-BCR+ ALL (4, 7–10) and early clinical evidence supports our observations (11). However, novel therapies in ALL including small molecule inhibitors such as dasatinib are limited by the development of resistance and short-lived responses (12). An understanding of drug sensitivity and resistance mechanisms to modern targeted small molecules will support the development of more rational combination therapies to improve efficacy and survival of patients and reduce adverse effects.
Using unbiased functional genomic screens and global transcriptomic analytic approaches, we studied intrinsic primary and acquired mechanisms of dasatinib sensitivity and resistance in pre-BCR+ ALL. Our studies identified CREB binding protein (CBP) as a major modulator of dasatinib sensitivity and demonstrate that its targeting by small molecule inhibitors shows promising preclinical efficacy increasing sensitivity to dasatinib in ALL.
Methods
Human cells and cell culture
Human leukemia cell lines were cultured in RPMI 1640 medium supplemented with 10% FBS, 100 U/ml penicillin/streptomycin, and 0.29 mg/ml L-glutamine. E2A-PBX1 positive cell lines (697, RCH-ACV and Kasumi-2) were authenticated using western blot for E2A-PBX1 fusion protein expression. E2A-PBX1 negative cell lines (SEM, REH, HAL-01) and not further authenticated. All cell lines were obtained from DSMZ in 2013. Cell lines were negatively tested for mycoplasma contamination by PCR. The REH cell line is found listed in the database of commonly misidentified cell lines maintained by ICLAC, and was used here as control. Pre-BCR status was assessed by flow cytometry for surface VPREB1. Fresh human umbilical cord blood was collected from newborn placenta (with informed consent and Stanford University Institutional Review Board [IRB] approval), CD34+ cells were enriched and cultured as described previously (13). Primary ALL cells were obtained from the Tissue Bank of the Department of Hematology/Oncology, University Medical Center of Freiburg and of the German cooperative ALL-BFM study group, University Medical Center Schleswig-Holstein, Campus Kiel. Experiments with human tissue samples were performed in accordance with the tenets of the declaration of Helsinki and were approved by the Ethics Commission from the University of Freiburg (ethical vote, approval No 279/17). All patients were informed by a physician and signed an informed consent form. Human cell lines, primary leukemia cells and cord blood CD34+ cells were treated with dasatinib (LC laboratories, Woburn, MA), saracatinib (Selleckchem, Houston,TX), bosutinib (Selleckchem), dexamethasone (Sigma-Aldrich, St. Louis, MO), XX-650–23 (14) and ICG-001 (Selleckchem) at the indicated concentrations. Titration curves were performed using increasing drug concentrations and cells were counted by trypan blue assay after 4 days.
shRNA screen for dasatinib sensitivity/resistance
The shRNA screen was performed as described elsewhere (15,16). Three shRNA lentivirus sublibraries containing a total of about 106,249 shRNA sequences and targeting 3,158 human genes (about 25 shRNAs for most genes and 50 shRNAs for kinases/phosphatases) and containing 13,774 negative control shRNAs were used to transduce RCH-ACV cells in four pools. After puromycin selection and expansion, RCH-ACV cells stably expressing pooled shRNAs were split and subjected to four rounds of treatment with dasatinib (20 nM) or vehicle, in two replicates. For each round, cells were treated with dasatinib for 72 hr. The dasatinib resistance/sensitivity conferred by an individual shRNA was calculated using Mann-Whitney U test and casTLE, a novel statistical framework for cas9 and shRNA screening technologies (ref. 17, v1.0 available at https://bitbucket.org/dmorgens/castle). The enrichment score for each shRNA was normalized by the distribution of enrichment scores of the negative control shRNAs. With this normalization, drug resistance phenotype is independent from growth phenotype (15). Candidate shRNA shown to confer sensitivity or resistance are shown in Supplementary Table S1.
shRNA knock-down and sgRNA knock-out vectors, lentiviral transduction and competition assays
Individual shRNA sequences (Supplementary Table S2) were cloned into the BstXI site of the p309 lentiviral vector (15), for stable transduction in human ALL cell lines. For mutagenesis studies, RCH-ACV cells were transduced with a modified vector expressing CRISPR/Cas9 (LentiCas9-Blast, Addgene 52962), and selected with blasticidin. Stably transduced cells were infected with a pU6-sgRNA EF1Alpha-puro-T2A-GFP lentiviral vector containing sgRNAs cloned into the BstXI/BlpI site (18). sgRNA sequences were selected for CBP from a human genome-wide library and from Gecko v2 database and are listed in Supplementary Table S3.
Lentivirus generation and transduction of human cells are described elsewhere (19). After puromycin selection, transduced cells were mixed 50/50 for competition assays using cells with control vectors and cells with shRNA knock-down or sgRNA knock-out for the gene of interest. Mixed cells were split for vehicle and drug treatment. Indel frequency was analyzed by Sanger sequencing and TIDE assay and performed as previously described (20).
Apoptosis and cell cycle analysis
Apoptosis and cell cycle analysis were described elsewhere (21). Briefly, apoptotic cells were quantified on the basis of annexin V staining (eBioscience, San Diego, CA) by flow cytometry. BrdU/7-AAD incorporation assays were performed according to the manufacturer’s protocol (cat. # 559619, BD Pharmingen, San Jose, CA).
Bone marrow transplantation assays and in vivo drug treatment
For xenograft bone marrow transplantation, sub-lethally (2 Gy) irradiated female NSG mice of 8–9 weeks of age were injected via tail vein with 1000 RCH-ACV leukemia cells. After randomization, mice were treated daily, intra-peritoneally, with dasatinib starting 7 days after transplantation with vehicle (30% PEG1500, 1% Tween 80, 2.5 % DMSO dissolved in PBS) or 2.5 mg/kg dasatinib for 20 days. For combination treatment with dasatinib and XX-650–23, mice were treated with dasatinib as previously described (9). Cohorts of mice treated with XX-650–23, received the drug dissolved in PBS daily, i.v., starting 7 days after transplantation with 1.25 mg/kg (14). The dose was increased every 4 days, in 1.25 mg/kg steps, to reach a maximal drug dose of 5 mg/kg daily. Mice were treated for 25 days. Analysis was performed in a non-blinded fashion, and statistical methods were not used to estimate sample size. All data collected from mice were included for analysis. Disease-free survival was defined by signs of illness including general lymphadenopathy, lethargy, weight loss and shivering. Moribund mice were euthanized.
RT-qPCR
RNA was isolated using the RNeasy Mini Kit (Qiagen, Hilden, Germany) and cDNA was synthesized using Superscript reverse transcriptase IV kit (Life Technologies) following the manufacturer’s recommendations. Relative expression was quantified using an ABI 7900HT Thermocycler. Taqman Master mix (Applied Biosystems, Carlsbad, CA) following Taqman gene expression assays from Life Technologies (Supplementary Table S4). All signals were quantified using the ΔΔCt method and normalized to ΔΔCt-values of the Actb gene expression levels.
Western blot analysis
Western blot for protein quantification was performed using a modified RIPA lysis buffer as previously described (9). The following antibodies were used for immunodetection: rabbit anti-CBP (clone C-20; Santa Cruz Biotechnologies, Dallas, TX), mouse anti-mouse AcH3K27 (cat # 17–683, EMD Millipore, Billerica, MA), rabbit anti histone H3 (#Ab1791, Abcam, Cambridge, UK), rabbit anti-Gapdh (#G9545, Sigma-Aldrich, St. Louis, MO), rabbit anti-HA (#Ab9110, Abcam), mouse anti-RAC1 (clone 23A8, #05–389, EMD Millipore). Secondary antibodies were rabbit anti-mouse (#816729, Life technologies, Carlsbad, CA) and goat anti-rabbit (#G21234, Life Technologies) coupled to HRP. Quantification by densitometry was performed using ImageJ software (NIH, Bethesda, MD).
RNA sequencing and bioinformatics analysis
RNA from dasatinib-sensitive and dasatinib-resistant RCH-ACV cells from two independent sublines were used for RNAseq analysis following isolation using an RNeasy Mini kit (Qiagen) and library preparation using the TruSeq Stranded mRNA Kit (#RS-122–2101, Illumina). Sequencing was performed in the Stanford Genome Center and Personalized Medicine using an Illumina HiSeq2500 (paired end 101bp). RNA sequences were aligned and analyzed in the web-based platform DNAnexus using following software tools (version): bedtools (2.19.0), bwa-aln (0.6.2-r126), bwa-mem (0.7.7-r441), fastqc (0.10.1), Picard Tools (1.107(1667)), samtools (0.1.19–44428cd).
Differential gene expression analysis was performed by DESeq2 in R platform using a p-value <0.05 as cut-off. Gene ontology enrichment of KEGG pathways was analyzed using DAVID bioinformatics resources (22) and row Z scores of were calculated and visualized in heat maps using CIMminer. For gene-set enrichment analysis (GSEA, ref. 23), RPKM values from expressed genes were plotted using Graphpad Prism software (Graphpad Inc., La Jolla, CA).
For SNV/indel calling, annotation and effect prediction, Varscan 2 (24) and SNPeff (25) software tools were employed using the following parameters: 50 transcripts minimum, p-value <0.05, heterozygosity defined as > 0.25 of transcripts and are shown in Supplementary Table S5. Germ line, non-coding and synonymous SNV/indels were filtered out. SNV/indels were confirmed and visualized by integrative genomics viewer (IGV, ref. 26). The CBP point mutation R1748H was validated after PCR amplification and Sanger sequencing using specific primers (Supplementary Table S6).
Cloning and CTNNB1 overexpression
cDNA of CTNNB1 with 4 phosphorylation site mutations in the N-terminal linked to hemagglutinin gene from mouse was amplified using following primers (5’ primer ATGGCTACTCAAGCTGACCTG and 3’ primer CAGGTCAGTATCAAACCAGGC). PCR products were isolated and cloned into the pMYS-IRES-GFP retroviral vector using the In-Fusion HD Cloning Kit (Clontech laboratories Inc, Mountain View, CA) as previously described (9).
Statistics
Statistical differences between two groups were analyzed by two-sided Mann Whitney U test or by Student’s t-test. Dose-response curves were compared using the extra sum of squares F test. The Bliss independence model (27) was used to evaluate synergy between drug combinations. A p-value <0.05 was considered statistically significant. Statistical differences from Kaplan-Meier curves were analyzed by log-rank (Mantel Cox) test. Statistical analysis and graphs were performed using Graphpad prism software (Graphpad Inc., La Jolla, CA).
Study approval
All experiments on mice were performed with the approval of and in accordance with Stanford’s Administrative Panel on Laboratory Animal Care (APLAC, Protocol 9839).
Results
Identification of key genes and pathways involved in dasatinib sensitivity.
A functional genomic screen was performed to prospectively identify novel genes and pathways involved in dasatinib sensitivity and resistance (15,16). The pre-BCR+ ALL cell line RCH-ACV was transduced using lentiviral vectors harboring pooled shRNA sub-libraries targeting in total 3,158 human genes (106,249 shRNAs) including negative controls (Fig 1A). The sub-libraries employed were selectively enriched for shRNAs that targeted either kinases, phosphatases, epigenetic factors or cancer-related pathways. Stably transduced cells were treated with dasatinib for 12 days (4 pulses) of in vitro growth (Fig. 1B). Dasatinib- and vehicle-treated cells were then analyzed by deep-sequencing to quantify shRNA relative abundance. Several candidate shRNAs were observed to confer dasatinib resistance or increase dasatinib sensitivity (Fig. 1C, Supplementary Table S1). Gene ontology analysis of shRNAs that increased dasatinib sensitivity identified the B-cell receptor signaling pathway as significantly enriched (p-value 5.7 × 10−9, FDR 6.9 × 10−6), including kinases ZAP70 and SYK, consistent with previous observations of their roles in pre-BCR+ ALL (10) and validating the shRNA screening approach. Notably, shRNAs targeting CREB binding protein (CBP) were markedly depleted in the dasatinib-treated population compared to control shRNAs (Supplementary Table S1), suggesting that depletion of the co-activator CBP increases dasatinib sensitivity in human pre-BCR+ ALL cells.
Figure 1. Identification of key genes and pathways involved in dasatinib sensitivity and resistance.
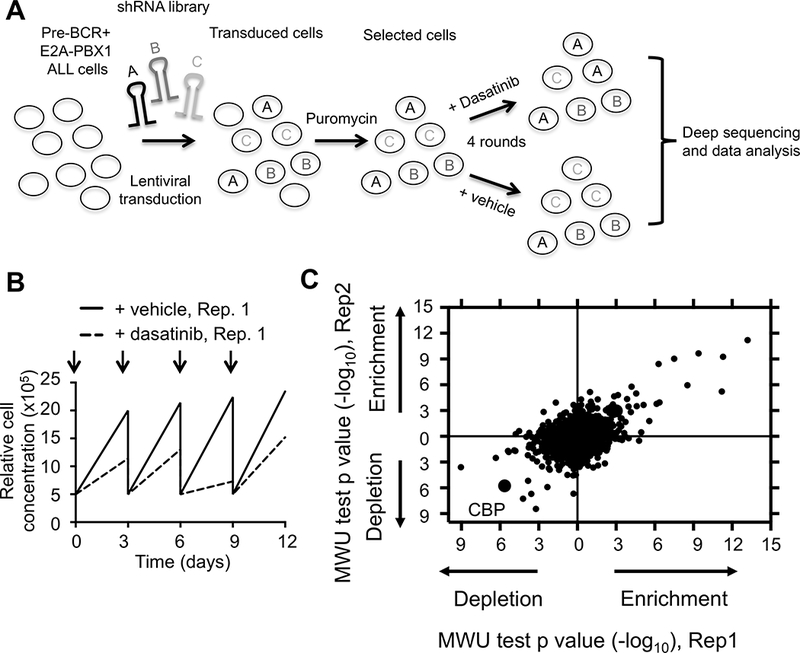
(A) Schematic representation of shRNA screen. RCH-ACV leukemia cells (E2A-PBX1+/pre-BCR+) were transduced with shRNA sub-libraries. After puromycin selection, cells were treated with dasatinib or vehicle every three days for four rounds. Frequency of individual shRNAs was quantified by deep sequencing. The experiment was performed in duplicate. (B) Cell proliferation curves from stably transduced RCH-ACV cells with shRNA sub-libraries in the presence of dasatinib (20nM) from replicate 1 (Rep.1). Arrows represent dasatinib pulses. (C) Dot plot represents Mann-Whitney U test p-values for enrichment (confer resistance) or depletion (increase sensitivity) of shRNAs targeting indicated genes by knock-down in dasatinib-treated cells from two replicates. Each dot represents a gene.
CBP modulates sensitivity to dasatinib.
Competition growth assays were performed to validate single shRNAs identified in the functional genomics screen (10). shRNA-mediated knockdown of CBP in RCH-ACV ALL cells (Fig. 2A) had significant stimulatory effects on cell growth in the absence of dasatinib (Fig. 2B), consistent with the previously described tumor suppressor function of CBP in ALL (28) and lymphoid malignancies (29). In contrast, CBP knockdown substantially inhibited proliferation of dasatinib-treated RCH-ACV cells compared to control transduced cells (Fig. 2B). The interactive effects of CBP knockdown and dasatinib treatment were observed in a second E2A-PBX1+/pre-BCR+ cell line, 697, but were not observed in representative pre-BCR-negative or non-E2A-PBX1 cells (Supplementary Fig. S1A-B). Mechanistically, shRNA-mediated repression of CBP results in decreased expression levels of pre-BCR associated genes, previously associated with dasatinib sensitivity and resistance including ZAP70, SYK and LCK (10) and novel identified genes in our shRNA screen as BTK and LYN (Fig. 2C-D). These results demonstrated a synthetic lethal interaction between CBP and dasatinib in two independent pre-BCR+ ALL cell lines consistent with the shRNA screen results, and reinforced the role of CBP as a master transcriptional regulator of pre-BCR/BCR associated genes (28–29).
Figure 2. CBP modulates sensitivity to dasatinib.
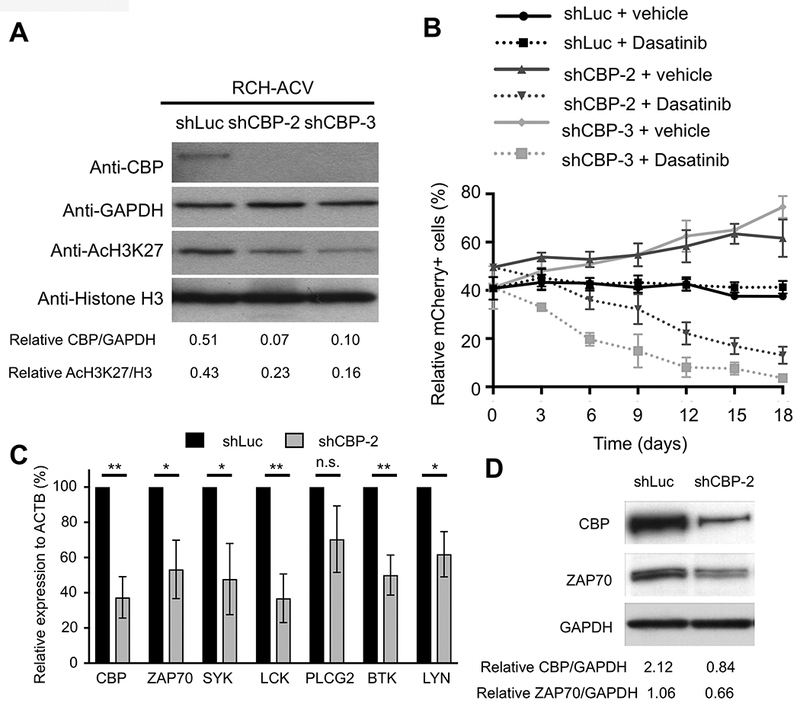
(A) Representative Western blot shows CBP levels following knock-down by two different shRNA constructs in RCH-ACV cells. Down-regulation of CBP correlates with global decreased levels of H3K27ac. GAPDH and histone H3 were used as loading controls, respectively. Densitometry values shown at the bottom of the panel were calculated using Image J software. Data representative of four independent Western blot experiments. (B) Diagram shows percentage of mCherry+ cells transduced with shRNAs for luciferase (control) or CBP in the presence of vehicle or dasatinib (20 nM) for 18 days. Data represent the mean ±SEM of three independent experiments. (C) Graph shows relative expression of pre-BCR –associated genes by RT-qPCR after shRNA-mediated knockdown of CBP. Data represent the mean ±SEM of three independent experiments. Statistical analysis by Student’s T-test, ** p-value <0.01, * p-value <0.05, n.s., not significant. (D) Western blot (representative of two independent experiments) shows decreased ZAP70 expression levels after shRNA-mediated knockdown of CBP. Densitometry values shown at the bottom of the panel were calculated using Image J software.
To further confirm a role for CBP in dasatinib sensitivity, mutagenesis studies were performed using a CRISPR/Cas9 approach in the context of competition growth assays. RCH-ACV cells expressing sgRNAs that efficiently targeted CBP (Supplementary Fig. S2A-B) showed a proliferative advantage in the presence of vehicle but considerably decreased competitive fitness in the presence of dasatinib consistent with the knockdown studies (Supplementary Fig. S2C). Thus, two different genetic approaches confirmed that CBP modulates dasatinib sensitivity in pre-BCR+ ALL cells.
Preclinical efficacy of pharmacologic therapy targeting CBP in combination with dasatinib.
Given the foregoing genetic results, a pharmacologic approach was employed to test the effects of CBP inhibitors in combination with dasatinib. The inhibitory compound XX-650–23, which antagonizes the interaction between the KIX domain of CBP and the KID domain of the transcription factor CREB (14), showed a synergistic effect in combination with dasatinib on cell growth inhibition of pre-BCR+ cell lines RCH-ACV (Fig. 3A-B) and 697 (Supplementary Fig. S3). Pharmacologic inhibition of CBP also increased dasatinib sensitivity in Kasumi-2 cells (which are E2A-PBX1+ pre-BCR-negative) and REH (TEL-AML1+ ALL) but not SEM, HAL-01 and SUP-B15, which lack E2A-PBX1 (Supplementary Fig. S3), suggesting some selectivity. Dasatinib in combination with XX-650–23 did not inhibit growth of human cord blood cells (IC50 >10 μM) cultured in vitro (Supplementary Fig. S4). These data suggest that the combination targeted therapy might be safe and effective in a subset of leukemias particularly pre-BCR+ ALLs.
Figure 3. Pharmacologic inhibition of CBP increases sensitivity to dasatinib.
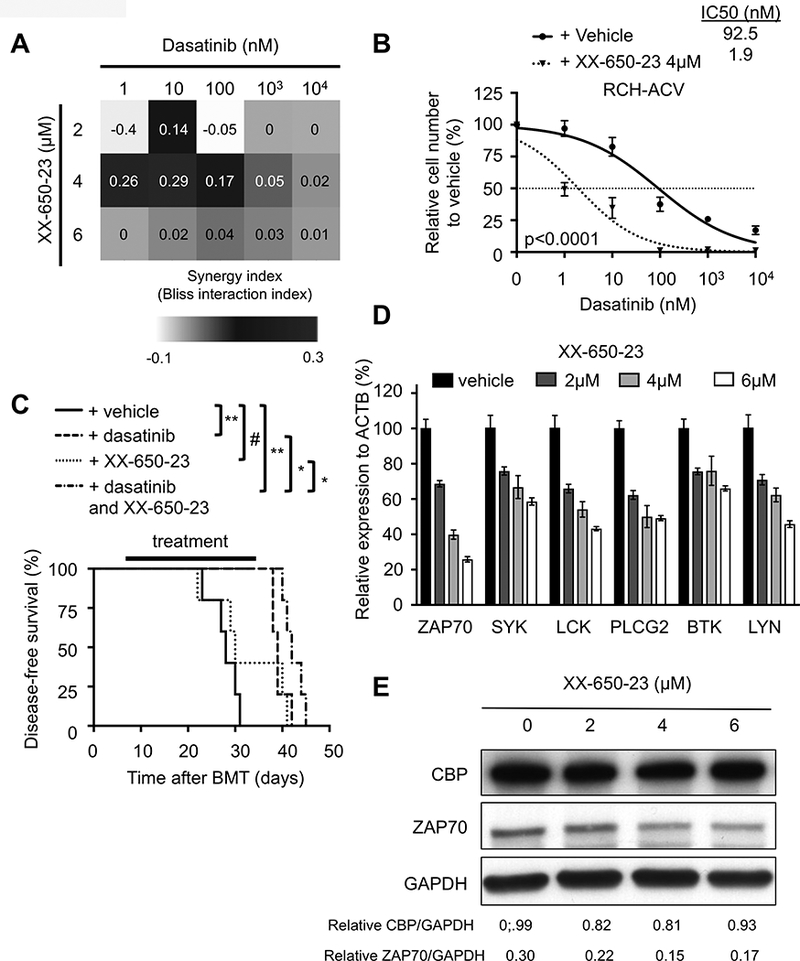
(A) Heat map representation of Bliss interaction index for RCH-ACV cells treated with dasatinib and the CBP-CREB inhibitor XX-650–23. Data represent the mean of three independent experiments. (B) Titration curves shown for RCH-ACV cells cultured with dasatinib in combination with vehicle or XX-650–23 (4 μM). Data represent mean ±SEM of three independent experiments. Statistical analysis was performed by F test. IC50, half maximal inhibitory concentration. (C) Kaplan-Meier curve represents disease-free survival of NSG mice after xenograft transplantation with RCH-ACV cells treated daily with vehicle, dasatinib (2.5 mg/kg/day/d), XX-650–23 (1.25 – 5 mg/kg/d, dose increased stepwise every 4 days with 1.25 mg/kg/d) or the combination of dasatinib and XX-650–23 for 25 days. Each cohort contains five mice. Statistical analysis was performed by log rank test. # not significant; * p<0.05, ** p<0.01. (D) Graph shows relative expression of pre-BCR –associated genes by RT-qPCR after pharmacologic inhibition of CBP in RCH-ACV cells with XX-650–23 at several concentrations. Data represent the mean ±SEM of three independent experiments. (E) Western blot (representative of two independent experiments) shows decreased ZAP70 expression levels after pharmacologic inhibition of CBP with XX-650–23. Densitometry values shown at the bottom of the panel were calculated using Image J software.
The preclinical efficacy of dasatinib in combination with XX-650–23 was tested in vivo after xeno-transplantation of RCH-ACV cells in NSG mice. As expected, in vivo treatment with dasatinib alone prolonged disease-free survival, consistent with previous observations using a conditional E2A-PBX1 transgenic mouse model (9). Notably, co-treatment with dasatinib and XX-650–23 modestly but significantly increased the disease-free survival of transplanted NSG mice compared to either drug alone (Fig. 3C, median disease-free survival for XX-650–23, 30 days; for dasatinib, 39 days; and for combination, 43 days). Consistent with the shRNA results, pharmacologic inhibition of CBP with XX-650–23 resulted in decreased expression levels of pre-BCR associated genes implicated in dasatinib sensitivity in RCH-ACV cells (Fig. 3D-E), and in a subset of primary ALL cells with selected karyotypes including E2A-PBX1+ ALL (Supplementary Fig. S5). These data further establish a novel combination therapy that inhibits a subset of human leukemias compared to normal hematopoietic progenitors and shows promising preclinical efficacy in vivo.
Acquired resistance to dasatinib in human pre-BCR+ ALL cells.
To prospectively define mechanisms of acquired dasatinib resistance in pre-BCR+ human leukemias, two independent dasatinib-resistant RCH-ACV sublines were generated by culture through multiple passages in the presence of increasing concentrations of dasatinib for several months. Acquired resistance to dasatinib was confirmed in vitro (Fig. 4A and Supplementary Fig. S6A) and in vivo in xeno-transplantation assays (Fig. 4B). Functionally, dasatinib-resistant cells showed reduced apoptosis/cell death (Supplementary Fig. S6B) and increased cycling cell frequencies compared to dasatinib-sensitive cells in the presence of dasatinib (Supplementary Fig. S6C). Dasatinib-resistant cells also displayed cross-resistance to the chemically distinct SRC-family kinase inhibitors bosutinib and saracatinib but not to the corticosteroid dexamethasone (Supplementary Fig. S7A-C). These data suggest that dasatinib-resistant cells developed a specific mechanism of drug resistance to SRC-family kinase inhibitors but not in general to other chemotherapeutics.
Figure 4. Generation of dasatinib resistant pre-BCR+ human ALL cells.
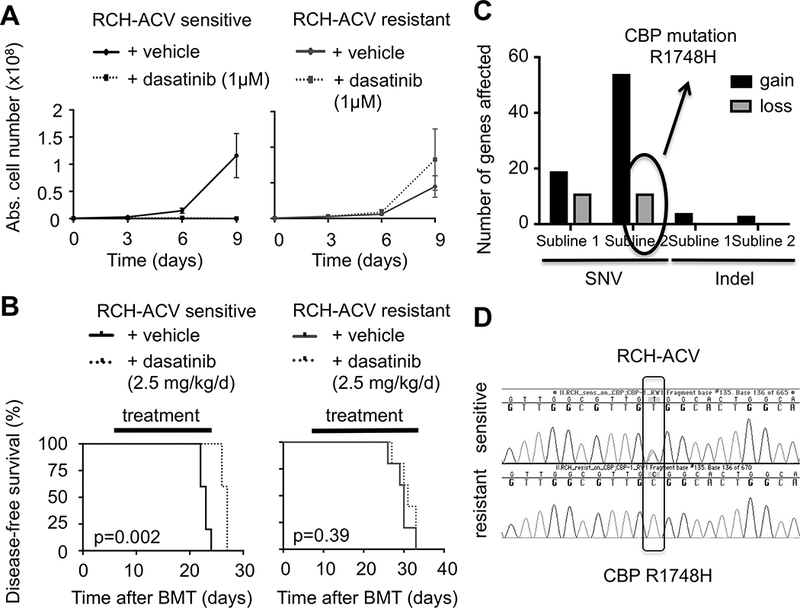
(A) Dasatinib-sensitive (left panel) and –resistant (right panel) RCH-ACV cells were cultured in the presence of vehicle or 1 μM dasatinib for nine days. Cell proliferation was assessed every three days by trypan blue assay. Data represent the mean ±SEM from four independent experiments. (B) Kaplan-Meier curve shows disease-free survival of NSG mice xeno-transplanted with dasatinib-sensitive (left panel) and –resistant (right panel) RCH-ACV cells. Recipients were treated with either vehicle or dasatinib for 20 days. Statistical analysis by log rank test. (C) Analysis of number of genes affected by SNV and indels in dasatinib-resistant compared to dasatinib-sensitive RCH-ACV sublines in RNA-seq data. Gain indicates presence and loss indicates absence of sequence aberrations in dasatinib-resistant sublines. (D) Chromatograms from Sanger sequencing show validation of CBP R1748H SNV identified by RNA-seq detected in dasatinib-sensitive but absent in dasatinib-resistant RCH-ACV cells.
Loss of CBP R1748H SNV in acquired dasatinib resistance.
The most frequent mechanism of resistance to small molecule inhibitors including dasatinib is point mutations in the targeted kinase domain (30,31). Therefore, RNA-seq was performed to detect SNVs and indels in transcribed genes in dasatinib-resistant compared to dasatinib-sensitive RCH-ACV cells. RNA-seq data showed that various SNVs and indels were acquired in the dasatinib-resistant sublines (Fig. 4C, Supplementary Table S5), however none affected SRC-family kinases or other dasatinib targets (HMS LINCS database, ref. 32), suggesting a possible bystander effect in the acquisition of dasatinib resistance.
Conversely, several SNVs originally present in the dasatinib-sensitive cells were unexpectedly absent in the dasatinib-resistant sublines (11 SNVs in each). One of these was a R1748H substitution in the CBP gene present in dasatinib-sensitive RCH-ACV cells (allele frequency of 12% in dasatinib-sensitive subline #1 and 58% in dasatinib-sensitive subline #2 in RNA-seq analysis) but not in dasatinib-resistant sublines as confirmed by Sanger sequencing (Fig. 4D, Supplementary Table S5). This SNV, which has not been previously described, is located in proximity to the histone acetyl-transferase domain of CBP. Mutations in this region can impair histone acetylation and transcriptional regulation of CBP target genes, including genes involved in signal transduction by the B-cell receptor and B-cell specific transcription factors (28,29). These data suggest that RCH-ACV cells heterozygous for R1748H may be partially insufficient for CBP tumor suppressor function, but this SNV was lost independently in both sublines during acquisition of dasatinib resistance likely due to selective pressure for restoration of wild-type CBP function.
Global transcriptome analysis identifies pathways involved in dasatinib resistance.
The observed selection on the CBP gene suggested that RCH-ACV cells might acquire dasatinib resistance through transcriptional plasticity by an epigenetic mechanism, as described for several enzymatic and epigenetic inhibitors (33–35). Because epigenetic states are dynamic and undergo reversal in the absence of selective conditions, we assessed for potential loss of resistance over time. After 2 months culture off-drug, dasatinib-resistant cells partially reversed their resistance phenotypes (Fig. 5A), suggesting transcriptional plasticity as a possible contributory cause of dasatinib-acquired resistance.
Figure 5. Global transcriptomic analysis highlights pathways involved in acquired dasatinib resistance.
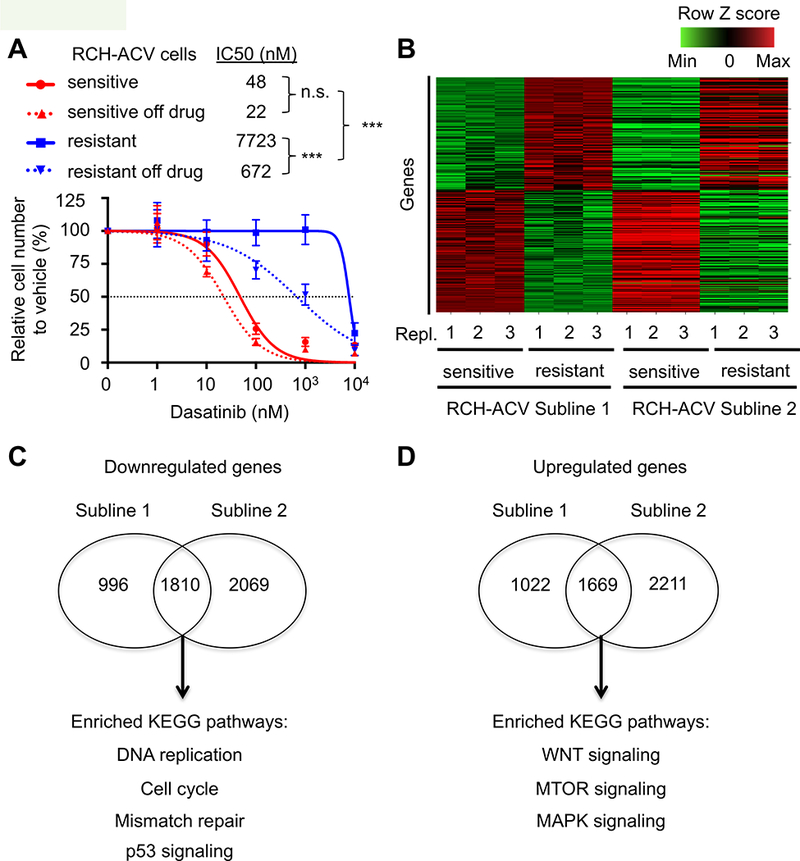
(A) Titration curves shown for RCH-ACV dasatinib-sensitive and -resistant cells treated with increasing concentrations of dasatinib. Titration experiments were also performed on cells after two months of culture following removal from dasatinib selection. Data represent mean ±SEM of three independent experiments. Statistical analysis by F test. n.s., not significant, *** p-value <0.001. IC50, half maximal inhibitory concentration. (B) Heat map represents row Z scores of common up- and downregulated genes analyzed by RNA-seq from two independently generated dasatinib-sensitive and –resistant RCH-ACV sublines. Each column represents a sample, sequenced in triplicate. Each row represents a gene. (C) Gene ontology analysis of KEGG pathways from common downregulated and (D) upregulated genes from generated dasatinib- sensitive and -resistant RCH-ACV sublines. Venn diagrams represent number of genes regulated (p-value<0.05) in common as well as specifically in each clone. Shown below are selected KEGG pathways enriched (p-value <0.05) in the gene ontology analysis from common regulated genes in dasatinib-resistant RCH-ACV sublines.
Whole transcriptome analysis of RNA-seq data from dasatinib-sensitive and resistant ALL cells from two independent sublines (Fig. 5B) identified 1810 down-regulated genes and 1669 up-regulated genes in common (p ≤ 0.05). Gene ontology analysis of common regulated genes identified several pathways involved in acquired dasatinib resistance (Fig. 5C-D, Supplementary Fig. S8A-B). Pathways depleted in dasatinib-resistant sublines included DNA replication/cell cycle, p53 signaling, and DNA repair pathways. Consistent with the transcriptional profile, dasatinib-resistant sublines showed a proliferative disadvantage in vitro in competition assays (Supplementary Fig. S9A-B) as well as in vivo after xenograft transplantation (Supplementary Fig. S9C). Depletion of the p53 signaling pathway was consistent with resistance to apoptosis/cell death (Supplementary Fig. S6B). These data suggest a mechanistic model in which dasatinib-resistant cells may bypass specific pathways inhibited by dasatinib through compensatory activation of alternative pathways.
The WNT signaling pathway contributes to acquired dasatinib resistance.
Gene ontology analysis identified the WNT, MTOR and MAPK signaling pathways enriched in dasatinib-resistant RCH-ACV cells (Fig. 5D and Supplementary Fig. S10A). Furthermore, RNAseq data showed that expression of several members of the WNT signaling pathway was increased in dasatinib-resistant RCH-ACV cells (Supplementary Fig. S10A-B), which was confirmed by RTqPCR (Supplementary Fig. S10C). To validate the WNT-pathway as a contributor to the dasatinib resistant phenotype, a stabilized β-catenin protein was expressed in RCH-ACV sensitive cells (Fig. 6A), which were subsequently treated with increasing dasatinib concentrations. Cells expressing exogenous β-catenin were significantly more resistant to dasatinib than cells transduced with empty vector (Fig. 6B). To further confirm the role of the WNT pathway in primary and acquired dasatinib resistance, we knocked down the GTPase RAC1, a positive regulator of the WNT signaling pathway (36) that was also identified as a sensitizer in our shRNA screen. Depletion of RAC1 in RCH-ACV sensitive and resistant cells (Supplementary Fig. S11A) caused decreased competitive fitness in the presence of dasatinib compared to control transduced cells (Supplementary Fig. S11B). These data suggest that the WNT-pathway is involved in primary and acquired dasatinib resistance in pre-BCR+ ALLs.
Figure 6. Involvement of the WNT signaling pathway in acquired dasatinib resistance.
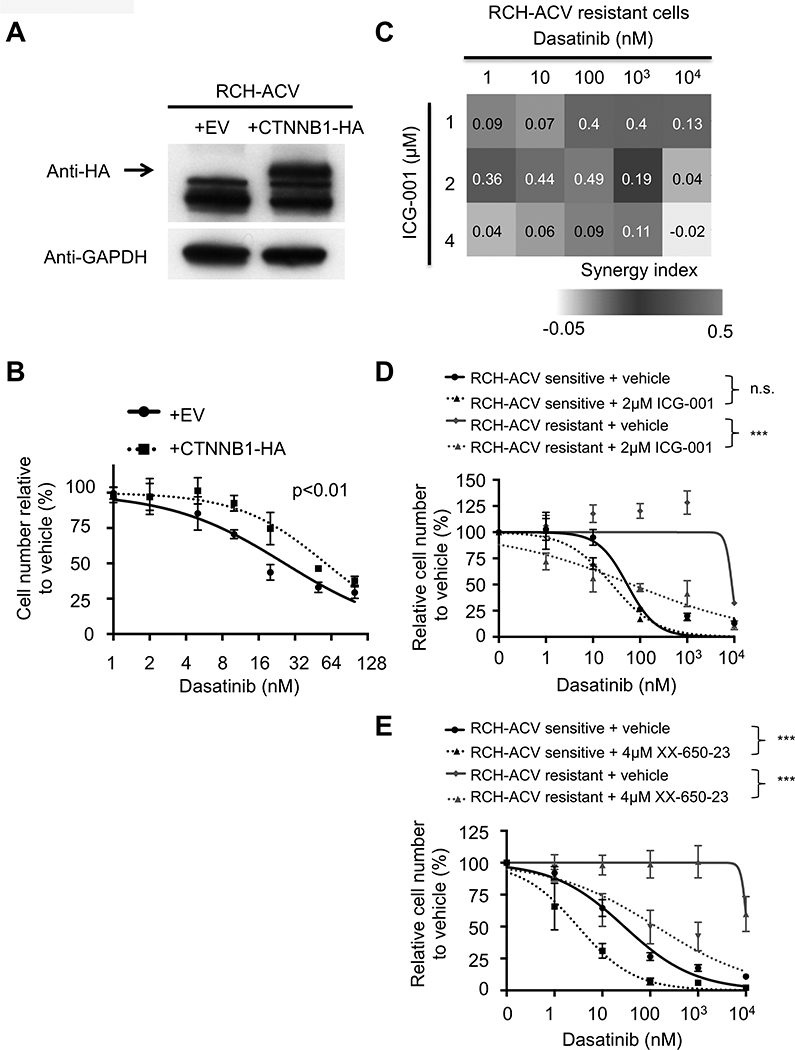
(A) RCH-ACV cells were transduced with retrovirus to ectopically express empty vector (EV) or β-catenin mutant with 4 phosphorylation site mutations in the N-terminal linked to hemagglutinin gene (CTNNB1+HA). Western blot (representative of three independent experiments) shows expression of β-catenin; GAPDH represents loading control. (B) Transduced RCH-ACV cells were cultured at increasing concentrations of dasatinib and cells were enumerated after 4 days. Titration curve data represent mean ±SEM of three independent experiments. Statistical analysis by F test. (C) Heat map representation of Bliss interaction index for dasatinib–resistant RCH-ACV cells treated with dasatinib and the CBP-β catenin inhibitor ICG-001. Data represent the mean of three independent experiments. (D) Dasatininb-sensitive and -resistant RCH-ACV cells were treated with increasing concentrations of dasatinib in combination with ICG-001 (2μM) or (E) XX-650–23 (4μM). Titration curve data represent mean ±SEM of three independent experiments. Statistical analysis by F test. *** p-value<0.001; n.s., not significant.
Pharmacological inhibition of CBP-β-catenin interaction restores dasatinib sensitivity.
Given the central role of CBP in dasatinib sensitivity and its interaction with β-catenin in regulation of gene expression (37), we tested the effect of ICG-001, an inhibitor of CBP interaction with β-catenin (38), in acquired dasatinib resistance. Combination treatment with ICG-001 and dasatinib displayed a synergistic interaction in two independent dasatinib-resistant RCH-ACV sublines (Fig. 6C-D, Supplementary Fig. S12A). The KIX domain inhibitor XX-650–23 also increased dasatinib sensitivity in the dasatinib-resistant sublines (Fig. 6E and Supplementary Fig. S12B), indicating that CBP can modulate intrinsic dasatinib sensitivity through multiple domains.
Discussion
Our studies using unbiased genetic approaches identified CBP as a major regulator of dasatinib sensitivity in a subset of acute leukemia. Pharmacologic inhibition of CBP enhances de novo sensitivity to dasatinib, overcomes acquired dasatinib resistance, and shows encouraging preclinical efficacy in vitro and in vivo.
Dasatinib is an FDA-approved drug for the treatment of BCR-ABL+ CML and ALL (39). Known mechanisms of dasatinib resistance in these clinical settings include somatic mutations in the ABL kinase domain and BCR-ABL amplifications (30,31). Novel mechanisms of dasatinib resistance in experimental models of BCR-ABL+ ALL have been proposed to involve upregulation of the anti-apoptotic oncogene BCL6 (40) as well as priming of mesenchymal stem cells (41). Preclinical studies demonstrate that dasatinib may also be an efficacious therapy for the subset of ALL that is pre-BCR+ through inhibition of SRC-family kinases including LCK (42). Functional studies suggested that depletion or inhibition of the kinases LCK, ZAP70 and SYK enhances dasatinib sensitivity in preBCR+ ALL. Interestingly, LCK, ZAP70 and SYK expression is directly regulated by E2A-PBX1, a common driver oncogene in pre-BCR+ leukemias (9,10). Notably, the dasatinib IC50 concentration for SYK was described at 2.9 μM (32), which is likely very similar to the IC50 concentration for its closely related tyrosine kinase family member ZAP70. This is not a physiologically achievable concentration, suggesting that the observed effect of dasatinib on pre-BCR+ ALL is likely mediated through inhibition of SRC-family kinases including BTK, LCK and LYN. Altogether, we hypothesized that the spectrum of drug resistance mechanisms would be different compared to BCR-ABL driven leukemias involving gene expression and transcriptional plasticity.
This was addressed through shRNA screens, which can identify synthetic lethal genetic interactions (43) and drug resistance mechanisms to targeted therapies (44), thus providing a powerful platform to identify effective drug combinations to overcome resistance. Our screen identified several genes, most notably CBP, involved in dasatinib sensitivity in human pre-BCR+ ALL cells. The role of CBP as a regulator of dasatinib sensitivity was validated using genetic and pharmacologic techniques. The observed increased sensitivity to dasatinib through pharmacological inhibition and genetic inactivation of CBP in RCH-ACV compared to 697 cells might be due to higher CBP expression in the former (Supplementary Fig. S1, Fig. 2). Our results support a crucial role in drug sensitivity mechanisms and intensify the focus on CBP as a potential drug target in ALL.
CBP is a large protein (265 kDa), with multiple domains amenable to potential drug targeting. It functions as a co-activator of gene transcription with histone acetyltransferase activity involved in many different cellular processes (45). CBP loss of function has been previously implicated in ALL relapsing disease and pathogenesis (28, 46) consistent with its role as a haplo-insufficient tumor suppressor in lymphoma (29). Conversely, CBP also serves a pro-oncogenic function in leukemia, and is a promising drug target in AML where the KIX domain inhibitor XX-650–23 shows anti-leukemic activity (14). Moreover, an inhibitor of CBP-β-catenin interaction showed efficacy as a single agent and in combination with chemotherapy in ALL (47). Mechanistically, CBP regulates pre-BCR/BCR-associated genes (28,29), involved in dasatinib sensitivity and resistance in our previous work (10) or identified in our shRNA screen. Genetic and pharmacologic inhibition of CBP results in downregulation of pre-BCR-pathway signaling members increasing synergistically sensitivity to dasatinib in pre-BCR+ ALLs. Dasatinib is known to inhibit BTK, LCK and LYN at the nanomolar range (32). Thus, there is some overlap between kinases inhibited by dasatinib and regulated by CBP, suggesting that shRNA-mediated knock-down of CBP is pulling out several signals with synthetic lethality. Our studies credential CBP as a target for combination therapy with dasatinib and establish its role to enhance drug sensitivity in a subset of ALL.
Global transcriptomic studies identified several pathways depleted in dasatinib-resistant ALL cells including DNA replication/cell cycle, p53 signaling and DNA repair machinery, which correlate with the resistance cellular phenotype. Conversely, several pathways are enriched in dasatinib-resistant cells, including WNT, MAPK and MTOR signaling, which are amenable to pharmacological inhibition and have been associated with drug resistance mechanisms. Due to the critical role of the CBP- β-catenin interaction in gene regulation, we focused on the WNT signaling pathway. Genetic perturbation of the pathway by overexpression of β-catenin or RAC1 knock-down confirmed an alternative pathway to overcome dasatinib resistance. Indeed, the CBP- β-catenin inhibitor ICG-001 showed a synergistic effect in combination with dasatinib further highlighting the role of CBP in modulating dasatinib sensitivity through bypass of targeted signaling pathways via WNT. Our observations extend recent studies of the role of the WNT signaling pathway in primary resistance to bromodomain inhibitors in AML (34,35) as well as acquired resistance to doxorubicin in neuroblastoma (48), suggesting a convergence to a common WNT pathway of resistance to multiple anti-cancer therapies. In these experiments, we used an in vitro system with human cell lines and therefore, the data should be interpreted with caution. Future studies are required to elucidate whether same or similar mechanisms occur in pre-BCR+ ALL patients treated with dasatinib.
Mutations of drug targets are a common mechanism of acquired resistance to kinase targeted therapies. Although mutational analysis showed acquisition of SNVs/indels in dasatinib-resistant cells, they did not affect known kinases targeted by dasatinib (HMS LINCS database, ref. 32), suggesting that they may be passenger mutations or have an indirect effect on dasatinib resistance through activation of alternative pathways. However, we observed strong selection for loss of the R1748H SNV in the CBP gene in dasatinib-resistant cells. Several loss-of-function variants affecting the histone acetyl-transferase domain of CBP or regions nearby have been described and well characterized in ALL (28). The preBCR+ ALL cell line RCH-ACV was established from bone marrow cells at relapse after chemotherapy, suggesting similar origin and probable loss-of-function as described mutations in relapsed ALL (49). We hypothesize, that preBCR+ ALL cells with impaired CBP function are more sensitive to dasatinib as further confirmed by pharmacologic studies. Collateral sensitivity of primary or relapsed ALL (28) and B-cell lymphomas with impaired dysfunctional CBP (50) might extend the indication for dasatinib treatment. Of note, the combination of XX-650–23 and dasatinib has a synergistic effect on cell growth inhibition not only of E2A-PBX1+/pre-BCR+ cells (RCH-ACV and 697) but also of E2A-PBX1+/pre-BCR- (Kasumi-2) and non-E2A-PBX1 cells (REH, THP-1). Hence, we observed a modest but statistically significant prolonged disease-free survival of dasatinib and XX-650–23 in xenograft transplantation experiments. The synergistic effect was observed at 2–4 μM concentration of XX-650–23 in vitro and these concentrations were likely not reached in vivo. Our data suggest that the development of small molecule inhibitors targeting CBP with improved pharmacodynamics and pharmacokinetics profile might provide a therapeutic option for a subset of ALLs particularly those that are pre-BCR+. Prospective studies and further experimentation beyond pre-BCR+ ALL are needed to establish how broad the indication is for combination therapy using dasatinib and CBP inhibitors.
In conclusion, using two different unbiased approaches, we have elucidated mechanisms of dasatinib sensitivity in human pre-BCR+ ALL and identified CBP as a master regulator to facilitate activation of alternative signaling pathways such as pre-BCR and WNT. Small molecules targeting CBP show promising preclinical efficacy modulating dasatinib sensitivity and should be considered in future clinical trials to prevent the emergence of resistant/relapsing ALL clones.
Supplementary Material
Significance:
Findings reveal mechanisms that modulate sensitivity to dasatinib and suggest therapeutic strategies to improve the outcome of patients with acute lymphoblastic leukemia.
Acknowledgments
We thank Maria Ambrus and Cita Nicolas for technical assistance, members of the Cleary laboratory for helpful discussions and Carlos Duque-Afonso for graphic design.
Financial support:
This work was supported in part by grants from the NIH (CA214888), the William Lawrence and Blanche Hughes Foundation (M.L. Cleary), the German Research Foundation (Deutsche Forschungsgemeinschaft, ref. DU 1287/2–1 and 1287/3–1) and Research Commission of the University of Freiburg Medical School (Forschungskommission, ref. DUQ1106/16) (J. Duque-Afonso), the Lucile Packard Foundation for Children’s Health, the Child Health Research Institute and the Stanford NIH-NCATS-CTSA grant #UL1 TR001085 (M.L. Cleary, J. Duque-Afonso, C.-H. Lin), Alex’s Lemonade Stand Foundation for Childhood Cancer (S. H. K. Wong). K. Han was supported by The Walter V. and Idun Berry Postdoctoral Fellowship Program and M.C. Bassik was supported by a grant from Stanford ChEM-H and an NIH Directors New Innovator Award (1DP2HD08406901). K.M. Sakamoto has been supported by the Maxfield Foundation, SPARK program and Child Health Research Institute Lucile Packard Foundation for Children’s Health, Leukemia and Lymphoma Society of America (SLP-8009–15), Pediatric Cancer Research Foundation, Hyundai Hope on Wheels (#02500CA), USC Parker Hughes Institute for Childhood Cancer Research/William Lawrence & Blanche Hughes Foundation, St. Baldrick’s Foundation, and Bear Necessities/Rally Foundation. E.E. Jeng was supported by a Hubert Shaw and Sandra Lui Stanford Graduate Fellowship. This material is based on work supported by the National Science Foundation (NSF) Graduate Research Fellowship (grant DGE-114747 to D.W.Morgens). Any opinions, findings and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the NSF.
Footnotes
Competing financial interests
The authors declare no competing financial interests.
Data availability
The RNA sequencing data that support the findings of this study have been deposited in the Gene Expression Omnibus (http://ncbi.nlm.nih.gov/geo) with the accession number GSE97352.
References
- 1.Inaba H, Greaves M, Mullighan CG. Acute lymphoblastic leukaemia. Lancet 2013; 381: 1943–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet 2008; 371: 1030–43. [DOI] [PubMed] [Google Scholar]
- 3.Bassan R, Hoelzer D. Modern therapy of acute lymphoblastic leukemia. J Clin Oncol. 2011; 29: 532–43. [DOI] [PubMed] [Google Scholar]
- 4.Geng H, Hurtz C, Lenz KB, Chen Z, Baumjohann D, Thompson S, et al. Self-enforcing feedback activation between BCL6 and pre-B cell receptor signaling defines a distinct subtype of acute lymphoblastic leukemia. Cancer Cell. 2015; 27: 409–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Köhrer S, Havranek O, Seyfried F, Hurtz C, Coffey GP, Kim E, et al. PreBCR signaling in precursor B-cell acute lymphoblastic leukemia regulates PI3K/AKT, FOXO1 and MYC, and can be targeted by SYK inhibition. Leukemia 2016; 30: 1246–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roberts KG, Mullighan CG. Genomics in acute lymphoblastic leukaemia: insights and treatment implications. Nat Rev Clin Oncol. 2015; 12: 344–57. [DOI] [PubMed] [Google Scholar]
- 7.Bicocca VT, Chang BH, Masouleh BK, Muschen M, Loriaux MM, Druker BJ, et al. Crosstalk between ROR1 and the Pre-B cell receptor promotes survival of t(1;19) acute lymphoblastic leukemia. Cancer Cell. 2012; 22: 656–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fischer U, Forster M, Rinaldi A, Risch T, Sungalee S, Warnatz HJ, et al. Genomics and drug profiling of fatal TCF3-HLF-positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat Genet. 2015; 47: 1020–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duque-Afonso J, Feng J, Scherer F, Lin CH, Wong SH, Wang Z, et al. Comparative genomics reveals multistep pathogenesis of E2A-PBX1 acute lymphoblastic leukemia. J Clin Invest. 2015; 125: 3667–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duque-Afonso J, Lin CH, Han K, Wei MC, Feng J, Kurzer JH, et al. E2A-PBX1 remodels oncogenic signaling networks in B-cell precursor acute lymphoid leukemia. Cancer Research 2016; 76: 6937–6949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eldfors S, Kuusanmäki H, Kontro M, Majumder MM, Parsons A, Edgren H, et al. Idelalisib sensitivity and mechanisms of disease progression in relapsed TCF3-PBX1 acute lymphoblastic leukemia. Leukemia 2017; 31: 51–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bernt KM, Hunger SP. Current concepts in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia. Front Oncol. 2014; 4: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buechele C, Breese EH, Schneidawind D, Lin CH, Jeong J, Duque-Afonso J, et al. MLL leukemia induction by genome editing of human CD34+ hematopoietic cells. Blood 2015; 126: 1683–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitton B, Chae HD, Hsu K, Dutta R, Aldana-Masangkay G, Ferrari R, et al. Small molecule inhibition of cAMP response element binding protein in human acute myeloid leukemia cells. Leukemia 2016; 30: 2302–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bassik MC, Kampmann M, Lebbink RJ, Wang S, Hein MY, Poser I, et al. A systematic mammalian genetic interaction map reveals pathways underlying ricin susceptibility. Cell. 2013; 152: 909–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matheny CJ, Wei MC, Bassik MC, Donnelly AJ, Kampmann M, Iwasaki M, et al. Next-generation NAMPT inhibitors identified by sequential high-throughput phenotypic chemical and functional genomic screens. Chem Biol. 2013; 20: 1352–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morgens DW, Deans RM, Li A, Bassik MC. Systematic comparison of CRISPR/Cas9 and RNAi screens for essential genes. Nat Biotechnol. 2016;34:634–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH,, et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014; 159: 647–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wong SH, Goode DL, Iwasaki M, Wei MC, Kuo HP, Zhu L et al. The H3K4-Methyl epigenome regulates leukemia stem cell oncogenic potential. Cancer Cell. 2015; 28: 198–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brinkman EK, Chen T, Amendola M, van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2015; 42: e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuo HP, Wang Z, Lee DF, Iwasaki M, Duque-Afonso J, Wong SH, et al. Epigenetic roles of MLL oncoproteins are dependent on NF-κB. Cancer Cell. 2013; 24: 423–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nature Protoc. 2009; 4: 44–57. [DOI] [PubMed] [Google Scholar]
- 23.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005; 102: 15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012; 22: 568–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012; 6: 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011; 29: 24–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bliss CI. The toxicity of poisons applied jointly. Ann Appl Biol. 1939; 26:585–615. [Google Scholar]
- 28.Mullighan CG, Zhang J, Kasper LH, Lerach S, Payne-Turner D, Phillips LA, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 2011; 471: 235–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang J, Vlasevska S, Wells VA, Nataraj S, Holmes AB, Duval R, et al. The Crebbp Acetyltransferase is a Haploinsufficient Tumor Suppressor in B Cell Lymphoma. Cancer Discov. 2017; 7: 322–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2002; 2: 117–25. [DOI] [PubMed] [Google Scholar]
- 31.Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006; 354: 2531–41. [DOI] [PubMed] [Google Scholar]
- 32.Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011; 29:1046–51. [DOI] [PubMed] [Google Scholar]
- 33.Knoechel B, Roderick JE, Williamson KE, Zhu J, Lohr JG, Cotton MJ, et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat Genet. 2014; 46: 364–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fong CY, Gilan O, Lam EY, Rubin AF, Ftouni S, Tyler D, et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature 2015; 525: 538–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rathert P, Roth M, Neumann T, Muerdter F, Roe JS, Muhar M, et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 2015; 525: 543–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu X, Tu X, Joeng KS, Hilton MJ, Williams DA, Long F. Rac1 activation controls nuclear localization of beta-catenin during canonical Wnt signaling. Cell 2008; 133: 340–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takemaru KI, Moon RT. The transcriptional coactivator CBP interacts with beta-catenin to activate gene expression. J Cell Biol. 2000; 149: 249–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Emami KH, Nguyen C, Ma H, Kim DH, Jeong KW, Eguchi M, et al. A small molecule inhibitor of beta-catenin/CREB-binding protein transcription. Proc Natl Acad Sci U S A. 2004; 101: 12682–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brave M, Goodman V, Kaminskas E, Farrell A, Timmer W, Pope S, et al. Sprycel for chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia resistant to or intolerant of imatinib mesylate. Clin Cancer Res. 2008; 14: 352–9. [DOI] [PubMed] [Google Scholar]
- 40.Duy C, Hurtz C, Shojaee S, Cerchietti L, Geng H, Swaminathan S, et al. BCL6 enables Ph+ acute lymphoblastic leukaemia cells to survive BCR-ABL1 kinase inhibition. Nature 2011; 473: 384–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mallampati S, Leng X, Ma H, Zeng J, Li J, Wang H, et al. Tyrosine kinase inhibitors induce mesenchymal stem cell-mediated resistance in BCR-ABL+ acute lymphoblastic leukemia. Blood 2015; 125: 2968–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lombardo LJ, Lee FY, Chen P, Norris D, Barrish JC, Behnia K et al. Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004; 47: 6658–61. [DOI] [PubMed] [Google Scholar]
- 43.Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 2009; 137: 835–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rudalska R, Dauch D, Longerich T, McJunkin K, Wuestefeld T, Kang TW, et al. In vivo RNAi screening identifies a mechanism of sorafenib resistance in liver cancer. Nat Med. 2014; 20: 1138–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vo N, Goodman RH. CREB-binding protein and p300 in transcriptional regulation. J Biol Chem. 2001; 276: 13505–8. [DOI] [PubMed] [Google Scholar]
- 46.Ma X, Edmonson M, Yergeau D, Muzny DM, Hampton OA, Rusch M, et al. Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia. Nat Commun. 2015; 6: 6604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gang EJ, Hsieh YT, Pham J, Zhao Y, Nguyen C, Huantes S, et al. Small-molecule inhibition of CBP/catenin interactions eliminates drug-resistant clones in acute lymphoblastic leukemia. Oncogene 2014; 33: 2169–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Flahaut M, Meier R, Coulon A, Nardou KA, Niggli FK, Martinet D, et al. The Wnt receptor FZD1 mediates chemoresistance in neuroblastoma through activation of the Wnt/beta-catenin pathway. Oncogene 2009; 28: 2245–56. [DOI] [PubMed] [Google Scholar]
- 49.Jack I, Seshadri R, Garson M, Michael P, Callen D, Zola H, et al. RCH-ACV: a lymphoblastic leukemia cell line with chromosome translocation 1;19 and trisomy 8. Cancer Genet Cytogenet. 1986;19:261–9. [DOI] [PubMed] [Google Scholar]
- 50.Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 2011; 471: 189–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.