

This is an exciting era of therapeutics development for sickle cell anemia (SCA). Yet, most resources have been focused upon single agents intended to target HbS polymerization or biology far along the pathobiologic vector. We suggest that, in between, there exists a “missing middle” that resides at the nexus of three opportunities: applying a multi‐agent regimen; using drugs already deployed in human general medicine; and strategically targeting a point of pathobiological vulnerability. In essence, we view this therapeutic space as one offering preventative benefit by targeting the sickle risk state rather than events.
We here develop this concept and describe a three‐drug example. Then, we address the practical challenges that face this approach, regardless of the targets or drugs one might favor. Our comments do reflect the belief that therapeutics development efforts should include agents that will be accessible to not only the ~15% of SCA patients who live in wealthier countries but also the ~85% who do not.1 To us this means agents having as many as possible of the characteristics listed in Table 1.
Table 1.
Characteristics of an ideal drug for global application in SCA
| Already used in human medicine Long track record Known and tolerable side effect profile Known and relevant mechanism of action Expectation of prophylactic effect Inexpensive Oral administration Easily shipped & stored & distributed Not requiring refrigeration Amenable to self‐administration |
Of course, this excludes emerging approaches dependent upon infusion and/or cell transfer and/or gene manipulation. We certainly do not argue that development of such advanced therapeutics be curtailed. Rather, we suggest that it only makes sense to concurrently devote resources to develop alternative approaches that would be better positioned for global application.
1. MULTI‐AGENT RATIONALE
As amply demonstrated by malignancies, agents having insufficient efficacy individually may still contribute benefit if deployed as a multi‐drug regimen. In SCA, a wealth of potential targets reside within a multi‐faceted pathobiology. Yet, its vast complexity could mean that therapeutic efficacy from multiple agents will be maximized if they converge upon the same “chokepoint” step in pathobiology. One of these, targeting HbS polymerization, already receives much attention. Here, as our example we propose targeting another, ischemia/reperfusion injury (I/R) pathophysiology.
2. I/R AS A TARGET
The physical chemistry of HbS dictates that most RBC cannot sickle within the microvasculature unless their transit time is slowed to overcome the two relevant delay times.2, 3 Such slowing from blood cell adhesion to the vascular well4, 5, 6, 7 requires an adhesion‐engendering endothelial surface, in turn requiring inflammation. It has been proposed—and supported experimentally—that I/R is what provokes the robust inflammatory state of SCA. This is reviewed thoroughly elsewhere.8
Briefly, I/R is the physiology of resolving vascular occlusion, triggered by the restoration of flow and, therefore, oxygen availability.9, 10 It is unlike hypoxia or unresolved occlusion. In general medicine, I/R underlies consequences of large vessel occlusive events, such as ischemic stroke and myocardial infarction.9, 10 In SCA, however, I/R probably arises from recurring, smaller, multi‐focal occlusive events.8 In particular, I/R in SCA establishes cyclicity to incessantly drive inflammation. In this sense, I/R assumes Janus‐like status, comprising both the beginning and the end of the core pathobiologic vector (Figure 1).
Figure 1.
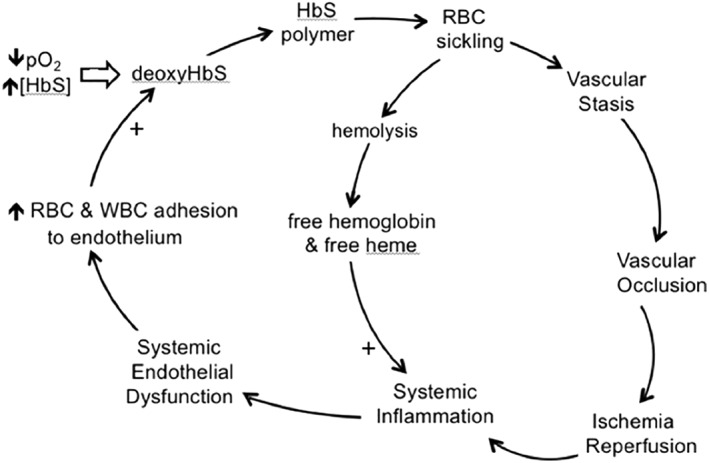
The vicious cycle of SCA, with ischemia/reperfusion pathophysiology as the driver of incessant, systemic inflammation
2.1. Initiation of I/R
Experimental models revealed that I/R initiation involves several prominent actors at the local site of the ischemic event: oxidant generation, NFκB activation, endothelial activation, TNF elaboration, sentinel cell activation (resident macrophages, mast cells), and leukocyte infiltration. After this, I/R pathobiology explosively spreads and arborizes, recruiting multiple biosystem aberrancies that, while uniquely identifiable, still exhibit substantial cross‐activating overlaps.8, 9, 10 It is the resulting, unwieldly complexity of established I/R that underlies our belief that the best therapeutic opportunity lies in targeting I/R initiation per se.
3. THREE‐DRUG TARGETING OF I/R INITIATION
For this specific goal, we choose a combination of three drugs: (a) allopurinol, because it inhibits xanthine oxidase, the enzyme producing an early burst of superoxide; (b) sulfasalazine, because it is a strong inhibitor of NFκB, the master on‐switch for inflammation; and (c) etanercept, because it blocks TNF, the “sentinel cytokine” residing at the top of inflammatory networks. Again, it is the combination of these drugs that may be helpful, even if none are individually sufficient.
Rationale for these specific drug choices is six‐fold. (a) Each drug's specific target is implicated in I/R initiation in non‐sickle models.8, 9, 10 (b) Each target is a participant in I/R in sickle mice.8 (c) Each drug ameliorates vascular wall abnormalities in sickle mice.8 (d) Each drug greatly improves microvascular blood flow in sickle mice.11, 12 (e) Each drug is already used in human medicine, with known targets, efficacies, track records, and side‐effect profiles. (f) A pilot test in SCA subjects documented oral sulfasalazine's efficacy for down‐regulating endothelial adhesion molecule expression (see below).3
Alternatives can be justified. Sulfasalazine can have unwelcome gastric side effects. Etanercept requires refrigeration and would require subcutaneous administration (although perhaps only monthly), and its inclusion raises the 3‐drug regimen's cost to $8.40 per day.1
Preserving the focus on I/R initiation, an alternative NFκB inhibitor can be found from the >700 known; among those shown to benefit sickle mice, curcumin and andrographolide were even drawn from Asian traditional medicine.14 Mast cell stabilization could be considered; cromolyn and imatinib benefited sickle mice.15 When oral agents become available, a TNF inhibitor or a P‐selectin blocker would be options. P‐selectin blockade not only improved microvascular flow but also eliminated the exaggerated leukocyte infiltration seen in sickle mice.6 Each of these possibilities preserves the goal of targeting I/R initiation.
Otherwise, targeting downstream from I/R initiation would increase the therapeutic challenge, but a drug exerting multiple relevant effects might be reasonable. Of these, statins stand out because of their pleiotropic, anti‐inflammatory and endothelial‐sparing effects.16 Indeed, for humans with elevated CRP (but normal blood lipids), rosuvastatin reduced incidence of stroke.17 In sickle mice lovastatin blocked I/R‐triggered endothelial activation,18 and in SCA patients simvastatin improved inflammation biomarkers and reduced pain.19 Substitution of lovastatin for etanercept would lower cost of our 3‐drug regimen to $1.67 per day.*
3.1. Precautionary note
We deliberately have not suggested specific dose‐schedules, as any regimen should be assessed for risks vs benefits before application. Doing so here is beyond the scope of this commentary. In our concluding section we will suggest vetting procedures that could be applied to new approaches to SCA.
4. CHALLENGES AND BARRIERS
These are several and substantial, and they pertain to any combination of drugs in the universe defined by the nexus we defined in our first paragraph. After delineating these challenges we suggest approaches to their solution.
4.1. Challenge: Our perceptions are limiting
The scope of therapeutics considered by our field has been limited by (at least) three perceptions. We suggest that modification of these is justifiable—and would broaden the universe of therapeutic targets and available drugs worth consideration.
4.2. Challenge: Defining severity independent of events
We suspect this can be done via the concept of “robustness.” That is, the robustness of the SCA underlying risk state may be quantifiable via the activity level of its causal core biologic process(es), as measured during steady state. Could this be developed and applied as a quantifiable predictor of forward trajectory? Would it enable risk stratification of SCA patients?
4.3. Events vs risk state
Although understandable, our traditional focus upon events (pain crisis, ACS, stroke) restricts the operational definition of efficacy to impacting event occurrence. It may be more helpful—and is pathobiologically more accurate—to emphasize that SCA is not events per se. Rather, it is a risk state from which events emerge—and substantial pathology progresses regardless of event occurrence. In these respects, SCA is analogous to atherosclerosis in the general population, for which disease management requires blunting the underlying risk state. As in atherosclerosis, the risk state in SCA is conceptually definable as the robustness of its core pathobiologic vector. Could it be operationally defined that way and thereby provide monitorable therapeutic endpoints?
4.4. Challenge: The steady state itself
Unfortunately, the SCA “steady state” is anything but steady. In Figure 2A we illustrate that even subjects with very mild SCA do not exhibit sufficient biomarker instability during (true) “steady state” for single‐timepoint measures to be informative. And combining single‐timepoint measures as an “index” simply compounds the potential error. Consequently, we do not even know what SCA steady state really looks like. Solving this is necessary to construct alternative, valid definitions of severity and to define pre‐therapy baselines.
Figure 2.
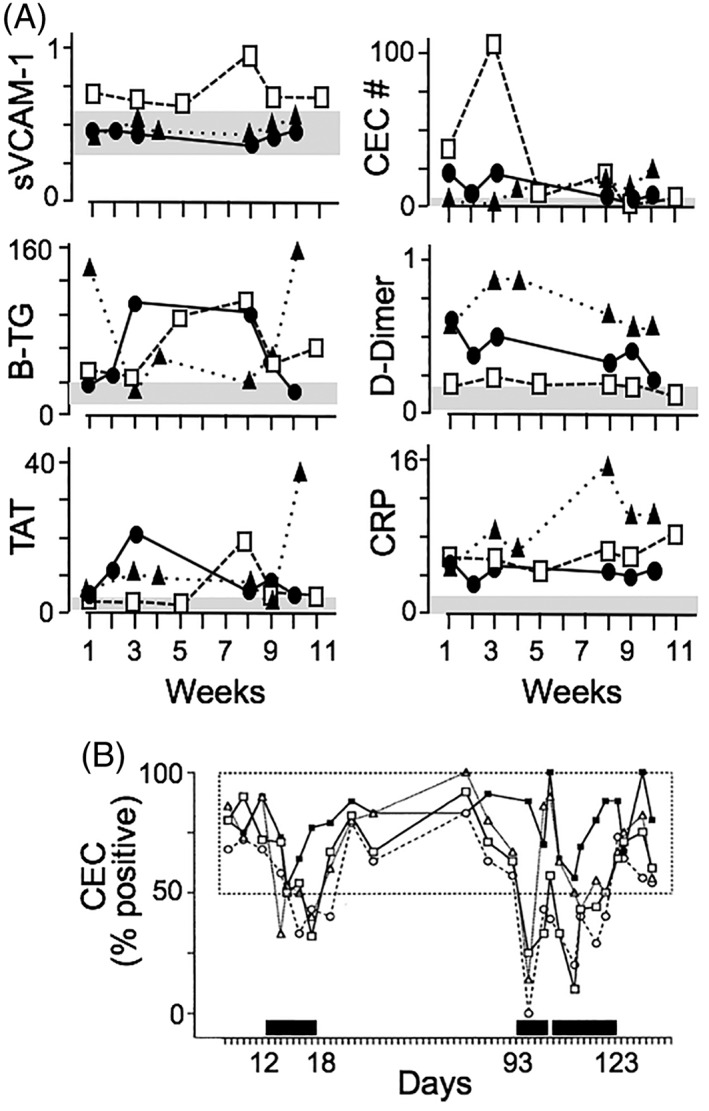
Taming the instability of “steady state.” (A) Instability of steady state is shown by biomarkers sampled longitudinally from three historically mild SCA subjects, without current symptoms, and no acute events >3 months before or after this study. Each symbol type indicates one of the subjects. For each biomarker, the shaded area indicates range for normal subjects. (B) Testing for efficacy while controlling for instability. Here, efficacy of oral sulfasalazine for downregulating endothelial adhesion molecule expression in one SCA subject was tested. Activation is expressed as % of circulating endothelial cells (CEC) positive for three adhesion molecules (open symbols) and tissue factor (closed symbol). Steady state baseline is established empirically by all values collected off‐drug (the box). Dark bars show days on‐drug; the indicated day numbers identify the start and stop dates for the mini‐trials. Figure reproduced with permission from Ref. 13
4.5. Challenge: Barriers to translational research
There are many. Opinions differ. There is unease about translating research data to human application in SCA, and there is no formal mechanism to facilitate this. Could/should a rationally designed regimen, in the sense used herein, be combined with other drugs already in use, such as hydroxyurea? How much pre‐clinical and early pilot study is needed? Even if likely, whether safe drugs will still be safe in SCA is unknown. In toto, these questions and apprehensions comprise a “stenosis” in what could be a more robust bench‐to‐beside pipeline. Can this be overcome?
4.6. Specificity vs biology
We tend to think in terms of “sickle specific” pathology. Is there such a thing? Clearly, there are unique (RBC sickling) plus other (hemolysis) disease‐causing inputs. Nonetheless, what we observe are, for the most part, “pre‐programmed” and understood patterns of tissue responses to injury. Are we taking full advantage of the vast trove of data residing within the general biomedical literature?
4.7. Pain vs severity
Therapeutics testing has emphasized interventional benefit for inferred markers of pain perception, the most challenging endpoint that could be devised. While pain frequency may correlate with mortality,19 this proves neither that pain causes mortality nor that treating pain will benefit mortality. Can we develop a more robust definition of severity that is both pathobiologically meaningful and quantifiable?
4.8. Challenge: Extant biomarker data are inadequate
An operational definition of robustness would rely, at least in part, on measured biomarkers. Unfortunately, biomarker data collected to date in SCA are of limited value because measurements usually have been obtained at a single time point, without stringent remoteness from acute influences, and often in subjects already experiencing a complication of interest.
Such associations can, with equal plausibility, be a measure of: (a) the activity level of a core pathogenic process; or (b) simple presence—but not cause—of the complication; or (c) concurrent presence of additional, but unrelated, complications—that may be known or unknown; or (d) adaptive or maladaptive responses that are not pathogenic; or (e) adaptive or maladaptive responses that do contribute to pathogenesis; or (f) fortuitous occurrence. This ambiguity does not foster utility.
5. AN ACTION PLAN
Addressing these challenges will require leadership by the sickle field itself, as finding solutions will require motivation, expertise, investment and effort. Further, before we can persuade potential funders that this approach is rational and possible, as a field we need to persuade ourselves. We offer the following suggestions.
5.1. Prevailing perspectives
The three challenges we began with could be approached via educational opportunities. Our field's journal(s) could solicit scholarly essays on the risk state concept per se and on ways it could be defined, identified and quantified. Our society meetings could include educational fora that compare and contrast pathobiology of a SCA feature vs its analogue in general medicine (and do so vis à vis therapeutics). Published essays and scientific fora could suggest and defend clinical definitions of severity that are not dependent upon perception of pain perception.
5.2. Define the steady state
Our funding agencies should support stand‐alone studies designed to expose the character of the steady state. The necessary data can only derive from longitudinally measuring candidate biomarkers for any given subject, over sufficient time to establish ranges, relationships, reliability, and redundancies. This is necessary if biomarkers are to be used for defining and measuring the robustness of core pathobiology.
5.3. Establish a definition of core pathobiology “robustness”
Our societies and funding agencies could co‐sponsor a “think tank” on this topic. Deliberations should be recurring, data‐based, thoroughly rationalized, include multiple perspectives, and involve experts from other fields. Work product could include: prioritizing biological processes as targets, in terms of their contributions to core pathophysiology; identifying the best available bio‐physiological measures (including beyond blood sampling) obtainable in humans; and identifying parameters most likely to quantify specific process activity levels. While admittedly unlikely that a consensus would be achieved, the deliberations could be disseminated via publication and by dedicated fora at national meetings. This would foster awareness and discussion and perhaps provide guidance.
5.4. Consider risks vs benefits of drug options
Availability of vetted guidance could only be helpful. A working‐group with multi‐disciplinary expertise (including pharmacology) could perhaps generate vetted risk‐to‐benefit estimates for candidate drugs applied in the sickle context. Society meetings could include joint research‐clinical sessions structured to foster discussions about applicability of emerging research data. Such efforts could ease apprehensions.
5.5. Demonstrating biological efficacy
Few subjects may be needed to demonstrate biological efficacy if longitudinal sampling for an appropriate, biologic, reporter biomarker is used. For example, documenting sulfasalazine's ability to have the effect we sought (downregulating expression of NFκB‐dependent endothelial adhesion molecules) in SCA required only three subjects,13 as illustrated for one in Figure 2B. This approach is analogous to monitoring HbF level as an indicator of desired response to hydroxyurea.
6. A FINAL CHALLENGE: FAITH‐BASED MEDICINE?
Some may believe that attempted targeting of the SCA risk state, in the manner described herein, would be “faith‐based medicine.” Not so, if we believe that clinical disease is actually caused by pathophysiology. Our 3‐drug example targeting I/R initiation is based on: abundant basic science data providing rationale; unambiguous demonstration of meaningful biologic efficacy in sickle mice; and prior regulatory approval, wide clinical use, and established track records. Arguably, this justifies a more favorable benefit‐to‐risk expectation than is available for newly emergent drugs.
As for all new approaches, whether this will translate into actual clinical benefit can only be answered via its clinical application. We suppose it remains possible that even successful targeting of the underlying SCA pathophysiology could exhibit another parallel to atherosclerosis. Statins target underlying pathophysiology, but it requires long time periods to see efficacy at the clinical level. With this possibility in mind, should a multi‐drug regimen targeting the SCA risk state be tried long‐term? This, of course, has ventured into “faith based medicine.” Yet, perhaps we must ask: should the approach be tried, or should anticipated benefits be foregone due to risk aversion? This too needs discussion.
CONFLICT OF INTEREST
Dr. Hebbel is a SAB member for Emmaus Life Sciences and has an interest in Vanguard Therapeutics; Dr. Kutlar is a consultant for Novartis, a DMC member for Bluebird‐Bio, and DSMB chair for Sancilio Parmaceuticals.
Hebbel RP, Elion J, Kutlar A. The missing middle of sickle therapeutics: Multi‐agent therapy, targeting risk, using biomarkers. Am J Hematol. 2018;93:1439–1443. 10.1002/ajh.25289
Cost from an online pharmacy in mid‐July, 2018.
Footnotes
Price from an on‐line, retail, Canadian pharmacy in mid‐July, 2018
REFERENCES
- 1. Piel FB, Hay S, Gupta S, Weatherall DJ, Williams TN. Global burden of sickle cell anaemia in children under five, 2010‐2050: modelling based on demographics, excess mortality, and interventions. PLoS Med. 2013;10(7):e1001484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mozzarelli A, Hofrichter J. EatonWA. Delaytime of hemoglobin S polymerization prevents most cells from sickling in vivo. Science. 1987;237(4814):500‐506. [DOI] [PubMed] [Google Scholar]
- 3. Ferrone FA. Polymerization and sickle cell disease: a molecular view. Microcirculation. 2004;11(2):115‐128. [DOI] [PubMed] [Google Scholar]
- 4. Hebbel RP. Adhesive interactions of sickle erythrocytes with endothelium. J Clin Invest. 1997;100(11 Suppl):S83‐S86. [PubMed] [Google Scholar]
- 5. Kaul DK, Fabry ME, Nagel RL. Microvascular sites and characteristics of sickle cell adhesion to vascular endothelium in shear flow conditions: pathophysiological implications. Proc Natl Acad Sci USA. 1989;86(9):3356‐3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kaul DK, Hebbel RP. Hypoxia/reoxygenation causes inflammatory response in transgenic sickle mice but not in normal mice. J Clin Invest. 2000;106(3):411‐420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Turhan A, Weiss LA, Mohandas N, Coller BS, Frenette PS. Primary role for adherent leukocytes in sickle cell vascular occlusion: a new paradigm. Proc Natl Acad Sci USA. 2002;99(5):3047‐3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hebbel RP. Ischemia‐reperfusion injury in sickle cell anemia: relationship to acute chest syndrome, endothelial dysfunction, arterial vasculopathy, and inflammatory pain. Hematol Oncol Clin North Am. 2014;28(2):181‐198. [DOI] [PubMed] [Google Scholar]
- 9. Eltzschig HK, Eckle T. Ischemia and reperfusion‐‐from mechanism to translation. Nat Med 201;17(11):1391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Granger DN, Kvietys PR. Reperfusion injury and reactive oxygen species: the evolution of a concept. Redox Biol. 2015;6:524‐551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kaul DK, Liu XD, Choong S, Belcher JD, Vercellotti GM, Hebbel RP. Anti‐inflammatory therapy ameliorates leukocyte adhesion and microvascular flow abnormalities in transgenic sickle mice. Am J Physiol Heart Circ Physiol. 2004;287(1):H293‐H301. [DOI] [PubMed] [Google Scholar]
- 12. Solovey A, Somani A, Belcher JD, et al. A monocyte‐TNF‐endothelial activation axis in sickle transgenic mice: therapeutic benefit from TNF blockade. Am J Hematol. 2017;92(11):1119‐1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Solovey AA, Solovey AN, Harkness J, Hebbel RP. Modulation of endothelial cell activation in sickle cell disease: a pilot study. Blood. 2001;97(7):1937‐1941. [DOI] [PubMed] [Google Scholar]
- 14. Kollander R, Solovey A, Milbauer LC, Abdulla F, Kelm RJ Jr, Hebbel RP. Nuclear factor‐kappa B (NFkappaB) component p50 in blood mononuclear cells regulates endothelial tissue factor expression in sickle transgenic mice: implications for the coagulopathy of sickle cell disease. Transl Res. 2010;155(4):170‐177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vincent L, Vang D, Nguyen J, et al. Mast cell activation contributes to sickle cell pathobiology and pain in mice. Blood. 2013;122:1853‐1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bu DX, Griffin G, Lichtman AH. Mechanisms for the anti‐inflammatory effects of statins. Curr Opin Lipidol. 2011;22(3):165‐170. [DOI] [PubMed] [Google Scholar]
- 17. Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C‐reactive protein. N Engl J Med. 2008;359(21):2195‐2207. [DOI] [PubMed] [Google Scholar]
- 18. Solovey A, Kollander R, Shet A, et al. Endothelial cell expression of tissue factor in sickle mice is augmented by hypoxia/reoxygenation and inhibited by lovastatain. Blood. 2004;1004(3):840‐846. [DOI] [PubMed] [Google Scholar]
- 19. Hoppe C, Jacob E, Styles L, Kuypers F, Larkin S, Vichinsky E. Simvastatin reduces vaso‐occlusive pain in sickle cell anaemia: a pilot efficacy trial. Br J Haematol. 2017;177(4):620‐629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330(23):1639‐1644. [DOI] [PubMed] [Google Scholar]