

Abstract
The (3+2) cycloaddition between azomethine ylides and alkenes is an efficient, convergent and stereocontrolled method for the synthesis of unnatural pyrrolidine and proline scaffolds. In this review, the application of this reaction to the synthesis of enantiopure organometallic ligands for asymmetric catalysis is presented first. These new EhuPhos ligands can participate in a second generation of 1,3‐dipolar reactions that generate an offspring of unnatural proline derivatives that behave as efficient organocatalysts. These densely substituted unnatural l‐proline derivatives exhibit distinct features, different to those described for natural l‐proline and its derivatives. Finally, several examples of biologically active proline derivatives obtained by means of (3+2) cycloadditions involving azomethine ylides are presented. These applications show the character of privileged structures of these polysubstituted pyrrolidine rings.
Keywords: 1,3‐Dipolar reactions; Cycloaddition; Asymmetric catalysis; Organocatalysis; Medicinal chemistry
1. Introduction
1,3‐Dipolar reactions are (3+2) cycloadditions that generate five‐membered rings in a single preparative step.1 In many cases, the reaction takes place with high or even complete regio‐, diastereo‐ and enantiocontrol. Among the different dipole/dipolarophile combinations,2 the 1,3‐dipolar reaction between azomethine ylides 1 and alkenes 2 constitutes a very efficient and convergent method for the synthesis of pyrrolidines 3 (Scheme 1).3 Given the fleeting nature of azomethine ylides, the rationalization of the different outcomes and the nature of the reaction mechanisms requires state‐of‐the‐art computational tools, thus providing a very fruitful interplay between theory and experiment.4 When the azomethine ylide is stabilized by an alkoxycarbonyl group the corresponding (3+2) cycloadduct is a proline scaffold 4 that can incorporate many different groups, thus permitting the synthesis of densely substituted and functionalized unnatural proline derivatives.
Scheme 1.
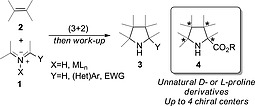
Synthesis of pyrrolidines 3 and, in particular, densely substituted prolines 4 by formal (3+2) cycloaddition between azomethine ylides 1 and alkenes 2.
Although the 1,3‐dipolar reaction depicted in Scheme 1 is formally a (3+2) cycloaddition, the actual mechanism can vary from a concerted (but not necessarily synchronous) [π4s+π2s] mechanism via supra‐supra5 transition structures TScn to a stepwise process (Scheme 2).6 This latter mechanism takes place via a Michael/Mannich sequence and involves zwitterionic intermediates 6 that in some cases can be detected.7 Therefore, this reaction does not follow a general mechanism and different reaction paths can be observed depending upon the substituents and the reaction conditions. In particular, N‐metallated azomethine ylides 5b (Scheme 2) in the presence of polarized π‐deficient alkenes 2 follow stepwise reaction paths. However, as it will be discussed below, this non‐concerted mechanism is compatible with an exquisite regio‐, diastereo‐ an enantiocontrol.
Scheme 2.
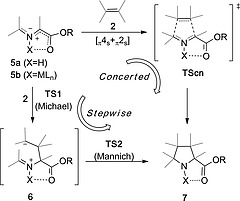
Alternative concerted (not necessarily synchronous) and stepwise mechanisms in the 1,3‐dipolar reaction between stabilized NH‐ or N‐metallated azomethine ylides (5a and 5b, respectively) and alkenes 2.
Stabilized azomethine ylides 9 can be generated by thermal isomerization8 or by metal‐assisted deprotonation9 of imines 8. Other methods such as electrocyclic ring opening of aziridines are also available.10 However, the metallic salt/base method possesses the advantage of allowing the development of a catalytic cycle that minimizes the introduction of additional reactants in the reaction medium. Although when doubly activated alkenes such as maleimides usually lead to concerted mechanisms, in the case of monoactivated dipolarophiles 10 the catalytic cycle leads to a N‐metallated zwitterionic intermediate 11 via a Michael‐like nucleophilic attack of 1,3‐dipole 9, which behaves as an enolate. Intramolecular Mannich‐like cyclization of this intermediate gives rise to the corresponding N‐metallated pyrrolidine 12. This latter polycyclic intermediate has a considerable strain that promotes the release of the catalyst and the formation of the substituted proline ester 13 (Scheme 3).
Scheme 3.
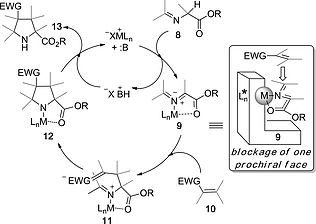
Catalytic cycle of a stepwise 1,3‐dipolar reaction between imines 8 mediated by N‐metallated azomethine ylides 9. EWG stands for an electron‐withdrawing group. MX is a metallic salt where M usually is CuI, CuII or AgI and the base B is a tertiary amine. The origin of the possible enantiocontrol by using chiral ligands in 9 is highlighted.
Another advantage of the catalytic methodology depicted in Scheme 3 is that, in the presence of chiral ligands, the N‐metallated azomethine ylide 9 only accepts the dipolarophile by the less hindered prochiral face, thus allowing the formation of Michael adducts 11 in which two chiral centers are generated with high or even complete regio‐ and stereocontrol. From these intermediates the ring closure gives rise to the two remaining chiral centers, also with very high stereocontrol. Therefore, the highly restricted structure of intermediates 9 relies on the nature of the chiral ligands L* (Scheme 3), which are ultimately responsible for the final enantiocontrol.
2. Synthesis of Catalysts by Means of 1,3‐Dipolar Reactions
In this section the synthesis of L* ligands shown in Scheme 2 by (3+2) cycloaddition reactions between azomethine ylides and alkenes will be discussed. Next, second‐generation (3+2) cycloadditions catalyzed by these ligands will be presented and discussed. Finally, the organocatalytic activity of the (3+2) cycloadducts formed in the latter process will be shown.
2.1. Ferrocenyl Prolines as Enantiopure Ligands
The activated azomethine ylides 9 shown in Scheme 3 require a rigid environment around the 1,3‐dipole to promote the preferential approach of the dipolarophile along the less hindered prochiral face. This conformationally restricted environment can be achieved by means of chiral ligands possessing donor atoms. Among the different possibilities, enantiopure ligands bearing phosphine and amine functional groups (P,N‐ligands)11 constitute the most efficient L* groups that can promote asymmetric catalytic (3+2) cycloadditions between azomethine ylides and alkenes. Within this context, ferrocenylphosphine groups incorporating five‐membered nitrogen‐containing heterocycles are especially relevant because of the interplay between the planar chirality of the ferrocenyl moiety and the central chirality of the heterocycle. These ferrocenylphosphine ligands include triazoles 14,12 1H‐pyrazoles 15,13 1H‐imidazoles 16,14 oxazolines 17[7b], [7f], 15 and pyrrolidines 18 16 (Figure 1). Other families of P,N‐ligands combining different kinds of chirality have been described, including spiro polycyclic systems and axially chiral aminophosphines.11 However, ferrocenylphosphine ligands benefit from a unique combination of the bulky organometallic group, the already mentioned interplay between central and planar chiralities and, last but not least, a robust chemistry that permits the synthesis of different families of ligands.
Figure 1.
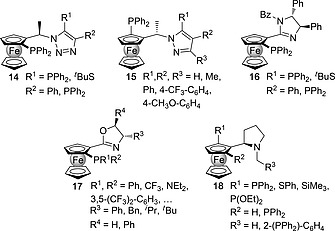
Different chiral ferrocenyl phosphines containing five‐membered nitrogen heterocycles.
We reasoned that ferrocenylphosphino pyrrolidines bearing four chiral centers after (3+2) cycloadditions could constitute promising P,N‐ligands11 in which the high density of chiral centers would in turn catalyze with high stereocontrol other (3+2) cycloadditions between azomethine ylides and nitroalkenes. The synthesis of these P,N‐ligands started with enantiomerically pure ferrocenyl carboxaldehyde 19 prepared according the method described by Kagan et al.17 (Scheme 4). Direct three‐component reaction between 19, sarcosine methyl ester hydrochloride 20 and (E)‐β‐nitrostyrene 21 yielded ligand NMe‐l‐EhuPhos 22 as the sole cycloadduct. Alternatively, imine 24 was formed from 19 and methyl glycinate hydrochloride 23. Subsequent (3+2) cycloaddition of 24 with dipolarophile 21 yielded cycloadduct NH‐d‐EhuPhos 25 in the presence of LiBr and triethylamine (Scheme 4).18
Scheme 4.
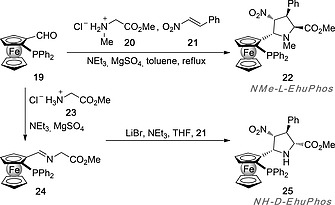
Synthesis of EhuPhos ligands 22 and 25.
The structures of both ligands were confirmed by X‐ray diffraction (Figure 2). It is noteworthy that both procedures yielded unnatural l‐ and d‐proline scaffolds, being the configurations of the remaining chiral centers identical. It is noteworthy that both ligands are stable and can be prepared in just one or two preparative steps at multigram scale from readily available materials.
Figure 2.
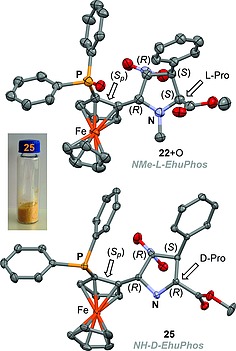
X ray diagrams (50 % probability) of ligands 22 (NMe‐l‐EhuPhos, CCDC 830011) and 25 (NH‐d‐EhuPhos, CCDC 830012). The different chirality descriptors are indicated. Crystal structure of 22 corresponds to the corresponding phosphine oxide. The L and D configurations of the respective proline moieties are highlighted. The macroscopic shape of a sample of 25 is also shown.
The behavior of these new ligands was tested in (3+2) cycloadditions between π‐deficient alkenes and N‐metallated azomethine ylides obtained in situ from the corresponding imines (Scheme 5).18 Among other results, it was observed that CuI‐N‐metallated azomethine ylides coordinated to NMe‐l‐EhuPhos ligand 22 produce endo‐l‐cycloadducts 28 with good yields and excellent ee's (Scheme 5), whereas the corresponding exo‐l‐pyrrolidines were obtained, also with excellent yields and enantiocontrol, in the presence of NH‐d‐EhuPhos ligand 25. It is noteworthy that, considering the proline motif transmitted from EhuPhos ligands to cycloadducts 28, the d‐proline moiety of the ligand generates an offspring of l‐proline derivatives exo‐l‐28, whereas the l‐proline motif present in NMe‐l‐EhuPhos 22 is transmitted with retention of l‐configuration to the endo‐l‐28 offspring. In addition, neither ligand 22 nor 25 were able to catalyze the formation of EhuPhos ligands. Therefore, the (3+2) processes gathered in Scheme 4 cannot be made autocatalytic.
Scheme 5.
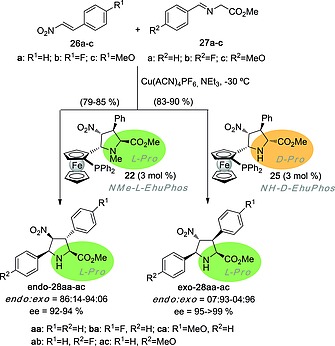
(3+2) Cycloaddition reaction between nitroalkenes 26 and imines 27 catalysed by EhuPhos ligands 22 and 25. The retention or inversion of configuration of the proline moieties in the ligands and cycloadducts are highlighted.
Computational studies18 on the origins of the distinct behavior of both ligands revealed transition structures closely related to the shape of azomethine ylides 9 (see Figure 3 and Scheme 3). However, in the case of NMe‐l‐EhuPhos 22 the CuI center cannot bind the nitrogen atom of the pyrrolidine moiety and therefore there is a vacant in the metallic center that can be filled by the nitro group of the alkene, thus giving rise to the corresponding endo‐cycloadduct 28. In contrast, NH‐d‐EhuPhos 25 behaves as a true N,P‐ligand and the nitro group of nitroalkene 26 can occupy the exo position to yield the corresponding exo‐cycloadduct. These calculations also provided a rationale for the enantioselectivity of the reaction. Thus, since both ligands 22 and 25 block the same prochiral (2Re, 5Si) face of the azomethine ylide, only the l‐configuration can be obtained in the corresponding cycloadduct 28.
Figure 3.
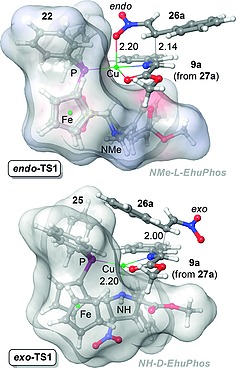
Transition structures endo‐TS1 and exo‐TS1 leading to (3+2) cycloadducts endo‐28aa and exo‐28aa, respectively, catalysed by EhuPhos ligands 22 and 25. The l‐shaped geometry of both ligands is highlighted (see Scheme 3). Both structures have been computed at the B3LYP/6‐31G(d)&LANL2DZ:PM8 level of theory. Bond lengths are given in Å. This Figure was made using Cartesian coordinates reported in ref.18.
NMe‐l‐EhuPhos ligand 22 was also used by Martin et al.19 in the diastereo‐ and enantioselective (3+2) cycloaddition between C60 and N‐metallated azomethine ylides derived from imines 29a–f. Also in this case, the l‐catalyst yields l‐cycloadducts (Scheme 6) with good chemical yields and excellent stereocontrol. It is interesting to note that in this series of experiments chiral quaternary centers at the α‐position of the unnatural proline ring could be generated with excellent enantiocontrol.
Scheme 6.
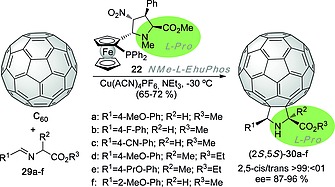
(3+2) Cycloaddition between C60 and imines 29 in the presence of NMe‐l‐EhuPhos ligand 22. The retention of configuration of the l‐proline scaffold in the ligand and cycloadduct is highlighted.
DFT calculations19 showed that the blockade of the (2Re, 5Re) prochiral side of the azomethine ylide is associated with the preferred conformation of ligand 22, whose rigidity stems from the interaction of the phosphine moiety and the methoxycarbonyl group of the proline moiety with the CuI metallic center. Therefore, in this case NMe‐l‐EhuPhos 22 behaves as a P,O‐ligand, which interacts with the azomethine ylide but not with the dipolarophile C60. This interaction model is compatible with the high enantioselectivity observed, since transition structure (2S, 5S)‐TS2 (Figure 4) leading to the experimentally observed major cycloadducts (2S, 5S)‐30‐af, is predicted to be 3–8 kcal/mol less energetic than the alternative (2R, 5R) saddle point.
Figure 4.
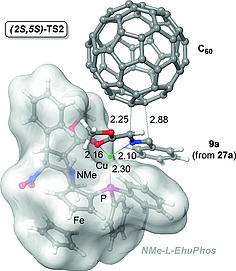
Transition structure (2S,5S)‐TS2, calculated at the M06/LANL2DZ//B3LYP/LANL2DZ:PM6 level, associated with the formation of (2S,5S) cycloadducts 30 (see Scheme 6), in the presence of NMe‐l‐EhuPhos ligand 22. Bond lengths are given in Å. This Figure was generated using Cartesian coordinates reported in ref.19.
In the preceding examples, DFT calculations showed highly asynchronous and stepwise reaction paths. Actually, in the reaction between imines and nitroalkenes in the presence of CuI salts (Scheme 5), the reaction takes place via zwitterionic intermediates of type 11 (Scheme 3). We reasoned that substituents that stabilize positive charges on the iminium moieties of these intermediates could frustrate the completion of the second step of the (3+2) cycloaddition, thus permitting the chemical synthesis of γ‐nitro‐α‐amino esters and their derivatives. In effect, we found20 that in the presence of 1 equiv. of NH‐d‐EhuPhos 25 and at –60 °C, reaction of nitroalkenes with imines 27 derived from 4‐methoxybenzaldehyde permitted to isolate syn‐γ‐nitro‐α‐amino esters 31 with excellent enantiocontrol (Scheme 7). Therefore, a simple electron donating group is enough to frustrate the (3+2) cycloaddition by stopping the stepwise process at the Michael‐like addition step. Alternatively, an additional phenyl group present in imines 31 derived from acetophenone is able to frustrate the cycloaddition but, in this case, only anti‐γ‐nitro‐α‐amino esters 32 were obtained in the presence of the same chiral ligand NH‐d‐EhuPhos 25 (Scheme 7). Also in this case, DFT calculations permitted to rationalize these different stereochemical outcomes.20 It is also noteworthy that, once again, NH‐d‐EhuPhos, an unnatural d‐Proline ester, promoted the formation of l‐amino esters via these frustrated (3+2) cycloadditions.
Scheme 7.
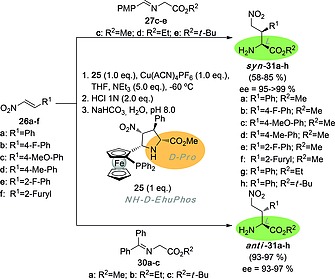
Synthesis of syn‐ and anti‐γ‐nitro‐α‐amino esters 32 via frustrated 1,3‐dipolar reaction between nitroalkenes 26 and amines 27, 31 with stoichiometric amounts of NH‐d‐EhuPhos ligands 25. The D and L configurations of ligand and products are highlighted.
In summary, l‐ and d‐proline‐based EhuPhos catalysts 22 and 25 are able to promote a second generation of unnatural proline derivatives via (3+2) cycloadditions. This process is compatible with many dipolarophiles including fullerenes. In addition, the stepwise nature of the associated mechanism permits the synthesis of other α‐l‐amino acid derivatives. It is expected that the catalytic scope of ligands 22 and 25 will be extended to other metal‐assisted reactions.
2.2. Densely Substituted Prolines as Organocatalysts
Natural l‐Proline and its derivatives constitute a privileged structure in the field of organocatalysis.21 However, most of these organocatalysts are based on naturally occurring proline or its post‐translational derivatives such as l‐4‐hydroxyproline or 4‐l‐aminoproline.22 We reasoned that densely substituted proline derivatives such (3+2) cycloadducts 28 described in the preceding section could exhibit organocatalytic properties and, even more interestingly, could provide new outcomes not accessible to organocatalysts generated from the naturally occurring amino acid.
The aldol reaction was explored first.23 Unnatural 4‐nitro‐l‐prolines 28 in their endo‐ and exo‐forms were tested in the aldol reaction between cyclic ketones 33 and aromatic aldehydes 34 (Scheme 8).18, 23 It was found that both l‐derivatives yielded opposite enantiomers. Thus, endo‐28aa yielded (2S,1′R)‐35 aldols, the same chiral induction generated by natural l‐Pro and its derivatives. In contrast, exo‐28aa gave rise to aldols possessing the (2R,1′S) stereochemistry. Therefore, since both unnatural organocatalysts 28 have l‐configuration, the stereochemistry of the distal positions with respect to the NH active site is responsible for the opposite enantioselectivities observed in these aldol reactions.
Scheme 8.
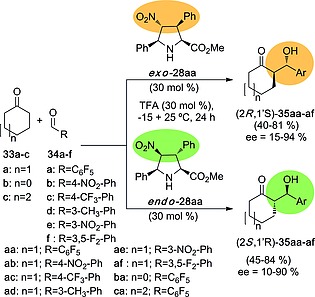
Aldol reactions between cyclic ketones 33 and aldehydes 34 catalysed by unnatural proline esters exo‐l‐28aa and endo‐l‐28aa. The different stereochemical outcomes generated by the distal substituents of the l‐cycloadducts 28 are highlighted.
DFT calculations showed that the stereochemical outcome of the aldol reactions gathered in Scheme 8 is determined by the equatorial/axial/isoclinal disposition of the four substituents of cycloadducts 28. As it can be seen by inspection of Figure 5, both minimum energy transition structures (2R,1′S)‐exo‐TS3 and (2S,1′R)‐endo‐TS3 adopt conformations of the organocatalysts that maximize the number of equatorial groups. In the case of (2R,1′S)‐exo‐TS3, the aryl group of the aldehyde adopts a distal disposition with respect to the 5‐phenyl group of the organocatalyst, whereas in the case of (2S,1′R)‐endo‐TS3 this disposition is proximal. The relative energies emerged from these geometric features are in good agreement with the observed enantioselectivities (Figure 5).
Figure 5.
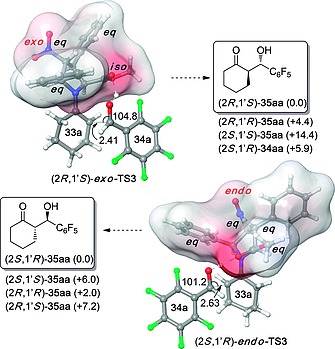
Optimized structures [M06–2X(PCM)/6‐31G(d,p)//B3LYP(PCM)/6‐31G(d) level of theory] of transition structures (2R,1′S)‐exo‐TS3 and (2S,1′R)‐endo‐TS3 leading to aldol adducts (2R,1′S)‐35aa and (2S,1′R)‐35aa, respectively. Numbers in parentheses correspond to the relative energies (in kcal/mol) of the transition structures leading to the respective stereoisomers. Bond lengths and angles are given in Å and deg, respectively. Cartesian coordinates were taken from ref.23.
When different substitution patterns were analyzed, it was observed that cis‐substituents at C3,C4 resulted in much less active organocatalysts than their C3,C4‐trans congeners (Figure 6). In addition, quaternary centers at C2 were found to be not convenient to generate good organocatalytic activity. These results can be rationalized in terms of the number of axial centers at the C2, C3 and C4 positions of the organocatalysts, since 3,4‐cis pyrrolidine scaffolds and C2 quaternized unnatural prolines must necessarily include axial substituents at the corresponding transition structures, thus raising the corresponding activation energies.
Figure 6.
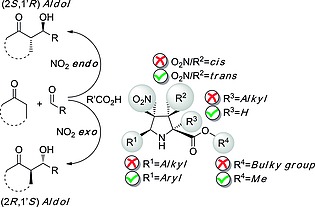
Effects of the substituents on the organocatalytic aldol activity of unnatural prolines 28.
Despite their ability to catalyze aldol reactions, 4‐nitroprolines 28 resulted to be unable to catalyze Michael additions of ketones and nitroalkenes. Instead of the expected conjugate addition, it was found that in the presence of equimolar amounts of acidic additives, a novel three‐component reaction was produced.24 This reaction, efficiently catalyzed by exo‐28aa, consists of the synthesis of 7a‐hydroxyoctahydro‐2H‐indol‐2ones 37 from ketones 33, acids 36 and nitroalkenes 26 (Scheme 9). This unexpected addition‐cyclization process involves the generation of three new chiral centers in one preparative step. In addition, it tolerates different heteroatoms at the starting ketone, as well as many substituents at the carboxylic acid and nitroalkene moieties.
Scheme 9.
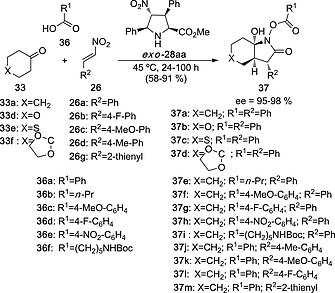
Three‐component reaction between ketones 33, acids 36 and nitroalkenes 26 to yield 7a‐hydrroxyoctahydro‐2H‐indol‐2‐ones 37, catalyzed by unnatural proline ester exo‐28aa.
Detailed experimental and computational work led to the catalytic cycle outlined in Figure 7A. After initial enamine formation and conjugate addition mediated by TS4 (Figure 7B), the key steps include an addition of the acid or the nitronate intermediate via saddle point TS5 (Figure 7C) and a rearrangement to generate the carboxamide moiety before the cyclization step. As far as the origins of the chiral induction of exo‐l‐28aa are concerned, transition structure TS4, whose chief geometric features are gathered in Figure 7B, shows a very efficient blockage of one prochiral face of the nitroalkene, thus generating the final bicyclic compound with virtually complete stereocontrol.
Figure 7.
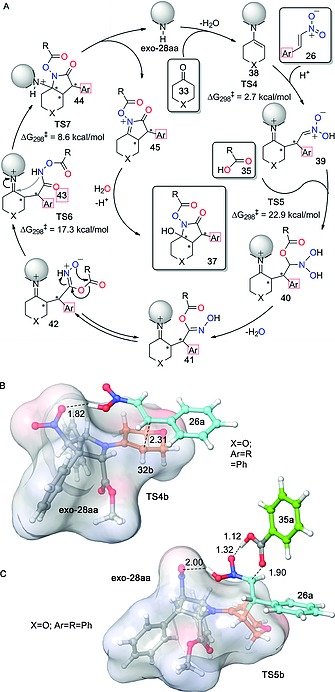
(A) Proposed catalytic cycle for the new reaction described in Scheme 9. Free activation energies calculated for the key steps of the 33b + 26a + 36a → 37a reaction, computed at the B3LYP‐D3(PCM = cyclohexanone)/6‐311+G*//B3LYP‐D3/6‐31G(d) level of theory, are also given. (B, C) Optimized structures for TS4b (B) and TS5b (C). Bond lengths are given in Å. This Figure was adapted from ref.24
The potential of this reaction was exemplified by its application to a concise synthesis of (+)‐pancracine.24, 25 In this synthesis, adduct 37n was obtained from readily available reactants 33f and 26h in satisfactory yield and virtually complete ee, despite working at a relatively high reaction temperature. It is interesting to note that nearly all the chiral information present in the alkaloid is generated in a single preparative step leading to adduct 37n (Scheme 10).24
Scheme 10.
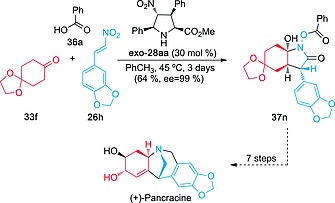
Synthesis of (+)‐pancracine initiated by the three‐component reaction between ketone 33f and nitroalkene 26h promoted by organocatalyst exo‐28aa. The origins of the scaffolds stemming from reactants 33f and 26h are highlighted in red and blue, respectively.
Although 4‐nitroprolines 28 were unable to catalyze Michael reaction between ketones and nitroalkenes, we found that the corresponding amino analogs 46, obtained by catalytic hydrogenation of their nitro precursors 28, catalyzed very efficiently this reaction.26 As it is shown in Scheme 11, organocatalyst exo‐46 generates Michael adducts (2R,1′S)‐47 with good to excellent ee's. In contrast, diastereomer endo‐46 gives rise to (2S,1′R)‐47aa enantiomer, similar to that produced by l‐Pro, with somewhat lower ee. Therefore, we found that a single transformation of the nitro group to the corresponding amino derivative results in a transformation of the organocatalytic ability of the unnatural proline scaffold. Other transformations on adducts 28 26 permitted to conclude that the exocyclic primary amino group in (3+2) cycloadducts 46 is essential for catalysis.
Scheme 11.
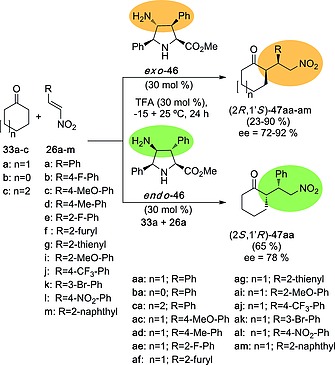
Michael addition of cyclic ketones 33 on nitroalkenes 26 in the presence of organocatalysts exo‐46 and endo‐46.
DFT calculations26 on the most relevant transition structures involved in these Michael additions reproduced correctly the trends in enantioselectivity found in the experimental studies. The shape of the transition structures associated with the behavior of exo‐47 and endo‐47 showed that the enamine derived from the primary amino group must adopt axial and isoclinal dispositions in (R,1′S)‐exo‐ TS6 and (S,1′R)‐endo‐TS6, respectively (Figure 8). We found that the stereocontrol is determined by the antiperiplanar conformation of the aryl substituent of the nitroalkene, by the optimal angle of attack associated with the formation of the new C–C bond and by the hydrogen bonding array between the organocatalyst and the nitro group of the Michael acceptor. Actually, chemical transformations that eliminated these hydrogen bonds, for instance via N‐acylation or N‐methylation, resulted in inactive pyrrolidine derivatives, as predicted by the computational studies.
Figure 8.
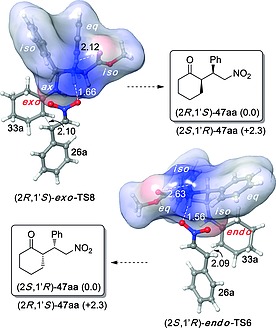
Optimized structures [M06–2X/6‐31+G(d,p)//B3LYP/6‐31G(d) level of theory] of transition structures (2R,1′S)‐exo‐TS6 and (2S,1′R)‐endo‐TS6 leading to Michael adducts (2R,1′S)‐47aa and (2S,1′R)‐47aa, respectively. Numbers in parentheses correspond to the relative energies (in kcal/mol) of the transition structures leading to the respective stereoisomers. Bond lengths are given in Å and deg, respectively. Cartesian coordinates were taken from ref.23.
The distinct organocatalytic behavior of catalysts 28 and 46 permitted to perform selective aldol and Michael additions.26 Thus, reaction of double electrophile 48 with cyclohexanone 33a in the presence of exo‐28aa yielded aldol 49 as the major isomer with good diastereo‐ and enantiocontrol. Subsequent reaction of this latter compound with cyclohexanone in the presence of exo‐46 gave rise to compound anti‐ 50 as the major diastereomer (Scheme 12). It Is interesting to note that the Michael addition took place with total diastereo‐ and enantiocontrol. However, a partial epimerization of the aldol moiety occurred and syn‐ 50 was obtained as a minor stereoisomer. In any case, this example indicates that unnatural organocatalysts can be used to achieve chemoselective transformations.
Scheme 12.
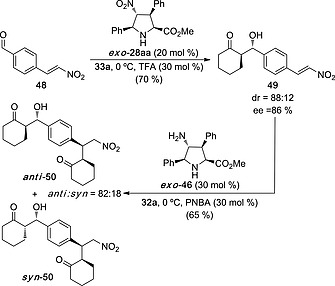
Selective aldol and Michael additions on double electrophile 48 catalyzed by unnatural prolines exo‐28aa and exo‐46, respectively. PNBA: para‐nitrobenzoic acid.
Highly substituted prolines formed from (3+2) cycloadditions can be used in selective polymerization reactions.27 Using catalysts endo‐ and exo‐52, it was found that N‐methylated unnatural proline exo‐l‐52 is able to catalyze the selective polymerization of l‐lactide 51 in a racemic mixture of 51. In contrast, endo‐l‐52 promoted the enantioselective polymerization of d‐lactide 51. In both cases, excellent stereoregularities (see the P m and [α] values in Scheme 13) were observed, as well as molecular weights that closely tracked the monomer‐to‐initiator ratio.
Scheme 13.
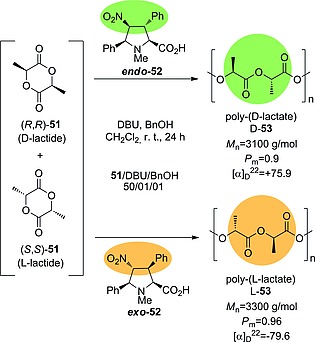
Polymerization of racemic d‐ and l‐lactide 51 in the presence of DBU and organocatalysts endo‐52 and exo‐52. Benzyl alcohol was used as initiator. M n: molecular weight. P m: probability of meso linkage between monomeric units.
Also in this case, DFT calculations27 permitted to explain the origins of this exceptional stereocontrol. The conformational flexibility of organocatalysts 52 allows different interaction patterns with the electrophilic and nucleophilic partners of the polymerization process (Figure 9). In this ring‐opening polymerization (ROP) process, organocatalysis is not covalent and consists of LUMO activation of the lactide by means of its interaction with the carboxylic acid, together with HOMO activation of the nucleophilic alcohol, which is benzyl alcohol in the first catalytic cycle (methanol in our computational model), or terminal alkoxy groups of the growing chain during the polymerization process. This latter activation takes place by means of the interaction of the hydroxy group with the amino group of the organocatalyst. When exo‐52 catalyzes the ROP the double carboxy/methylamine interaction takes place via transition structure (S,S)‐exo‐TS7, in which the N‐methyl group occupies an equatorial position and is anti with respect to the carboxy group. In contrast, in the case of endo‐52 these groups are syn to each other, which results in an axial disposition of the N‐methyl group (Figure 9). These preferred interactions, aside the maximization of the equatorial groups of the catalysts, account for the selectivity of exo‐52 for l‐53 via preferred formation of first intermediate ester l‐54 and subsequent incorporation of additional equivalents of (S,S)‐51 (l‐lactide). Similarly, these calculations resulted to be compatible with the selectivity of endo‐52 towards the ROP of (R,R)‐51 (d‐lactide) to yield highly stereoregular polymer d‐53, although with slightly lower stereocontrol.
Figure 9.
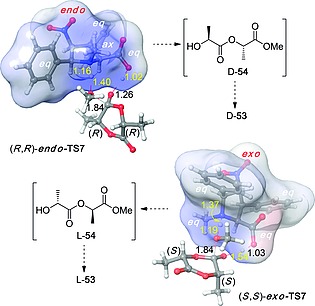
Optimized geometries [B3LYP/6‐31G(d) level of theory] of preferred transition structures (R,R)‐endo‐TS7 and (S,S)‐exo‐TS7 associated with the interaction between methanol (as a model primary alcohol) and lactides (R,R)‐51 and (S,S)‐51 in the presence of endo‐52 and exo‐52 (endo,syn‐TS). Distances are given in Å. Data taken from ref.27.
Chiral dipolarophiles can be used for the synthesis of enantiopure pyrrolidines than in turn are suitable organocatalysts. Sarotti, Suarez et al.28 described the stereocontrolled synthesis of unnatural tricyclic proline derivatives by means of the (3+2) cycloaddition between imines 56 and levoglucosenone 55 to yield l‐cycloadducts 57 via intermediate N‐Ag azomethine ylides. These compounds isomerized under microwave irradiation to yield the thermodynamically more stable unnatural tricyclic l‐proline esters 58 (Scheme 14).
Scheme 14.
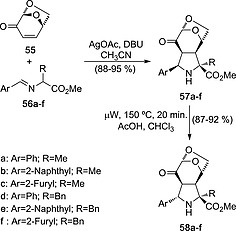
Synthesis of tricyclic unnatural l‐proline esters 58 via (3+2) cycloaddition between levoglucosenone 55 and imines 56.
Tricyclic unnatural l‐proline esters 58 resulted to be efficient organocatalysts of Diels–Alder (DA) reactions between cyclopentadiene 59 and α,β‐unsaturated aldehydes such as cinnamaldehyde 60. For instance, 58a promoted the formation of norbonenes 61 with moderate exo‐selectivity and ee's (Scheme 15). Intensive computational analysis of the DA reactions led to Sarotti et al.29 to predict that organocatalyst 58g should be more efficient in terms of enantiocontrol. This prediction was successfully corroborated by the experiment (Scheme 15).
Scheme 15.
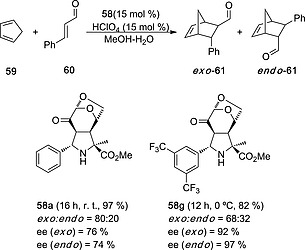
Diels–Alder reaction between cyclopentadiene 59 and cinnamaldehyde 60 catalysed by (3+2) l‐cycloadducts 58a and 58g.
In summary, the examples shown in this section demonstrate that densely substituted unnatural proline derivatives possess interesting covalent organocatalytic properties mediated by enamine intermediates. These unusual properties range from the reverse enantiocontrol induced from distal configurations with respect to the active sites, to different scopes by simple change of functional groups, including the ability to catalyze an unexpected complex three‐component cyclization reaction. In addition, the above discussed example of enantioselective polymerization permits to expect further developments in non‐covalent organocatalysis induced by this kind of synthetic organocatalysts.
3. Unnatural Synthetic Prolines as Biologically Active Compounds
Pyrrolidines are considered privileged structures in medicinal chemistry.30 According to the definition proposed by Evans,31 these saturated heterocycles are capable of providing useful ligands for more than one receptor. Therefore, “judicious modifications of such structures could be a viable alternative in the search for new receptor agonists and antagonists”.31 More recently, an analysis of privileged structures based on Shannon entropy formalism32 suggests that “heterocyclic, sp3‐rich frameworks are particularly suited for target‐focused library design.” Actually, an analysis of the structural diversity and substitution patterns of nitrogen heterocycles among U.S. FDA approved pharmaceuticals33 revealed that the pyrrolidine ring is the fifth heterocycle present in approved drugs. Among the 37 pyrrolidine‐containing pharmaceuticals, eight of them incorporate the proline scaffold, with 1–3 additional substituents (Scheme 16). All these compounds are angiotensin converting enzyme (ACE) inhibitors,34 with the exception of Clindamycin, an antibiotic for the treatment of several bacterial infections.35
Scheme 16.
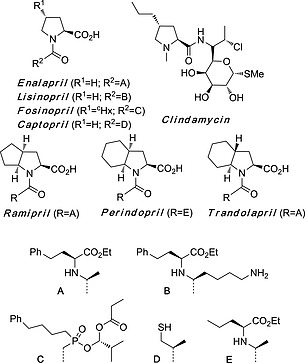
U.S. FDA approved pharmaceuticals containing proline scaffolds.
The above indicated pharmaceuticals can be obtained by derivatization of l‐proline or other post‐translational proline derivatives. Therefore, as far as the chemical synthesis of highly prescribed drugs is concerned, to date (3+2) cycloadditions have been used for the synthesis of other aromatic five‐membered nitrogen containing pharmaceuticals36 such as celecoxib.37 One patented synthesis of prescription drug atorvastatin38 is an exception to this statement (vide infra). In the following sections the synthesis of biologically active substituted prolines or their aromatized 1H‐pyrrole derivatives is presented. Although these compounds are not in clinical use so far, most likely the role of these compounds (or their analogs) in Medicinal chemistry will be more relevant in the years to come.
3.1. Synthesis of Biologically Active Racemic Prolines via (3+2) Cycloadditions
Racemic proline derivatives are useful intermediates in the synthesis of pharmaceuticals when aromatization processes occur during or after the (3+2) cycloaddition to yield substituted 1H‐pyrroles. A relevant example consists of the (3+2) cycloaddition/(4+2) cycloreversion of azomethine ylide 63 (derived from amino acid 62) with alkyne 64 to yield cycloadduct 65 that in situ decarboxylates to yield pentasubstituted 1H‐pyrrole 66,38 a key precursor of atorvastatin (Scheme 17), a prescription drug used to lower blood cholesterol.
Scheme 17.
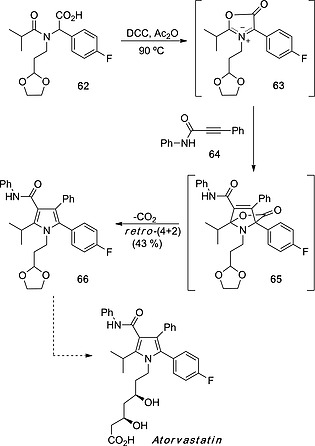
Synthesis of atorvastatin by (3+2) cycloaddition between azomethine ylide 63 and alkyne 64.
Another example of synthesis of biologically active 1H‐pyrroles by aromatization of pyrrolidines is the synthesis of histone deacetylase (HDAC) inhibitors via (3+2) cycloaddition between imines 27 and nitroalkenes 26.39 Under thermal conditions, mixtures of endo‐ and exo‐cycloadducts 67 were formed, whose aromatization under thermal heating or microwave irradiation[8a] gave mixtures of methyl 3,5‐diaryl‐1H‐pyrrole‐2‐carboxylates 68 and 69. Subsequent functionalization of these latter aromatic cycloadducts led to hydroxamic acids 70, which in some cases such as of compound 70a, resulted to be very potent HDAC inhibitors, with IC50 values within the picomolar range40 (Scheme 18). This latter molecule has shown strong growth‐inhibitory effect in Burkitt's cell lymphoma, follicular lymphoma and, especially, mantle cell lymphoma (MCL).[40b]
Scheme 18.
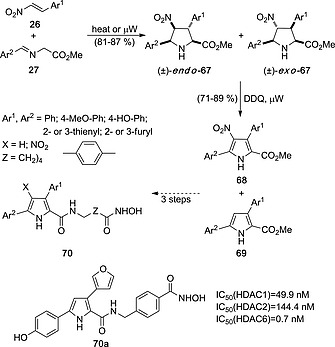
Synthesis of HDAC inhibitors via consecutive (3+2) cycloaddition/aromatization reactions.
Racemic dispiropyrrolidines constitute potentially useful anti‐inflammatory agents.41 Reaction of isatins 71 with sarcosine 20 led to the formation of fleeting azomethine ylides 72, whose reaction with dipolarophile 73 yielded racemic tricyclic dispiropyrrolidines 74 in good yields (Scheme 19). These racemic compounds showed interesting in vivo anti‐inflammatory activity, measured as aedema inhibition.
Scheme 19.
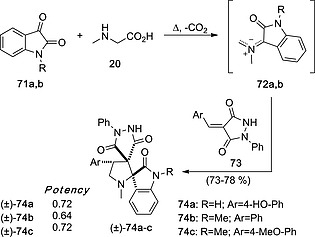
Synthesis of anti‐inflammatory racemic dispiropyrrolidines (±)‐74 by reaction of isatins 71a,b with sarcosin 20. Potencies were measured with respect to reference standard indometracin.
A similar approach was followed to synthesize inhibitors of advanced glycation end product, which is a relevant target in the treatment of diabetes mellitus.42 Reaction of isatin 71a with benzylamine led to highly substituted racemic spiroprolines 77 with excellent diastereocontrol and chemical yields via reaction of azomethine ylide 75 with α,β‐unsaturated ketones 76. (Scheme 20) .
Scheme 20.
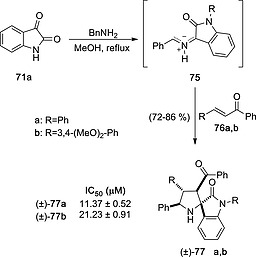
Synthesis of inhibitors of advanced glycation end products by (3+2) cycloaddition between azomethine ylide 75 and α,β‐unsaturated ketones 76.
3.2. Synthesis of Biologically Active Prolines via Asymmetric Catalysis on (3+2) Cycloadditions
Nájera and Sansano43 have demonstrated the usefulness of 1,3‐dipolar reactions with N‐metallated azomethine ylides for the synthesis of drug candidates for hepatitis C treatment. Reaction of tert‐butyl acrylate 78 with imines 29i,j led to substituted l‐prolines 81a,b with high chemical yield and diastereo‐ and enantiocontrol in the presence of chiral ligands 79 44 and 80 45 and using AgI and AuI metallic salts, respectively (Scheme 21). N‐Acylation and deprotection of both ester groups led to the inhibitors of hepatitis C virus 82a,b in good overall yields and excellent enantioselectivities.
Scheme 21.
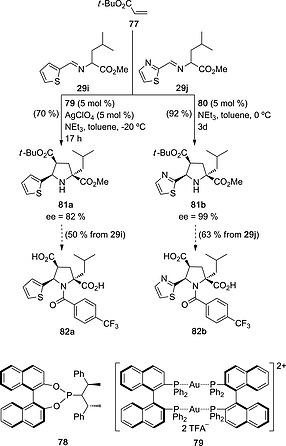
Asymmetric synthesis of hepatitis C virus polymerase inhibitors 82a,b from imines 29i,j.
Using (R)‐Fesulphos 83 as chiral ligand, Antonchick and Waldmann46 described the enantioselective synthesis of iridoid‐inspired bicyclic proline derivatives (Scheme 22) by means of a (3+2) cycloaddition between imines 27 and racemic 2H‐pyran‐3(6H)‐one derivatives 84. Under the reaction conditions gathered in Scheme 22, the authors obtained bicyclic (3+2) cycloadducts with good yields and stereoselectivities. In all the reported examples, endo diastereomers were obtained with diastereomeric excesses of ca. 90 % and ee's up to 96 %, l‐proline configuration being the major one. Since only the (S)‐enantiomer of dipolarophiles 84 was able to react with the corresponding imine 27, the method provides a kinetic resolution of racemic 2H‐pyran 3(6H)‐ones 84. Among the 28 cycloadducts thus synthesized, five of them showed promising inhibitory activity of the Hedgehog (Hh) and Wingless/integration (Wnt) signaling pathways (Scheme 22).46
Scheme 22.
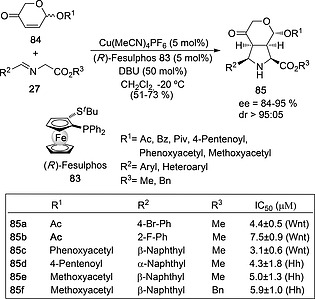
Synthesis of Hedgehog (Hh) and Wingless/integration (Wnt) signaling pathways inhibitors 85 from 2H‐pyran‐3(6H)‐one dipolarophiles 84 and imines 27.
The same research group47 reported the synthesis of functionalized tropanes by means of (3+2) cycloaddition between nitroalkenes 86 and azomethine ylides derived from tryptophan derivatives 87 (Scheme 23). Using CuI as source of the N‐metallated azomethine ylide and (R)‐Fesulphos 83 as chiral ligand, the authors obtained tetracyclic proline esters 88 in good yields and excellent ee's. In this case, exo cycloadducts were obtained. The structure of amino esters 88 is closely related to that of tropanes, whose biological activity in naturally occurring compounds is well known. The authors prepared 22 (3+2) cycloadducts 88. In three cases, significant Hh activity was observed (Scheme 23). Interestingly, the chiral configuration of these (3+2) cycloadducts was found to be relevant for biological activity, since in the case of racemic 88a no Hh inhibitory activity was observed.47
Scheme 23.
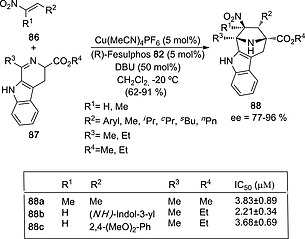
Synthesis of Hedgehog (Hh) signaling pathway inhibitors 88 from nitroalkenes 86 and tryptophan‐derived imines 87.
3.3. Synthesis of Biologically Active Prolines via (3+2) Cycloadditions Involving Enantiopure Dipolarophiles
1,3‐Dipolar reactions between azomethine ylides usually require π‐deficient alkenes incorporating electron‐withdrawing groups such as carboxamido, ester or nitro groups. A relevant example of dipolarophiles incorporating chiral esters consists of the reaction between azomethine ylide 90 and enantiopure alkene 89 to yield tricyclic (3+2) adduct 91. Treatment of this latter compound with AgOTf led to 3,4‐dihydro‐2H‐pyrrole 92 together with two additional diastereomers in a diastereomeric excess of 66 %. Compound 92 was converted to (2S,4R)‐93 (Scheme 24). This proline derivative proved to be a potent inhibitor of CNS l‐glutamate neurotransmitter transporters. Interestingly, (2S,4R)‐93 showed non‐substrate inhibitory activity, in contrast with the biological behavior observed for its non‐methylated analogue.48
Scheme 24.
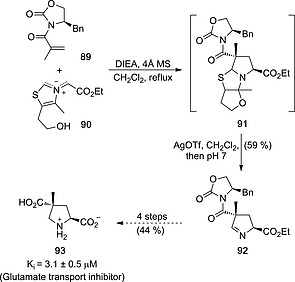
Synthesis of glutamate transport inhibitors 93 by reaction between azomethine ylide 90 and chiral alkene 89.
Nájera, Sansano et al.49 used acrylates derived from (R) and (S)‐lactate to synthesize hepatitis C inhibitors 82 (Scheme 25). Reaction between imine 29i and (R)‐acrylate 94 [prepared from acrylic acid and methyl (R)‐lactate] in the presence of silver acetate yielded unnatural l‐proline 95 with excellent diastereomeric excess. Subsequent derivatization of this latter cycloadduct permitted to obtain compound 82a with high yield and enantiocontrol. A similar route starting from (S)‐lactic acid led to ent‐82b.
Scheme 25.
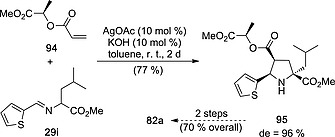
Synthesis of hepatitis C virus inhibitor 82a from chiral acrylate 94.
The 1,3‐dipolar reaction between azomethine ylides and α,β‐unsaturated carbonyl compounds can be extended to intramolecular processes that actually involve a common chiral precursor. For instance, in situ formation of azomethine ylides 97 (Scheme 26) from amine 96 led to a tricyclic cycloadduct 98.50 Compound 99, which incorporates a highly substituted polycyclic l‐proline scaffold, is a cyclic analogue of antibacterial agent cethromycin and was obtained from 98 after a deprotection/oxidation sequence.
Scheme 26.
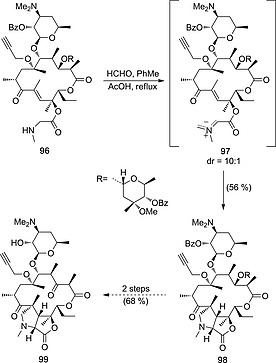
Synthesis of an analogue of antibacterial agent cethromycin 99 by intramolecular (3+2) cycloaddition of azomethine ylide 97.
Enantiopure nitroalkenes can also be used for the synthesis of biologically active compounds. We prepared chiral nitroalkenes 100 from l‐Leu and l‐Ile (Scheme 27). These latter dipolarophiles reacted with N‐Ag azomethine ylides derived from imines 29 to yield, after a (3+2) cycloaddition/ester hydrolysis sequence, l‐prolines 101 with excellent endo diastereocontrol.51 Subsequent amide coupling with linear amino esters and hydrolysis led to compounds 103. When the coupling reactions were carried out with glycine methyl ester (n = 0), the corresponding adducts showed excellent inhibitory activity of Very Late Antigen‐4 (VLA‐4), an α4β1 integrin involved in murine models of hepatic melanoma metastasis.51 On the other hand, when n = 1,4 adducts 103 showed excellent antagonists ability of Leucocyte Function Associated Antigen‐1 (LFA‐1) αLβ2 integrin,52 which is involved in early and late stages of cancer development. Alternatively, N‐acylation of compounds 102 followed by deprotection of the phthalimido group yielded compounds 105, which also showed LFA‐1 antagonist activity in a murine model of colon cancer52 (Scheme 27).
Scheme 27.
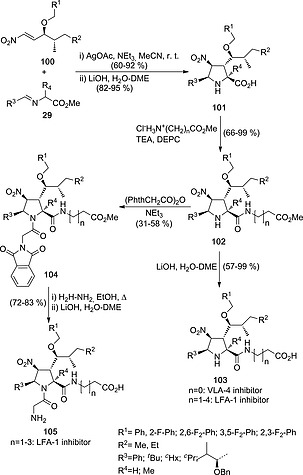
Synthesis of VLA‐4 and LFA‐1 integrin inhibitors 103 and 105 via (3+2) cycloadditions involving enantiopure nitroalkenes 100.
3.4. Synthesis of Biologically Active Prolines via (3+2) Cycloadditions Involving Enantiopure Azomethine Ylides
In Section 2.1, we have shown that NH‐d‐EhuPhos 25 can react with imines 27c–e and 31a–c to yield syn‐ and anti‐γ‐nitro‐α‐amino esters 32 by an interrupted stepwise 1,3‐dipolar reaction that stops at the Michael addition step.20 From these amino esters we generated cis‐ and trans‐γ‐lactams 106 after a catalytic hydrogenation/imine formation sequence (Scheme 28). In situ formation of the N‐Ag azomethine ylides 107 and reaction with suitable dipolarophiles yielded spiro‐l‐proline derivatives 108 with excellent endo stereocontrol. It is noteworthy that the chiral information of α‐carbon atoms of γ‐lactams 106 is lost after formation of the corresponding azomethine ylides. Therefore, the chirality of the Cβ atom of the 1,3‐dipoles is the only responsible for the formation of one given enantiomer of (3+2) cycloadducts 108, which incorporate up to four additional chiral centers.
Scheme 28.
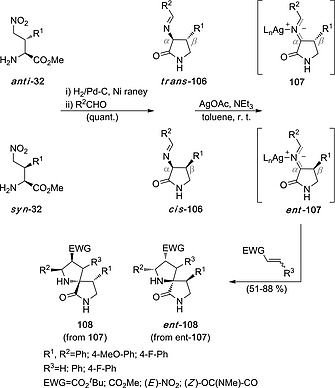
Formation of spiro‐l‐prolines 108 from γ‐nitro‐α‐amino esters 32.
Some of these spiro‐l‐proline lactams showed interesting biological activity.20 For example, spiro compound 108 with R1 = R2 = Ph and EWG‐R3 = N‐methyl phthalimido is an inhibitor of proprotein convertase subtilisin/kexin type 9 (PCSK9) enzyme with IC50 = 5 µm. In addition, conversion of tert‐butyl ester 108a into acid 109, followed by N‐acylation, deprotection of the methyl ester and N‐hydroxyamidation led to hydroxamic acid 110 with good yield (Scheme 29). This latter compound showed antiproliferative activity in HeLa nuclear extracts via HDAC inhibition.20
Scheme 29.
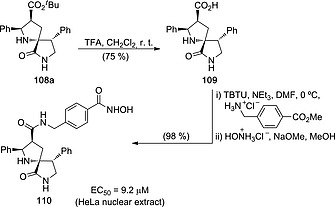
Synthesis of HDAC inhibitor 110 from spiro‐l‐proline derivative 108a.
4. Conclusions and Outlook
The pyrrolidine ring present in l‐Pro represents a good compromise between preorganization and a certain conformational flexibility. Highly substituted unnatural proline derivatives allow for the incorporation of chirality sources and additional functional groups. These features confer convenient properties upon densely substituted unnatural proline scaffolds. Fortunately, (3+2) cycloaddition between azomethine ylides and π‐deficient alkenes is a powerful chemical tool to generate the pyrrolidine ring in a highly regio‐, diastereo‐ and enantiocontrolled manner. This stereochemical control can be achieved by using enantiopure reactants and by means of asymmetric catalysis.
The (3+2) cycloaddition between enantiopure ferrocenyl imines and nitroalkenes leads to the synthesis of chiral EhuPhos ligands, in which the interplay between planar and central chiralities results in highly efficient and selective P‐, P,O‐ and P,N‐ligands. These ligands can be used as chiral catalysts in other chemical transformations, including asymmetric (3+2) cycloaddition between similar reactants, to generate an offspring of unnatural l‐proline derivatives.
These latter cycloadducts are excellent organocatalysts (offspring catalysts) in different reactions that include aldol and Michael additions, as well as enantioselective ring opening polymerizations. In one case, an unexpected three‐component reaction has been observed. The behavior of these unnatural l‐prolines is different to that observed for organocatalysts derived from the natural amino acid. In particular, the origins of the stereocontrol are related to the chirality of the distal substituents with respect to the active site. Thus, endo and exo cycloadducts show different stereochemical outcomes. The possibilities of this kind of organocatalysts in both enamine (HOMO raising) and non‐covalent (LUMO lowering) catalysis remain largely unexplored.
The above‐mentioned tradeoff between preorganization and flexibility present in the sp3‐rich pyrrolidine ring can be useful in the chemical synthesis of novel biologically active unnatural proline derivatives. A relatively reduced number of molecules of this kind of compounds has been reported so far. However, we are convinced that the favorable features of the (3+2) cycloaddition between azomethine ylides and alkenes will be increasingly used in the synthesis of proline derivatives as privileged structures for drug discovery.
Acknowledgements
Financial support was provided by the Ministerio de Economía y Competitividad (MINECO) of Spain and FEDER (projects CTQ2016‐80375‐P and Red de Excelencia Consolider CTQ2014‐51912‐REDC) and the Basque Government (GV/EJ, grant IT‐324‐07). The authors thank the SGI/IZO‐SGIker UPV/EHU and the DIPC for generous allocation of computational and analytical resources.
Biographies
Iosune Arrastia studied Chemistry at the Universidad del País Vasco/Euskal Herriko Unibertsitatea (UPV/EHU) and received her PhD in 2001 (Prof. Fernando P. Cossío). She is an Associate Researcher at the Donostia International Physics Center (DIPC). Her current research interests focus on the computational study of the origins of stereocontrol in organocatalysis.

Ana Arrieta studied Chemistry at the Universidad del País Vasco/Euskal Herriko Unibertsitatea (UPV/EHU) and received her PhD in 1986 (Prof. Claudio Palomo). She joined the UPV/EHU as Profesor Titular in 1991. She has served as Dean of the Faculty of Chemistry and Vice Rector of the Campus of Gipuzkoa of the UPV/EHU. Her current research interests focus on the computational study of the reaction mechanisms and origins of stereocontrol in organocatalysis and (n+2) thermal cycloadditions, as well as on the mode of action and origins of selectivity of epigenetic enzymes.

Fernando P. Cossio (San Martin de Villafufre, Cantabria, Spain, 1960) studied Chemistry at the Universidad de Zaragoza (1977–1981) and in 1986 obtained his PhD in the Universidad del País Vasco/Euskal Herriko Unibertsitatea (UPV/EHU) under the supervision of Prof. Claudio Palomo. After a postdoctoral stage at the Université de Bordeaux I‐CNRS (Dr. Jean‐Paul Picard) he was promoted to associate professor in 1988. In 1993 he went to UCLA as visiting scholar (Prof. Kendall N. Houk). In 2002 he was promoted to full professor at the UPV/EHU. He has served as Dean of the Faculty of Chemistry and Vice Rector for Research and International Relations of the UPV/EHU. He is currently the scientific Director of Ikerbasque, the Basque foundation for science. His main research interests cover cycloaddition chemistry, asymmetric catalysis and medicinal chemistry. The methodology used in these studies is based on the synergistic interplay between computational and experimental chemistry.

Dedicated to Professor Dr. Luis A. Oro.
References
- 1. Harwood L. M. and Vickers R. J. in Synthetic Applications of 1,3‐Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products (Eds.: Padwa A. and Pearson W. H.), J. Wiley & Sons, Howoken, NJ, 2002, pp. 169–252. [Google Scholar]
- 2.a) Sankararaman S. in Pericyclic Reactions ‐ A Textbook, Wiley‐VCH, 2005, pp. 169–191; [Google Scholar]; b) Held F. E. and Tsogoeva S. B., Catal. Sci. Technol., 2016, 6, 645. [Google Scholar]
- 3.a) Pandey G., Banerjee P. and Gadre S. R., Chem. Rev., 2006, 106, 4484–4517; [DOI] [PubMed] [Google Scholar]; b) Stanley L. M. and Sibi M. P., Chem. Rev., 2008, 108, 2887–2902; [DOI] [PubMed] [Google Scholar]; c) Adrio J. and Carretero J. C., Chem. Commun., 2011, 47, 6784–6794; [DOI] [PubMed] [Google Scholar]; d) Najera C., Sansano J. M. and Yus M., J. Braz. Chem. Soc., 2010, 21, 377–412; [Google Scholar]; e) Narayan R., Potowski M., Jia Z.‐J., Antonchick A. P. and Waldmann H., Acc. Chem. Res., 2014, 47, 1296–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) de Cozar A. and Cossio F. P., Phys. Chem. Chem. Phys., 2011, 13, 10858–10868; [DOI] [PubMed] [Google Scholar]; b) Arrieta A., de la Torre M. C., de Cozar A., Sierra M. A. and Cossio F. P., Synlett, 2013, 24, 535–549. [Google Scholar]
- 5.a) Fleming I. in Molecular Orbitals and Organic Chemical Reactions, J. Wiley & Sons, Chichester, U. K. 2010, pp. 258–260; [Google Scholar]; b) Woodward R. B. and Hoffmann R. in The Conservation of Orbital Symmetry, Verlag Chemie, Weinheim, 1970. [Google Scholar]
- 6.a) Di Valentin C., Freccero M., Gandolfi R. and Rastelli A., J. Org. Chem., 2000, 65, 6112–6120; [DOI] [PubMed] [Google Scholar]; b) Lan Y. and Houk K. N., J. Am. Chem. Soc., 2010, 132, 17921–17927; [DOI] [PubMed] [Google Scholar]; c) Jin G., Sun J., Yang R.‐Y. and Yan C.‐G., Sci. Rep., 2017, 7, 46470; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Emamian S., RSC Adv., 2016, 6, 75299–75314. [Google Scholar]
- 7.a) Vivanco S., Lecea B., Arrieta A., Prieto P., Morao I., Linden A. and Cossio F. P., J. Am. Chem. Soc., 2000, 122, 6078–6092; [Google Scholar]; b) Li Q., Ding C.‐H., Hou X.‐L. and Dai L.‐X., Org. Lett., 2010, 12, 1080–1083; [DOI] [PubMed] [Google Scholar]; c) Kim H. Y., Li J.‐Y., Kim S. and Oh K., J. Am. Chem. Soc., 2011, 133, 20750–20753; [DOI] [PubMed] [Google Scholar]; d) Imae K., Konno T., Ogata K. and Fukuzawa S., Org. Lett., 2012, 14, 4410–4413; [DOI] [PubMed] [Google Scholar]; e) Castello L. M., Najera C., Sansano J. M., Larranaga O., de Cozar A. and Cossio F. P., Org. Lett., 2013, 15, 2902–2905; [DOI] [PubMed] [Google Scholar]; f) Yan X.‐X., Peng Q., Li Q., Zhang K., Yao J., Hou X.‐L. and Wu Y.‐D., J. Am. Chem. Soc., 2008, 130, 14362–14363. [DOI] [PubMed] [Google Scholar]
- 8.a) Arrieta A., Otaegui D., Zubia A., Cossio F. P., Diaz‐Ortiz A., de la Hoz A., Herrero M. A., Prieto P., Foces‐Foces C., Pizarro J. L. and Arriortua M. I., J. Org. Chem., 2007, 72, 4313–4322; [DOI] [PubMed] [Google Scholar]; b) Grigg R., Chem. Soc. Rev., 1987, 16, 89–121. [Google Scholar]
- 9.a) Ayerbe M., Arrieta A., Cossio F. P. and Linden A., J. Org. Chem., 1998, 63, 1795–1805; [Google Scholar]; b) Kanemasa S. and Tsuge O. in Advances in Cycloadditions, Vol. 3 (Ed. D. P. Curran), Jai Press, Greenwich, 1993, pp. 99–159. [Google Scholar]
- 10. Banks H. D., J. Org. Chem., 2010, 75, 2510–2517. [DOI] [PubMed] [Google Scholar]
- 11.a) Carroll M. P. and Guiry P. J., Chem. Soc. Rev., 2014, 43, 819–833; [DOI] [PubMed] [Google Scholar]; b) Guiry P. J. and Saunders C. P., Adv. Synth. Catal., 2004, 346, 497–537. [Google Scholar]
- 12.a) Oura I., Shimizu K., Ogata K. and Fukuzawa S., Org. Lett., 2010, 12, 1752–1755; [DOI] [PubMed] [Google Scholar]; b) Shimizu K., Ogata K. and Fukuzawa S., Tetrahedron Lett., 2010, 51, 5068–5070; [Google Scholar]; c) Fukuzawa S. and Oki H., Org. Lett., 2008, 10, 1747–1750. [DOI] [PubMed] [Google Scholar]
- 13.a) Abbenhuis H. C. L., Burckhardt U., Gramlich V., Togni A., Albinati A. and Mueller B., Organometallics, 1994, 13, 4481–4493; [Google Scholar]; b) Burckhardt U., Hintermann L., Schnyder A. and Togni A., Organometallics, 1995, 14, 5415–5425. [Google Scholar]
- 14. Smilovic I. G., Casas‐Arce E., Roseblade S. J., Nettekoven U., Zanotti‐Gerosa A., Kovacevic M. and Casar Z., Angew. Chem. Int. Ed., 2012, 51, 1014–1018; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2012, 124, 1038. [Google Scholar]
- 15.a) Schuecker R., Zirakzadeh A., Mereiter K., Spindler F. and Weissensteiner W., Organometallics, 2011, 30, 4711–4719; [Google Scholar]; b) Hu Z., Li Y., Liu K. and Shen Q., J. Org. Chem., 2012, 77, 7957–7967; [DOI] [PubMed] [Google Scholar]; c) Yan X.‐X., Peng Q., Zhang Y., Zhang K., Hong W., Hou X.‐L. and Wu Y.‐D., Angew. Chem. Int. Ed., 2006, 45, 1979–1983; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2006, 118, 2013; [Google Scholar]; d) Liu W., Chen D., Zhu X.‐Z., Wan X.‐L. and Hou X.‐L., J. Am. Chem. Soc., 2009, 131, 8734–8735; [DOI] [PubMed] [Google Scholar]; e) Zeng W. and Zhou Y.‐G., Org. Lett., 2005, 7, 5055–5058; [DOI] [PubMed] [Google Scholar]; f) Gao W., Zhang X. and Raghunath M., Org. Lett., 2005, 7, 4241–4244; [DOI] [PubMed] [Google Scholar]; g) Zhang W., Yoneda Y.‐I., Kida T., Nakatsuji Y. and Ikeda I., Tetrahedron: Asymmetry, 1998, 9, 3371–3380; [Google Scholar]; h) Park J., Quan Z., Lee S., Han Ahn K. and Cho C.‐W., J. Organomet. Chem., 1999, 584, 140–146; [Google Scholar]; i) Deng W.‐P., Hou X.‐L., Dai L.‐X., Yu Y.‐H. and Xia W., Chem. Commun., 2000, 285–286; [Google Scholar]; j) Deng W.‐P., You S.‐L., Hou X.‐L., Dai L.‐X., Yu Y.‐H., Xia W. and Sun J., J. Am. Chem. Soc., 2001, 123, 6508–6519; [DOI] [PubMed] [Google Scholar]; k) You S.‐L., Hou X.‐L., Dai L.‐X., Yu Y.‐H. and Xia W., J. Org. Chem., 2002, 67, 4684–4695; [DOI] [PubMed] [Google Scholar]; l) Manoury E., Fossey J. S., Aiet‐Haddou H., Daran J.‐C. and Balavoine G. G. A., Organometallics, 2000, 19, 3736–3739; [Google Scholar]; m) Sammakia T. and Latham H. A., J. Org. Chem., 1995, 60, 6002–6003; [Google Scholar]; n) Richards C. J. and Locke A. J., Tetrahedron: Asymmetry, 1998, 9, 2377–2407; [Google Scholar]; o) Jones G. and Richards C. J., Tetrahedron Lett., 2001, 42, 5553–5555; [Google Scholar]; p) You S.‐L., Zhu X.‐Z., Luo Y.‐M., Hou X.‐L. and Dai L.‐X., J. Am. Chem. Soc., 2001, 123, 7471–7472; [DOI] [PubMed] [Google Scholar]; q) Dai L., Xu D., Tang L.‐W. and Zhou Z.‐M., ChemCatChem, 2015, 7, 1078–1082; [Google Scholar]; r) McManus H. A. and Guiry P. J., Chem. Rev., 2004, 104, 4151–4202. [DOI] [PubMed] [Google Scholar]
- 16. Delaye P.‐O., Ahari M., Vasse J.‐L. and Szymoniak J., Tetrahedron: Asymmetry, 2010, 21, 2505–2511. [Google Scholar]
- 17. Riant O., Samuel O., Flessner T., Taudien S. and Kagan H. B., J. Org. Chem., 1997, 62, 6733–6745. [Google Scholar]
- 18. Conde E., Bello D., de Cozar A., Sanchez M., Vazquez M. A. and Cossio F. P., Chem. Sci., 2012, 3, 1486–1491. [Google Scholar]
- 19. Maroto E. E., Filippone S., Suarez M., Martinez‐Alvarez R., de Cozar A., Cossio F. P. and Martin N., J. Am. Chem. Soc., 2014, 136, 705–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Conde E., Rivilla I., Larumbe A. and Cossio F. P., J. Org. Chem., 2015, 80, 11755–11767. [DOI] [PubMed] [Google Scholar]
- 21.a) Dondoni A. and Massi A., Angew. Chem. Int. Ed., 2008, 47, 4638–4660; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2008, 120, 4716; [Google Scholar]; b) Dalko P. I. and Moisan L., Angew. Chem. Int. Ed., 2004, 43, 5138–5175; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2004, 116, 5248; [Google Scholar]; c) Mukherjee S., Yang J. W., Hoffmann S. and List B., Chem. Rev., 2007, 107, 5471–5569; [DOI] [PubMed] [Google Scholar]; d) Scheffler U. and Mahrwald R., Synlett, 2011, 1660–1667; [Google Scholar]; e) Albrecht L., Jiang H. and Jorgensen K. A., Angew. Chem. Int. Ed., 2011, 50, 8492–8509; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2011, 123, 8642. [Google Scholar]
- 22.a) Trost B. M. and Brindle C. S., Chem. Soc. Rev., 2010, 39, 1600–1632; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bertelsen S. and Jorgensen K. A., Chem. Soc. Rev., 2009, 38, 2178–2189; [DOI] [PubMed] [Google Scholar]; c) MacMillan D. W. C., Nature, 2008, 455, 304–308. [DOI] [PubMed] [Google Scholar]
- 23. Retamosa M. d. G., de Cozar A., Sanchez M., Miranda J. I., Sansano J. M., Castello L. M., Najera C., Jimenez A. I., Sayago F. J., Cativiela C. and Cossio F. P., Eur. J. Org. Chem., 2015, 2503–2516. [Google Scholar]
- 24. de Gracia Retamosa M., Ruiz‐Olalla A., Bello T., de Cozar A. and Cossio F. P., Angew. Chem. Int. Ed., 2018, 57, 668–672; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130 [Google Scholar]
- 25.a) Overman L. E. and Shim J., J. Org. Chem., 1993, 58, 4662–4672; [Google Scholar]; b) Chen X.‐Y. and Enders D., Chem, 2018, 4, 21–23. [Google Scholar]
- 26. Ruiz‐Olalla A., Retamosa M. d. G. and Cossio F. P., J. Org. Chem., 2015, 80, 5588–5599. [DOI] [PubMed] [Google Scholar]
- 27. Sanchez‐Sanchez A., Rivilla I., Agirre M., Basterretxea A., Etxeberria A., Veloso A., Sardon H., Mecerreyes D. and Cossio F. P., J. Am. Chem. Soc., 2017, 139, 4805–4814. [DOI] [PubMed] [Google Scholar]
- 28. Sarotti A. M., Spanevello R. A., Suarez A. G., Echeverria G. A. and Piro O. E., Org. Lett., 2012, 14, 2556–2559. [DOI] [PubMed] [Google Scholar]
- 29. Gerosa G. G., Spanevello R. A., Suarez A. G. and Sarotti A. M., J. Org. Chem., 2015, 80, 7626–7634. [DOI] [PubMed] [Google Scholar]
- 30. Müller G. in Chemogenomics in Drug Discovery. (Eds.: Kubinyi H. and Müller G.), Wiley‐VCH, Weinheim, 2004, pp. 18–21. [Google Scholar]
- 31. Evans B. E., Rittle K. E., Bock M. G., DiPardo R. M., Freidinger R. M., Whitter W. L., Lundell G. F., Veber D. F., Anderson P. S. and et a., J. Med. Chem., 1988, 31, 2235–2246. [DOI] [PubMed] [Google Scholar]
- 32. Schneider P. and Schneider G., Angew. Chem. Int. Ed., 2017, 56, 7971–7974; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem., 2017, 129, 8079. [Google Scholar]
- 33. Vitaku E., Smith D. T. and Njardarson J. T., J. Med. Chem., 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- 34. Acharya K. R., Sturrock E. D., Rirodan J. F. and Ehlers M. R. W., Nat. Rev. Drug Discovery, 2003, 2, 891–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Thomas C., Stevenson M. and Riley T. V., J. Antimicrob. Chemother., 2003, 51, 1339–1350. [DOI] [PubMed] [Google Scholar]
- 36. Baumann M., Baxendale I. R., Ley S. V. and Nikbin N., Beilstein J. Org. Chem., 2011, 7, 442–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Oh L. M., Tetrahedron Lett., 2006, 47, 7943–7946. [Google Scholar]
- 38. Roth B. D., Trans‐6‐[2‐(3‐ or 4‐ carboxamido‐substituted pyrrol‐1‐yl)alkyl]‐4‐hydroxypyran‐2‐one Inhibitors of Cholesterol Synthesis US Patent 4, 681, 893 July 21 1987.
- 39. Zubia A., Ropero S., Otaegui D., Ballestar E., Fraga M. F., Boix‐Chornet M., Berdasco M., Martinez A., Coll‐Mulet L., Gil J., Cossio F. P. and Esteller M., Oncogene, 2009, 28, 1477–1484. [DOI] [PubMed] [Google Scholar]
- 40.a) Cossío F. P., Zubia A., Vara Y. I., San Sebastián E., Otaegui D., Masdeu M. d. C., Aldaba E. New Histone Deacetylase Inhibitors Based Simultaneously on Trisubstituted 1H‐Pyrroles and Aromatic and Heteroaromatic Spacers. PCT/EP2010/064653 7 April 2011;; b) Perez‐Salvia M., Aldaba E., Vara Y., Fabre M., Ferrer C., Masdeu C., Zubia A., San Sebastian E., Otaegui D., Llinàs‐Arias P., Rosselló‐Tortella A., Berdasco M., Moutinho C., Setien F., Villanueva A., González‐Barca E., Muncunill J., Navarro J.‐T., Piris M. A., Cossío F. P. and Esteller M., Haematologica 2018, ( 10.3324/haematol.2018.189241) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hussein E. M. and Abdel‐Monem M. I., ARKIVOC, 2011, 85–98. [Google Scholar]
- 42. Kaur A., Singh B., Vyas B. and Silakari O., Eur. J. Med. Chem., 2014, 79, 282–289. [DOI] [PubMed] [Google Scholar]
- 43. Najera C. and Sansano J. M., Actual. Chim., 2013, 370, 28–30. [Google Scholar]
- 44. Najera C., Retamosa M. d. G., Martin‐Rodriguez M., Sansano J. M., de Cozar A. and Cossio F. P., Eur. J. Org. Chem., 2009, 5622–5634. [Google Scholar]
- 45. Martin‐Rodriguez M., Najera C., Sansano J. M., de Cozar A. and Cossio F. P., Beilstein J. Org. Chem., 2011, 7, 988–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Takayama H., Jia Z.‐J., Kremer L., Bauer J. O., Strohmann C., Ziegler S., Antonchick A. P. and Waldmann H., Angew. Chem. Int. Ed., 2013, 52, 12404–12408; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2013, 125, 12630. [Google Scholar]
- 47. Narayan R., Bauer J. O., Strohmann C., Antonchick A. P. and Waldmann H., Angew. Chem. Int. Ed., 2013, 52, 12892–12896; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2013, 125, 13130. [Google Scholar]
- 48. Esslinger C. S., Titus J., Koch H. P., Bridges R. J. and Chamberlin A. R., Bioorg. Med. Chem., 2002, 10, 3509–3515. [DOI] [PubMed] [Google Scholar]
- 49. Najera C., Retamosa M. d. G., Sansano J. M., de Cozar A. and Cossio F. P., Eur. J. Org. Chem., 2007, 5038–5049. [Google Scholar]
- 50. Gu Y. G., Zhang X., Clark R. F., Djuric S. W. and Ma Z., Tetrahedron Lett., 2004, 45, 3051–3053. [Google Scholar]
- 51. Zubia A., Mendoza L., Vivanco S., Aldaba E., Carrascal T., Lecea B., Arrieta A., Zimmerman T., Vidal‐Vana‐clocha F. and Cossio F. P., Angew. Chem. Int. Ed., 2005, 44, 2903–2907; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2005, 117, 2963. [Google Scholar]
- 52. San Sebastian E., Zimmerman T., Zubia A., Vara Y., Martin E., Sirockin F., Dejaegere A., Stote R. H., Lopez X., Pantoja‐Uceda D., Valcarcel M., Mendoza L., Vidal‐Vanaclocha F., Cossio F. P. and Blanco F. J., J. Med. Chem., 2013, 56, 735–747. [DOI] [PubMed] [Google Scholar]