

Abstract
Parasite recognition by Toll-like receptors (TLRs) contributes to macrophage activation and subsequent control of Leishmania infection through the coordinated production of pro-inflammatory and microbicidal effector molecules. The modulation of microRNA (miRNA) expression by Leishmania infection potentially mediates the post-transcriptional regulation of the expression of genes involved in leishmanicidal activity. Here, the contribution of TLR signaling to the miRNA profile and gene expression was evaluated in Leishmania amazonensis-infected murine macrophages. The infectivity of L. amazonensis was higher in murine bone marrow-derived macrophages from mice knockout for myeloid differentiation factor 88 (MyD88−/−), TLR2 (TLR2−/−), or TLR4 (TLR4−/−) than wild type C57BL/6 (WT). L. amazonensis infection of WT macrophages modulated the expression of 32% of the miRNAs analyzed, while 50% were upregulated. The absence of MyD88, TLR2, and TLR4 altered the percentage of miRNAs modulated during L. amazonensis infection, including the downregulation of let-7e expression. Moreover, the absence of signals mediated by MyD88, TLR2, or TLR4 reduced nitric oxide synthase 2 (Nos2) mRNA expression during infection. Indeed, the inhibition of let-7e increased levels of the Nos2 mRNA and NOS2 (or iNOS) protein and nitric oxide (NO) production in L. amazonensis-infected macrophages (4–24 h). The absence of TLR2 and inhibition of let-7e increased the expression of the arginase 1 (Arg1) mRNA but did not alter the protein level during infection. However, higher levels of the L-arginine transporters Cat2B and Cat1 were detected in the absence of Myd88 signaling during infection but were not altered following let-7e inhibition. The inhibition of let-7e impacted the global expression of genes in the TLR pathway by upregulating the expression of recognition and adaptors molecules, such as Tlr6, Tlr9, Ly96, Tirap, Traf 6, Ticam1, Tollip, Casp8, Map3k1, Mapk8, Nfkbib, Nfkbil1, Ppara, Mapk8ip3, Hspd1, and Ube2n, as well as immunomodulators, such as Ptgs2/Cox2, Csf 2, Csf 3, Ifnb1, Il6ra, and Ilr1, impacting NOS2 expression, NO production and parasite infectiveness. In conclusion, L. amazonensis infection alters the TLR signaling pathways by modulating the expression of miRNAs in macrophages to subvert the host immune responses.
Keywords: microRNA, MyD88, NOS2, NO, bone marrow-derived macrophages, post-transcriptional regulation
Introduction
Protozoan parasites of the genus Leishmania are the causative agents of leishmaniases, diseases that affect more than 12 million people worldwide (1). Cutaneous leishmaniasis is widespread in Brazil, the country reporting the greatest number of cases in the Americas, with an estimated incidence of 100,000 cases per year (1). L. amazonensis induces cutaneous and/or diffuse cutaneous manifestations (2, 3).
Host-parasite interactions during the innate immune response to Leishmania are mediated by the recognition of pathogen-associated molecules (PAMPs) by pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs) that are mainly expressed in phagocytes and antigen-presenting cells, as neutrophils, macrophages and dendritic cells (4, 5). The surface of Leishmania spp is covered with molecules that are recognized by TLRs (6), which play central roles in macrophage activation, signaling to induce phagocytosis, parasite healing or persistence and in the control of infections by innate and adaptive immunity (7–11).
The interaction of TLR2 or TLR4 starts the signal transduction cascade through the activation of adaptor molecules, such as MyD88 and Toll-like receptor adapter-inducing interferon β (TRIF/TICAM1) (12), mediating Leishmania recognition and modulation of infectivity in macrophages (13–15). The activation of the TLR2 or TLR4 signaling cascade by LPG stimulation and L. donovani, L. major or L. amazonensis infections modulates the expression of transcription factors, such as nuclear factor kappa B (NF-κB), interferon regulatory factors (IRFs) and mitogen-activated protein kinase (MAPK) that induce the transcription of pro-inflammatory cytokines, such as tumor necrosis factor (TNF), and NO and superoxide production (14, 16–21). However, the mechanism regulating the signal required for the activation of TLR signaling is not completely understood.
The NO produced during the initial steps of the inflammatory response may exhibit leishmanicidal activity (14, 22–24). The outcome of Leishmania infection depends on the balance between the induction of NOS2 expression to produce NO and subsequently kill the parasite (25–31) and the induction of ARG1 expression to produce polyamines that promote Leishmania survival (28, 32–35). Interestingly, L-arginine is a substrate for both NOS2 and ARG1. Depending on the environmental conditions and immune response, the prevalence of one activity over the other results in the killing of the parasite or its survival. The expression of cationic amino acid transporters (CATs) and their roles in L-arginine uptake into macrophages determines the fate of the infectiveness (36–40).
The immune response is subverted by post-transcriptional regulation mediated by microRNAs (miRNAs), leading to the survival of Leishmania (31, 41, 42). The miRNAs are evolutionary conserved small (18–25 nucleotides) non-coding RNAs that fine-tune gene expression by complementary base pairing to the 3′ untranslated region (3′UTR) of a target mRNA, blocking target mRNA translation or inducing its degradation (43–48). The miRNAs are divided into several families, including the lethal-7 (let-7) family, which was first described in the nematode Caenorhabditis elegans as a group of miRNAs responsible for the regulating development in a time-dependent manner (49) and is conserved among various species, including Drosophila melanogaster (fruit fly), Mus musculus (mouse), and humans (Homo sapiens) (50, 51). The let-7e isoform comprises the let-7 family in mice (50–52) and regulates pro- and anti-inflammatory responses during infection or TLR/PAMP stimulation by inducing NF-κB activation and cytokine production (53–57).
In this study, we analyzed the activation of the TLR signaling cascade and the miRNA-mRNA interactions to understand the outcome of L. amazonensis infection in C57BL/6 macrophages. TLR2 and TLR4 mediated L. amazonensis recognition and infectivity resistance in macrophages. Other MyD88-dependent receptors may participate in macrophage activation in response to L. amazonensis. L. amazonensis infection altered the expression of genes involved in TLR signaling, transcription factors and pro-inflammatory molecules. MyD88, TLR2, and TLR4 regulated miRNA expression, such as let-7e, during the course of infection. In addition, the functional inhibition of let-7e altered the activation of the TLR pathway, Nos2/NOS2 expression and NO production, impacting the infectiveness of the parasite.
Materials and methods
Ethics statement
The protocol used for the animal experiments was approved by the Comissão de Ética no Uso de Animais (CEUA) from Instituto de Biociências of the Universidade de São Paulo (the approval number CEUA-IB: 233/2014). This study was performed in strict accordance with the recommendations in the Guide and Policies for the Care and Use of Laboratory Animals of the São Paulo State (Lei Estadual 11.977, de 25/08/2005) and Brazilian government (Lei Federal 11.794, de 08/10/2008).
Parasite culture
L. amazonensis (MHOM/BR/1973/M2269) promastigotes were maintained in culture in M199 medium (Gibco™, Grand Island, NY, USA) supplemented with 10% heat-inactivated fetal bovine serum (Gibco™), 5 ppm hemin, 100 μM adenine (Gibco™), 100 U penicillin, 100 μg/mL streptomycin (Gibco™), 40 mM HEPES-NaOH (Promega, Madison, WI, USA) and 12 mM NaHCO3 (Sigma-Aldrich, St Louis, MO, USA), pH 7.0, for 1 week at 25°C at a low passage (P1 to P5).
in vitro macrophage infections
All experiments were performed using female (6–8 weeks old) C57BL/6 wild type (WT), MyD88−/−, TLR2−/−, or TLR4−/− mice from Biotério do Departamento de Imunologia, Instituto de Ciências Biomédicas—Universidade de São Paulo and maintained in the Biotério do Departamento de Fisiologia, Instituto de Biociências—Universidade de São Paulo. The animals were sacrificed in a CO2 chamber and femurs were collected to obtain bone marrow-derived macrophages (BMDMs). The femurs were washed with 2 mL of cold PBS, and the cells were collected by centrifugation at 1,500 × g for 10 min at 4°C and resuspended in RPMI 1640 medium (LGC Biotecnologia, São Paulo, SP, Brazil), supplemented with penicillin (100 U/mL (Gibco™), streptomycin (100 μg/mL) (Gibco™), 5% heat-inactivated FBS (Gibco™) and 20% L9-29 supernatant. The cells were cultivated for 7–8 days at 34°C in 5% CO2 atmosphere. After differentiation, cellular viability was evaluated using flow cytometry (FlowSight® Amnis, MerckMillipore, Darmstadt, Germany), as determined by the presence of 95% F4/80- and CD11b-positive cells.
Approximately 2 × 105 BMDMs were plated into 8-well glass chamber slides (Lab-Teck Chamber Slide; Nunc, Naperville, IL, USA) and incubated at 34°C in 5% CO2 atmosphere for the infectivity analysis. Approximately 3 × 106 BMDMs were plated into 6-well plates (SPL, Lifescience, Pocheon, Korea) and incubated at 34°C in 5% CO2 atmosphere for analyses of miRNA and mRNA expression. After 18 h of incubation, non-adherent cells were removed by washes with PBS and the infection was performed with promastigotes in the stationary growth phase (MOI 5:1). After 4 h of incubation, non-phagocytosed promastigotes were removed by washes with fresh medium. Thereafter, the infection course was followed for 4, 24 and 48 h. Non-infected macrophages maintained in culture under the same conditions were used as controls. The infectivity was microscopically analyzed after cells were fixed with acetone/methanol (1:1, v:v, Merck, Darmstadt, Germany) for 20 min at −20°C, followed by washes with PBS and staining with DAPI (Invitrogen). The infectivity indexes (rate of infected macrophages multiplied by the average number of amastigotes per infected macrophage) were calculated by randomly counting at least 600 macrophages per slide. For the analysis of RNA expression, cells were washed with PBS, resuspended in QIAzol (Qiagen, Germantown, MD, USA) and stored at −20°C until use.
RNA extraction, reverse transcription, and miRNA PCR array
RNA was extracted using the miRneasy® Mini kit (Qiagen, Hilden, Germany), according to the manufacturer's instructions. The cDNA templates were produced from mature miRNAs using the miScript II RT kit (Qiagen, Hilden, Germany), according to the manufacturer's instructions. Briefly, 250 ng of total RNA were added to 2 μL of 5X miScript HiSpec Buffer, 1 μL of 10X Nucleic Acids Mix, 1 μL of miScript Reverse Transcriptase Mix and RNAse-free water to a final volume of 10 μL. The mixture was incubated at 37°C for 60 min to insert the poly-A+ sequence at the end of the miRNA sequence (downstream) and anneal a T-tail-tag for the elongation of the cDNA. The enzyme was inactivated at 95°C for 5 min. The reaction was performed in a Thermocycler Mastercycler gradient (Eppendorf, Hamburg, Germany). The cDNAs were stored at −20°C until use.
The miRNA PCR array was performed with 10-fold diluted cDNAs as the template using Mouse Inflammatory Response and Autoimmunity miRNA PCR Array: MIMM-105Z (Qiagen, Germantown, MD, USA) and the miScript SYBR PCR Kit (Qiagen, Hilden, Germany), as previously described (58). Briefly, the pooled mixture was prepared with 1X QuantiTect SYBR Green PCR Master Mix, 1X miScript Universal Primer, 105 μL of cDNA, and RNAse-free water to a final volume of 2,625 μL, and 25 μL was aliquotted into each well. The specific amplification of let-7e-5p by RT-qPCR was performed with 1X QuantiTect SYBR Green PCR Master Mix, 1X miScript Universal Primer, 1X Specific Primer, 5 μL of cDNAs, and RNAse-free water to a final volume of 25 μL. The reactions consisted of the activation of the HotStart Polymerase for 15 s at 95°C and 40 cycles of 15 s at 94°C, followed by 30 s at 55°C and 30 s at 70°C, and were performed in the Thermocycler ABI Prism 7300 instrument (Applied Biosystems, Carlsbad, CA, USA). The relative Ct value was analyzed using the miScript miRNA PCR Array Data Analysis software (www.qiagen.com). Triplicate samples were analyzed for each condition. The average Ct value was calculated to represent the gene expression variation with good accuracy. The geometric average Ct values for the miRNAs were normalized based on the average of SNORD95A. The controls for PCR and RT-qPCR were performed according to the manufacturer's instructions and determined based on efficiency reactions. A negative control containing all reaction components except the reverse transcriptase enzyme was included and subjected to RT-qPCR to confirm the absence of DNA contamination in RNA samples. The Fold Regulation (FR) value is defined as the negative inverse of the fold change [function = −1*(1/fold change value)]. FR levels ≥1.2 were considered to indicate upregulation, and levels ≤ 1.2 were considered to indicate downregulation. A Venn diagram was designed Venny 2.1 tool (http://bioinfogp.cnb.csic.es/tools/venny/index.html) (59) and modified to include miRNA or mRNA names inside the areas.
Reverse transcription and RT-qPCR for analysis of mRNA expression
The cDNA templates for the analysis of mRNA expression were synthesized using 2 μg of total RNA, 20 nmol of random primers (Applied Biosystems, Carlsbad, CA, USA) and water to a final volume of 28 μL. The RNA was incubated at 65°C for 5 min and then cooled to 10°C. Thereafter, mix 2 containing 8 μL of 5X buffer, 2 μL of 10 mM dNTPs and 2 μL of RevertAid™ Reverse Transcriptase (200 U/μL) (Fermentas Life Sciences, Burlington, Ontario, Canada) were added to the reaction, followed by an incubation at 25°C for 5 min and 42°C for 60 min. The enzyme was inactivated at 70°C for 10 min, and the reaction was stored at −20°C until further use. A negative control containing all reaction components except the reverse transcriptase enzyme was included and subjected to RT-qPCR to confirm the absence of DNA contamination in the RNA samples.
For mRNA quantification, we used 100-fold diluted cDNAs as templates. The reaction was performed with 1X SYBR Green PCR Master Mix (Applied Biosystems), 0.4 μM each corresponding primers pair, 5 μL of cDNAs, and RNAse-free water to a final volume of 25 μL. The PCR reaction consisted of 40 cycles of 30 s at 94°C followed by 30 s at 60°C and was performed in the Exicycler™ 96 Real-Time Quantitative Thermal Block (Bioneer, Daejeon, Korea). Triplicate samples were analyzed for each condition. Target gene expression was quantified based on a standard curve prepared from 10-fold serial dilutions of a quantified and linearized plasmid containing the target DNA. The following primers pairs were used for the analysis of mammalian mRNA expression: Nos2: 5′-agagccacagtcctctttgc-3′ and 5′-gctcctcttccaaggtgctt-3′; Arg1: 5-agcactgaggaaagctggtc-3′ and 5′-cagaccgtgggttcttcaca-3′; Cat-2b: 5′-tatgttgtctcggcaggctc-3′ and 5′-gaaaagcaacccatcctccg-3′; Cat1: 5′-cgtaatcgccactgtgacct-3′ and 5′-ggctggtaccgtaagaccaa-3′; and Gapdh: 5′-ggcaaattcaacggcacagt-3′ and 5′-ccttttggctccacccttca-3′.
Reverse transcription and TLR PCR array
The cDNA templates were produced from 1 μg of total RNA using RT2 First Strand Kit (Qiagen, Hilden, Germany), according to the manufacturer's instructions. The reactions were performed in a Thermocycler Mastercycler gradient instrument (Eppendorf, Hamburg, Germany) and stored at −20°C until use.
The TLR PCR array was performed with 10-fold diluted cDNAs as templates using RT2 Profiler™ PCR Array Mouse Toll-Like Receptor Signaling Pathway (PAMM-018Z) (Qiagen, Germantown, MD, USA) and RT2 SYBR Green qPCR Mastermix (Qiagen, Hilden, Germany). The reactions were performed with 1X RT2 SYBR Green Master Mix and 100 μL of cDNAs and RNAse-free water to a final volume of 2,700 μL (25 μL/well). The PCR consisted of the activation of the HotStart Polymerase for 10 s at 95°C and 40 cycles of 15 s at 95°C, followed by 1 min at 60°C, and was performed in Thermocycler ABI Prism 7300 (Applied Biosystems, Carlsbad, CA, USA). The relative Ct value was calculated using the RT PCR Array Data Analysis software (www.qiagen.com). Quadruplicate samples were analyzed for each condition. The average Ct value was calculated to represent the gene expression variation with good accuracy. The geometric average Ct values for the mRNAs were normalized based on the average value for the housekeeping gene B2 microglobulin. The PCR and RT-PCR controls were performed according to the manufacturer's instructions and determined using efficiency reactions. A negative control containing all reaction components except the reverse transcriptase enzyme was included and subjected to real-time PCR to confirm the absence of DNA contamination in the RNA samples. The Fold Regulation (FR) value is defined as the negative inverse of the fold change [function = −1*(1/fold change value)]. FR levels ≥1.2 were considered to indicate upregulation, and levels ≤ −1.2 were considered to indicate downregulation.
Transfection of miRNA inhibitors
BMDMs were collected, plated and incubated as described in a previously study (58). Thereafter, macrophages were incubated with 100 nM of let-7e-5p inhibitor or the negative control (Ambion, Carlsbad, CA, USA) after a previous incubation with 3 μL of the FugeneHD transfection reagent (Roche, Madison, WI, USA) in 500 μL of RPMI 1640 medium (LGC Biotecnologia, São Paulo, SP, Brazil), supplemented as described above, for 20 min at room temperature. After 24 h of transfection, the cells were infected with L. amazonensis promastigotes, as described above.
Flow cytometry to detect NOS2 and ARG1 expression
Infected BMDMs were fixed with 1% paraformaldehyde (1 h at 4°C) and permeabilized with 0.05% Tween20 for 30 min at 4°C, followed by blocking with Odyssey blocking buffer (LI-COR, Bad Homburg, Germany) for 1 h at room temperature. Then, samples were incubated with 1:200 dilutions of rabbit anti-NOS2 (sc651) or anti-ARG1 (sc20150) antibodies (Santa Cruz, CA, USA) for 16 h at 4°C. Samples were then incubated with 1:300 dilution of FITC-conjugated goat anti-rabbit IgG (F7512, Sigma-Aldrich) antibody for 1 h at room temperature. The fluorescence signal from single cells was measured in channel 2 using FlowSight® Amnis (MerckMillipore, Darmstadt, Germany) and analyzed using the Ideas® Software (Amnis Corporation, Seattle, WA, USA).
Quantification of NO production
BMDMs were seeded in 24-well plates (SPL) (1 × 106 cells/well) and incubated for 18 h at 34°C in 5% CO2 atmosphere. Then, the macrophages were infected for 4 and 24 h, as described above. Cells were detached with 0.5 M EDTA in PBS for 10 min at 37°C, scraped and washed with PBS. Cells were incubated with 5 μM DAF-FM (for NO quantification) (Molecular Probes, Life Technologies, Darmstadt, Germany) in PBS for 30 min at 34°C in 5% CO2 atmosphere, as described previously (31, 58). Fluorescence signals from single cells were acquired in channel 2 using FlowSight® Amnis (MerckMillipore, Darmstadt, Germany) and analyzed using the Ideas® Software (Amnis Corporation, Seattle, WA, USA).
in silico analysis
We used the databases miRecords and Dianna tools to determine the interactions between miRNAs and target mRNAs. The miRecords platform (http://c1.accurascience.com/miRecords/) is based on predicted mRNA targets and integrates the predicted targets from various prediction tools: DIANA-microT, MicroInspector, miRanda, MirTarget2, miTarget, NBmiRTar, PicTar, PITA, RNA22, RNAhybrid, and TargetScan/TargertScanS. The Dianna platform (http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=site/index) is based on validated mRNA targets.
Statistical analysis
Statistical analyses were performed with Student's t or ANOVA tests using the GraphPad Prism Software 7 (GraphPad Software, Inc., La Jolla, CA, USA). The obtained p-values are indicated throughout the Results section.
Results
Absence of the MyD88, TLR2, or TL4 gene increased L. amazonensis infectivity
The roles of MyD88, TLR2 and TLR4 in the recognition of the parasite and defining the fate of L. amazonensis infection were evaluated by calculating the percentage of infected macrophages, number of amastigotes per infected macrophage and infectivity index in BMDMs from WT, MyD88−/−, TLR2−/−, or TLR4−/− mice, after 4, 24, and 48 h of infection (Figure 1). We observed increased numbers of infected macrophages during the course of infection in TLR4−/− macrophages, and an increased rate only after 48 h of infection of TLR2−/− macrophages (Figure 1A). The number of amastigotes per infected macrophage appeared to increase in all knockout mouse strains compared to WT macrophages during the course of infection (Figure 1B). Interestingly, the infectivity index appeared to increase in the MyD88−/−, TLR2−/−, and TLR4−/− mice during the course of infection compared to infected WT macrophages (Figure 1C), confirming the involvement of TLR2, TLR4, and MyD88 in the recognition of the parasite and defining the fate of L. amazonensis infection.
Figure 1.
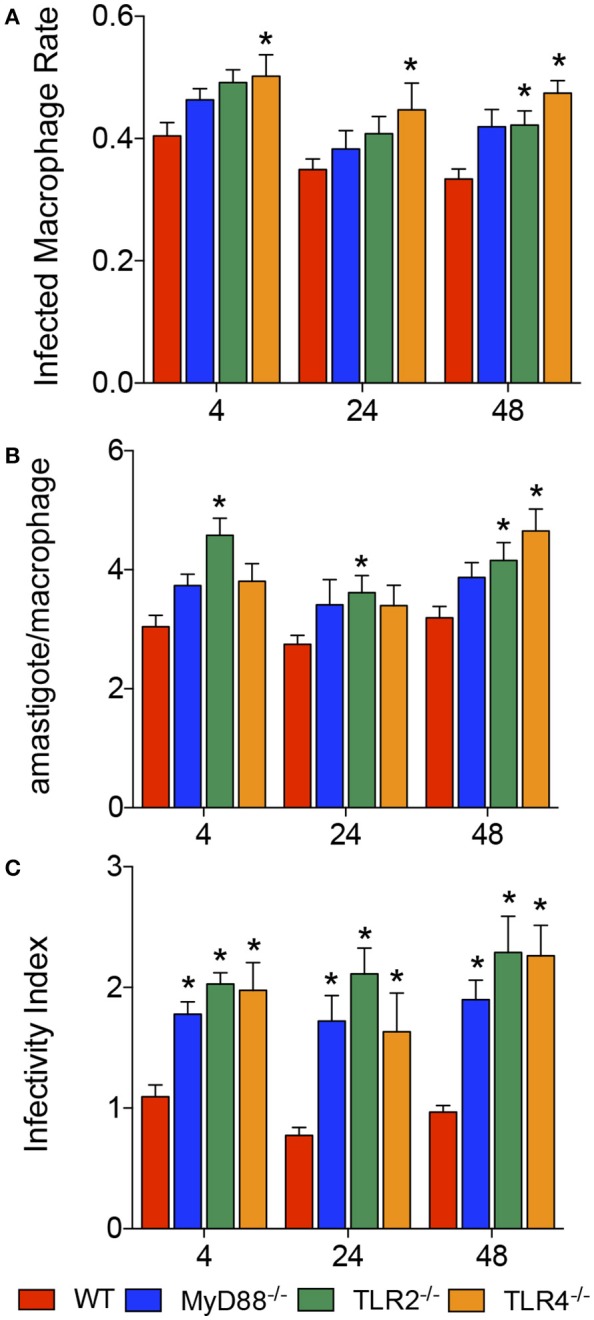
Infectivity of L. amazonensis in macrophages from WT, MyD88−/−, TLR2−/−, and TLR4−/− mice. Results from evaluations of the percentages of infected macrophages (A), numbers of amastigotes per infected macrophage (B), and infectivity index (C) in BMDMs from WT (red), MyD88−/− (blue), TLR2−/− (green), and TLR4−/− (orange) mice. Each bar represents the mean ± SEM of the values obtained from three independent experiments (n = 6–9). Statistical significance was determined using non-parametric two-way ANOVA. *p < 0.05 compared to WT macrophages.
Modulation of the expression of genes involved in the NO/polyamine pathway depends on TLR signaling
We revealed the roles of MyD88, TLR2, and TLR4 in the L. amazonensis infection in vitro. We next analyzed how TLR signaling interferes with L-arginine metabolism to produce polyamines for parasite replication or to produce NO for parasite killing. First, we analyzed the basal levels of the Cat1, Cat2B, Arg1, and Nos2 mRNAs in each mouse strain and compared the values to the WT strain. TLR4−/− macrophages expressed lower basal levels of the Cat1, Cat2B, Arg1, and Nos2 transcripts (Figures 2A–D). MyD88−/− macrophages showed lower basal levels of the Arg1 and Nos2 transcripts (Figures 2C,D). TLR2−/− macrophages did not show differences in the basal levels of any transcript (Figures 2A–D).
Figure 2.

Lack of TLR signaling leads to the differential expression of genes involved in polyamine and NO production. The BMDMs (5 × 106) from WT (red), MyD88−/− (blue), TLR2−/− (green), and TLR4−/− (orange) mice were infected with L. amazonensis (MOI 5:1). Four, 24 and 48 h after infection, the copy numbers of the Cat2B (A), Cat1 (B), Nos2 (C), and Arg1 (D) mRNAs in these macrophages were quantified by RT-qPCR. Uninfected macrophages (0 h) served as the control. Each bar represents the average ± SEM of the values obtained from three independent experiments (n = 6–8). Statistical significance was determined using two-tailed Student's t-tests. *p < 0.05 all knockout macrophages compared to WT macrophages, **p < 0.05 compared to uninfected macrophages.
We analyzed the levels of those transcripts 4, 24, and 48 h after the L. amazonensis infection. The only transcript whose expression was modulated during the course of infection was Nos2, which was increased at 24 h and decreased at 48 h after infection (Figure 2D). Compared to WT macrophages, the level of the Cat2B transcript was increased in MyD88−/− macrophages 48 h after infection and increased levels of the Cat1 transcript were observed in MyD88−/− macrophages 4, 24, and 48 h after infection (Figures 2A,B, respectively). In contrast, MyD88−/− macrophages showed decreased levels of Arg1 and Nos2 transcripts in all infection periods analyzed (Figures 2C,D, respectively). The comparison between WT and TLR2−/− macrophages showed increased levels of the Arg1 transcript 4 h after infection and decreased levels of Nos2 transcript in all infection periods analyzed (Figures 2C,D, respectively). The comparison between WT and TLR4−/− macrophages revealed decreased levels of the Arg1 and Nos2 transcripts throughout the course of infection (Figures 2C,D, respectively). Based on these data, MyD88, TLR2, and TLR4 signaling altered the expression of genes involved in polyamine/NO production in L. amazonensis-infected macrophages.
TLR modulates the miRNA profile in L. amazonensis-infected macrophages
We evaluated the miRNA profile of infected macrophages and the impact of the absence of TLR signaling on miRNA expression by analyzing the miRNA profiles of macrophages from WT, MyD88−/−, TLR2−/−, or TLR4−/− mice infected with L. amazonensis for 4, 24, and 48 h and comparing them to uninfected macrophages (Figures 3, 4, S1; Table S1) to investigate the importance of miRNA-mediated post-transcriptional regulation of gene expression.
Figure 3.

Volcano plot of the miRNA profiles of macrophages from WT, MyD88, TLR2 and TLR4 null mice infected with L. amazonensis. Each dot represents the level of one miRNA in BMDMs from WT, MyD88−/−, TLR2−/−, or TLR4−/− mice infected with L. amazonensis for 4, 24, and 48 h. The red dots indicate upregulated miRNAs and green dots indicate downregulated miRNAs. The blue dotted line corresponds to p = 0.05, log 10. The relative upregulation and downregulation of miRNAs are presented as boundaries of 1.2 or −1.2 of Fold Regulation, respectively. P-values were determined using two-tailed Student's t-tests. Representative data from three independent experiments are presented.
Figure 4.

Venn diagram depicting unique and common miRNAs that were significantly differentially expressed in macrophages from WT or MyD88, TLR2 and TLR4 null mice infected with L. amazonensis. The miRNA expression levels in infected WT (purple), MyD88−/− (yellow), TLR2−/− (green), or TLR4−/− (red) macrophages were examined. The upregulated (A) and downregulated (B) miRNAs in each group were compared to uninfected macrophages. The relative up- and downregulation of miRNAs are presented as boundaries of 1.2 or −1.2 of Fold Regulation, respectively. P-value < 0.05.
The miRNA profile of infected WT macrophages revealed the differential regulation of the expression of 32% (27/84) of the analyzed miRNAs during the course of infection compared to uninfected WT macrophages. Fourteen of the 27 miRNAs were upregulated. Notably, let-7e, miR-182, miR-294, miR-30b, miR-369, miR-429, miR-466d, miR-495, miR-497a, miR-699h, and miR-721 were significantly upregulated 4 h after infection. The upregulation of let-7e, mir-294, and miR-721 was maintained for 24–48 h after infection. The miRNAs let-7f and let-7g appeared to be upregulated 24 and 48 h after infection. Moreover, miR-369 only appeared to be upregulated 24 h after infection, while miR-712 only appeared to be upregulated 48 h after infection (Figure 3; Table S1). Interestingly, we did not detect downregulation of miRNA expression 4 h after infection. On the other hand, 24 and 48 h after infection, we observed downregulation of miR-130a, miR-15a, miR-221, miR-27b, miR-295, miR-30a, miR-340, miR-350 and miR-497a; and miR-17, miR-19a, miR-221, miR-27b, miR-29a, miR-30a, miR-340, miR-350, and miR-98, respectively (Figure 3).
The miRNA profile of infected MyD88−/− macrophages showed the differential regulation of the expression of 35% (30/84) of analyzed miRNAs: 50% of were upregulated and 50% were downregulated (Figure 3). Unexpectedly, the miRNA profile of infected TLR2−/− macrophages revealed low levels of miRNA modulation during the course of infection. Only 12% (10/84) of miRNAs were differentially regulated, and 80% were downregulated. No miRNA modulation was observed 48 h after infection. The miRNA profile of infected TLR4−/− macrophages showed the differential regulation of the expression of 28% (24/84) of the analyzed miRNAs, 92% of these were downregulated (Figure 3).
We constructed a Venn diagram showing exclusively and commonly expressed miRNAs to elucidate the relationships between miRNAs expression among the groups (Figure 4). Notably, miR-294 and miR-721 were both upregulated in all four groups (Figure 4A). In addition, the miRNAs let-7e, let-7f and let-7g were exclusively upregulated in WT cells compared to MyD88−/−, TLR2−/−, and TLR4−/− cells (Figure 4A). Additionally, let-7e was commonly downregulated miRNA in infected MyD88−/− and TLR2−/− macrophages, and its expression was reduced in TLR4−/− macrophages, but the difference was not significant (p = 0.055, Table S1).
The expression of miR-30b, miR-369, miR-429, miR-466d, miR-495, miR-497a, miR-669h, and miR-712 was commonly upregulated in infected WT and MyD88−/− macrophages. Additionally, miR-130a, miR-16, miR-338, and miR-9 were uniquely expressed in infected MyD88−/− macrophages.
Moreover, the Venn diagram showed the following exclusively downregulated miRNAs: miR-182, miR-186 and miR-497a in infected WT macrophages; miR-155, miR-23b, miR-26b, and miR-322 in infected MyD88−/− macrophages; let-7a, miR-135a, miR-466d, miR-495 and miR-9-5p and miR-126a, miR-128, miR-130b, miR-181b, miR-181c, miR-181d, miR-195a, miR-19b, miR-20a, miR-27a, miR-29b, miR-29c, and miR-93-5p in infected TLR4−/− macrophages (Figure 4B). The following miRNAs were commonly downregulated: in WT and MyD88−/− macrophages: miR-221, miR-295, miR-29a, miR-30a, miR-350, and miR-98-5p; in WT and TLR2−/− macrophages: miR-130a; in WT and TLR4−/− macrophages: miR-15a, miR-17, and miR-27b; and in WT, MyD88−/−, and TLR4−/− macrophages: miR-144-3p, miR-19a, and miR-340-5p. Notably, miR-301a and miR-301b-3p were commonly downregulated in infected MyD88−/− and TLR4−/− macrophages. Additionally, miR-712 was commonly downregulated in infected TLR2−/− and TLR4−/− macrophages (Figure 4B).
Altogether, our data revealed the differential regulation of the miRNA profiles in L. amazonensis-infected macrophages via MyD88, TLR2, and TLR4 signaling.
in silico identification of let-7e target mRNAs
Since we observed the modulation of let-7e expression in the MyD88- and TLR2-deficient mice infected with L. amazonensis, we decided to assess the effects of this miRNA on modulating the expression of target mRNAs in the TLR pathway. Based on the results from an in silico analysis using miRecords tool, we identified 3,357 predicted interactions for let-7e (Table S2). On the other hand, based on a search using the Dianna tool for experimentally validated targets, let-7e targeted Tlr4, Tbk1, Map2k4, Map3k1, Chuk, Tnf , Tnfpaip3, Tnfrsf 1a, and Il10 (60).
Let-7e affects L. amazonensis infectivity by regulating L-arginine metabolism
Next, we analyzed the impact of let-7e expression on L. amazonensis-infected macrophages. First, macrophages were transfected with 100 nM let-7e inhibitor or negative-control (NC). Then, macrophages were infected with L. amazonensis for 4 and 24 h. Expression assays were used to validate let-7e inhibition, and the inhibition of this miRNA decreased the expression of the miRNA let-7e 4 and 24 h after infection compared to negative control-infected macrophages (Figure S2). The impact of let-7e inhibition on L. amazonensis infectivity was also evaluated. As shown in Figures 5A–C, the percentage of infected macrophages, number of amastigotes per infected macrophage and infectivity index at 4 h of infection were reduced by ~10, 30, and 35%, respectively, compared to NC (Figures 5A–C).
Figure 5.

Inhibition of let-7 increases differential gene expression and reduces L. amazonensis infectivity. BMDMs were transiently transfected with 100 nM negative control (NC—filed patterned bar) or let-7e-5p inhibitor (let-7e—blue bar). Twenty-four hours after transfection, the cells were co-cultivated with L. amazonensis (MOI 5:1) for 4 h, and the cultures were then washed. Four and 24 h after infection, the samples were analyzed for infectivity under the microscope by counting the numbers of infected macrophages (A), amastigotes per infected macrophage (B), and infectivity index (C) (n = 1,000 macrophages/treatment). RT-qPCR was used to determine the total levels of the Arg1 (D) and Nos2 (G) mRNAs. The percentage of cells expressing ARG1 (E) or NOS2 (H) and fluorescence intensity of ARG1 (F) or NOS2 (I) staining were determined using flow cytometry. The percentage of NO-producing cells (DAF-FM+ cells—K) and fluorescence intensity of NO production per cell (DAF-FM mean—J) were also determined using flow cytometry. The values were normalized to the average values of NC-transfected and infected macrophages (100%). Each bar represents the average ± SEM of the values obtained from three independent experiments (n = 4–8). Statistical significance was determined using two-tailed Student's t-test. *p < 0.05 compared to NC-transfected, infected macrophages.
We measured the levels of the Cat2B, Cat1, Arg1, and Nos2 transcripts to determine whether let-7e and the subsequent decrease in infectivity required L-arginine metabolism (Figures 5, S2). We did not observe modulation of the levels of the Cat2 and Cat1 transcripts among the samples (Figure S2). The level of the Arg1 transcript was increased in cells transfected with the let-7e inhibitor 24 h after infection (35%, Figure 5D), but the percentage of cells expressing ARG1 (Figure 5E) and mean intensity of ARG1 (Figure 5F) were not noticeably modified. The level of the Nos2 transcript was reduced following let-7e inhibition 4 h after infection, but Nos2 levels were increased 24 h after infection (Figure 5G). Moreover, the percentage of cells expressing NOS2 (Figure 5H), mean intensity of NOS2 (Figure 5I) and NO production were increased in let-7e inhibitor-transfected infected macrophages compared to NC-transfected macrophages, as observed by the percentage of DAF-FM+ cells (Figure 5J) and mean intensity of the DAF-FM signal (Figure 5K). Thus, let-7e inhibition indirectly impacted the expression of genes involved in L-arginine metabolism, promoting NO production and subsequent infectiveness.
Let-7e impacts global variations in the expression of TLR pathway genes in L. amazonensis-infected macrophages
We evaluated and compared the transcript levels of molecules involved in recognition/binding and downstream signaling, such as adapters, transcription factors, cytokines/chemokines and cytokine receptors, in infected WT macrophages or negative control (NC)- and let-7e-inhibited macrophages 4 and 24 h after L. amazonensis infection and compared the results to uninfected macrophages to determine the role of let-7e in regulating the expression of molecules in the TLR pathway involved in pathogen recognition and the establishment of an infection (Figures 6, S3; Table S3).
Figure 6.

Venn diagram depicting unique and common mRNAs in the TLR pathway and cytokines that were significantly differentially expressed in response to let-7e inhibition in L. amazonensis-infected macrophages. BMDMs were transiently transfected with 100 nM negative control (NC) or let-7e-5p inhibitor (let-7e). Twenty-four hours after transfection, cells were co-cultivated with L. amazonensis (MOI 5:1) for 4 h, and the cultures were then washed. After 4 and 24 h of infection, cells were analyzed for the expression of mRNAs in the TLR signaling cascade, using RT-qPCR. The upregulated (A) and downregulated (B) mRNAs were compared at 4 and 24 h after infection. Comparison of mRNA expression in NC- and let-7e inhibitor-transfected cells: upregulated at 4 h (C) or 24 h (D) or downregulated at 4 h (E) or 24 h (F). The relative up- and downregulation of miRNAs are presented as boundaries of 1.2 or −1.2 of Fold Regulation, respectively. P-value < 0.05.
First, we compared the expression of genes in the TLR signaling cascade in infected macrophages 4 and 24 h after infection (Figures 6A,B, S3). A substantial number of genes were upregulated 4 h after infection: Tlr1, Tlr2, C-type lectin domain family 4-member e (Clec4e/Mincle), heat shock protein 1A (Hspa1a/HSP70), interferon regulatory factor 1 (Irf 1), receptor-interacting protein kinase 2 (Ripk2), nuclear factor kappa B subunit 1 (Nfkb1), NF-kB inhibitor alpha (Nfkbia/Ikbalpha), CD86 antigen (Cd86), interleukin 1 alpha (Il1a), prostaglandin-endoperoxide synthase 2 (Ptgs2/Cox-2), tumor necrosis factor (Tnf ), and TNF alpha-induced protein 3 (Tnfaip3). Of these genes, only the upregulation of Hspa1a and Il1a was maintained 24 h after infection, and the expression of the heat shock protein 1 (Hspd1) gene was upregulated (Figure 6A).
A few genes were downregulated 4 h after infection: caspase 8 (Casp8), nuclear receptor subfamily 2, group C, member 2 (Nr2c2/TAK1), mitogen-activated protein kinase 9 (Mapk9/JNK2) and interleukin 6 receptor-alpha (Il6ra). The following genes were downregulated after 24 h: Tlr2, Tlr5, Nfkb1 (p105/p50), Nfkb2 (p100/p52), Nfkbia/Ikbalpha, chemokine CXC-motif ligand 10 (Cxcl10), interleukin 1 beta (Il1b), and interleukin 10 (Il10) (Figures 6B, S3; Table S3).
In the infected macrophages transfected with the let-7e inhibitor we detected the following upregulated genes 4 h after infection compared to infected NC-transfected macrophages: Tlr6, Tlr9, Toll-interleukin 1 receptor (TIR) domain-containing adaptor protein (Tirap), Tnf receptor-associated factor 6 (Traf 6), Toll-like receptor adaptor molecule 1 (Ticam1), Tollip, Nfkbib/TRIP9, Nfkbil1, peroxisome proliferator activated receptor alpha (Ppara), Mapk8ip3/Jip3, Ptgs2/Cox2, colony-stimulating factor 2 (Csf 2/GMCSF), Hspd1, Csf3, interferon beta 1 (Infb1), Il6ra and Ilr1 (Figure 6C). A few genes were downregulated: Tlr1, CD14 antigen (Cd14), lymphocyte antigen 86 (Ly86/MD-1), conserved helix-loop-helix ubiquitous kinase (Chuk/IKBKA), and Il10 (Figure 6D).
At 24 h after infection, the following genes were upregulated in infected macrophages transfected with the let-7e inhibitor compared to macrophages transfected with NC: Tlr7, Ly96/MD-2, Casp8, Map3k1, Mapk8, Ptgs2/Cox2, Csf 3/GCSF, and the ubiquitin-conjugating enzyme E2N (Ube2n). Indeed, let-7e inhibition did not alter the downregulated genes compared to NC-transfected infected macrophages.
Additionally, let-7e inhibition increased the levels of predicted target mRNAs, including Traf 6, Ppara, Mapk8ip3/Jip3, Map3k1, and Ube2n, in infected macrophages (Tables S2, S3). Based on these data, the expression of genes involved in TLR pathways is altered during L. amazonensis infection. Indeed, let-7e inhibition during L. amazonensis infection impacts global variations in the expression of genes TLR pathways (Figure 6) and infectiveness (Figures 5A–C).
Discussion
The importance of Leishmania-TLR interactions in enabling the activation of macrophage during healing and protection against leishmaniases has been extensively studied; however, the findings are very controversial. Macrophages have been reported to recognize the parasite, inducing a pro-inflammatory response and microbicidal mechanisms. TLR4 induces TIRAP-MyD88 signaling and then endocytosis, whereas TRIF-TRAM is activated in early endosomes (12). The trafficking of TLRs 3, 7, 8, and 9 from the endoplasmic reticulum to the endolysosomal compartment collaborates in the recognition of PAMPS and DAMPs (12). Here, we reported the roles of TLR2, TLR4, and the adaptor molecule MyD88 in mediating the phagocytosis of L. amazonensis and conferring resistance to infection, in contrast to the higher infectivity index observed in the absence of those molecules.
The recognition of virulence factors, such as LPG and αGalβ1, 4Manα-PO(4)-containing phosphoglycans from L. major, by TLR2 requires the adaptor MyD88 (14, 61). Stimulation of TLR2 induces the production of pro-inflammatory cytokines, such as TNF and IL-6, reactive oxygen species (ROS) (61, 62), and suppressors of the cytokine signaling family members SOCS-1 and SOCS-3 (15, 61, 62). P8 proteoglycolipid complex (P8 PGLC) from L. pifanoi induces the production of TNF and IL-12 in macrophages via TLR4/MD2-CD14 signaling. Additionally, TNF production and the parasite burden depend on TLR2 and MyD88 (20). Furthermore, MyD88 assists in the recognition of L. donovani and maturation of dendritic cells during infection (63). Indeed, TLR2, TLR3, MyD88, and IRAK-1 are involved in the phagocytosis of L. donovani and production of TNF-α and NO, although only IFN-γ-primed macrophages present leishmanicidal activity (14).
Several studies have described the participation of transcription factors and miRNAs in the transcriptional and post-transcriptional regulation of gene expression, respectively (64). L. major (65), and L. donovani (41, 42) have been reported to modulate miRNA expression in infected human macrophages. In BALB/c macrophages infected with L. amazonensis, the miRNA profile was altered and the involvement of miR-294, miR-497a and miR-721 in infection was confirmed (31). The results presented in this study corroborate these data, showing that L. amazonensis infection regulated the miRNA profiles and let-7e expression in infected C57BL/6 WT, MyD88−/−, TLR2−/−, and TLR4−/− macrophages. The miRNAs let-7e, let-7f, let-7g miR-30b, miR-369, miR-495, and miR-712 were only upregulated in infected C57BL/6 WT macrophages, while TLR2-, TLR4-, or MyD88-deficient mice exhibited alterations in the expression of other miRNAs during L. amazonensis infection. Based on this evidence, the expression of let-7e, let-7f, and let-7g require MyD88, TLR2, and TLR4 signaling during L. amazonensis infection, and our findings reveal the role of the TLR pathway in the transcriptional and post-transcriptional regulation of gene expression during Leishmania infection.
The miRNA let-7 family is highly conserved from bacteria to mammals and modulates the expression of some anti-inflammatory effector molecules, such as IL-10 (66), and pro-inflammatory effector molecules, such as IL-6 (let-7a) (67) and IL-13 (let-7d) (68). Infections with Mycobacterium and Neisseria induce let-7e expression in macrophages (56, 57). The expression of let-7e modulates p65 NF-κB activation (54) and phosphoinositide-3 kinase/serine-threonine protein kinase (Pi3k/Akt) (53, 57). Indeed, let-7e downregulated TLR4 and reduced pro-inflammatory signals, such as TNF, IFN-alpha, IL-6, IL-17, MCP-1, MIP-1, and IP-10 (53, 55, 57). Moreover, let-7e targeted the suppressor of cytokine signaling 4 (SOCS4) and regulated IL-13 expression in an allergic process (69).
Here, L. amazonensis infection in C57BL/6 WT macrophages increased the levels of recognition molecules, such as Tlr1, Tlr2, Tlr5, and Clec4e/Mincle, supporting the involvement of TLRs in the identification and phagocytosis of the parasite. Moreover, the increased expression of adaptor and effector molecules, such as Hspa1a, Hspd1, Irf 1, Ripk2, Nfkb1, Nfkb2, Nfkbia/Ikbalpha, as well as immunomodulators, such as Cxcl10, Cd86, Il1a, Il1b, Tnf , Il10, and Tnfaip3, and the decrease in the expression of Casp8, Nr2c2/TAK1, Mapk9/JNK2, and Il6ra during infection indicates that L. amazonensis modulated macrophage activation.
Interestingly, functional inhibition of let-7e during L. amazonensis infection reduced the infectivity and impacted the global expression of mRNAs involved in the TLR signaling cascade. As shown in Figure 7, the levels of predicted targets, such as Traf 6, Ppara, Mapk8ip3/Jip3, Map3k1, and Ube2n (represented in red), and the following indirect targets (represented in orange) were increased: recognition molecules: Tlr9 and Ly96/MD-2; adaptors and effectors: Tirap, Ticam1, Tollip, Casp8, Map3k1, Peli1, Nfkbib/TRIP9, Nfkbil1, and Hspd1; and immunomodulators: Ptgs2/Cox2, Csf 2/GMCSF, Csf 3/GCSF, and Ilr1. Nevertheless, the previously validated let-7e targets Tnfpaip3, Map2k4, Tbk1, and Tnf were upregulated during L. amazonensis infection, whereas the decrease in Chuk/IKBKA and Il10 expression during infection was not reversed by let-7e inhibition.
Figure 7.

Schematic of the TLR pathway and let-7e target mRNAs. TLR activation is regulated by let-7e-mediated post-transcriptional mechanisms acting directly (predicted targets in red) or indirectly (molecules in orange) on mRNA/protein expression by modulating the expression of molecules involved in recognition, adaptors and effector molecules, as well as transcription factors (green) that regulate the expression of cytokines, chemokines and other immunomodulators.
The roles of TLR/IL-1R signaling pathways in the ubiquitin-dependent NF-κB activation and subcellular trafficking have been extensively studied. The ubiquitination system is based on the ubiquitin-activating E1 enzyme, ubiquitin-conjugating E2 enzymes, and ubiquitin E3 ligases. The E2 UBE2n dimerizes with UBE2v, forming K63-linked ubiquitin chains to polyubiquitinate adaptor molecules, such as TRAF6, which is recruited to the TAB2/TAK1 complex and activates NF-κB, as shown in Figure 7 (70–73). However, the role of ubiquitination in the immune response to Leishmania has not been studied. Another molecule, PPARα was not previously reported to be involved in Leishmania infection. However, TLR4 transcription and signaling are negatively regulated via PPARα, which inhibits the NF-κB signaling pathway (74). PPARα also regulates the expression of genes involved in free fatty acid metabolism, could bind to TLR4 and interfere with the inflammatory response. In addition, Tollip is a negative regulator of the TLR signaling pathway, regulates the production of the pro-inflammatory cytokines IL-6 and TNF (75) and potentially influences the response to cutaneous L. guyanensis infection (76). Mapk8ip3/JIP3 helps the TLR4 signaling cascade enhance the activation of the JNK and MEK1/2 pathways (77, 78).
Indeed, an early step in the infection of human macrophages with L. amazonensis and L. major occurs the upregulation of pro-inflammatory cytokine genes, such as Il1b, Tnf , and Il6, as well as other molecules involved in tissue growth and repair, such as Csf 2 and Ptgs2/Cox2 (79). Similarly, L. major infection of murine macrophages increases the levels of Tnf, Il1, Il6, Nos2, I1rap, and Csf 3, as well as molecules in the NF-κB and MAPK signaling pathways (21).
Here, TLR2 and TLR4 induced the expression of the Cat2B and Cat1 mRNAs in L. amazonensis-infected macrophages, while a MyD88 deficiency induced their expression, helping control L-arginine uptake. L-arginine uptake mediated by Cat2B helps control L. amazonensis infectiveness (40). The regulation of polyamine production by Arg1 mRNA expression depended on TLR4 and MyD88. Thus, Nos2 expression depends on TLR2, TLR4, and MyD88 molecules during infection and its activation in C57BL/6 mouse macrophages may enhance the resistance to infection, as previously shown in murine macrophages lacking Nos2 (80). Notably, let-7e inhibition regulated Arg1 and Nos2 expression during infection, corroborating the importance of the TLR signaling cascade in the mechanism regulating Arg1 and Nos2 expression at the transcriptional level. The NF-κB pathway regulates Nos2 expression at the transcriptional level (81). In cells infected with L. major and L. amazonensis increasing in the nuclear translocation of the NF-κB p50/p50 dimer correlates with the decrease in the levels of the Nos2 and Cat2B transcripts (16). In response to an L. amazonensis infection, miR-294-3p and miR-721 post-transcriptionally degrade the Nos2 mRNA in BALB/c macrophages (31). Leishmania also induces L-arginine uptake mediated by amino acid permease 3 (82–84), amino acid metabolism via arginase activity (85, 86), and NOS-like activity (87). L-arginine is essential for parasite survival and interferes with macrophage activation during infection (88). Indeed, parasite arginase competes with L-arginine and reduces Nos2 expression and NO production in L. amazonensis-infected hosts (31).
NOS2 activity induces NO production to trigger the macrophage-mediated inflammatory response and kill the parasites (39, 89–91). The balance between polyamine production via ARG1 and NO production via NOS2 influences the fate of infectivity in different mouse models. IL-12 production and NOS2 expression reduce the infectivity in C57BL/6 mice. In contrast, IL-4 production and Arg1 expression increase the infectivity in BALB/c mice (37, 38, 88). The cytokines IL-12 and IL-4 induce CAT2B expression and the uptake of L-arginine, a substrate of ARG1 and NOS2, altering outcome of infection, while the negative regulation of CAT2B expression also reduces the infectivity of L. amazonensis (40).
Interestingly, TLR4 contributes to the control of parasite growth in the initial and later steps of L. major infection (13). In addition, L. major infection induces NOS2 expression by activating TLR4, although ARG1 expression is independent of TLR4 during infection in vivo and in vitro (13). TLR9 induces NOS2, IL-12 and IFN-gamma expression and helps control the parasite burden in L. major-infected BALB/c and C57BL/6 mice (92). However, MyD88 signaling increases the ratio of IL-12/IL-10 in L. amazonensis- and L. braziliensis-infected dendritic cells (15). MyD88 mediates the resistance to L. braziliensis infection in C57BL/6 mice, but TLR2 enhances regulation of the immune response by reducing the ratio of IL-12/IL-10 (15).
L. amazonensis infection has been reported to regulate the activation of MAPK in BALB/c macrophages by regulating IL-10 production (28). However, p38-MAPK activation reduces the infectivity of L. donovani (93). On the other hand, L. mexicana glycosylphosphatidylinositol-anchored lipophosphoglycan (LPG)/TLR stimulation regulates the activity of the MAPK and NF-κB pathways to alter IL-12 production (94, 95). The chaperone HSP60 serves as danger signal for stressed or damaged cells and its effects are potentially mediated by its anti-apoptotic activity induced by the formation of the HSP60/pC3 complex (96). TNF or TLR signaling leads to the formation of a Fas-associated protein with death domain (FADD)/pro-caspase 8 complex and activation of caspase 8, which induces apoptosis via direct cleavage of downstream caspases, caspase-3/7 (97). In response to an L. major infection, the inhibition of caspase 8 increases the production of Th1 and Th2 cytokines and NO (98), in contrast to the antiapoptotic signaling mediated by caspase 8 during L. infantum infection (99). The modulation of the expression of mRNAs involved in the TLR pathway impacted the infectivity of L. amazonensis.
Our data corroborated the idea that Leishmania infection may regulate the TLR pathway and subsequent cytokine and chemokine production, as well as NO/polyamine production to control macrophage activation and infectiveness. We emphasize the importance of TLR signaling and miRNA expression during infection and the integration of these molecules in the mechanism regulating gene expression required to determine the fate Leishmania infection, as well as the identification of potentially new targets to control infectivity and pathogenesis.
Author contributions
SM, JA, and LF-W conceived and designed the experiments. SM, JA, RZ, and SA performed the experiments. SM, JA, and LF-W wrote the original article. SM designed the figures. SM, JA, RZ, SA, and LF-W revised the article.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This work was supported by grants from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, www.cnpq.br: 307587/2014-2) and Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, www.fapesp.br: 2016/19815-2 and 2014/50717-1), as well as FAPESP fellowships: SA (2017/23519-2) and JA (2016/03273-6). LF-W received a research fellowship from CNPq (307587/2014-2). We would like to thank the “Central de Aquisição de Imagens e Microscopia” (CAIMI) at Instituto de Biociências of the Universidade de São Paulo for the use of FlowSight Amnis (grant number FAPESP Temático Multiusuário 2014/20809-1).
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2018.02792/full#supplementary-material
Scatter plots of the miRNA profiles of C57BL/6 (WT), MyD88−/−, TLR2−/−, and TLR4−/− murine BMDMs infected with L. amazonensis. Each dot represents one miRNA in BMDMs infected with L. amazonensis for 4, 24, and 48 h. The red dots indicate upregulated miRNAs and green dots indicate downregulated miRNAs in both comparisons. Blue dots indicate upregulated miRNAs in WT macrophages compared to knockout cells and orange dots indicate downregulated miRNAs in WT macrophages compared to knockout cells. The relative up- and downregulation of miRNAs are reported as boundaries of 1.2 or −1.2 of Fold Regulation, respectively. Only significant (p < 0.05) Fold Regulation values are presented.
Inhibition of let-7 functions. BMDMs were transiently transfected with 100 nM negative control (NC) or let-7e-5p inhibitor. Twenty-four hours after transfection incubation, cells were co-cultivated with L. amazonensis (MOI 5:1) for 4 h, and the cultures were then washed. After 4 and 24 h of infection, the samples were analyzed for let-7e levels normalized to SNORD95 (A), and levels of the Cat2B (B), and Cat1 (C) mRNAs using RT-qPCR. The values were normalized to the average values of NC-transfected, infected macrophages (100%). Each bar represents the average ± SEM of the values obtained from three independent experiments (n = 3–6). Statistical significance was determined using two-tailed Student's t-tests. *p < 0.05 compared to negative control-transfected, infected macrophages.
Expression of mRNAs encoding proteins in the TLR pathway molecules and cytokines in L. amazonensis-infected macrophages. The BMDMs (5 × 106) from WT mice were infected with L. amazonensis (MOI 5:1). Each dot represents one mRNA in BMDMs from WT mice infected with L. amazonensis for 4 and 24 h. Blue dots indicate upregulated mRNAs and orange dots indicate downregulated mRNAs. The blue dotted line corresponds to p = 0.05, log 10. The relative up- and downregulation of miRNAs are presented as boundaries of 1.2 or −1.2 of Fold Regulation, respectively. P-values were determined using two-tailed Student's t-test. Representative data from three independent experiments are shown.
C57BL/6 (WT), MyD88−/−, TLR2−/−, and TLR4−/− murine macrophages during L. amazonensis infection. FR (Fold Regulation) represents the fold change results at a biological magnitude. Values >1.2 indicate positive regulation and values < -1.2 indicate negative regulation.
Predicted target mRNAs of the let-7e miRNA. The miRNA-mRNA interactions predicted using miRecord tools (http://c1.accurascience.com/miRecords/) were based on the integration of the predicted targets obtained from at least three of the following prediction tools: DIANA-microT, MicroInspector, miRanda, MirTarget2, miTarget, NBmiRTar, PicTar, PITA, RNA22, RNAhybrid, and TargetScan/TargertScanS.
Let-7e inhibition alters the levels of mRNAs in the TLR pathway in WT macrophages during L. amazonensis infection. FR (Fold Regulation) represents the fold change results at a biological magnitude. Values >1.2 indicates positive regulation and values < -1.2 indicate negative regulation.
References
- 1.Alvar J, Vélez ID, Bern C, Herrero M, Desjeux P, Cano J, et al. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE (2012) 7:e35671. 10.1371/journal.pone.0035671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bogdan C. Mechanisms and consequences of persistence of intracellular pathogens: leishmaniasis as an example. Cell Microbiol. (2008) 10:1221–34. 10.1111/j.1462-5822.2008.01146.x [DOI] [PubMed] [Google Scholar]
- 3.Liese J, Schleicher U, Bogdan C. The innate immune response against Leishmania parasites. Immunobiology (2008) 213:377–87. 10.1016/j.imbio.2007.12.005 [DOI] [PubMed] [Google Scholar]
- 4.Liu D, Uzonna JE. The early interaction of Leishmania with macrophages and dendritic cells and its influence on the host immune response. Front Cell Infect Microbiol. (2012) 2:83. 10.3389/fcimb.2012.00083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carlsen ED, Liang Y, Shelite TR, Walker DH, Melby PC, Soong L. Permissive and protective roles for neutrophils in leishmaniasis. Clin Exp Immunol. (2015) 182:109–18. 10.1111/cei.12674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tuon FF, Amato VS, Bacha HA, Almusawi T, Duarte MI, Amato Neto V. Toll-like receptors and leishmaniasis. Infect Immun. (2008) 76:866–72. 10.1128/IAI.01090-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akira S, Sato S. Toll-like receptors and their signaling mechanisms. Scand J Infect Dis. (2003) 35:555–62. 10.1080/00365540310015683 [DOI] [PubMed] [Google Scholar]
- 8.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. (2003) 21:335–76. 10.1146/annurev.immunol.21.120601.141126 [DOI] [PubMed] [Google Scholar]
- 9.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell (2006) 124:783–801. 10.1016/j.cell.2006.02.015 [DOI] [PubMed] [Google Scholar]
- 10.Gazzinelli RT, Denkers EY. Protozoan encounters with Toll-like receptor signalling pathways: implications for host parasitism. Nat Rev Immunol. (2006) 6:895–906. 10.1038/nri1978 [DOI] [PubMed] [Google Scholar]
- 11.Uematsu S, Akira S. Toll-like receptors and innate immunity. J Mol Med (Berl). (2006) 84:712–25. 10.1007/s00109-006-0084-y [DOI] [PubMed] [Google Scholar]
- 12.Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat Immunol. (2008) 9:361–8. 10.1038/ni1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kropf P, Freudenberg MA, Modolell M, Price HP, Herath S, Antoniazi S, et al. Toll-like receptor 4 contributes to efficient control of infection with the protozoan parasite Leishmania major. Infect Immun. (2004) 72:1920–8. 10.1128/IAI.72.4.1920-1928.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flandin JF, Chano F, Descoteaux A. RNA interference reveals a role for TLR2 and TLR3 in the recognition of Leishmania donovani promastigotes by interferon-gamma-primed macrophages. Eur J Immunol. (2006) 36:411–20. 10.1002/eji.200535079 [DOI] [PubMed] [Google Scholar]
- 15.Vargas-Inchaustegui DA, Tai W, Xin L, Hogg AE, Corry DB, Soong L. Distinct roles for MyD88 and Toll-like receptor 2 during Leishmania braziliensis infection in mice. Infect Immun. (2009) 77:2948–56. 10.1128/IAI.00154-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dixit V, Mak TW. NF-kappaB signaling. Many roads lead to madrid. Cell (2002) 111:615–9. 10.1016/S0092-8674(02)01166-2 [DOI] [PubMed] [Google Scholar]
- 17.Janeway CA Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. (2002) 20:197–216. 10.1146/annurev.immunol.20.083001.084359 [DOI] [PubMed] [Google Scholar]
- 18.Becker I, Salaiza N, Aguirre M, Delgado J, Carrillo-Carrasco N, Kobeh LG, et al. Leishmania lipophosphoglycan (LPG) activates NK cells through toll-like receptor-2. Mol Biochem Parasitol. (2003) 130:65–74. 10.1016/S0166-6851(03)00160-9 [DOI] [PubMed] [Google Scholar]
- 19.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature (2007) 449:819–26. 10.1038/nature06246 [DOI] [PubMed] [Google Scholar]
- 20.Whitaker SM, Colmenares M, Pestana KG, Mcmahon-Pratt D. Leishmania pifanoi proteoglycolipid complex P8 induces macrophage cytokine production through Toll-like receptor 4. Infect Immun. (2008) 76:2149–56. 10.1128/IAI.01528-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dillon LA, Suresh R, Okrah K, Corrada Bravo H, Mosser DM, El-Sayed NM. Simultaneous transcriptional profiling of Leishmania major and its murine macrophage host cell reveals insights into host-pathogen interactions. BMC Genom. (2015) 16:1108. 10.1186/s12864-015-2237-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nathan C, Shiloh MU. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc Natl Acad Sci USA. (2000) 97:8841–8. 10.1073/pnas.97.16.8841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gregory DJ, Olivier M. Subversion of host cell signalling by the protozoan parasite Leishmania. Parasitology (2005) 130 Suppl.:S27–35. 10.1017/S0031182005008139 [DOI] [PubMed] [Google Scholar]
- 24.Lima-Junior DS, Costa DL, Carregaro V, Cunha LD, Silva AL, Mineo TW, et al. Inflammasome-derived IL-1beta production induces nitric oxide-mediated resistance to Leishmania. Nat Med. (2013) 19:909–15. 10.1038/nm.3221 [DOI] [PubMed] [Google Scholar]
- 25.Ghalib HW, Whittle JA, Kubin M, Hashim FA, El-Hassan AM, Grabstein KH, et al. IL-12 enhances Th1-type responses in human Leishmania donovani infections. J Immunol. (1995) 154:4623–9. [PubMed] [Google Scholar]
- 26.Vieira LQ, Goldschmidt M, Nashleanas M, Pfeffer K, Mak T, Scott P. Mice lacking the TNF receptor p55 fail to resolve lesions caused by infection with Leishmania major, but control parasite replication. J Immunol. (1996) 157:827–35. [PubMed] [Google Scholar]
- 27.Wilhelm P, Ritter U, Labbow S, Donhauser N, Rollinghoff M, Bogdan C, et al. Rapidly fatal leishmaniasis in resistant C57BL/6 mice lacking TNF. J Immunol. (2001) 166:4012–9. 10.4049/jimmunol.166.6.4012 [DOI] [PubMed] [Google Scholar]
- 28.Yang Z, Mosser DM, Zhang X. Activation of the MAPK, ERK, following Leishmania amazonensis infection of macrophages. J Immunol. (2007) 178:1077–85. 10.4049/jimmunol.178.2.1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ben-Othman R, Dellagi K, Guizani-Tabbane L. Leishmania major parasites induced macrophage tolerance: implication of MAPK and NF-kappaB pathways. Mol Immunol. (2009) 46:3438–44. 10.1016/j.molimm.2009.05.337 [DOI] [PubMed] [Google Scholar]
- 30.Srivastava A, Singh N, Mishra M, Kumar V, Gour JK, Bajpai S, et al. Identification of TLR inducing Th1-responsive Leishmania donovani amastigote-specific antigens. Mol Cell Biochem. (2012) 359:359–68. 10.1007/s11010-011-1029-5 [DOI] [PubMed] [Google Scholar]
- 31.Muxel SM, Laranjeira-Silva MF, Zampieri RA, Floeter-Winter LM. Leishmania (Leishmania) amazonensis induces macrophage miR-294 and miR-721 expression and modulates infection by targeting NOS2 and L-arginine metabolism. Sci Rep. (2017) 7:44141. 10.1038/srep44141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nasseri M, Modabber FZ. Generalized infection and lack of delayed hypersensitivity in BALB/c mice infected with Leishmania tropica major. Infect Immun. (1979) 26:611–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bacellar O, D'oliveira A Jr, Jeronimo S, Carvalho EM. IL-10 and IL-12 are the main regulatory cytokines in visceral leishmaniasis. Cytokine (2000) 12:1228–31. 10.1006/cyto.2000.0694 [DOI] [PubMed] [Google Scholar]
- 34.Bhattacharyya S, Ghosh S, Jhonson PL, Bhattacharya SK, Majumdar S. Immunomodulatory role of interleukin-10 in visceral leishmaniasis: defective activation of protein kinase C-mediated signal transduction events. Infect Immun. (2001) 69:1499–507. 10.1128/IAI.69.3.1499-1507.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kane MM, Mosser DM. The role of IL-10 in promoting disease progression in leishmaniasis. J Immunol. (2001) 166:1141–7. 10.4049/jimmunol.166.2.1141 [DOI] [PubMed] [Google Scholar]
- 36.Liew FY, Millott S, Parkinson C, Palmer RM, Moncada S. Macrophage killing of Leishmania parasite in vivo is mediated by nitric oxide from L-arginine. J Immunol. (1990) 144:4794–7. [PubMed] [Google Scholar]
- 37.Iniesta V, Gomez-Nieto LC, Corraliza I. The inhibition of arginase by N(omega)-hydroxy-l-arginine controls the growth of Leishmania inside macrophages. J Exp Med. (2001) 193:777–84. 10.1084/jem.193.6.777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iniesta V, Gomez-Nieto LC, Molano I, Mohedano A, Carcelen J, Miron C, et al. Arginase I induction in macrophages, triggered by Th2-type cytokines, supports the growth of intracellular Leishmania parasites. Parasite Immunol. (2002) 24:113–8. 10.1046/j.1365-3024.2002.00444.x [DOI] [PubMed] [Google Scholar]
- 39.Wanasen N, Soong L. L-arginine metabolism and its impact on host immunity against Leishmania infection. Immunol Res. (2008) 41:15–25. 10.1007/s12026-007-8012-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Laranjeira-Silva MF, Zampieri RA, Muxel SM, Floeter-Winter LM, Markus RP. Melatonin attenuates Leishmania (L.) amazonensis infection by modulating arginine metabolism. J Pineal Res. (2015) 59:478–87. 10.1111/jpi.12279 [DOI] [PubMed] [Google Scholar]
- 41.Geraci NS, Tan JC, Mcdowell MA. Characterization of microRNA expression profiles in Leishmania-infected human phagocytes. Parasite Immunol. (2015) 37:43–51. 10.1111/pim.12156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singh AK, Pandey RK, Shaha C, Madhubala R. MicroRNA expression profiling of Leishmania donovani-infected host cells uncovers the regulatory role of MIR30A-3p in host autophagy. Autophagy (2016) 12:1817–31. 10.1080/15548627.2016.1203500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature (2001) 409:363–6. 10.1038/35053110 [DOI] [PubMed] [Google Scholar]
- 44.Schwarz DS, Hutvagner G, Du T, Xu Z, Aronin N, Zamore PD. Asymmetry in the assembly of the RNAi enzyme complex. Cell (2003) 115:199–208. 10.1016/S0092-8674(03)00759-1 [DOI] [PubMed] [Google Scholar]
- 45.Vaucheret H, Vazquez F, Crete P, Bartel DP. The action of ARGONAUTE1 in the miRNA pathway and its regulation by the miRNA pathway are crucial for plant development. Genes Dev. (2004) 18:1187–97. 10.1101/gad.1201404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bagga S, Bracht J, Hunter S, Massirer K, Holtz J, Eachus R, et al. Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell (2005) 122:553–63. 10.1016/j.cell.2005.07.031 [DOI] [PubMed] [Google Scholar]
- 47.Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature (2005) 433:769–73. 10.1038/nature03315 [DOI] [PubMed] [Google Scholar]
- 48.Wang B, Li S, Qi HH, Chowdhury D, Shi Y, Novina CD. Distinct passenger strand and mRNA cleavage activities of human argonaute proteins. Nat Struct Mol Biol. (2009) 16:1259–66. 10.1038/nsmb.1712 [DOI] [PubMed] [Google Scholar]
- 49.Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature (2000) 403:901–6. 10.1038/35002607 [DOI] [PubMed] [Google Scholar]
- 50.Pasquinelli AE, Reinhart BJ, Slack F, Martindale MQ, Kuroda MI, Maller B, et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature (2000) 408:86–9. 10.1038/35040556 [DOI] [PubMed] [Google Scholar]
- 51.Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol. (2002) 12:735–9. 10.1016/S0960-9822(02)00809-6 [DOI] [PubMed] [Google Scholar]
- 52.Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell (2007) 129:1401–14. 10.1016/j.cell.2007.04.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Androulidaki A, Iliopoulos D, Arranz A, Doxaki C, Schworer S, Zacharioudaki V, et al. The kinase Akt1 controls macrophage response to lipopolysaccharide by regulating microRNAs. Immunity (2009) 31:220–31. 10.1016/j.immuni.2009.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.De La Rica L, Garcia-Gomez A, Comet NR, Rodriguez-Ubreva J, Ciudad L, Vento-Tormo R, et al. NF-kappaB-direct activation of microRNAs with repressive effects on monocyte-specific genes is critical for osteoclast differentiation. Genome Biol. (2015) 16:2 10.1186/s13059-014-0561-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dong G, Fan H, Yang Y, Zhao G, You M, Wang T, et al. 17beta-Estradiol enhances the activation of IFN-alpha signaling in B cells by down-regulating the expression of let-7e-5p, miR-98-5p and miR-145a-5p that target IKKepsilon. Biochim Biophys Acta (2015) 1852:1585–98. 10.1016/j.bbadis.2015.04.019 [DOI] [PubMed] [Google Scholar]
- 56.Elizabeth MC, Hernandez De La Cruz ON, Mauricio CA. Infection of J774A.1 with different Mycobacterium species induces differential immune and miRNA-related responses. Microbiol Immunol. (2016) 60:356–63. 10.1111/1348-0421.12380 [DOI] [PubMed] [Google Scholar]
- 57.Kalantari P, Harandi OF, Agarwal S, Rus F, Kurt-Jones EA, Fitzgerald KA, et al. miR-718 represses proinflammatory cytokine production through targeting phosphatase and tensin homolog (PTEN). J Biol Chem. (2017) 292:5634–44. 10.1074/jbc.M116.749325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Muxel SM, Laranjeira-Silva MF, Zampieri RA, Aoki JI, Acuña SM, Floeter-Winter LM. Functional validation of miRNA-mRNA interactions in macrophages by inhibition/competition assays based in transient transfection. Protocol Exchange (2017) 34:1–14. 10.1038/protex.2017.034 [DOI] [Google Scholar]
- 59.Oliveros JC. Venny. An Interactive Tool for Comparing Lists With Venn's Diagrams (2007–2015). Available online at: http://bioinfogp.cnb.csic.es/tools/venny/index.html (Accessed).
- 60.Karagkouni D, Paraskevopoulou MD, Chatzopoulos S, Vlachos IS, Tastsoglou S, Kanellos I, et al. DIANA-TarBase v8: a decade-long collection of experimentally supported miRNA-gene interactions. Nucleic Acids Res. (2018) 46:D239–45. 10.1093/nar/gkx1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.De Veer MJ, Curtis JM, Baldwin TM, Didonato JA, Sexton A, Mcconville MJ, et al. MyD88 is essential for clearance of Leishmania major: possible role for lipophosphoglycan and Toll-like receptor 2 signaling. Eur J Immunol. (2003) 33:2822–31. 10.1002/eji.200324128 [DOI] [PubMed] [Google Scholar]
- 62.Kavoosi G, Ardestani SK, Kariminia A. The involvement of TLR2 in cytokine and reactive oxygen species (ROS) production by PBMCs in response to Leishmania major phosphoglycans (PGs). Parasitology (2009) 136:1193–9. 10.1017/S0031182009990473 [DOI] [PubMed] [Google Scholar]
- 63.De Trez C, Brait M, Leo O, Aebischer T, Torrentera FA, Carlier Y, et al. Myd88-dependent in vivo maturation of splenic dendritic cells induced by Leishmania donovani and other Leishmania species. Infect Immun. (2004) 72:824–32. 10.1128/IAI.72.2.824-832.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lin Y, Sibanda VL, Zhang HM, Hu H, Liu H, Guo AY. MiRNA and TF co-regulatory network analysis for the pathology and recurrence of myocardial infarction. Sci Rep. (2015) 5:9653. 10.1038/srep09653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lemaire J, Mkannez G, Guerfali FZ, Gustin C, Attia H, Sghaier RM, et al. MicroRNA expression profile in human macrophages in response to Leishmania major infection. PLoS Negl Trop Dis. (2013) 7:e2478. 10.1371/journal.pntd.0002478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Swaminathan S, Suzuki K, Seddiki N, Kaplan W, Cowley MJ, Hood CL, et al. Differential regulation of the Let-7 family of microRNAs in CD4+ T cells alters IL-10 expression. J Immunol. (2012) 188:6238–46. 10.4049/jimmunol.1101196 [DOI] [PubMed] [Google Scholar]
- 67.Meng F, Henson R, Wehbe-Janek H, Smith H, Ueno Y, Patel T. The MicroRNA let-7a modulates interleukin-6-dependent STAT-3 survival signaling in malignant human cholangiocytes. J Biol Chem. (2007) 282:8256–64. 10.1074/jbc.M607712200 [DOI] [PubMed] [Google Scholar]
- 68.Kumar M, Ahmad T, Sharma A, Mabalirajan U, Kulshreshtha A, Agrawal A, et al. Let-7 microRNA-mediated regulation of IL-13 and allergic airway inflammation. J Allergy Clin Immunol. (2011) 128:1077–1085 e1071–e1010. 10.1016/j.jaci.2011.04.034 [DOI] [PubMed] [Google Scholar]
- 69.Li L, Zhang S, Jiang X, Liu Y, Liu K, Yang C. MicroRNA-let-7e regulates the progression and development of allergic rhinitis by targeting suppressor of cytokine signaling 4 and activating Janus kinase 1/signal transducer and activator of transcription 3 pathway. Exp Ther Med. (2018) 15:3523–9. 10.3892/etm.2018.5827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ikeda F, Dikic I. Atypical ubiquitin chains: new molecular signals. ‘Protein Modifications: Beyond the Usual Suspects' review series. EMBO Rep. (2008) 9:536–42. 10.1038/embor.2008.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.David Y, Ziv T, Admon A, Navon A. The E2 ubiquitin-conjugating enzymes direct polyubiquitination to preferred lysines. J Biol Chem. (2010) 285:8595–604. 10.1074/jbc.M109.089003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Roh YS, Song J, Seki E. TAK1 regulates hepatic cell survival and carcinogenesis. J Gastroenterol. (2014) 49:185–94. 10.1007/s00535-013-0931-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fletcher AJ, Mallery DL, Watkinson RE, Dickson CF, James LC. Sequential ubiquitination and deubiquitination enzymes synchronize the dual sensor and effector functions of TRIM21. Proc Natl Acad Sci USA. (2015) 112:10014–9. 10.1073/pnas.1507534112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shen W, Gao Y, Lu B, Zhang Q, Hu Y, Chen Y. Negatively regulating TLR4/NF-kappaB signaling via PPARalpha in endotoxin-induced uveitis. Biochim Biophys Acta (2014) 1842:1109–20. 10.1016/j.bbadis.2014.03.015 [DOI] [PubMed] [Google Scholar]
- 75.Liew FY, Xu D, Brint EK, O'neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol. (2005) 5:446–58. 10.1038/nri1630 [DOI] [PubMed] [Google Scholar]
- 76.De Araujo FJ, Da Silva LD, Mesquita TG, Pinheiro SK, Vital Wde S, Chrusciak-Talhari A, et al. Polymorphisms in the TOLLIP gene influence susceptibility to cutaneous Leishmaniasis caused by Leishmania guyanensis in the Amazonas State of Brazil. PLoS Negl Trop Dis. (2015) 9:e0003875 10.1371/journal.pntd.0003875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ito M, Yoshioka K, Akechi M, Yamashita S, Takamatsu N, Sugiyama K, et al. JSAP1, a novel jun N-terminal protein kinase (JNK)-binding protein that functions as a Scaffold factor in the JNK signaling pathway. Mol Cell Biol. (1999) 19:7539–48. 10.1128/MCB.19.11.7539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Matsuguchi T, Masuda A, Sugimoto K, Nagai Y, Yoshikai Y. JNK-interacting protein 3 associates with Toll-like receptor 4 and is involved in LPS-mediated JNK activation. EMBO J. (2003) 22:4455–64. 10.1093/emboj/cdg438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fernandes MC, Dillon LA, Belew AT, Bravo HC, Mosser DM, El-Sayed NM. Dual transcriptome profiling of Leishmania-infected human macrophages reveals distinct reprogramming signatures. MBio (2016) 7:e00027-16. 10.1128/mBio.00027-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wei XQ, Charles IG, Smith A, Ure J, Feng GJ, Huang FP, et al. Altered immune responses in mice lacking inducible nitric oxide synthase. Nature (1995) 375:408–11. 10.1038/375408a0 [DOI] [PubMed] [Google Scholar]
- 81.Beck KF, Sterzel RB. Cloning and sequencing of the proximal promoter of the rat iNOS gene: activation of NFkappaB is not sufficient for transcription of the iNOS gene in rat mesangial cells. FEBS Lett. (1996) 394:263–7. 10.1016/0014-5793(96)00966-0 [DOI] [PubMed] [Google Scholar]
- 82.Castilho-Martins EA, Laranjeira Da Silva MF, Dos Santos MG, Muxel SM, Floeter-Winter LM. Axenic Leishmania amazonensis promastigotes sense both the external and internal arginine pool distinctly regulating the two transporter-coding genes. PLoS ONE (2011) 6:e27818. 10.1371/journal.pone.0027818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Castilho-Martins EA, Canuto GA, Muxel SM, Da Silva MF, Floeter-Winter LM, Del Aguila C, et al. Capillary electrophoresis reveals polyamine metabolism modulation in Leishmania (Leishmania) amazonensis wild type and arginase knockout mutants under arginine starvation. Electrophoresis (2015) 38:2314–23. 10.1002/elps.201500114 [DOI] [PubMed] [Google Scholar]
- 84.Aoki JI, Muxel SM, Zampieri RA, Acuna SM, Fernandes JCR, Vanderlinde RH, et al. L-arginine availability and arginase activity: characterization of amino acid permease 3 in Leishmania amazonensis. PLoS Negl Trop Dis. (2017) 11:e0006025. 10.1371/journal.pntd.0006025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Laranjeira-Silva MF, Zampieri RA, Muxel SM, Beverley SM, Floeter-Winter LM. Leishmania amazonensis arginase compartmentalization in the glycosome is important for parasite infectivity. PLoS ONE (2012) 7:e34022 10.1371/journal.pone.0034022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Aoki JI, Muxel SM, Zampieri RA, Laranjeira-Silva MF, Muller KE, Nerland AH, et al. RNA-seq transcriptional profiling of Leishmania amazonensis reveals an arginase-dependent gene expression regulation. PLoS Negl Trop Dis. (2017) 11:e0006026. 10.1371/journal.pntd.0006026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Acuna SM, Aoki JI, Laranjeira Da Silva MF, Zampieri RA, Fernandes JC, Muxel SM, et al. Arginase expression modulates nitric oxide production in Leishmania (Leishmania) amazonensis. PLoS ONE (2017) 12:e0187186. 10.1371/journal.pone.0187186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Muxel SM, Aoki JI, Fernandes JCR, Laranjeira-Silva MF, Zampieri RA, Acuna SM, et al. Arginine and polyamines fate in Leishmania infection. Front Microbiol. (2017) 8:2682. 10.3389/fmicb.2017.02682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Green SJ, Crawford RM, Hockmeyer JT, Meltzer MS, Nacy CA. Leishmania major amastigotes initiate the L-arginine-dependent killing mechanism in IFN-gamma-stimulated macrophages by induction of tumor necrosis factor-alpha. J Immunol. (1990) 145:4290–7. [PubMed] [Google Scholar]
- 90.Green SJ, Meltzer MS, Hibbs JB, Jr, Nacy CA. Activated macrophages destroy intracellular Leishmania major amastigotes by an L-arginine-dependent killing mechanism. J Immunol. (1990) 144:278–83. [PubMed] [Google Scholar]
- 91.Liew FY, Li Y, Moss D, Parkinson C, Rogers MV, Moncada S. Resistance to Leishmania major infection correlates with the induction of nitric oxide synthase in murine macrophages. Eur J Immunol. (1991) 21:3009–14. 10.1002/eji.1830211216 [DOI] [PubMed] [Google Scholar]
- 92.Liese J, Schleicher U, Bogdan C. TLR9 signaling is essential for the innate NK cell response in murine cutaneous leishmaniasis. Eur J Immunol. (2007) 37:3424–34. 10.1002/eji.200737182 [DOI] [PubMed] [Google Scholar]
- 93.Junghae M, Raynes JG. Activation of p38 mitogen-activated protein kinase attenuates Leishmania donovani infection in macrophages. Infect Immun. (2002) 70:5026–35. 10.1128/IAI.70.9.5026-5035.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cameron P, Mcgachy A, Anderson M, Paul A, Coombs GH, Mottram JC, et al. Inhibition of lipopolysaccharide-induced macrophage IL-12 production by Leishmania mexicana amastigotes: the role of cysteine peptidases and the NF-kappaB signaling pathway. J Immunol. (2004) 173:3297–304. 10.4049/jimmunol.173.5.3297 [DOI] [PubMed] [Google Scholar]
- 95.Argueta-Donohue J, Carrillo N, Valdes-Reyes L, Zentella A, Aguirre-Garcia M, Becker I, et al. Leishmania mexicana: participation of NF-kappaB in the differential production of IL-12 in dendritic cells and monocytes induced by lipophosphoglycan (LPG). Exp Parasitol. (2008) 120:1–9. 10.1016/j.exppara.2008.04.002 [DOI] [PubMed] [Google Scholar]
- 96.Chandra D, Choy G, Tang DG. Cytosolic accumulation of HSP60 during apoptosis with or without apparent mitochondrial release: evidence that its pro-apoptotic or pro-survival functions involve differential interactions with caspase-3. J Biol Chem. (2007) 282:31289–301. 10.1074/jbc.M702777200 [DOI] [PubMed] [Google Scholar]
- 97.Feltham R, Vince JE, Lawlor KE. Caspase-8: not so silently deadly. Clin Transl Immunol. (2017) 6:e124. 10.1038/cti.2016.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pereira-Manfro WF, Ribeiro-Gomes FL, Filardy AA, Vellozo NS, Guillermo LV, Silva EM, et al. Inhibition of caspase-8 activity promotes protective Th1- and Th2-mediated immunity to Leishmania major infection. J Leukoc Biol. (2014) 95:347–55. 10.1189/jlb.0912463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cianciulli A, Porro C, Calvello R, Trotta T, Panaro MA. Resistance to apoptosis in Leishmania infantum-infected human macrophages: a critical role for anti-apoptotic Bcl-2 protein and cellular IAP1/2. Clin Exp Med. (2018) 18:251–61. 10.1007/s10238-017-0482-1 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Scatter plots of the miRNA profiles of C57BL/6 (WT), MyD88−/−, TLR2−/−, and TLR4−/− murine BMDMs infected with L. amazonensis. Each dot represents one miRNA in BMDMs infected with L. amazonensis for 4, 24, and 48 h. The red dots indicate upregulated miRNAs and green dots indicate downregulated miRNAs in both comparisons. Blue dots indicate upregulated miRNAs in WT macrophages compared to knockout cells and orange dots indicate downregulated miRNAs in WT macrophages compared to knockout cells. The relative up- and downregulation of miRNAs are reported as boundaries of 1.2 or −1.2 of Fold Regulation, respectively. Only significant (p < 0.05) Fold Regulation values are presented.
Inhibition of let-7 functions. BMDMs were transiently transfected with 100 nM negative control (NC) or let-7e-5p inhibitor. Twenty-four hours after transfection incubation, cells were co-cultivated with L. amazonensis (MOI 5:1) for 4 h, and the cultures were then washed. After 4 and 24 h of infection, the samples were analyzed for let-7e levels normalized to SNORD95 (A), and levels of the Cat2B (B), and Cat1 (C) mRNAs using RT-qPCR. The values were normalized to the average values of NC-transfected, infected macrophages (100%). Each bar represents the average ± SEM of the values obtained from three independent experiments (n = 3–6). Statistical significance was determined using two-tailed Student's t-tests. *p < 0.05 compared to negative control-transfected, infected macrophages.
Expression of mRNAs encoding proteins in the TLR pathway molecules and cytokines in L. amazonensis-infected macrophages. The BMDMs (5 × 106) from WT mice were infected with L. amazonensis (MOI 5:1). Each dot represents one mRNA in BMDMs from WT mice infected with L. amazonensis for 4 and 24 h. Blue dots indicate upregulated mRNAs and orange dots indicate downregulated mRNAs. The blue dotted line corresponds to p = 0.05, log 10. The relative up- and downregulation of miRNAs are presented as boundaries of 1.2 or −1.2 of Fold Regulation, respectively. P-values were determined using two-tailed Student's t-test. Representative data from three independent experiments are shown.
C57BL/6 (WT), MyD88−/−, TLR2−/−, and TLR4−/− murine macrophages during L. amazonensis infection. FR (Fold Regulation) represents the fold change results at a biological magnitude. Values >1.2 indicate positive regulation and values < -1.2 indicate negative regulation.
Predicted target mRNAs of the let-7e miRNA. The miRNA-mRNA interactions predicted using miRecord tools (http://c1.accurascience.com/miRecords/) were based on the integration of the predicted targets obtained from at least three of the following prediction tools: DIANA-microT, MicroInspector, miRanda, MirTarget2, miTarget, NBmiRTar, PicTar, PITA, RNA22, RNAhybrid, and TargetScan/TargertScanS.
Let-7e inhibition alters the levels of mRNAs in the TLR pathway in WT macrophages during L. amazonensis infection. FR (Fold Regulation) represents the fold change results at a biological magnitude. Values >1.2 indicates positive regulation and values < -1.2 indicate negative regulation.