

Purification of yeast exosome, RNA polymerase II and proteasome in native conditions allowed detection of novel PTMs using high resolution MS. The MD-based computational workflow developed here for prediction of PTM’s effect on subunit’s binding outperforms and is more widely applicable than Mechismo and FoldX. The predictions suggest a locally stabilizing role of acetylation, and destabilizing role of phosphorylation sites in protein complexes, while MD further reveals short and long range effects of PTMs on amino acids interaction patterns.
Keywords: Phosphorylation, Acetylation, Yeast, Structural Biology, Computational Biology, Binding Affinity, Exosome, Proteasome, Proteomics, RNA polymerase II
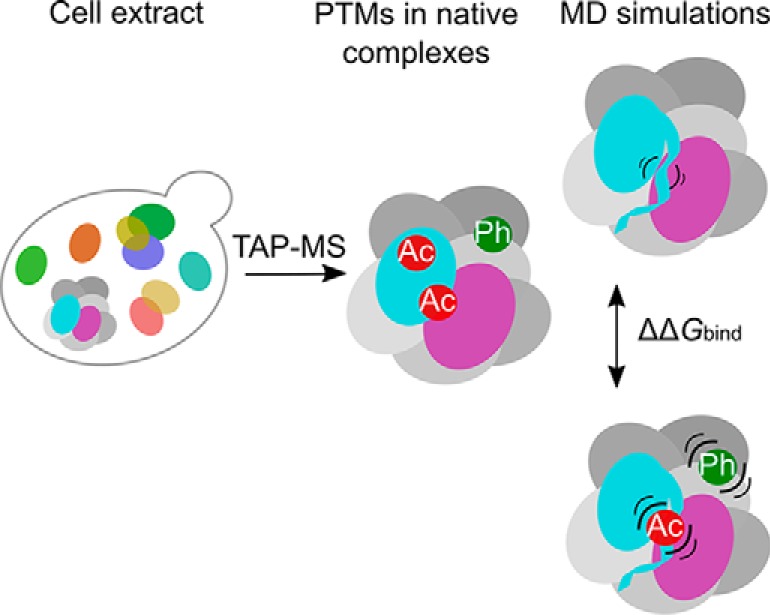
Graphical Abstract
Highlights
Novel PTMs detected in native yeast exosome, RNA polymerase II and proteasome.
MD based approach outperforms published tools in predicting PTMs stability effect.
Dynamical approach reveals long range effects of PTMs on subunits binding.
Acetylations may have a role in local stabilization of protein complex formation.
Abstract
Protein post-translational modifications (PTMs) have an indispensable role in living cells as they expand chemical diversity of the proteome, providing a fine regulatory layer that can govern protein-protein interactions in changing environmental conditions. Here we investigated the effects of acetylation and phosphorylation on the stability of subunit interactions in purified Saccharomyces cerevisiae complexes, namely exosome, RNA polymerase II and proteasome. We propose a computational framework that consists of conformational sampling of the complexes by molecular dynamics simulations, followed by Gibbs energy calculation by MM/GBSA. After benchmarking against published tools such as FoldX and Mechismo, we could apply the framework for the first time on large protein assemblies with the aim of predicting the effects of PTMs located on interfaces of subunits on binding stability. We discovered that acetylation predominantly contributes to subunits' interactions in a locally stabilizing manner, while phosphorylation shows the opposite effect. Even though the local binding contributions of PTMs may be predictable to an extent, the long range effects and overall impact on subunits' binding were only captured because of our dynamical approach. Employing the developed, widely applicable workflow on other large systems will shed more light on the roles of PTMs in protein complex formation.
A protein's functional engagement with other molecules in the cell is finely regulated by an array of post-translational modifications (PTMs)1 (1, 2) that locally change the chemical properties of a protein, and alter its activity, localization and stability (3–5). PTMs affect a significant part of eukaryotic and prokaryotic proteins, and different types were studied to different extents, with phosphorylation receiving most of the attention (4) and becoming the first PTM studied at a proteome-wide level (6, 7). In contrast, it has been appreciated only more recently that lysine acetylation is also a wide-spread modification (8, 9). Except its well-described roles in histones (10), it was also found to regulate the activities of other enzymes, such as tubulin (11).
Because of technological developments (7, 12), PTMs have been identified on a proteome-wide scale for several organisms, with the resulting data of more than 600,000 individual experimentally found modification sites stored in the freely available on-line database dbPTM (13). Availability of PTMs data enabled investigation of their functional roles on a large scale. For instance, the PTMfunc (14) resource was made, in which the predictions of PTM's functional relevance for 200,000 sites from 11 eukaryotic species is based on the conservation analysis. Moreover, the Mechismo tool (15) was developed, which estimates the effect of an interface located mutation or modification on interaction stability, based on the observed amino acids interaction patterns, but without the explicit 3D modeling of neither the query, nor the mutated/modified complex. In a recent expansion of the Mechismo work, the accent was placed on detecting phosphorylation sites that act as protein interaction switches (16) by additionally considering the similarity of the protein with its template, as well as conservation of the phosphorylation site. However, in all these tools the dynamic information is missing, so a mechanistic understanding of PTMs effect is still lacking.
Various efforts have also been undertaken to understand the functional roles of PTMs in specific (sets of) proteins, using not only experimental, but also computational approaches. For instance, Nishi et al. (17) examined the effect on energetics of binding caused by interface phosphorylations in human protein complexes. The calculations were performed using the empirical force field FoldX(18), in which side chains of the residues surrounding the phosphorylation site are optimized before binding energy calculation is performed. The obtained distribution was very much shifted toward destabilization, and for approximately one third of the sites destabilization effect was larger than 2 kcal/mol, which was taken as a threshold for a site to have a strong effect on interaction. Several studies also applied molecular dynamics (MD) simulations to assess the effect of PTMs on proteins, e.g. Narayanan et al. (19) investigated actin-related protein 2/3 complex, and Kumar et al. (20) cytoplasmic linker-associated protein 2 binding to end-binding protein 1. Their conclusions were of qualitative nature, for instance they observed the reorientation of proteins, formation and breaking of salt bridges and hydrogen bonds in the respective complexes. Additionally, an MD-based method employing nine physicochemical parameters extracted from the trajectories was recently proposed to predict the impact of phosphorylation on protein-protein interactions (21).
Until now, PTMs have been identified in whole cell lysates and mapped to protein structures, but it was unclear if they were actually present in native protein complexes. In this study, we aimed to elucidate the roles of phosphorylation and acetylation sites in three large yeast complexes that are essential for life - exosome, RNA polymerase II and 26S proteasome - by combining experimental and computational approaches (Fig. 1). Exosome catalyzes 3′-5′ ribonucleic acid (RNA) degradation in eukaryotes, which is involved in regulating the amount of transcripts, as well as their maturation and quality control (22). The core of the exosome consists of a hexameric ring (subunits Rrp41, Rrp42, Rrp43, Rrp45, Rrp46 and Mtr3) and a trimeric cap (Rrp4, Rrp40 and Csl4) (22, 23). In the cytoplasm, this nonamer recruits the catalytically active Rrp44 subunit (23), whereas nuclear exosome additionally has Rrp6 subunit and its obligate partner C1D (24). The second complex that we investigated, RNA polymerase II, is responsible for synthesis of all messenger RNA molecules, as well as several noncoding ones in eukaryotic cells (25, 26). Although 10 of its 12 subunits are conserved across species and identical or homologous to those in RNA polymerases I and III (26, 27), the remaining subunits Rpb4 and Rpb7 are specific for RNA polymerase II and are not important for the elongation process (28). Till date, the investigation of PTMs' function in RNA polymerase II was mainly focused on phosphorylation of the C-terminal domain (CTD) of its largest subunit, Rpb1, an important regulatory element not found in other RNA polymerases (28). CTD is composed of the consensus sequence Tyr-Ser-Pro-Thr-Ser-Pro-Ser repeats (29), known to change their phosphorylation status during the transcription cycle, and therefore dictate CTD's shape and binding of specific factors (28). Other PTM types—OGlcNAcylation, ubiquitylation, methylation, proline isomerization—have also been reported for CTD (25, 29), as well as acetylation of Lys from the non-consensus repeats found in some organisms (30). Finally, the proteasome is the major protein degradation machinery present in all three domains of life. Its substrates differ from other proteins in the cell by an attached chain of small proteins, ubiquitins. In eukaryotes, the 26S proteasome contains the proteolytically active 20S core particle (CP), composed of α and β subunits, and the 19S regulatory particle (RP), which together count 33 different protein subunits (31). Acetylation of CP and phosphorylation of both CP and RP subunits were found to affect proteasome activity (31), while phosphorylation of the Rpt6 ATPase subunit of RP was found to have a role in proteasome assembly (31, 32). Recently, more than 345 PTMs of 11 different types were detected on the 26S proteasome (33), however because most of the obtained PTM data is quite novel and originates from large proteomics studies, their roles are still predominantly unknown (34).
Fig. 1.
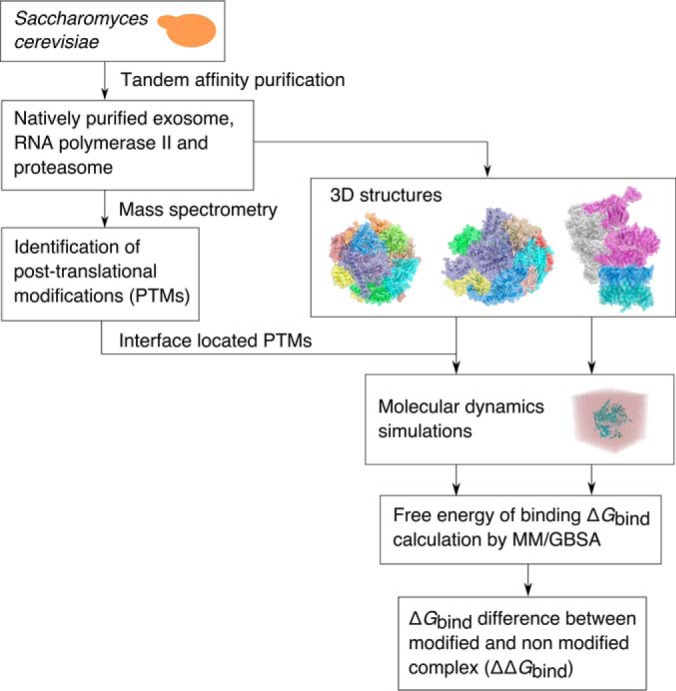
Overview of the workflow. The strategy applied for prediction of the effect of post-translational modifications on binding of subunits in yeast exosome, RNA polymerase II and proteasome 19S regulatory particle.
In this work, we first employ tandem affinity purification (TAP) followed by high resolution mass spectrometry (MS) in order to obtain the high fidelity information about PTM sites in the three natively purified complexes. Our data set contains a total of 129 acetylation and 41 phosphorylation sites detected within the complexes, almost all of which are novel. Secondly, we employ the available high-resolution 3D data to map the detected PTMs on the protein structures. Our focus is then placed on PTMs that are located at the subunits' interfaces, as such locations are generally more conserved, and therefore more likely functionally important and involved in the regulation of binding affinities (14, 17). Thirdly, in order to elucidate the effects of interface located novel PTMs on binding of subunits, we employ a computational approach consisting of meticulous conformational sampling via molecular dynamics simulations, followed by calculation of the Gibbs energy of binding by MM/GBSA method. We first test the robustness of this approach on yeast Skp1:Met30 system, benchmark it against Mechismo and FoldX on a set of mammalian protein complexes, apply it on the three large complexes, and experimentally validate our results. Finally, we compare the results for yeast complexes with predictions of the Mechismo tool and look into conservation of the PTM sites. Our predictions suggest the locally stabilizing role of the interface located acetylated lysines, and a locally destabilizing one for the phosphorylated residues. Moreover, our approach based on protein dynamics allowed us to capture global effects of PTMs on binding, with even binding of the chains that are not in a direct vicinity of PTMs being affected by their presence.
EXPERIMENTAL PROCEDURES
Purification of Protein Complexes, Reverse Phase Chromatography and Mass Spectrometry
Exosome, RNA polymerase II and 26S proteasome were purified from yeast Saccharomyces cerevisiae in native conditions, using tandem affinity purification (TAP). TAP was performed using one bait for each protein complex: YHR069C (Rrp4, exosome), YOR151C (Rpb2, RNA polymerase II) and YKL145W (Rpt1, 26S proteasome). Purified proteins were separated by SDS-PAGE followed by staining with Coomassie Brilliant Blue. Gel lanes were cut into slices, and subjected to in-gel digestion, using two different proteolytic enzymes (trypsin and chymotrypsin) in parallel, to increase the coverage. Obtained peptides were identified using high-resolution mass spectrometry (MS), as previously described (35).
In brief, peptides were subjected to reversed phase nLC-MS/MS analysis using an Agilent (Santa Clara, CA) 1290 Infinity UHPLC system, coupled to a TripleTOF 5600 (AB Sciex, Framingham, MA) mass spectrometer. The UHPLC was equipped with a double frit trapping column (Agilent Zorbax SB-C18, 1.8 μm material, 0.5 cm x 100 μm) and a single frit analytical column (Agilent Zorbax SB-C18, 1.8 μm material, 40 cm x 50 μm). Trapping was performed for 10 min in solvent A (0.1% FA in water) at 5 μl/min, whereas chromatographic separation was performed with a gradient of 23 min from 13% to 44% of solvent B (80% ACN/0.1% FA). Total analysis time was 45 min. The mass spectrometer was operated in data-dependent mode. After the initial survey scan in high resolution mode, the 20 most intense precursors were selected for subsequent HCD fragmentation using rolling collision energy, and tandem mass spectra were acquired in high sensitivity mode with a resolution of 20,000.
Experimental Design and Statistical Rationale
The acquired raw files were first recalibrated based on two background ions with m/z values of 371.1012 and 445.1200, and then converted to peak lists by the AB Sciex MS Data Converter tool version 1.1. Database search was performed with Mascot (Matrix Science, Boston, MA, version 2.3.2) using Proteome Discoverer (Thermo Scientific, Waltham, MA, version 1.2). The spectra were searched individually against the Saccharomyces cerevisiae SwissProt database (version 02_2012 - 6,619 sequences). All the results belonging to the same purification and enzyme digestion were grouped together in Proteome Discoverer, to get the final list of identifications. Trypsin or chymotrypsin were set as the enzyme accordingly, and up to two missed cleavages were allowed. Cysteine carbamidomethylation was set as the fixed modification. Methionine oxidation, serine, threonine, and tyrosine phosphorylation, protein n-term acetylation, as well as lysine acetylation were set as the variable modifications. Peptide tolerance was set to 50 ppm, whereas the MS/MS tolerance was 0.15 Da. Peptides identification where filtered for: (1) minimum ion score of 20, (2) position rank 1, (3) minimum peptide length of 6 amino acids, and (4) peptide tolerance 10 ppm. The phosphorylation site localization of the identified phosphopeptides was performed by the phosphoRS algorithm(36), implemented in Proteome Discoverer. A site localization probability of at least 0.75 was used as the threshold for confident localization. The MS proteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository (37) with the data set identifier PXD008324.
Protein Structures
The following experimentally obtained structures deposited in PDB (38) (www.rcsb.org/) were used: 4OO11 for the exosome (23), 1I3Q2 for the RNA polymerase II (26) and 4CR23 for the 26S proteasome (39). We worked with the exosome structure 4OO1 which does not contain Rrp44 subunit, even though the complete 11 subunits structure is also deposited under ID 4IFD4 (22). None of the Rrp44 PTMs found in this study are located at its interface with other subunits in 4IFD, so the 4OO1 structure that does not contain this 1003 amino acid long chain was used to make calculations less demanding.
The structure of yeast Skp1:Met30 complex was obtained from Genome Wide Docking Database GWIDD (40) as the homology model GWD368CM. For obtaining the model, chains A and B of 1NEX5 PDB structure (41), corresponding to the yeast Skp1 and Cdc4 proteins, were used as templates. However, the N-terminal part of Met30 remained unstructured because of lack of a Cdc4 region that would serve as a template. The first 179 amino acids of the modeled Met30 were therefore removed, and such a modified structural model was used in further steps. Structures of the protein complexes from the benchmark data set were acquired from PDB, using the identifiers listed in the corresponding Table in Betts et al. 2017 (16).
Bio.PDB module of Biopython (42) was employed to parse the PDB files, i.e. to remove the headers, undesired chains and hetero atoms, and to correct possible mistakes in the files. Nucleic acid (chain S) was removed from the exosome structure, and the entire 20S core particle (chains A - G and 1 - 7) from the 26S proteasome structure. 20S proteasome core particle was removed to obtain a smaller and computationally less demanding, while still biologically relevant system, and was possible because all PTMs at the interfaces were located in the 19S proteasome regulatory particle. Finally, an exception regarding hetero atoms removal was the RNA polymerase II structure, in which one Mg2+ and eight Zn2+ ions were retained, as they are either catalytically or structurally important (26).
Benchmarking
We have benchmarked our approach for prediction of PTM's effect, based on molecular dynamics for conformational sampling and Gibbs energy calculation (described below), against FoldX (18) and an extended Mechismo method by Betts et al. 2017 for identification of phosphorylation switches (16). Our benchmark data set consists of mammalian, predominantly human, protein-protein interactions whose stability was experimentally shown to be affected by an interface phosphorylation, and is a subset of the “Phosphoswitch benchmark set” of Betts et al. 2017, where we selected the systems that do not require modeling, i.e. those where the structure of the exact protein pair affected by phosphorylation is available in the PDB (supplemental Table S4.2). This selection was made to allow for a direct comparison of predictions obtained by our MD-based method with the existing data on FoldX and Mechismo performance presented in the mentioned work. In cases where the structure retrieved from PDB contained phosphorylated sites, the respective modifications were removed prior to analysis.
Preparation of PDB Files
pdb4amber from Amber16 software package (43) was used for addition of hydrogen atoms by reduce (44), detection of disulfide bonds, and overall adjustment of atoms and residues names in the PDB file to make them suitable for work with this package. Some protein parts remained unresolved in the structures of both benchmarking and yeast complexes deposited in PDB. pdb4amber gives a warning about such gaps, however this list is based on analysis of spatial distance of atoms and is sometimes incomplete, so we made a parser that detects all the gaps based on residue numeration in original PDB file. Gaps were then treated by addition of the C-terminal cap (N-methyl group attached to the carbon atom of the backbone amide group) and N-terminal cap (acetyl group attached to the nitrogen atom of the backbone), using PyMOL (45). This ensured that the residues flanking the gaps are not interpreted as chain ends and are therefore not charged. A total of 17 gaps were treated in this fashion in the exosome structure, 14 in the RNA polymerase II and 14 in the proteasome regulatory particle (details in supplemental Tables S1.5, 2.5, 3.5).
Addition of Post-translational Modifications to the Structures
Interchain contacts in PDB structures of three yeast complexes were analyzed using the designated WHAT IF (46) tool, which reports a contact when the distance of Van der Waals surfaces is less than 1 Å. Only the PTM sites located on the subunit interfaces were used in further analysis, as they are more likely to have a direct detectable effect on subunits' interactions. Phosphorylation sites that were unlocalized in out data set were excluded from mapping to 3D structures. The PTMs were added to the structures with the capped gaps using the PyTMs plugin (47) of PyMOL. A total of 20 PTMs were added to the exosome structure, 18 to the RNA polymerase II, and 6 to proteasome 19S regulatory particle. Ser162 of Skp1 was phosphorylated in the Skp1:Met30 complex, and each benchmarking system had modifications denoted in supplemental Table S4.2. Finally, all PDB files saved after editing in PyMOL were parsed in order to make final corrections (such as fixing positions and numeration of caps, and adding missing “TER”s between them), using Python.
Parametrization
Program teLeap from Amber16 package was used for parametrization of the systems. The force field ff14SB was used for proteins, and tip3p for water. Parameters for phosphorylated amino acids are also available within the Amber16 package (phosaa10) (48). For acetylated lysines, Amber force field parameters for PTMs developed by Khoury et al. (49) were used. Joung/Cheatham parameters for monovalent and Li/Merz parameters for +2 to +4 charged ions (12–6 normal usage set) optimized for TIP3P water were employed. Magnesium and zinc ions in RNA polymerase II were treated in non-bonded fashion. The disulfide bonds detected by pdb4amber were retained. Each system was neutralized by addition of the required number of counter charged ions (Na+ or Cl−, as needed), and surrounded by a rectangular box of explicit TIP3P water spanning 10 Å from the protein.
Optimization
Optimization of structures was done using sander from Amber16. The optimization was done in 25,000 steps divided in 5 cycles. In each cycle, the first 1000 steps were performed by the steepest descent, and the following 4000 by the conjugated gradient method. In the first three cycles, there was a constraint on (1) the entire protein, (2) heavy protein atoms, and (3) backbone atoms, using force constant of 100 kcal/mol/Å2. The constraint on backbone atoms was reduced to 50 kcal/mol/Å2 in the fourth cycle, whereas in the fifth cycle no constraints were applied.
Molecular Dynamics Simulations
Each system was equilibrated during the first 500 ps, using pmemd from Amber16 package. The time step during the entire equilibration phase was 1 fs. SHAKE algorithm was used to apply constraints on hydrogen-containing bonds, as TIP3P water model requires. The cutoff distance for non-bonded interactions was 15 Å. During the first 300 ps, the canonical NVT ensemble was simulated. The temperature was increased from 0 K to 300 K during the first 250 ps. In the final 50 ps of the first equilibration phase, the temperature was kept constant at 300 K. During these 300 ps, the constraint was applied to all protein atoms using the force constant of 25 kcal/mol/Å2. In the following 200 ps of equilibration, the isothermal-isobaric NpT ensemble was simulated. The values of temperature and pressure were kept at 300 K and 1.0 bar, and no additional constraints were applied to protein atoms.
Production phase of MD simulation was done using Gromacs 5 software (50–57) as a 19.5 ns continuation of 500 ps equilibration. ParmEd 2.7 tool (58) was used to convert the files from Amber to Gromacs formats. The time step was 2 fs and coordinates were written each 1 ps. The LINCS algorithm (55) was used to apply constraints on hydrogen-containing bonds only. The cutoff distance for non-bonded interactions was 12 Å, and the neighbor list was updated each 20 steps. For electrostatic interactions, the particle mesh Ewald method was employed. Temperature and pressure were kept constant at 300 K and 1.0 bar, respectively. Modified Berendsen thermostat was used for temperature coupling and Parrinello-Rahman barostat for pressure coupling. Throughout the simulation, periodic boundary conditions were applied. The infrastructure of the Flemish Supercomputer Centre (VSC) was used to perform the calculations.
Visualization and Analysis
For each of the three large yeast complexes, trajectories of non modified and fully modified (all inter-subunits PTMs added) complex were obtained. Additionally, the exosome complex containing only one modification, acetylated Lys69 in Rrp46, was simulated. For Skp1:Met30 system, nonmodified complex and the one with phosphorylated Ser162 in Skp1 were simulated. The analogous was done for 47 systems from the benchmarking data set. Visual Molecular Dynamics (VMD) program (59) was used for visualization of the trajectories. Tools from the Gromacs package were used for the statistical analysis, such as calculation of root mean square deviation (RMSD), as well as for manipulation of trajectories (correction for periodic boundary conditions, and extraction of the conformational snapshots into a new trajectory file for Gibbs energy calculation). Matplotlib plotting library (60) was used to visualize the analysis results.
Gibbs Energy of Binding
Computational methods of different accuracies have been developed for Gibbs energy calculation of protein-ligand binding (61). The end-point methods use only the initial and final binding states: receptor, ligand and complex. The most commonly employed ones are MM/PBSA and MM/GBSA (Molecular Mechanics energies with Poisson-Boltzmann (or Generalized Born) and Surface Area continuum solvation). GB approximates the theory-based, but computationally expensive, PB equation. While PB describes electrostatics in a solution with ions, GB approximates solute by spheres of dielectric constant that is different from the solvent's.
In both of these methods, the free energy is calculated as the following sum (61).
| (Eq. 1) |
where bonded, electrostatic and Van der Waals interactions energy terms are calculated via molecular mechanics, the polar solvation term Gpol either by solving Poisson-Boltzmann equation (in MM/PBSA) or employing generalized Born (in MM/GBSA), and the non-polar term Gnp usually from a linear relation to the solvent accessible surface area. The entropy term is oftentimes omitted (61) as it does not improve the final result (62), while being the most computationally demanding one.
If the system of interest is a protein complex, the sum of these contributions is calculated for each protein component, as well as for the complex. Furthermore, if the conformational sampling is done e.g. by MD simulations, the average is made for each molecular species, using conformational snapshots (from the stabilized trajectory part). The expression for Gibbs energy of binding of proteins A and B, calculated from a single trajectory of the AB complex, is:
| (Eq. 2) |
with each G term calculated according to the equation (1).
In this work, Amber MMPBSA.py.MPI (63) was used for calculation of Gibbs energy by MM/GBSA using trajectories of protein complexes corrected for periodic boundary conditions. As the backbone RMSD appeared to be quite stable in the last 10 ns of 20 ns long trajectories, a total of 100 conformational snapshots was typically taken by extracting each 100-th frame from this second half of trajectory. Variations of the method were tested on Skp1:Met30 (using last 5 ns or 7.5 ns of simulation; using 100 and 1000 snapshots; using MM/PBSA method). Topology files stripped of water and ions were prepared with Amber ante-MMPBSA.py for protein complex and each binding component individually. Linear Combination of Pairwise Overlaps was used to calculate the surface area for the non-polar solvation term. Entropy term was not calculated (61), and salt concentration of 0.15 mol/dm3 was used.
To assess information about the PTMs' influence on subunits' binding, ΔGbind values of modified and non modified systems are compared. In multimeric complexes, each subunit was treated as a ligand in a separate calculation, with the rest of the complex acting as a receptor. ΔΔGbind is then calculated as:
| (Eq. 3) |
with each term calculated according to the equation (2). The negative ΔΔGbind value for subunit's binding to the remaining of the complex indicates a stabilizing effect of PTMs on its binding, and vice versa for the positive sign.
Finally, MM/GBSA allows per-residue decomposition and therefore getting an insight into the contributions of specific residues to ΔΔGbind, with PTM sites being of special interest. For instance, local binding contribution of an acetylation site in Rpb1 subunit is obtained by decomposition of binding energies of nonmodified and fully modified RNA polymerase II, when Rpb1 is treated as a ligand.
Experimental Validation
Yeast-2-Hybrid assays were carried out by Next Interactions, Inc (Richmond, CA). RRP45 and RRP41 ORF's were PCR amplified using Q5 polymerase (NEB, Ipswich, MA) from plasmids encoding RRP45 and RRP41 cDNA and cloned into pGBKT7-BD and pGAD-AD, respectively, by Gibson assembly. RRP45 mutants, K250R and K250Q were generated by site-directed mutagenesis with Platinum SuperFi DNA polymerase (Invitrogen, Carlsbad, CA). The plasmids were transformed into the yeast Y2HGold and Y187 (Clontech, Mountain View, CA) to generate Y2HGold-pGBKT7-RRP45-WT, K250R, and K250Q and Y187-pGAD-RRP41. Cells were mated on YPAD media and transferred to media lacking Ade, His, Leu and Trp (Sunrise, CA). A quantitative measure of binding was obtained by counting clones after proper dilutions. Tests were done in triplicates. Significance was obtained by Student t test.
Residue Conservation
For each subunit of the yeast exosome, RNA polymerase II and 26S proteasome that was found to contain PTM(s) in this study, a multiple sequence alignment of a corresponding group of orthologous eukaryotic proteins was retrieved from the EggNOG database (64). The conservation was determined as the percentage of sequences that contain the given amino acid at the corresponding position. For the acetylation site, both conservation of the strict position and the one allowing plus-minus one positional variation were calculated. This is based on findings of a recent study, showing that when the conservation of the Lys modification site is lacking, Lys residues appear one position upstream or downstream in the sequence, likely preserving the function independent on the strict position (65). For Ser and Thr phosphorylation sites, either of these two amino acids at the strict position was allowed, while only Tyr was allowed at the strict position of the Tyr modification site.
Conservation was determined in the same way for Lys, Ser, Thr and Tyr sites that do not undergo post-translational modification according to our data, separately for subunits of each of the three complexes. Additionally, the calculations were done taking into account the PTMs data for three complexes available in databases (supplemental Tables S1.6, S2.6, S3.6). To assess the statistical significance of the differences of conservations, the unpaired t test was applied.
RESULTS
Acetylation and Phosphorylation Sites Detected In Purified Yeast Complexes
Our method of choice for obtaining the PTMs data set was tandem affinity purification followed by mass spectrometry (TAP-MS), where two consecutive chromatographic steps are applied in order to purify complexes in their native conditions before MS analysis (Fig. 2). Using TAP-purified proteins for identification of PTMs is much cleaner when compared with the commonly used whole-cell lysates, where it is uncertain whether identified PTMs were indeed part of native complexes. Our method therefore allowed us to obtain the high-fidelity information about PTMs present in the native complexes.
Fig. 2.

Post-translational modifications in native yeast complexes. Individual subunits of the TAP-purified exosome, RNA polymerase II and proteasome separated on the gel, with number of acetylation (Ac) and phosphorylation (P) sites detected by mass spectrometry denoted on the right side from each band.
Very high sequence coverage of up to 98% could be obtained in our MS data because of the combination of used digestion enzymes trypsin and chymotrypsin (supplemental Tables S1.1, S2.1, S3.1). The number of identified subunits in the complexes was 12 in the exosome, 12 in the RNA polymerase II and 34 in the 26S proteasome (2 of which are not among the 33 unique proteasome chains). All exosome subunits were identified with a sequence coverage ranging from 62% to 87%. Core subunits of RNA polymerase II were identified with a coverage ranging from 47% to 98%, whereas the dissociable heterodimer Rpb4/Rpb7 was identified with 25 and 39% sequence coverage, respectively. For the proteasome, a subunit of the base of the 19S regulatory particle was used as bait and, accordingly, proteins of the 19S regulatory particle are observed with a higher sequence coverage (73% on average) than members of the 20S core particle (18% on average). Moreover, the core α subunits that bind directly to the regulatory particle are covered more (25%) than the β subunits (10%). All seven α subunits were identified, whereas one of the seven β subunits (Pre4) is missing.
We identified a total of 385 acetylated and 68 phosphorylated peptides on three purified yeast protein complexes (197 identifications for the exosome, 193 for the RNA polymerase II and 63 for the 26S proteasome; supplemental Tables S1.2, S2.2, S3.2). After merging the redundantly detected sequences and confidentially localizing the positions of the phosphorylated residues, a total of 53 lysine acetylation, 12 localized and 6 unlocalized (serine/threonine/tyrosine) phosphorylation sites were detected on the exosome; 50 lysine acetylation, 10 localized and 1 unlocalized (serine/threonine/tyrosine) phosphorylation sites were detected on the RNA polymerase II; 26 lysine acetylation, 6 localized and 6 unlocalized (serine/threonine) phosphorylation sites were detected on the 26S proteasome (supplemental Tables S1.3, S2.3, S3.3). According to dbPTM (13) and phosphoGRID (66), only 15 localized and 6 unlocalized (serine/threonine/tyrosine) phosphorylation sites from our data set have previously been identified. Furthermore, only 18 lysine acetylation sites from our data set are listed in the Protein Lysine Modification Database (PLMD) (67) (supplemental Tables S1.6, S2.6, S3.6).
Our computational analysis of PTM roles was focused on a subset of PTMs that are located at the interfaces of subunits in the respective complexes. This included a subset of 20 PTMs in the exosome, 18 in the RNA polymerase II and 6 in the proteasome 19S regulatory particle (supplemental Files S1.5, S2.5, S3.5; Fig. 3B–3D); the unlocalized phosphorylation sites were excluded from the analysis.
Fig. 3.

Structures of the simulated complexes. Interface located modification sites are emphasized by the space filled representation. Each site carries a label denoting the subunit, amino acid type, and its position within the respective chain. A, In Skp1:Met30 complex, Skp1 is shown in magenta and Met30 in cyan. B, Exosome and (C) RNA polymerase II subunits are each shown in different color, while (D) parts instead of individual 33 subunits of 26S proteasome are differently colored: 20S core particle β subunits in cyan and α in blue, and 19S regulatory particle base subunits in magenta and lid in gray.
Robustness of Gibbs Energy Calculation
We first tested the robustness of our pipeline for prediction of PTM's effect on interaction stability using the yeast Skp1:Met30 complex (Fig. 3A). Skp1 is an essential protein component of the SCF (Skp1, Cullin, F-box protein) E3 ubiquitin ligase, and it binds the F-box motif, present in amino acids 180 - 225 in the yeast Met30. These amino acids were found as necessary and enough for Met30 to bind Skp1(68). By means of in vivo complementation and immunoprecipitation, Beltrao et al. (14) found that phosphorylation of the highly conserved Ser162 site in Skp1 weakens the Skp1:Met30 interaction.
In brief, our computational approach consists of conformational sampling of the complex via MD simulations, followed by calculation of Gibbs energy of binding ΔGbind by MM/GBSA method. Employing MD ensures the insight into the conformational changes that are the result of PTMs' introduction into the protein, and therefore the altered amino acids interaction patterns even in the protein regions that are distant from the PTM site. This kind of information cannot be obtained by looking into a static conformation, or even by using just optimization as it does not allow overcoming of the energetic barriers. Finally, performing simulation and calculation for both nonmodified and post-translationally modified complex allows the comparison of their Gibbs energies of binding, and therefore assessment of the PTM effect on binding stability through the ΔΔGbind value (details in the Methods section).
The workflow was designed bearing in mind that the systems of interest are significantly larger than Skp1:Met30 (2775 residues in exosome, 3570 in RNA polymerase II and 6996 in 19S proteasome regulatory particle, compared with 655 in Skp1:Met30). In the recent studies that employed MM/GB(PB)SA, different MD production times (from 5 ns to 100 ns, or even more) and numbers of conformational snapshots were used. Here, the MD simulation time of 20 ns was chosen in order to allow for system stabilization, while not being computationally overly demanding. After obtaining the trajectories for Skp1:Met30 system, the root mean square deviation (RMSD) of backbone showed that the complex was fairly stabilized in the last 10 ns (supplemental Fig. S1A). This trajectory part was then used for extraction of the conformational snapshots. Similar predictions of ΔΔGbind were obtained by analysis of 100 and 1000 snapshots using MM/GBSA, and independent on whether the snapshots were taken from the last 5, 7.5 or 10 ns of the trajectories (supplemental Table S4.1), showing the robustness of the method. Using 1000 snapshots reflects in smaller standard error when compared with 100 snapshots, however average ΔGbind values were not significantly affected. Moreover, using either MM/PBSA or MM/GBSA yielded the positive value of ΔΔGbind, which indicates the destabilization effect of Ser162 phosphorylation on Skp1:Met30 binding. MM/GBSA applied on 100 conformational snapshots from the last 10 ns of MD was therefore chosen for further benchmarking, as it is both 3 times faster than MM/PBSA and allows per residue decomposition of binding energy, which is not possible when e.g. PBSA surface terms are present. The decomposition allows identification of residues with large predicted effects on binding (supplemental Fig. S2A), and here it suggested that the modified Ser162 also has a slight destabilization effect. This possibility of decomposing ΔGbind offers further insight into effects of PTMs' introduction on the distant amino acids interaction patterns, as exemplified below.
Benchmarking
We benchmarked our method against FoldX and an extended Mechismo method from Betts et al. 2017, using a set of 24 mammalian protein-protein complexes known to be stabilized by an interface-located phosphorylation, as well as 23 complexes in which phosphorylation has the opposite effect. As mentioned above, FoldX uses an empirical force field for its predictions and allows only the optimization of the side chains surrounding the phosphorylation site before calculating the binding energy, while the prediction by Betts et al. relies on amino acid interaction patterns and the conservation of the site. Differently from our MD-based approach, these methods have notable limitations regarding type, number and location of PTMs (details in the Discussion section).
The benchmarking results (supplemental Table S4.2) show that FoldX has the lowest accuracy of three methods that does not improve by increasing the prediction threshold; Mechismo can reach 75% (destabilizing/disabling) and 72% (stabilizing/enabling) accuracy at the cost of coverage (sensitivity) (13 and 33%) (Fig. 4), with our method having a much higher coverage. It is interesting to note that the phosphorylation sites' contribution to the binding calculated by our method, where majority of them contribute to ΔGbind in a destabilizing fashion, does not necessarily coincide with the overall effect indicated by the ΔΔGbind of the complex. While FoldX and method by Betts et al. perform similar or a bit better than ours in predicting the stabilizing switches, our method performs better in predictions of the destabilizing effects. Another difference is in the number of predictions that are above the threshold of what is considered to indicate a significant effect on binding in the given method (±1.7 for Mechismo from Betts et al. 2017, ±2 kcal/mol for ΔΔG methods), where the numbers are quite low for two opponent methods especially for the destabilizing systems. Overall, these results justify our application of MD and MM/GBSA for assessing the effects of PTMs in large yeast complexes.
Fig. 4.
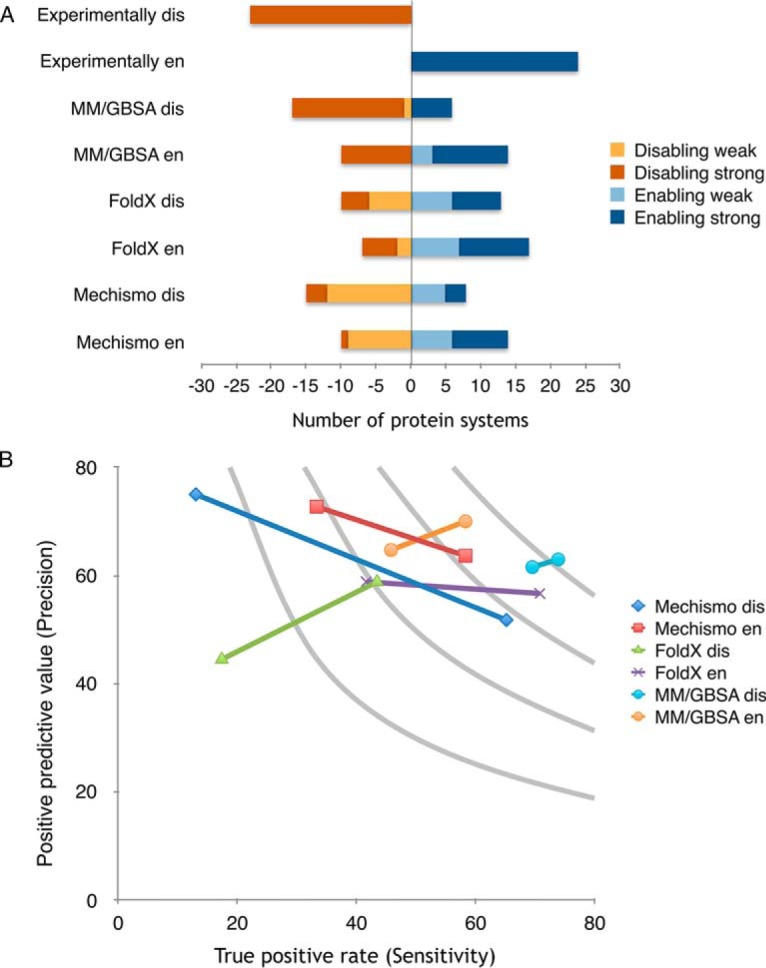
Benchmarking of molecular dynamics based approach for prediction of PTMs effect on binding. A, Performance of Mechismo (Betts et al. 2017), FoldX and the MM/GBSA method presented in this work in predicting the effect of interface located phosphorylation sites on binding. Destabilizing (disabling, short dis) cases are shown in blue and stabilizing (enabling, short en) in orange, with darker shades representing predictions above the threshold for the given method. B, Precision and sensitivity of the predictions of enabling and disabling effects of interface phosphorylation sites by the three methods. Two data points for each case refer to the predicted values above/below threshold. As an ideal method has high values of both precision and sensitivity, MM/GBSA outperforms the other methods, especially in predictions of disabling effects.
Effect of Interface PTMs on Binding of Subunits
For the 10 subunits of the exosome, with the exception of Rrp46 we predict the stabilizing roles of PTMs introduction on subunits binding in the fully modified complex based on the negative ΔΔGbind values (supplemental Table S4.3; Fig. 5A). To see how these predictions compare with a singly modified exosome, we introduced a single acetylation at the Lys69 in Rrp46. This residue was chosen as it had the highest local contribution among the 20 PTMs in the fully modified exosome. Although the local binding contribution of this acetylation site is comparable between the fully and singly modified exosome (−13.95 and −11.45 kcal/mol), the overall effect on subunits' binding is largely different, with only 4 out of 10 subunits showing better binding in singly modified than in non modified complex, as compared with 9 out of 10 in the fully modified. This suggests that there is (1) a long range effect of a single modification on binding of subunits in multimeric complex, captured because of dynamics, and (2) an optimal combination of PTMs that offers a complex-wide stabilization of binding.
Fig. 5.

Predicted effects of phosphorylations and acetylations on binding affinities in yeast exosome, RNA polymerase II and exosome. A, ΔΔGbind for all subunits of three fully modified yeast complexes and the singly modified exosome, as obtained by MM/GBSA while the respective subunit is treated as a ligand. B, Individual contributions of the interface modification sites to the overall ΔΔGbind of the respective subunit. Phosphorylation sites are depicted in blue, and acetylation in red. As in (A), negative values indicate stabilizing effect on the binding after the modifications were introduced in the structure of the complex, while the opposite is true for positive values.
Comparison of individual residues' contributions to binding in nonmodified and fully modified exosome further revealed residues with large predicted effects on ΔΔGbind (supplemental Table S4.6; supplemental Fig. S2C). Along with some PTM sites, these are predominantly charged residues located either in the proximity of PTM sites or influenced by PTMs introduction indirectly. For instance, acetylation of Lys101 in Rrp4 is accompanied by the change of the interaction pattern in its surrounding, majorly affecting Asp149 and Arg239 residues in Rrp41 (Fig. 6A, 6B). Therefore, these Rrp41 residues have large and opposing effects on Rrp4:Rrp41 binding. Other residues located near the PTM sites that were predicted to have large effect on subunits ΔΔGbind are Glu202 in Rrp41 (close to Lys110 of Rrp45), Glu130 in Rrp41 (close to Lys48 from the same subunit), and Lys154 in Rrp43 and Asp181 in Rrp46 (close to Lys69 in Rrp46). On opposite, the major destabilizing residues in Rrp46:Rrp40 interaction are Arg210 and Asp9, which are not in the proximity of any PTM site but are stably involved in a salt bridge at the interface of subunits in the non modified, but not in the fully modified complex (Fig. 6C).
Fig. 6.
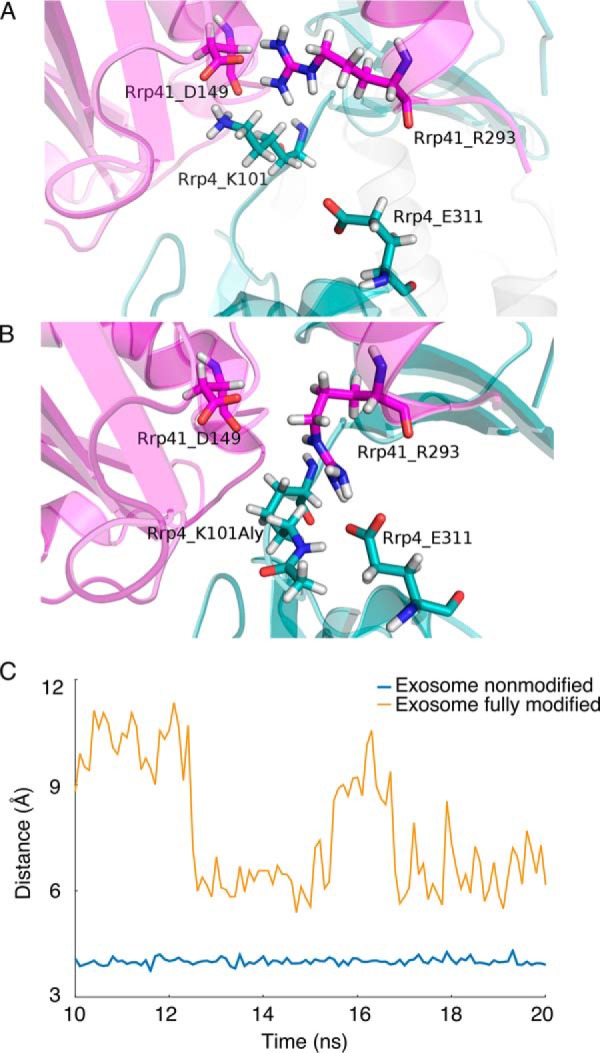
Molecular dynamics revealed significant conformational changes after introduction of post-translational modifications. Changes of the interaction pattern at the interface of Rrp4 and Rrp41 exosome subunits between (A) non modified and (B) fully modified complex. Spatial re-orientation of the acetylation site Rrp4 Lys101 is accompanied by a change of interaction partner of the Rrp41 Arg293 residue from the Asp149 in its own chain, to Glu311 in the opposing subunit. Because of this change, Rrp41 Asp149 has a destabilizing effect on ΔΔGbind, while the opposite is true for Rrp41 Arg293. This can also be seen from per residue decomposition (supplemental Fig. S2C). C, Distance of salt bridge forming residues Arg210 in Rrp46 and Asp9 in Rrp40 subunit at the respective interface, measured in 100 snapshots used for MM/GBSA analysis as a distance between arginine Cζ and Asp Cγ atoms. Although these residues are distant from the modified sites in the fully modified exosome, the presence of the PTMs in the structure affected their interaction.
Differently from the fully modified exosome, for 8 out of 10 RNA polymerase II subunits we predict the binding to become less favorable after the addition of all 18 inter-subunits located PTMs, reflected in the positive ΔΔGbind values (Fig. 5A). The most prominent destabilizing effect among all subunits is observed for the phosphorylation site Tyr88 of Rpb6, which directly affects binding of Rpb1:Rpb6 (supplemental Fig. 2D). This example shows how a single modification site of the interface residue might significantly influence binding. As in the exosome, other residues predicted to majorly affect ΔΔGbind are predominantly the charged ones, however here most of them are not near the PTM sites, e.g. the destabilizing residues Glu1342 and Asp1373 in Rpb1, Glu1004 and Glu1206 in Rpb2, Asp60 and Glu177 in Rpb3, and the stabilizing Asp1009 and Asp1100 in Rpb2, and Asp2 and Arg11 in Rpb5. For instance, Arg11 in Rpb5 forms salt bridge with Glu1315 in Rpb1 only in the fully modified complex, therefore contributing in a stabilizing manner to the Rpb1:Rpb5 interaction. In general, visualization of the residues in the obtained trajectories shows obvious differences in the positions of the neighboring, interacting amino acids. Residues with prominent contributions that are located near the PTM sites in RNA polymerase II are Lys452 in Rpb1 (while non-phosphorylated Tyr88 interacts with nonacteylated neighboring Lys87 in Rpb6, once they are modified the phosphorylated Tyr88 involves in interaction with the mentioned Lys452) and Asp266 and Asp268 at the C terminus of Rpb3 (whereas Asp266 interacts with side chains of non modified Lys84 and Lys88, in the fully modified complex its carboxyl group is close to that of Asp268).
In the simulated proteasome 19S regulatory particle, 2 out of 5 subunits containing modified residues were predicted to have overall positive, but small ΔΔGbind values. In the entire complex, 12 out of 19 subunits bound more stably to the remaining of the complex, indicating a predominantly stabilizing influence of 6 interface PTMs on the level of the entire 19S regulatory particle. Examples of highly destabilizing residues are Asp285 in Rpt2, Glu389 in Rpt3 and Glu293 in Rpt4, while Glu196 in Rpt2 was predicted to have a pronounced stabilizing effect (supplemental Fig. S2E). None of these residues are located close to a PTM site. Again, for all these negatively charged residues, interaction pattern with the nearby positively charged residues changes after PTMs addition, consequently affecting their contribution to ΔGbind. This example, together with those from exosome and RNA polymerase II, underscores the importance of looking into dynamics when studying effect of PTMs on protein interactions.
Local Binding Contributions of PTM Sites
The results of MM/GBSA energy decomposition indicate the locally destabilizing effect of phosphorylations on the subunits binding for all examined phosphorylation sites: three in the exosome, two in the RNA polymerase II and one in the proteasome 19S regulatory particle (supplemental Tables S4.3, S4.4, S4.5; Fig. 5B). Opposite from that, the acetylations are predicted to contribute to the binding in a locally stabilizing manner with almost no exceptions. Two cases of destabilizing acetylation sites arose, one in the exosome and another in the RNA polymerase II, however their ΔΔGbind contributions are predicted to be small. Even at a local level, predicted ΔΔGbind contributions for all phosphorylation and majority of acetylation sites are larger than 2 kcal/mol, which is often used as a threshold that defines a residue with significant impact on protein interactions (17). Notably, the global effect that these PTMs have on the ΔΔGbind of subunits largely exceeds 2 kcal/ mol with practically no exceptions.
Experimental Validation
We have next experimentally validated our computational predictions by yeast 2-hybrid (Y2H). After screening for pairs of exosome subunits whose interactions were previously successfully captured by Y2H, as listed in the SGD database (69), we identified the PTMs from our fully modified exosome that are located on interfaces of those subunits (supplemental Table S5.1). From those, we tested the effect of Rrp45_Lys250 acetylation on interaction with Rrp41 subunit, as i) we predict the local contribution of this site to be highly stabilizing, and ii) binding of Rrp45 is predicted to be stabilized for ∼50% when compared with the ΔΔGbind in the nonmodified exosome. We do not predict a switch from non-binding to binding, but rather an affinity change (ΔGbind values of both nonmodified and modified subunit are negative).
The Rrp41 interaction with Rrp45_Lys250Arg, in which acetylation is prevented, was found to be significantly weaker compared with the wild-type. Lys250Arg had around 50% less Y2H reporter activation in both experiments (supplemental Tables S5.2, S5.3), and when using both selection for histidine and adenine reporter activation. The experimental results are therefore consistent with our predictions that lysine acetylation stabilizes this interaction.
Mechismo Predictions of PTMs Effect in Three Complexes
The Mechismo tool developed by Betts et al. 2015 (15), available as a web-server, enables the prediction of the effect on the protein interaction because of mutation or modification of an interface located residue. We made predictions for each of the 44 inter-subunit PTM sites from yeast exosome, RNA polymerase II and proteasome 19S regulatory particle using Mechismo, to make comparison with our results. However, it did not make predictions for all 44 sites (supplemental Tables S4.3, S4.4, S4.5). This reflected either in the output related to nucleic acids and nucleotides binding, or in the score equal to zero, which suggests that the PTM sites were not interpreted as interface-located residues. For the sites that Mechismo did recognize as interface residues in the respective complexes, its predictions coincided with our predicted binding contributions in 14 out of 18 cases.
Conservation of the Modified Sites
Finally, we wanted to obtain insight into conservation of the interface modification sites examined in the present study. In proteasome, the interface located acetylation sites from our data set were found to be statistically significantly more conserved than the nonmodified Lys (p value < 0.05), as well as than the noninterface located sites (supplemental Table S6.2). Differently from that, Ser phosphorylation sites turned out as less conserved when compared with nonmodified Ser in both RNA polymerase II and proteasome (supplemental Table S6.3). Similar comparisons for other sites returned either as statistically insignificant, or were impossible to do because of lack of data (e.g. only one Thr was identified as a PTM site in proteasome in this study).
As PTM sites were previously reported as more conserved than other residues (14), the statistically insignificant differences might be the result of our incomplete picture of PTM sites present in these proteins. We have therefore additionally considered PTMs that were previously found and are reported in on-line databases, even though they dominantly originate from large proteomics studies where it is uncertain whether PTMs are part of native complexes. Performing the conservation analysis with this extended data set (supplemental Tables S1.6, S2.6, S3.6) did not return significantly different average values for nonmodification sites when compared with non PTM sites from this study only, and the same is true for modification sites. The only exception are Ser phosphorylation sites in RNA polymerase II, which now appeared to be less conserved. However, this might be the bias of our small data set containing only 3 Ser phosphorylation sites in RNA polymerase II.
We found the average conservation of all sites to be the lowest in the exosome and the highest in the proteasome for each of the four amino acids separately, with average conservation of Tyr in proteasome going beyond 50%. In each of the three protein complexes, 5 to 6 interface located PTM sites are conserved more than 50%, with the most conserved residue being Lys87 in Rpb6 of RNA polymerase II reaching 98.5% (supplemental Table S6i.1; Fig. 7; supplemental Fig. S4). Conservation of all interface located PTMs in proteasome is above the average. Values of conservation for both interface and noninterface located acetylation sites are spread from below to above the average conservation, while majority of the noninterface phosphorylation sites appears to be less conserved than average nonmodified site. Interestingly, in the RNA polymerase II system we can also see a trend of conserved interface PTM sites having larger local effect on the stability of binding (supplemental Fig. S3B).
Fig. 7.
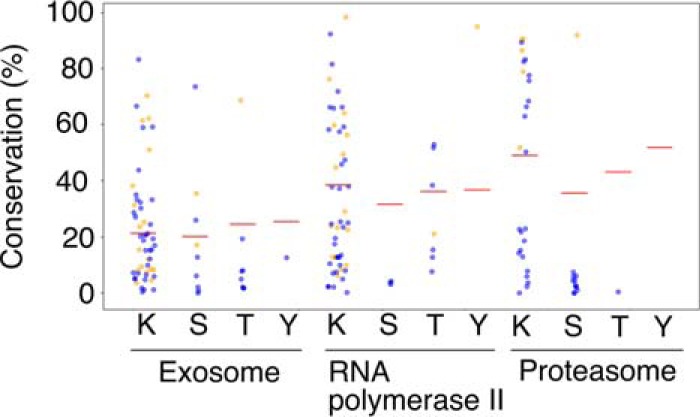
Conservation of modification sites identified in this study. Interface positioned residues are shown in orange, while the others are depicted in blue. Conservation with taking into account plus-minus one positions is shown for lysines. Red horizontal lines represent average conservations of non-modified residues of the same type in the investigated subunits of the respective complexes.
DISCUSSION
In this work, we have extended the knowledge on post-translational modifications in yeast Saccharomyces cerevisiae by identifying novel phosphorylation and acetylation sites, as well as predicted their roles in interactions within three large and essential complexes - exosome, RNA polymerase II and proteasome. The latter was never done in a dynamic fashion and with simultaneously taking into account multiple PTMs in protein complexes of these sizes (up to 7000 amino acid residues in explicit solvent). Finally, we have experimentally tested our computational predictions on the Rrp45:Rrp41 exosome interactions, affected in a stabilizing manner by Rrp45_Lys250 acetylation.
Employing TAP-MS allowed us to identify the modifications that are indeed present in complexes in their native states. The number of detected acetylation sites is approximately five times larger than the number of phosphorylation sites within each complex. While we successfully captured all subunits of exosome and RNA polymerase II, this was not the case for 26S proteasome, probably because of its size. Employing more baits at different proteasome subunits would likely result in a more complete data set for this complex. Given the high coverage, our PTMs data set is likely comprehensive.
While it was argued that introduction of point mutations causes only local conformational changes that do not extend on the tertiary structure (70), numerous examples show that PTMs, such as phosphorylation and acetylation, can cause major conformational changes with great impact on protein function. For instance, comparison of crystal structures of nonphosphorylated and phosphorylated rabbit muscle glycogen phosphorylase nicely demonstrates how introduction of modification can cause long range effects through the conformational changes. More specifically, phosphorylation of Ser14 in this protein induces adoption of α-helical structure of N terminus, and this change gets transmitted as far as 30 Å further to the active site, resulting in T-R transition (71). Furthermore, a well-known example of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase enzyme demonstrates how phosphorylation can act as a trigger between two functions of a bifunctional enzyme, again through the conformational changes it induces (72). Finally, phosphorylation of intrinsically disordered protein domains, e.g. the AF1 domain of glucocorticoid receptor, can cause the formation of secondary and tertiary structure, providing a mechanism for functional activation of such domains (73). Although it received less research attention than phosphorylation, lysine acetylations also induce changes that play role in many different regulatory events. For example, acetylation-induced allosteric conformational changes can provide a regulatory switch between two functions of a protein, as was shown for heat shock protein 70 (Hsp70), where acetylation status of Lys77 determines the co-chaperone that will preferably bind to the protein, and consequently whether Hsp70 will take place in protein degradation or refolding (74). Lysine acetylation can also induce conformational changes in a crosstalk with other post-translational modification, such as methylation in p53 protein (75). The notion that PTMs can cause such large changes in protein structure underlines the need for a dynamic approach in prediction of their effects on the protein and its interactions.
A notable difference also exists between mutations and modifications data sets used in publications. While the benchmark data set of Betts et al. 2017 (16) had a bias toward stabilizing phosphorylation sites (238 out of 335 sites were experimentally found to enable interactions), mutations data sets dominantly consist of destabilizing sites. Examples include the benchmarking data set of Betts et al. 2015 (15) based on UniProt data for human site-directed mutations and disease variants, in which 79% of mutations and variants have disabling, 3.2% enabling effect, and the remaining are neutral. Furthermore, in their prediction of affinity changes for interface-located protein mutations, Dourado et al. (70) employed a subset of SKEMPI (76), the data set of heterodimeric protein complexes mutants with experimentally determined ΔΔG, in which 987 out of 1,254 mutations are destabilizing. The difference in biases is likely the result of fundamental difference between post-translational modifications and mutations—while the first have evolved to appear at specific locations, often to modulate protein function and interactions, the appearance of the latter works against what the nature has optimized. Taken together with the fact that PTMs in our data set were detected in native complexes, a more frequent occurrence of stabilizing than destabilizing effects on binding is not surprising.
In the present work, we have computationally explored the influence that interface located PTMs, majority of which we have newly discovered, have on the subunits' interaction, where we were mainly interested in direction of the change (stabilization versus destabilization). Although the TAP-MS approach allowed the PTMs detection on purified native complexes, from these data it is not possible to distinguish which combination of PTMs was present in which natively purified complex molecule. Ideally, we would simulate structures of complexes with different combinations of PTMs that co-occur in the yeast cell. However, as such data is unavailable, and because proteins are oftentimes modified on multiple sites (65), in the present study we decided to add all intersubunits located PTMs on the structure simultaneously. The next step to tackle the question of PTM combinations co-existing within native complexes would be the top-down proteomics.
On a local level, we predict interface phosphorylation sites to be destabilizing, presumably because the negative charge could not be locally compensated because of absence of positively charged groups. On opposite, acetylation of a site dominantly caused stabilization of the binding, usually because of diminishing lysine's positive charge that is not involved in a salt bridge. Thanks to dynamics, we could also capture broader effects that these modifications have on subunits' binding in multimeric complexes, which do not necessarily coincide with the local effect of PTM sites for a subunit in which they are located. For instance, while presence of 20 interface PTMs in exosome was predicted to cause a more stable binding of 9 out of 10 subunits, 18 PTMs in RNA polymerase II appeared to destabilize binding of 8 out of 10 subunits, even though both systems predominantly contained acetylations that are locally stabilizing the binding. As the PTMs are detected in natively purified complexes, we would expect the overall effects to be mainly stabilizing. This result might therefore suggest that those 18 PTMs are likely not simultaneously present in the RNA polymerase II, but were instead present in different RNA polymerase II sub-populations. Similarly, if the predicted effects on the subunits' binding are compared between fully and singly modified exosome, the complex stabilization is achieved by the presence of multiple PTMs, rather than a single one. The magnitude of ΔΔGbind compared with the ΔGbind of nonmodified subunit ranged from 3% for Rpb1 in the RNA polymerase II, to as high as 55% in the case of Rrp41 in the exosome. Therefore, although we can assume the individual effect of a PTM site based on the chemistry of a modification, the overall consequences on subunits' binding is captured thanks to the freedom of residues to reposition in space.
Compared with Mechismo and FoldX, which produce results in a matter of minutes, our MD-based approach is computationally more intensive, mainly because of MD simulations component. For instance, the 500 ps equilibration phase took 38 h of computational time on a single compute node (CPU, 20 cores, Flemish Supercomputer Center infrastructure) for exosome in explicit water, 56 h for RNA polymerase II, and 180 h for proteasome regulatory particle. Moreover, the 19.5 ns production phase of MD required 58 h for exosome and 102 h for RNA polymerase II using 2 nodes, and 71 h for proteasome using 8 compute nodes. Notably, these simulated complexes are very large, containing 2775, 3570, and 6996 amino acids, respectively. Protein complexes of smaller sizes demand less time, e.g. Dynll1 homodimer from the benchmarking data set (system with a total of 170 amino acids) was equilibrated in 3.5 h on 1 node, while the production phase required 6 h of time on 2 nodes. Certain modifications of the workflow could be made with the aim of reducing the time and/or computational resources needed. For example, time step in equilibration phase could be set to 2 fs instead of 1 fs, which would halve the time required for this step. Moreover, the production phase could be of shorter total duration, especially for systems that stabilize quickly; in a recent study, MD simulations of only 1 ns duration were used for conformational sampling before MM/PBSA (77). Furthermore, more computational resources could be employed for the time saving purposes, especially when dealing with larger systems. If our MD-based approach were to be used for predictions on a larger set of protein complexes (e.g. on a proteome wide level), the above changes would be a good way to reduce the overall analysis time, making such an analysis feasible.
An important advantage of our computational approach is obtaining a dynamic view of a protein after a PTM is introduced. Although it is known that PTMs can cause conformational changes as exemplified above, alternative approaches such as Mechismo and FoldX do not take dynamics into account. Instead, Mechismo uses static picture, if the amino acids interaction pattern stays unchanged when a PTM is introduced, whereas FoldX optimizes surrounding side chains, but does not allow for any major perturbations of the structure. Our result show that, on opposite, interactions patterns can be changed not only at short, but also at long distances from the PTM site (Fig. 6), which is nicely demonstrated by ΔΔGbind values different from zero for subunits in the singly modified exosome in our study. Further limitations of Mechismo method originate from its dependence on the local rules and the need for training sets, as shown in Results for the three yeast complexes where it failed to make predictions for majority of PTMs. Mechismo also cannot assess what would be the overall effect if multiple PTMs are present in the structure simultaneously. This is rather different from our approach, which requires knowledge of a structure and PTMs' positions only and has no difficulty in making predictions of multiple PTMs' effect to binding in a complex. On the other side, although it supports predictions with multiple PTMs present in complex, FoldX has only a very narrow set of modifications that can be introduced with its “PositionScan” functionality—phosphorylated Ser, Thr and Tyr, and hydroxyproline. On contrary, PyTMs plugin that we use for preparation of the structures for MD enables introduction of 11 different post-translational modifications, including Ser/Thr/Tyr phosphorylation and Lys acetylation. Moreover, Amber force field parameters by Khoury et al. (49) are developed for 32 common post-translation modifications. Finally, although these tools are limited on predictions of the effect of interface located modifications, our approach is applicable independent of PTM site's position. In conclusion, the approach we show here is easily applicable to both known and PTMs that are yet to be discovered, independent of their position within the complex.
The functional roles of the post-translational modifications in the examined complexes remain to be clarified in the cellular conditions. For instance, if the combination of 18 interface PTMs actually is present in a single RNA polymerase II molecule in the cell, the destabilization of subunits' binding might serve to prime this complex for a specific function it has to perform at a certain moment. Finally, it would be very interesting to get a dynamic insight into the PTMs effect on proteome level, especially if it would be accompanied by the knowledge of PTMs regulation. With the pipeline that we developed in this work, more analyses can be done on large complexes in the future to shed more light on the role of post-translational modifications.
DATA AVAILABILITY
The MS proteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository (37) with the data set identifier PXD008324.
Supplementary Material
Acknowledgments
We would like to thank the anonymous reviewers for useful comments to improve the manuscript.
Footnotes
* This work is supported by Onderzoeksraad, KU Leuven (KU Leuven Research Fund) (to V.N.). N.S. is a doctoral fellow (1112318N) of Fonds Wetenschappelijk Onderzoek (the Research Foundation - Flanders) (FWO). P.G. and A.J.R.H. acknowledge support from the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (Netherlands Organization for Scientific Research) (NWO) funding the large-scale proteomics facility Proteins@Work (project 184.032.201) embedded in the Netherlands Proteomics Centre. ACG acknowledges support from the CellNetworks (Excellence Initiative of the University of Heidelberg). The computational resources and services used in this work were provided by the VSC (Flemish Supercomputer Center), funded by the Research Foundation - Flanders (FWO) and the Flemish Government - department EWI.
 This article contains supplemental material.
This article contains supplemental material.
Nikolina Šoštarić1 (nikolina.sostaric@kuleuven.be), Francis J. O'Reilly2,$ (oreilly@tu-berlin.de), Piero Giansanti3,4,# (piero.giansanti@tum.de), Albert J. R. Heck3,4 (a.j.r.heck@uu.nl), Anne-Claude Gavin2 (gavin@embl.de), Vera van Noort1,5 (vera.vannoort@kuleuven.be).
Footnotes: 1PDB Number 4OO1, 2PDB Number 1I3Q, 3PDB Number 4CR2, 4PDB Number 4IFD, 5PDB Number 1NEX.
1 The abbreviations used are:
- PTM
- post-translational modification
- CP
- 20S yeast proteasome core particle
- CTD
- C-terminal domain of Rpb1 subunit of RNA polymerase II
- MD
- molecular dynamics
- MM/GBSA
- molecular mechanics energies combined with Generalized Born and surface area continuum solvation
- MM/PBSA
- molecular mechanics energies combined with Poisson-Boltzmann and surface area continuum solvation
- RMSD
- root mean square deviation
- RP
- 19S yeast proteasome regulatory particle
- TAP-MS
- tandem affinity purification followed by mass spectrometry
- Y2H
- yeast 2-hybrid.
REFERENCES
- 1. Seet B. T., Dikic I., Zhou M.-M., and Pawson T. (2006) Reading protein modifications with interaction domains. Nat. Rev. Mol. Cell Biol. 7, 473–483 [DOI] [PubMed] [Google Scholar]
- 2. Benayoun B. A., and Veitia R. A. (2009) A post-translational modification code for transcription factors: sorting through a sea of signals. Trends Cell Biol. 19, 189–197 [DOI] [PubMed] [Google Scholar]
- 3. Kim S. C., Sprung R., Chen Y., Xu Y., Ball H., Pei J., Cheng T., Kho Y., Xiao H., Xiao L., Grishin N. V., White M., Yang X.-J., and Zhao Y. (2006) Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 23, 607–618 [DOI] [PubMed] [Google Scholar]
- 4. Cohen P. (2001) The role of protein phosphorylation in human health and disease. Eur. J. Biochem. 268, 5001–5010 [DOI] [PubMed] [Google Scholar]
- 5. Audagnotto M., and Dal Peraro M. (2017) Protein post-translational modifications: In silico prediction tools and molecular modeling. Comput. Struct. Biotechnol. J. 15, 307–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beausoleil S. A., Jedrychowski M., Schwartz D., Elias J. E., Villen J., Li J., Cohn M. A., Cantley L. C., and Gygi S. P. (2004) Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. 101, 12130–12135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., and Mann M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
- 8. Wang Q., Zhang Y., Yang C., Xiong H., Lin Y., Yao J., Li H., Xie L., Zhao W., Yao Y., Ning Z.-B., Zeng R., Xiong Y., Guan K.-L., Zhao S., and Zhao G.-P. (2010) Acetylation of Metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327, 1004–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Choudhary C., Kumar C., Gnad F., Nielsen M. L., Rehman M., Walther T. C., Olsen J. V., and Mann M. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 [DOI] [PubMed] [Google Scholar]
- 10. Bannister A. J., and Kouzarides T. (2011) Regulation of chromatin by histone modifications. Cell Res. 21, 381–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hammond J. W., Cai D., and Verhey K. J. (2008) Tubulin modifications and their cellular functions. Curr. Opin. Cell Biol. 20, 71–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhou H., Watts J. D., and Aebersold R. (2001) A systematic approach to the analysis of protein phosphorylation. Nat. Biotechnol. 19, 375–378 [DOI] [PubMed] [Google Scholar]
- 13. Huang K.-Y., Su M.-G., Kao H.-J., Hsieh Y.-C., Jhong J.-H., Cheng K.-H., Huang H.-D., and Lee T.-Y. (2016) dbPTM 2016: 10-year anniversary of a resource for post-translational modification of proteins. Nucleic Acids Res. 44, D435–D446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Beltrao P., Albanèse V., Kenner L. R., Swaney D. L., Burlingame A., Villén J., Lim W. A., Fraser J. S., Frydman J., and Krogan N. J. (2012) Systematic functional prioritization of protein posttranslational modifications. Cell 150, 413–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Betts M. J., Lu Q., Jiang Y., Drusko A., Wichmann O., Utz M., Valtierra-Gutiérrez I. A., Schlesner M., Jaeger N., Jones D. T., Pfister S., Lichter P., Eils R., Siebert R., Bork P., Apic G., Gavin A.-C., and Russell R. B. (2015) Mechismo: predicting the mechanistic impact of mutations and modifications on molecular interactions. Nucleic Acids Res. 43, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Betts M. J., Wichmann O., Utz M., Andre T., Petsalaki E., Minguez P., Parca L., Roth F. P., Gavin A.-C., Bork P., and Russell R. B. (2017) Systematic identification of phosphorylation-mediated protein interaction switches. PLOS Comput. Biol. 13, e1005462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nishi H., Hashimoto K., and Panchenko A. R. (2011) Phosphorylation in protein-protein binding: effect on stability and function. Structure 19, 1807–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schymkowitz J., Borg J., Stricher F., Nys R., Rousseau F., and Serrano L. (2005) The FoldX web server: an online force field. Nucleic Acids Res. 33, W382–W388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Narayanan A., LeClaire L. L., Barber D. L., and Jacobson M. P. (2011) Phosphorylation of the Arp2 subunit relieves auto-inhibitory interactions for Arp2/3 complex activation. PLoS Comput. Biol. 7, e1002226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kumar P., Chimenti M. S., Pemble H., Schönichen A., Thompson O., Jacobson M. P., and Wittmann T. (2012) Multisite phosphorylation disrupts arginine-glutamate salt bridge networks required for binding of cytoplasmic linker-associated protein 2 (CLASP2) to end-binding protein 1 (EB1). J. Biol. Chem. 287, 17050–17064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chiappori F., Mattiazzi L., Milanesi L., and Merelli I. (2016) A novel molecular dynamics approach to evaluate the effect of phosphorylation on multimeric protein interface: the αB-Crystallin case study. BMC Bioinformatics 17, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Makino D. L., Baumgärtner M., and Conti E. (2013) Crystal structure of an RNA-bound 11-subunit eukaryotic exosome complex. Nature 495, 70–75 [DOI] [PubMed] [Google Scholar]
- 23. Wasmuth E. V., Januszyk K., and Lima C. D. (2014) Structure of an Rrp6-RNA exosome complex bound to poly(A) RNA. Nature 511, 435–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zinder J. C., and Lima C. D. (2017) Targeting RNA for processing or destruction by the eukaryotic RNA exosome and its cofactors. Genes Dev. 31, 88–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hsin J.-P., and Manley J. L. (2012) The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev. 26, 2119–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cramer P., Bushnell D. A., and Kornberg R. D. (2001) Structural Basis of Transcription: RNA Polymerase II at 2.8 Ångstrom Resolution. Science 292, 1863–1876 [DOI] [PubMed] [Google Scholar]
- 27. Vannini A., and Cramer P. (2012) Conservation between the RNA Polymerase I, II, and III transcription initiation machineries. Mol. Cell 45, 439–446 [DOI] [PubMed] [Google Scholar]
- 28. Hahn S. (2004) Structure and mechanism of the RNA polymerase II transcription machinery. Nat. Struct. Mol. Biol. 11, 394–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fuchs S. M., Laribee R. N., and Strahl B. D. (2009) Protein modifications in transcription elongation. Biochim. Biophys. Acta 1789, 26–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schröder S., Herker E., Itzen F., He D., Thomas S., Gilchrist D. A., Kaehlcke K., Cho S., Pollard K. S., Capra J. A., Schnölzer M., Cole P. A., Geyer M., Bruneau B. G., Adelman K., and Ott M. (2013) Acetylation of RNA polymerase II regulates growth-factor-induced gene transcription in mammalian cells. Mol. Cell 52, 314–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Livneh I., Cohen-Kaplan V., Cohen-Rosenzweig C., Avni N., and Ciechanover A. (2016) The life cycle of the 26S proteasome: from birth, through regulation and function, and onto its death. Cell Res. 26, 869–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Satoh K., Sasajima H., Nyoumura K. I., Yokosawa H., and Sawada H. (2001) Assembly of the 26S proteasome is regulated by phosphorylation of the p45/Rpt6 ATPase subunit. Biochemistry 40, 314–319 [DOI] [PubMed] [Google Scholar]
- 33. Hirano H., Kimura Y., and Kimura A. (2016) Biological significance of co- and post-translational modifications of the yeast 26S proteasome. J. Proteomics 134, 37–46 [DOI] [PubMed] [Google Scholar]
- 34. Im E., and Chung K. C. (2016) Precise assembly and regulation of 26S proteasome and correlation between proteasome dysfunction and neurodegenerative diseases. BMB Rep. 49, 459–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cristobal A., Hennrich M. L., Giansanti P., Goerdayal S. S., Heck A. J. R., and Mohammed S. (2012) In-house construction of a UHPLC system enabling the identification of over 4000 protein groups in a single analysis. Analyst 137, 3541–3548 [DOI] [PubMed] [Google Scholar]
- 36. Taus T., Köcher T., Pichler P., Paschke C., Schmidt A., Henrich C., and Mechtler K. (2011) Universal and confident phosphorylation site localization using phosphoRS. J. Proteome Res. 10, 5354–5362 [DOI] [PubMed] [Google Scholar]
- 37. Vizcaíno J. A., Côté R. G., Csordas A., Dianes J. A., Fabregat A., Foster J. M., Griss J., Alpi E., Birim M., Contell J., O'Kelly G., Schoenegger A., Ovelleiro D., Pérez-Riverol Y., Reisinger F., Ríos D., Wang R., and Hermjakob H. (2012) The Proteomics Identifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41, D1063–D1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., and Bourne P. E. (2000) The Protein Data Bank. Nucleic Acids Res. 28, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Unverdorben P., Beck F., Śledź, Schweitzer P. A., Pfeifer G., Plitzko J. M., Baumeister W., and Förster F. (2014) Deep classification of a large cryo-EM dataset defines the conformational landscape of the 26S proteasome. Proc. Natl. Acad. Sci. U.S.A. 111, 5544–5549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kundrotas P. J., Zhu Z., and Vakser I. A. (2010) GWIDD: Genome-wide protein docking database. Nucleic Acids Res. 38, D513–D517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Orlicky S., Tang X., Willems A., Tyers M., and Sicheri F. (2003) Structural Basis for Phosphodependent Substrate Selection and Orientation by the SCFCdc4 Ubiquitin Ligase. Cell 112, 243–256 [DOI] [PubMed] [Google Scholar]
- 42. Hamelryck T., and Manderick B. (2003) PDB file parser and structure class implemented in Python. Bioinformatics 19, 2308–2310 [DOI] [PubMed] [Google Scholar]
- 43. Salomon-Ferrer R., Case D. A., and Walker R. C. (2013) An overview of the Amber biomolecular simulation package. WIREs Comput. Mol. Sci. 3, 198–210 [Google Scholar]
- 44. Word J. M., Lovell S. C., Richardson J. S., and Richardson D. C. (1999) Asparagine and glutamine: using hydrogen atom contacts in the choice of side-chain amide orientation. J. Mol. Biol. 285, 1735–1747 [DOI] [PubMed] [Google Scholar]
- 45. The PyMOL Molecule Graphics System, Version 2.0 Schrödinger, LLC.
- 46. Vriend G. (1990) WHAT IF: a molecular modeling and drug design program. J. Mol. Graph. 8, 52–56 [DOI] [PubMed] [Google Scholar]
- 47. Warnecke A., Sandalova T., Achour A., and Harris R. A. (2014) PyTMs: a useful PyMOL plugin for modeling common post-translational modifications. BMC Bioinformatics 15, 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Steinbrecher T., Latzer J., and Case D. A. (2012) Revised AMBER Parameters for Bioorganic Phosphates. J. Chem. Theory Comput. 8, 4405–4412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Khoury G. A., Thompson J. P., Smadbeck J., Kieslich C. A., and Floudas C. A. (2013) Forcefield_PTM: Ab initio charge and AMBER forcefield parameters for frequently occurring post-translational modifications. J. Chem. Theory Comput. 9, 5653–5674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lindahl E., Hess B., and van der Spoel D. (2001) GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 7, 306–317 [Google Scholar]
- 51. Hess B., Kutzner C., van der Spoel D., and Lindahl E. (2008) GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 [DOI] [PubMed] [Google Scholar]
- 52. Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A. E., and Berendsen H. J. C. (2005) GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 [DOI] [PubMed] [Google Scholar]
- 53. Berendsen H. J. C., van der Spoel D., and van Drunen R. (1995) GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91, 43–56 [Google Scholar]
- 54. Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., and Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 [Google Scholar]
- 55. Hess B. (2008) P-LINCS: A parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 4, 116–122 [DOI] [PubMed] [Google Scholar]
- 56. Miyamoto S., and Kollman P. A. (1992) Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 13, 952–962 [Google Scholar]
- 57. Bussi G., Donadio D., and Parrinello M. (2007) Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 14101. [DOI] [PubMed] [Google Scholar]
- 58.Swails J., Hernandez C., Mobley D. L., Nguyen H., Wang L.-P., and Janowski P. ParmEd. https://github.com/ParmEd/ParmEd.
- 59. Humphrey W., Dalke A., and Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–8:27–8 [DOI] [PubMed] [Google Scholar]
- 60. Hunter J. D. (2007) Matplotlib: A 2D graphics environement. Comput. Sci. Eng. 9, 90–95 [Google Scholar]
- 61. Genheden S., and Ryde U. (2015) The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 10, 449–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yang T., Wu J. C., Yan C., Wang Y., Luo R., Gonzales M. B., Dalby K. N., and Ren P. (2011) Virtual screening using molecular simulations. Proteins 79, 1940–1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Miller B. R., McGee T. D., Swails J. M., Homeyer N., Gohlke H., and Roitberg A. E. (2012) MMPBSA.py : An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 8, 3314–3321 [DOI] [PubMed] [Google Scholar]
- 64. Huerta-Cepas J., Szklarczyk D., Forslund K., Cook H., Heller D., Walter M. C., Rattei T., Mende D. R., Sunagawa S., Kuhn M., Jensen L. J., von Mering C., and Bork P. (2016) eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 44, D286–D293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. van Noort V., Seebacher J., Bader S., Mohammed S., Vonkova I., Betts M. J., Kühner S., Kumar R., Maier T., O'Flaherty M., Rybin V., Schmeisky A., Yus E., Stülke J., Serrano L., Russell R. B., Heck A. J., Bork P., and Gavin A.-C. (2012) Cross-talk between phosphorylation and lysine acetylation in a genome-reduced bacterium. Mol. Syst. Biol. 8, 571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Stark C., Su T.-C., Breitkreutz A., Lourenco P., Dahabieh M., Breitkreutz B.-J., Tyers M., and Sadowski I. (2010) PhosphoGRID: a database of experimentally verified in vivo protein phosphorylation sites from the budding yeast Saccharomyces cerevisiae. Database (Oxford). 2010, bap026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Xu H., Zhou J., Lin S., Deng W., Zhang Y., and Xue Y. (2017) PLMD: An updated data resource of protein lysine modifications. J. Genet. Genomics 44, 243–250 [DOI] [PubMed] [Google Scholar]
- 68. Brunson L. E., Dixon C., Kozubowski L., and Mathias N. (2003) the amino-terminal portion of the F-box protein Met30p mediates its nuclear import and assimilation into an SCF complex. J. Biol. Chem. 279, 6674–6682 [DOI] [PubMed] [Google Scholar]
- 69. Cherry J. M., Hong E. L., Amundsen C., Balakrishnan R., Binkley G., Chan E. T., Christie K. R., Costanzo M. C., Dwight S. S., Engel S. R., Fisk D. G., Hirschman J. E., Hitz B. C., Karra K., Krieger C. J., Miyasato S. R., Nash R. S., Park J., Skrzypek M. S., Simison M., Weng S., and Wong E. D. (2012) Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res. 40, D700–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Dourado D. F. A. R., and Flores S. C. (2014) A multiscale approach to predicting affinity changes in protein-protein interfaces. Proteins Struct. Funct. Bioinforma. 82, 2681–2690 [DOI] [PubMed] [Google Scholar]
- 71. Barford D., Hu S. H., and Johnson L. N. (1991) Structural mechanism for glycogen phosphorylase control by phosphorylation and AMP. J. Mol. Biol. 218, 233–260 [DOI] [PubMed] [Google Scholar]
- 72. Rider M. H., Bertrand L., Vertommen D., Michels P. A., Rousseau G. G., and Hue L. (2004) 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase: head-to-head with a bifunctional enzyme that controls glycolysis. Biochem. J. 381, 561–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Garza A. M. S., Khan S. H., and Kumar R. (2010) Site-specific phosphorylation induces functionally active conformation in the intrinsically disordered N-terminal activation function (AF1) domain of the glucocorticoid receptor. Mol. Cell. Biol. 30, 220–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Seo J. H., Park J.-H., Lee E. J., Vo T. T. L., Choi H., Kim J. Y., Jang J. K., Wee H.-J., Lee H. S., Jang S. H., Park Z. Y., Jeong J., Lee K.-J., Seok S.-H., Park J. Y., Lee B. J., Lee M.-N., Oh G. T., and Kim K.-W. (2016) ARD1-mediated Hsp70 acetylation balances stress-induced protein refolding and degradation. Nat. Commun. 7, 12882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tong Q., Mazur S. J., Rincon-Arano H., Rothbart S. B., Kuznetsov D. M., Cui G., Liu W. H., Gete Y., Klein B. J., Jenkins L., Mer G., Kutateladze A. G., Strahl B. D., Groudine M., Appella E., and Kutateladze T. G. (2015) An acetyl-methyl switch drives a conformational change in p53. Structure 23, 322–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Moal I. H., and Fernández-Recio J. (2012) SKEMPI: a Structural Kinetic and Energetic database of Mutant Protein Interactions and its use in empirical models. Bioinformatics 28, 2600–2607 [DOI] [PubMed] [Google Scholar]
- 77. Li M., Petukh M., Alexov E., and Panchenko A. R. (2014) Predicting the impact of missense mutations on protein–protein binding affinity. J. Chem. Theory Comput. 10, 1770–1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MS proteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository (37) with the data set identifier PXD008324.