

Phenotypic drug screening identified the clinical PKC inhibitor midostaurin to have potent activity in several lung cancer cells independently of inhibiting PKC. Network-based integration of chemo- and phosphoproteomics data highlighted multiple midostaurin targets and signaling pathways. Functional validation determined a complex polypharmacology mechanism involving the midostaurin targets TBK1, PDPK1 and AURKA and the downstream node PLK1. Combination of midostaurin with the potent PLK1 inhibitor BI2536 further enhanced the cells’ network vulnerability and resulted in strong synergy.
Keywords: Cancer Biology, Chemoproteomics, Phosphoproteome, Chemical biology, Mechanism of action
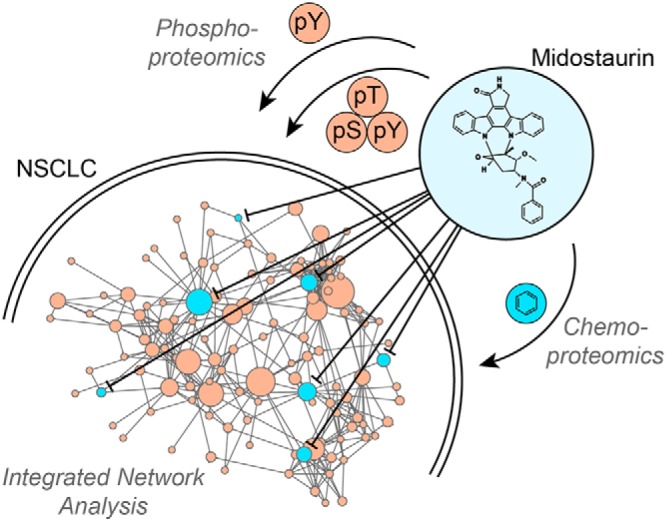
Graphical Abstract
Highlights
Anticancer activity of midostaurin but not other PKC inhibitors in NSCLC cells.
Mechanism of action by network-based integration of chemo- and phosphoproteomics.
Midostaurin polypharmacology by simultaneous inhibition of TBK1, PDPK1 and AURKA.
Design of synergistic drug combination with PLK1 inhibitor by pathway validation.
Abstract
Lung cancer is associated with high prevalence and mortality, and despite significant successes with targeted drugs in genomically defined subsets of lung cancer and immunotherapy, the majority of patients currently does not benefit from these therapies. Through a targeted drug screen, we found the recently approved multi-kinase inhibitor midostaurin to have potent activity in several lung cancer cells independent of its intended target, PKC, or a specific genomic marker. To determine the underlying mechanism of action we applied a layered functional proteomics approach and a new data integration method. Using chemical proteomics, we identified multiple midostaurin kinase targets in these cells. Network-based integration of these targets with quantitative tyrosine and global phosphoproteomics data using protein-protein interactions from the STRING database suggested multiple targets are relevant for the mode of action of midostaurin. Subsequent functional validation using RNA interference and selective small molecule probes showed that simultaneous inhibition of TBK1, PDPK1 and AURKA was required to elicit midostaurin's cellular effects. Immunoblot analysis of downstream signaling nodes showed that combined inhibition of these targets altered PI3K/AKT and cell cycle signaling pathways that in part converged on PLK1. Furthermore, rational combination of midostaurin with the potent PLK1 inhibitor BI2536 elicited strong synergy. Our results demonstrate that combination of complementary functional proteomics approaches and subsequent network-based data integration can reveal novel insight into the complex mode of action of multi-kinase inhibitors, actionable targets for drug discovery and cancer vulnerabilities. Finally, we illustrate how this knowledge can be used for the rational design of synergistic drug combinations with high potential for clinical translation.
The landscape of cancer therapy has changed dramatically over the last few years because of novel precision medicine and immunotherapy approaches, which provide significant benefit to many cancer patients (1, 2). Precision medicine has been enabled by major advances in next-generation sequencing-based genomics technologies. These allow matching of targeted therapies to mutated oncogene drivers, such as BRAF and EGFR in melanoma and non-small cell lung cancer (NSCLC),1 respectively. Although there is an entire array of driver mutations and gene fusions in NSCLC (3), most of these are rare events and the majority of lung cancers currently does not show actionable mutations illustrating the urgent need for new anticancer targets. However, some of these needed targets may not be detectable using genomic methods as they promote oncogenicity without being genetically altered (4, 5). It is becoming increasingly appreciated that targeted drugs, particularly kinase inhibitors, which make up most targeted therapeutics, can have broadly varying target profiles (6). Thus, using multi-targeted compounds with unexplained anticancer activity as research tools to identify previously unrecognized cancer vulnerabilities constitutes an intriguing novel modality for drug development. This strategy can lead to new therapeutic approaches via drug repurposing, if these compounds are already approved therapeutics, or to new drug discovery efforts to develop inhibitors for the responsible targets. Particularly in the latter case, it is essential to understand the underlying mechanism of action (MoA) and identify the most relevant target(s). Although there are multiple approaches with different strengths and weaknesses (7, 8), the unbiased identification of targets and MoAs often is still a major challenge, particularly if several targets are involved, a phenomenon referred to as “polypharmacology” (9). A viable approach to capture the correct cellular context and dynamic crosstalk between targets and pathways is to interrogate the proteome, which represents the cell's first responder to a drug challenge. Specifically, the integration of phosphoproteomics, which can describe proteome-wide drug effects on the oncogenic signaling network (10, 11), and chemical proteomics, which can identify direct drug targets that serve as entry points into this network (12–14), allows for deep network mining and is a powerful method to dissect complex kinase inhibitor MoAs (15, 16).
Midostaurin (PKC412), a structural derivative of the multi-kinase inhibitor staurosporine, has been developed as an inhibitor of protein kinase C (PKC) (17) and is of specific interest because it has recently gained approval by the FDA for the treatment of acute myeloid leukemia (AML) because of its ability to potently inhibit FLT3 (18). Interestingly, in NSCLC cells midostaurin has been found to have unexpected, but beneficial off-target activity against the drug-resistant EGFR gatekeeper mutant, but not wild-type EGFR (19, 20). We and others furthermore observed that midostaurin displayed potent antiproliferative activity in several other NSCLC cell lines not driven by mutant EGFR or other shared genomic aberrations (17), which could indicate new drug repurposing opportunities. As NSCLC cell lines generally do not express FLT3 and other potent PKC inhibitors were inactive in the same cell lines, the underlying MoA of midostaurin in these cells was unclear, but likely involves underappreciated off-targets that could constitute novel actionable targets for lung cancer. Applying a layered functional proteomics approach consisting of chemical proteomics, tyrosine and global phosphoproteomics and subsequent data integration through comprehensive network analysis, we here describe the elucidation of the complex polypharmacology MoA of midostaurin in NSCLC cells, identify a new mix of actionable targets and rationally design a synergistic drug combination.
EXPERIMENTAL PROCEDURES
Cell Culture and Reagents
A427, A549, H2170, HCC4006, and PC9 cells were provided by the Moffitt Lung Cancer Center of Excellence Cell Line Core. Cells were tested negative for mycoplasma and were authenticated via short tandem repeat (STR) analysis. Cells were cultivated in RPMI 1640 media with 10% FBS (RP10). All drug dilutions were carried out in RP10. Midostaurin and staurosporine (LCLabs, Woburn, MA), sotrastaurin, ruboxistaurin (Axon Medchem, Reston, VA), BX795 (MedChem Express, Monmouth Junction, NJ), alisertib and BI2536 (Selleckchem, Houston, TX), STO-609 (Cayman, Ann Arbor, MI), GSK2334470 (Chemietek, Indianapolis, IN), and nocodazole (Sigma, St. Louis, MO) were dissolved in DMSO (10 mm stock) and diluted in RP10 for use.
Cell Viability Assays
Cells were plated at 1000 cells/well in black, clear bottom 384 well microtiter plates and incubated at 37 °C with 5% CO2. After 24 h, cells were treated at indicated concentrations and incubated for another 72 h before Celltiter-Glo reagent (Promega, Madison, WI) was added according to the manufacturer's instructions. Plates were read using an M5 Spectramax plate reader (Molecular Devices, San Jose, CA). Data were analyzed using GraphPad Prism 7 and R.
Western Blotting
Cells were lysed using 0.2% NP-40, 50 mm Tris pH 7.5, 5% glycerol, 1.5 mm MgCl2, 100 mm NaCl, 25 mm NaF, 1 mm Na3VO4, 1 mm PMSF, 1 mm DTT, 30 μm TLCK, 30 μm Tosyl phenylalanyl chloromethyl ketone (TPCK), 0.1 μg/ml leupeptin, 0.1 μg/ml aprotinin, 10 μg/ml trypsin inhibitor lysis buffer containing phosphatase inhibitor mixture (Sigma P5726). Proteins were resolved on SDS-PAGE gels, transferred to activated PVDF membranes using the TransBlot Turbo system (BioRad, Hercules, CA) and incubated with primary antibodies. Antibodies were obtained from Sigma: actin (#A5441), CAMKK2 (#HPA017389), ERK1/2 (#M567) and Cell Signaling (Danvers, MA): AKT (#9272), AMPKa1 (#2795), Aurora A (#3092), cleaved Caspase 3 (#9661), Histone 3 (#3013), p44/42 MAPK ERK1/2 T202 Y204 (#9106), AKT S473 (#9271), AKT T308 (#D25E6), AMPKa1 T172 (#2535), PARP1 (#9542), PDPK1 (#3062), Histone 3 S10 (#9062), PLK1 (#4513), PLK1 T210 (#9062), TBK1 (#3013). Secondary antibodies were HRP-conjugated anti-rabbit (NA934, GE Healthcare, Chicago, IL) or anti-mouse (NA931, GE Healthcare). Images were obtained on an Odyssey Fc Imaging System (LI-COR, Lincoln, NE) and analyzed using Image Studio Lite (LI-COR) software.
RNA Interference
The following siRNAs were obtained from Dharmacon as ON-TARGETplus SMARTpools: PDPK1 (L003017-00-0005), TBK1 (l-003788-00-0005), CAMKK2 (l-004842-00-0005), AURKA (l-003545-01-0005), non-targeting control (d-001810-10−20). RNA was re-suspended in 1x siRNA buffer (diluted from Thermo Scientific 5x siRNA Buffer (B-002000-UB-100)) with RNase-free water (B-002000-WB-100) to a final concentration of 20 μm. Stocks were diluted to a final concentration of 20 nm in OptiMEM (31985062, Fisher Scientific, Hampton, NH) and mixed with lipofectamine RNAiMax (Invitrogen, Carlsbad, CA). For double knockdowns, siRNAs were combined to a final concentration of 20 nm, the corresponding single knockdowns were combined with non-targeting siRNA accordingly. Drug dilutions were added 24 h after transfection. Knockdown efficiency was monitored using Western blotting. Cells were counted in triplicates with trypan blue staining using a hemocytometer. Data were analyzed using Microsoft Excel and R.
Fluorescence Microscopy Analysis of Caspase 3/7 Activity
Cells were plated at 1000 cells/well in a 384-well plate. After 24 h, appropriate drug dilutions containing IncuCyte Caspase 3/7 reagent for apoptosis (4440, Essen BioScience, Ann Arbor, MI) at a final 5 μm dilution in RP10 was added to the cells. Experiments were conducted in technical triplicates. Live-cell imaging in the IncuCyte was conducted every 3 h for 72 h. Data was analyzed using the green object count of every well and normalized using the phase object confluence. Normalized data were further analyzed using GraphPad Prism 7 and R.
Cell Cycle Analysis Via Flow Cytometry
Cells were treated for the indicated times with the appropriate drug dilution, harvested and fixed with ice-cold 70% ethanol until use. Cell stage was determined using DAPI (4′, 6-diamidino-2-phenylindole, Sigma D9542)/0.1% Triton X-100/PBS solution at a final concentration of 1 μg/ml. Cells were analyzed using a FACSCanto II benchtop analyzer (BD Biosciences, Franklin Lakes, NJ) at 340 to 380 nm. Data were analyzed using ModFitLT V3.2.1 and R.
Clonogenic Assays
Cells were plated in a 6-well plate at 4000 and 2500 cells/well for A427 and A549, respectively, and treated with the appropriate drug dilutions for a total of 9 days. Cells were fixed with methanol and stained with 0.1% crystal violet solution. Cells were imaged using a tabletop scanner. Crystal violet was quantified using methanol extraction and analyzed at 540 nm on an M5 Spectramax plate reader (Molecular Devices). Data were analyzed using GraphPad Prism 7 and R.
In Vitro Kinase Assay
In vitro kinase inhibition and IC50 determinations were performed by Eurofins on the KinaseProfiler platform using 10 μm ATP. Values were obtained from a nine dose, 3-fold dilution series starting at 10 μm.
i-Midostaurin Synthesis
Immobilizable midostaurin (i-midostaurin) was synthesized by amidification of staurosporine with N-Boc-protected m-aminomethylbenzoic acid in presence of (1-[Bis(dimethylamino)methylene]1H-1,2,3-triazolo[4,5-b]pyridinum 3-oxid hexafluorophosphate) (HATU) and N, N-diisopropylethylamine (DIPEA) and subsequent deprotection using trifluoroacetic acid according to the method published previously (21). i-Midostaurin was dissolved in DMSO for a 10 mm stock and stored at −20 °C until use.
Experimental Design and Statistical Rationale
Protein quantification was performed using either chemical labeling (TMT 6-plex) or label free mass spectrometry-based approaches. Drug affinity pulldowns were performed as two technical replicates representing independent drug affinity purifications on separate days using different lysate aliquots from a single cell pellet. Competition control was performed by pre-incubating the lysate with varying concentrations of “free” drug for 30 min (3-fold dilution series from 30 μm to 3 nm) to determine specificity and relative binding affinity (22). Flow through of the i-midostaurin pulldown was reapplied to new i-midostaurin affinity resin for pull down of pull down (PDoPD) control samples. Drug cross-competition was performed by 30 min pre-incubation with 20 μm sotrastaurin. Global and tyrosine phosphoproteomics experiments were performed as three biological replicates. Midostaurin treatment of 1 μm for 3 h was compared with vehicle control (DMSO). Reproducibility of all proteomics datasets was evaluated through correlation analysis using the PerformanceAnalytics and corrplot R (3.3.0) environment. All Western blots were performed as three biological replicates and representative images are shown. siRNA mediated knockdowns and cell viability assays were performed as three biological replicates with three technical replicates for each biological replicate.
Chemical Proteomics
Drug affinity pulldowns and proteomics analysis were performed as described previously (23, 24). Briefly, i-midostaurin was immobilized on NHS-activated Sepharose beads for fast flow resin (GE Healthcare). Coupling of the drug to the bead was monitored using LC-MS analysis (14 min 5–95% MeOH gradient). Beads were blocked with ethanolamine overnight. An aliquot of protein (5 mg) from the total cell lysate was added to the beads. Experiments were conducted as two technical replicates representing independent drug affinity purifications on separate days using different lysate aliquots from a single large cell pellet. After in-gel tryptic digestion, peptides were analyzed using a nanoflow ultra-high performance liquid chromatograph (RSLC, Dionex, Sunnyvale, CA) coupled to a nanoelectrospray source-equipped bench top hybrid orbitrap mass spectrometer (QExactive Plus, Thermo, San Jose, CA). Samples were loaded on a pre-column (2 cm x 100 μm ID packed with C18 reversed-phase resin, 5 μm particle size, 100 Å pore size) and washed with 2% ACN/0.04% TFA for 8 min. Peptides were eluted onto an analytical column (C18, 75 μm ID x 50 cm, 2 μm particle size, 100 Å pore size, Dionex) applying a 90 min gradient using LC-MS solvent A (2% ACN, 0.1% FA) and LC-MS solvent B (90% ACN, 0.1% FA). Solvent B was held at 5% for 8 min, increased from 5% to 38.5% over 60 min and from 38.5% to 90% in 7 min where it was held for 5 min to wash the column. Solvent B was decreased from 90% to 5% in 1 min and the column was re-equilibrated for 10 min. The flow rate on the analytical column was set to 300 nL/minute. In each cycle, 16 MS/MS spectra were acquired in a data-dependent manner using a 60 s exclusion window. A lock mass correction was applied using a background ion (m/z 445.12003). Data were searched with MaxQuant against annotated protein sequences in the UniProt human database (2018_05, 20350 entries) using the embedded search engine Andromeda (for search parameters see supplemental information) (25). Carbamidomethylation of cysteine and oxidation of methionine were included as variable modifications. Trypsin/P was specified as the proteolytic enzyme and maximum 2 missed cleavages were allowed. Precursor and fragment ion tolerance was set to 20 ppm and 0.05 Da, respectively. Label free quantification and data match between runs was enabled within MaxQuant (26). MaxQuant output was filtered for minimum peptide length of 7 amino acids, 1% protein and PSM FDR using reverse sequences, which are then removed, plus common contaminants.
Phosphoproteomics
Cells were lysed with urea lysis buffer (20 mm HEPES pH 8, 9 m urea, 1 mm sodium orthovanadate, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate), sonicated at 15 W with 3 bursts of 15 s each using a microtip sonicator, digested and extracted using the Cell Signaling Sep-Pak C18 peptide purification protocol (Cell Signaling Technology). Phosphotyrosine peptides were enriched using the PTMScan Phospho-Tyrosine Rabbit mAb (P-Tyr-1000, Cell Signaling #8803) according to the manufacturer's instructions. For global phosphoproteomics, samples were labeled using TMT reagents (Thermo Fisher TMTsixplex™ Isobaric Mass Tagging Kit, #90064). pSTY peptides were enriched using an IMAC kit (Sigma I1408) adapted from the manufacturer's instructions (Sigma-Aldrich). After lyophilization, peptides were redissolved in 400 μl of 20 mm ammonium formate (pH 10.0). The basic pH reversed phase liquid chromatography separation (bRPLC) was performed on a XBridge column (4.6 mm ID x 100 mm length) packed with BEH C18 resin (3.5 μm particle size, 130 Å pore size) (Waters, Milford, MA). The solvent system used bRPLC A (aqueous 2% acetonitrile with 5 mm ammonium formate, pH 10.0) and bRPLC B (aqueous 90% acetonitrile with 5 mm ammonium formate, pH 10.0). After loading at 1% B, the peptides were eluted using 5% B for 10 min, 5–15% B in 5 min, 15–40% B in 47 min, 40–100% B in 5 min and 100% B held for 10 min, followed by re-equilibration at 1% B. The flow rate was 600 μl/minute collecting 12 concatenated fractions (27). Peptide fractions were dried by vacuum. Samples were re-suspended in appropriate volumes of LC-MS Solvent A (described above). Samples were spiked with Thermo Scientific Pierce Retention Time Calibration Mixture (PRTC) to confirm consistent performance of the LC-MS analyses. Data acquisition parameters for LC-MS/MS were as described above. LC-MS/MS data were searched with MaxQuant 1.2.2.5 (pY) against all protein sequences annotated in the human UniProt database (downloaded 12/2015, 20198 entries) and MaxQuant 1.5.2.8 (pSTY) with the human UniProt database (downloaded 2018_5, 20350 entries)(28) using the embedded search engine Andromeda (for search parameters see supplemental information) (25). Carbamidomethylated cysteines was set as fixed modification and oxidation of methionine, N-terminal protein acetylation and phosphorylation of serine, threonine, and tyrosine as variable modifications. Precursor and fragment ion tolerance was set to 20 ppm and 0.05 Da, respectively, and for label free quantification match between consecutive runs was enabled within MaxQuant (26). Data were filtered for 2% protein FDR and 5% peptide and site FDR, plus common contaminants. For quantification of the label free pY data set, peak apexes from extracted ion chromatograms were used; for the TMT 6-plex global pSTY data, the reporter ion intensity was used for relative quantification of each peptide.
Data Processing and Proteomics Analysis
All data sets were normalized using iterative rank order normalization (IRON) (29), then further filtered for a PEP score ≤ 0.1 and mass error ≤ 10 ppm. Competition in the i-midostaurin pulldowns with either “free” midostaurin or sotrastaurin was calculated. IC50 values were calculated using GraphPad Prism 7. The depletion factor (r) was calculated for every relevant kinase (at least 8 unique peptides across all pulldowns and 2 unique peptides in uncompeted pulldown) as described by Medard et al. and the Cheng-Prusoff equation was used to calculate the Ki indicating the inhibition constant for binding of the inhibitor to the kinase (22). Phosphorylation site quantification was set to the least modified peptide. Identification and quantification of phosphopeptides directly connected to the most relevant kinases determined in Fig. 4C and displayed in Fig. 4D were manually confirmed using extracted ion chromatograms via Scaffold 4.8.7 and Skyline daily 4.1.0.18169 (30). Log2 fold change and Student's t test values were calculated for both phosphoproteomics datasets. Cut-offs of p value ≤ 0.05 for both datasets, a log2 fold change of 1.5 (representing an actual 2.82-fold change) for the phosphotyrosine and a log2 fold change of at least 2 times the standard deviation for the global phosphoproteomics datasets were applied. Subsequently, the drug affinity dataset was filtered for kinases and combined with all significantly modulated proteins from the phosphoproteomics data in a STRING (version 10.5) (31) network analysis, as described previously (32). The minimum required interaction score was set to a medium confidence of 0.4 (scale: 0–1), which is calculated for each protein-protein association by STRING and indicates the probability that the given interaction is biologically meaningful, specific and reproducible (33). Interactions were imported into GEPHI for visualization and non-randomized agglomerative clustering-based community detection (modularity) with a resolution of 1 was used to determine smaller subcommunities within the network (34). All ten communities were included in a pathway analysis of the Kyoto Encyclopedia of Genes and Genomes (KEGG) using DAVID (35). To prioritize kinase targets for further analysis the betweenness centrality was calculated for the network. Betweenness centrality ranks nodes according to their position in the network indicating how often a node appears on the shortest path between nodes in the network (36). Data were displayed using R. To prepare MaxQuant files for upload to PRIDE, the database search results were imported into Scaffold to export mzIdentML files by choosing PRIDE/Scaffold re-analysis option.
Fig. 4.

Network-based integration of chemical and phosphoproteomics datasets. A, STRING network of midostaurin's kinase targets and significantly modulated phosphosignals. Only kinases observed by chemical proteomics are labeled. Node size correlates with betweenness centrality and sub-networks are colored according to communities determined via modularity. B, KEGG pathway analysis of STRING sub-networks. Nodes are colored according to modules in A and labeled based on the pathway with the most significant p value. Node size correlates with the number of proteins present in each module. C, Kinase target prioritization using the -log10 Ki derived from the chemical proteomics dataset and log2 betweenness centrality values from the network. Node colors reflects binding selectivity for midostaurin over sotrastaurin (see Fig. 3E). D, Phosphorylation changes and phosphosites of proteins directly connected to selected kinases in the network displayed in A including both potential kinase substrates as well as functionally associated phosphoproteins. Node size correlates with log2 fold change and color indicates up (red)- or down-regulation (blue). All phosphosites shown were manually validated.
RESULTS
Midostaurin Shows Beneficial Off-target Activity In NSCLC Cells
We have previously reported on the development of a customized in-house library of 180 targeted compounds, which are mostly represented in either clinical trials or FDA-approved and include several drug classes such as kinase, PARP and HSP90 inhibitors, and its screening against 24 non-small cell lung cancer (NSCLC) cell lines for antiproliferative effects (37). Following up on this dataset, we now expanded this analysis to 240 targeted compounds and 33 lung cancer cell lines. The screen revealed potent activity of the protein kinase C (PKC) inhibitor midostaurin compared with other PKC inhibitors, such as sotrastaurin and ruboxistaurin, in several NSCLC cell lines (Fig. 1A). This outcome suggested a PKC-independent MoA and the presence of other, potentially novel and actionable targets in NSCLC. In addition, comparing sensitive and insensitive cell lines, no clear association with known oncogenic driver mutations was observed (e.g. A427 and A549 KRAS mutant, HCC4006 EGFR mutant, and H2170 KRAS/EGFR wild-type were all sensitive). Specific clustering of selected compound classes included in the screen was investigated by performing a principal component analysis of the reduction of viability at 2.5 μm by each compound across all screened cell lines, incorporating several compounds per target class in the analysis. HSP90, Aurora kinase (AURK) and the majority of PI3K inhibitors all grouped with their target classes (Fig. 1B). However, the PKC and FLT3 inhibitor midostaurin did not group with other PKC or FLT3 inhibitors implying distinct cellular efficacy and target profiles. As midostaurin has been recently approved by the FDA for treatment of AML with FLT3 mutations (18), this observation might indicate an interesting drug repurposing opportunity. Consistently, tissue-specific sensitivity analysis of previously reported cellular midostaurin activity suggested efficacy not only in hematopoietic cell lines, but also across several lung cancer cell lines (supplemental Fig. S1A) (38).
Fig. 1.

Viability screen reveals differential activity profiles of PKC inhibitors in NSCLC cell lines. A, Viability of 33 NSCLC cell lines upon 72 h of 2.5 μm midostaurin, sotrastaurin and ruboxistaurin treatment was evaluated using CellTiterGlo. Relevant genetic alterations are displayed for respective cell lines. NA: Not acquired. B, Principal component analysis for the phenotypic drug screen including all 33 NSCLC cell lines based on reduction of cell viability at 2.5 μm. PC1 and PC2 with an explained variance of 29 and 20%, respectively are displayed. Multiple compounds per target class were included in the analysis.
PKC-independent Cellular Effects of Midostaurin
The most sensitive cell lines to midostaurin in our screen were selected for in-depth analysis. KRAS-mutant A427 and A549, KRAS-wild type H2170 as well as EGFR-mutant HCC4006 and PC9 were investigated using 10-point dose-response curves (Fig. 2A). Consistent with the screening results and in contrast to sotrastaurin and ruboxistaurin, midostaurin showed potent activity in all five NSCLC cell lines with similar maximal cellular efficacy (Emax) and submicromolar half-maximal effective concentration (EC50) values, which were within clinically achievable plasma concentration ranges. Furthermore, midostaurin, but not sotrastaurin or ruboxistaurin, caused potent and rapid induction of apoptosis in A427 cells as apparent by immunoblot analysis of PARP1 and Caspase 3 cleavage (Fig. 2B), as well as live cell imaging for Caspase 3/7 cleavage (Fig. 2C, supplemental Fig. S1B). Similarly, midostaurin potently inhibited colony formation of A427 cells (Emax = 0% at 500 nm) whereas ruboxistaurin and sotrastaurin had no effect (Fig. 2D). Finally, midostaurin elicited a significant, dose-dependent accumulation of A427 cells in G2 phase (Fig. 2E, supplemental Fig. S1C). In summary, these results suggest high potency of midostaurin in NSCLC cells as well as a distinct MoA from ruboxistaurin and sotrastaurin, which is independent of PKC inhibition and elucidation of which may reveal new actionable targets for NSCLC.
Fig. 2.

Differential anticancer effects of PKC inhibitors in midostaurin-sensitive NSCLC cell lines. A, Dose-response curves for inhibition of cell viability by midostaurin, sotrastaurin and ruboxistaurin of A427, A549, H2170, HCC4006 and PC9 cells upon 72 h of treatment evaluated using CellTiterGlo. Data represents biological triplicates, each performed in technical triplicate. Concentrations and EC50 values are given in μm. The gray bar indicates clinically observed midostaurin plasma concentration range. B, Western blotting of Caspase 3 and PARP1 cleavage upon 24 h of midostaurin, sotrastaurin or ruboxistaurin treatment in A427 cells. Staurosporine was used as a positive control. Image is representative of three biological replicates. Concentrations are given in μm. C, Induction of apoptosis determined by Caspase 3/7 cleavage in A427 cells upon 1 μm of midostaurin, sotrastaurin or ruboxistaurin treatment over a period of 72 h using IncuCyte live cell analysis. Data was acquired every 3 h. Data points represent biological triplicates each performed in technical triplicate (mean). D, Colony formation assay of A427 cells upon 9 days of midostaurin, sotrastaurin and ruboxistaurin treatment at indicated concentrations (μm). Image is representative of three biological replicates. E, Cell cycle analysis of A427 cells after midostaurin treatment for 6 h at indicated concentrations (μm). Data represents three biological replicates (mean ± S.D.). Mido: midostaurin; sotra: sotrastaurin; ruboxi: ruboxistaurin; stauro: staurosporine; DMSO: vehicle control.
Chemical and Phosphoproteomics Analysis
In order to elucidate the MoA of midostaurin in these NSCLC cells, we applied an unbiased multi-pronged proteomics approach consisting of chemical proteomics, as well as tyrosine and global phosphoproteomics (Fig. 3A). To understand the network-wide signaling effects of midostaurin in these cells, we performed quantitative phosphotyrosine (pY) and global phosphoserine/threonine/tyrosine (pSTY) phosphoproteomics analysis of A427 cells comparing midostaurin versus vehicle control (DMSO). Global enrichment of phosphopeptides was done by immobilized metal ion affinity chromatography (IMAC). Quantification of pSTY peptides was achieved by introduction of chemical labels using a tandem mass tag (TMT) 6-plex approach, bRPLC off-line fractionation and subsequent LC-MS/MS analysis, which after strict filtering detailed in Experimental procedures yielded a total of 12,861 unique pSTY phosphopeptides (supplemental Table S1). In parallel, antibody-based immunoprecipitation of pY peptides and label-free LC-MS/MS analysis resulted in the identification of 307 unique pY phosphopeptides (supplemental Table S2). Both phosphoproteomics data sets showed good correlation between biological replicates and similar intensity distributions (supplemental Fig. S2). All pY phosphopeptides with a log2 fold change ≥ 1.5 (i.e. an actual fold change of 2.82) or a p value of ≤ 0.05 (Fig. 3B) and all pSTY phosphopeptides with a fold change ≥ 2 times the standard deviation greater or lower than the average value for the whole dataset and a p value of ≤ 0.05 (Fig. 3C) were selected for subsequent analysis, constituting two sets of 82 (pY) and 180 (pSTY) peptides, respectively.
Fig. 3.

Layered functional proteomics strategy to elucidate midostaurin's MoA in A427 NSCLC cells. A, Mass spectrometry-enabled combination of direct target identification via affinity-based chemical proteomics (label-free) and midostaurin-induced signaling changes via global (TMT 6-plex) and tyrosine (label-free) phosphoproteomics. B, Volcano plot of all identified phosphopeptides of the pY dataset, displaying log2 fold change and p value. Peptides selected for network analysis are marked in blue. C, Volcano plot of all identified phosphopeptides in the global phosphoproteomics dataset, displaying log2 fold change and p value. Peptides selected for network analysis are marked in coral/red. D, Chemical structures of midostaurin and its immobilizable analogue i-midostaurin. E, Kinome tree of midostaurin's direct kinase targets. Color intensity indicates specific targets of midostaurin (dark blue) and targets shared between midostaurin and sotrastaurin (partial overlap: light blue; strong overlap: white). Kinome phylogenetic tree adapted courtesy of Cell Signaling Technology, Inc. (www.cellsignal.com).
To determine direct binding partners of midostaurin and define its target profile in A427 cells, a drug affinity-based chemical proteomics approach was used (14). For that purpose, we prepared an immobilizable derivative of midostaurin (i-midostaurin, Fig. 3D), which has been previously validated and employed successfully (21). i-Midostaurin was tethered to NHS-activated Sepharose beads and pulldown experiments were performed from A427 total cell lysates with dose-dependent midostaurin competition (nine doses in 3-fold dilutions from 30 μm to 3 nm) and a sotrastaurin cross-competition (20 μm) control. Furthermore, pulldowns from pulldown flow through were performed. Subsequent LC-MS/MS analysis of these samples revealed a set of 1883 proteins (complete dataset can be found in supplemental Table S3), including multiple protein kinases binding to i-midostaurin beads. Data showed high reproducibility across replicates (supplemental Fig. S3). Filtering for competing proteins and calculating the Ki for inhibition of kinase binding (for details see Data processing and proteomics analysis) led to identification of 35 high confidence protein kinase targets of midostaurin in A427 cells. The majority of these kinases has been validated as midostaurin targets in previous studies, which employed in vitro binding assays or chemical proteomics with different cell types (21, 39, 40), and our findings here generally agree well with these reports. Mapping the identified kinase targets of midostaurin on the phylogenetic kinome tree illustrated that midostaurin predominantly interacted with serine/threonine kinases in these cells (Fig. 3E). Importantly, whereas several well-known midostaurin targets were either weakly (e.g. CAMK2B/D/G, GAK) or strongly (e.g. PRKCD/G [PKCδ/γ], GSK3A/B) competed by sotrastaurin, this analysis also highlighted many midostaurin targets not bound by sotrastaurin (e.g. TBK1, CAMKK2). In summary, functional proteomics successfully identified multiple midostaurin kinase targets and midostaurin-modulated signals in sensitive NSCLC cells.
Proteomics Data Integration
To identify the signaling pathways that were most prominently affected by midostaurin and their associated kinase targets, we next integrated the chemical and phosphoproteomics data using a network-based approach. Direct kinase targets identified in the chemical proteomics data set were connected with proteins of the selected phosphopeptides using STRING-based network analysis (confidence > 0.4), which queries both physical and functional protein-protein interactions (detailed in Data Processing and Proteomics Analysis) (31). To further enhance confidence and use of the network, textmining was disabled and disconnected nodes were excluded from the analysis, which resulted in a total of 151 nodes (proteins) and 399 edges (interactions). This network was analyzed for sub-communities using agglomerative clustering-based community detection (modularity = 0.616) (34), generating ten distinct modules (Fig. 4A). Based on the local hypothesis of biological networks, these topological modules correspond to particular biological functions (functional modules) (41). To identify the underlying biological processes, we performed a pathway analysis for each distinct module using the Kyoto Encyclopedia of Genes and Genome (KEGG) database (Fig. 4B, supplemental Table S4). Modules 1, 2, and 6 were most strongly associated with cancer-related pathways. Module 2, which included the known midostaurin targets TBK1, AAK1, BMP2K, FER, GSK3A/B, and IKBKE, is enriched for infection-associated pathways (Chemokine signaling) and cancer-related pathways (Proteoglycans in cancer; Focal adhesion; Ras signaling pathway). MAPK and mTOR signaling pathways are highly enriched in module 6 comprising PDPK1, PKN1 and RPS6KA6. Conversely, module 1 encompassing the known midostaurin targets PRKAA1 (AMPKα1), CAMKK2, MAP3K11 and MARK2/3 is enriched for the tumor suppressive AMPK signaling pathway. To prioritize the kinases within the network that are most relevant for midostaurin's activity, we next calculated the betweenness centrality, which is based on the shortest paths between nodes and thus is a metric for the relative importance of a node within the network, for each kinase (42). Because STRING incorporates both physical and functional interactions, better characterized kinases are prone to appear more important than others that have not been studied extensively. In order to balance this bias, we performed target prioritization considering also the apparent interaction strength between midostaurin and its kinase targets as represented by the Ki values derived from the chemical proteomics data set (Fig. 4C). This analysis indicated that TBK1 interacted most potently with midostaurin while displaying strong betweenness centrality. In contrast, within the group of the most potent midostaurin-specific binders (i.e. not competed by sotrastaurin) AURKA exhibited the strongest betweenness centrality whereas PDPK1 ranked second in both categories. Corroborating the relative contributions for the midostaurin effector network, analysis of the immediate neighboring nodes in the network, which include direct substrates as well as functionally associated phosphoproteins and kinases, furthermore revealed markedly more phosphosites associated with AURKA and PDPK1 than with TBK1 or AMPKα1 (Fig. 4D). Taken together, integrated network-based analysis of kinases and phosphorylation sites engaged by midostaurin suggested TBK1, PDPK1 and AURKA as targets with potential relevance for midostaurin's MoA in NSCLC cells.
Cellular Target Validation and Mechanism of Action
To interrogate the functional relevance of TBK1, PDPK1, and AURKA in NSCLC cells we used the chemical probes BX795 (TBK1 IC50 = 2.3 nm) (43), GSK2334470 (PDPK1 IC50 = 10 nm) (44), and alisertib (AURKA IC50 = 1 nm) (45). As TBK1 and particularly PDPK1 are important upstream signaling molecules of AKT and mTOR and this pathway is furthermore affected by the midostaurin targets CAMKK2 and AMPKα1, we also interrogated these kinases using the dual inhibitor STO-609 (CAMKK2 IC50 = 10 nm, AMPK IC50 = 160 nm) (43). Unexpectedly, single pharmacological inhibition of TBK1, PDPK1 or AURKA resulted only in a partial decrease of A427 and A549 cell viability whereas inhibition of CAMKK2 or AMPK showed no effect even at relatively high STO-609 concentrations of 4 μm (Fig. 5A, 5B, supplemental Fig. S4A). However, combined inhibition of TBK1, PDPK1 and AURKA elicited potent antiproliferative effects (Fig. 5A, 5B). In contrast, probe combinations with STO-609 were essentially limited to the effects caused by the respective partner compounds themselves (supplemental Fig. S4A). This suggested that neither CAMKK2 nor AMPKα1 meaningfully contribute to midostaurin's MoA in these cells even though AMPKα1/α2 phosphorylation at T172/T183, known CAMKK2 target sites, was inhibited by midostaurin (supplemental Fig. S4B). This result was consistent with observations from parallel gene silencing in combination with alisertib, where siRNA-mediated knockdown of CAMKK2 only marginally enhanced the effects from combined targeting of AURKA together with knockdown of either TBK1 or PDPK1 (supplemental Fig. S4C). However, triple targeting of AURKA, PDPK1, and TBK1 with siRNA and probe compounds led to potent decreases in cell viability of A427 and A549 cells (Fig. 5C, 5D). Finally, in vitro kinase activity assays confirmed potent functional inhibition of AURKA (IC50 = 4 nm), TBK1 (IC50 = 9 nm) and PDPK1 (IC50 = 72 nm) by midostaurin in addition to physical binding (supplemental Fig. S4D). In contrast and consistent with the chemical proteomics data, sotrastaurin did not meaningfully inhibit AURKA (IC50 > 10,000 nm), TBK1 (IC50 > 10,000 nm) or PDPK1 (IC50 = 9,530 nm). In summary, these results suggested a complex polypharmacology mechanism of midostaurin in NSCLC cells that involves concurrent targeting of AURKA, TBK1, and PDPK1.
Fig. 5.

Cellular validation of midostaurin targets using RNAi and small molecule probes. A, Viability of A427 cells upon 72 h treatment with increasing concentrations of GSK2334470 (PDPK1i), 0.5 μm BX795 (TBK1i) and/or 0.5 μm alisertib (AURKAi) determined by CellTiterGlo. Data depicts three biological replicates each performed in technical triplicate (mean ± S.D.). Colors represent specific drug combinations as indicated. B, Viability of A549 cells upon 72 h treatment with increasing concentrations of GSK2334470 (PDPK1i), 0.75 μm BX795 (TBK1i) and/or 0.5 μm alisertib (AURKAi) determined by CellTiterGlo. Data depicts three biological replicates each performed in technical triplicate (mean ± S.D.). Colors represent specific drug combinations as indicated. C, Relative A427 cell counts upon 96 h siRNA-mediated knockdown of PDPK1 and/or AURKA and/or 72 h treatment with 250 nm BX795. Data depicts three biological replicates each performed in technical triplicate (mean ± S.D.). Knockdown efficiency was determined using immunoblotting. Color is according to target inhibition displayed in A and B. D, Relative A549 cell counts upon 96 h siRNA-mediated knockdown of TBK1 and/or PDPK1 and/or 72 h treatment with 50 nm alisertib. Data depicts three biological replicates each performed in technical triplicate (mean ± S.D.). Knockdown efficiency was determined using immunoblotting. Color is according to target inhibition displayed in A and B. NT, non-targeting siRNA; DMSO: vehicle control.
Synergistic Drug Combination
We aimed to identify potential intervention points for designing more effective drug combinations by investigating signaling events downstream of the functionally validated midostaurin targets. Therefore, we generated a condensed signaling network that included relevant direct kinase targets of midostaurin and significantly modulated phosphorylation sites identified by phosphoproteomics and Western blotting (Fig. 6A). Querying this midostaurin effector network, we found two signaling nodes, AKT and PLK1, which seemed to be major points of convergence for several modulated pathway arms. Immunoblot analysis confirmed that midostaurin inhibits phosphorylation of PLK1 at T210 and AKT at T308, but not S473 (Fig. 6B), which was consistent with our previous observations (supplemental Fig. S4B). Notably, both GSK2334470 (PDPK1i) and to somewhat lesser extent BX795 (TBK1i) reduced phosphorylation of both AKT sites. Conversely, only BX795, but not GSK2334470, slightly decreased PLK1 phosphorylation in these cells. Inhibition of AURKA by alisertib decreased phosphorylation of PLK1 at T210 more prominently and potently inhibited histone H3 phosphorylation at S10, a canonical AURKA downstream signal, as expected. Consistently, histone H3 S10 phosphorylation was also strongly inhibited by midostaurin treatment. We hypothesized that enhanced inhibition of PLK1 may increase midostaurin-induced anticancer effects considering that midostaurin alone did not completely inhibit PLK1 T210 phosphorylation. PLK1 is an essential mitotic regulator as well as a validated cancer target and phosphorylation on T210 is associated with kinase activity. Indeed, combination of midostaurin with the potent PLK1 inhibitor BI2536 (IC50 = 0.83 nm), which is a close analogue of the drug candidate volasertib that has been under evaluation in several clinical trials for various cancers itself (46, 47), showed strong synergy with regard to inhibition of A427 and A549 cell viability as determined by the established combination index (CI) model (Fig. 6C, 6E). Importantly, this effect was apparent even at midostaurin concentrations ∼10 times lower than reported plasma levels. Likewise, using clinically relevant drug concentrations, we observed pronounced combination effects in clonogenic assays with A427 and A549 cells (Fig. 6D, 6F). Taken together, the observed alterations in the downstream signaling pathways confirmed cellular inhibition of the midostaurin targets AURKA, PDPK1, and TBK1 and suggested complex functional interplay between these targets in NSCLC cells. This result led to identification of the strongly synergistic drug combination of midostaurin with the potent PLK1 inhibitor BI2536.
Fig. 6.

Rational design and validation of synergistic midostaurin drug combination. A, Condensed schematic of midostaurin's effector signaling network. Direct midostaurin targets are depicted in blue, MS-detected phosphosites in purple. Gray nodes complement the signaling network and were not detected via MS, dark gray phosphosites were validated by immunoblotting. B, Western blot analysis of downstream signaling nodes of critical midostaurin targets. A427 cells were treated for 3 h with 0.5 and 1 μm of alisertib (AURKAi), BX795 (TBK1i), GSK2334470 (PDPK1i) or midostaurin. Representative images of three biological replicates are shown. C/E, Viability of A427 (C) and A549 (E) cells upon midostaurin and the PLK1i BI2536 (C: 15 nm; E: 20 nm) treatment determined by CellTiterGlo. Data is representative of three biological replicates (mean ± S.D.). CI values were calculated using CompuSyn. D/F, Colony formation assay of A427 (D) and A549 (F) cells after 9 days of treatment with midostaurin and BI2536 at indicated concentrations. Data is representative of three biological replicates. Mido, midostaurin; BI, BI2536; DMSO: vehicle control.
DISCUSSION
Despite tremendous advances in genomics and the associated success of precision medicine, the majority of cancers, including many NSCLC tumors, currently does not display actionable mutations thus illustrating the urgent need for new anticancer targets (3). Guided by the unexplained cellular activity of the FDA-approved multikinase inhibitor midostaurin in NSCLC cells, we applied an integrated functional proteomics approach, comprised of chemical proteomics as well as global and tyrosine phosphoproteomics. This strategy led to the identification of new actionable targets for lung cancer through elucidation of midostaurin's network-wide signaling effects and complex MoA. Importantly, by comparison with other potent and selective inhibitors, midostaurin's activity in these cells was independent of its canonical targets PKC and FLT3. Moreover, we found that midostaurin's MoA depends on the concurrent inhibition of at least three different non-canonical targets. This observation not only constitutes a rare and intriguing case of polypharmacology (9), but also powerfully demonstrates the need for applying an unbiased, multi-layered proteomics approach and network-based analysis to capture the complexity of the underlying mechanisms. This strategy was particularly important as the relevant midostaurin targets in these cells were not mutated and therefore “hidden” from most genomics approaches, and as their contribution was rather based on their widespread impact in the signaling network (4). Our findings furthermore illustrate the power of combining these orthogonal proteomics technologies as neither chemical proteomics nor phosphoproteomics analysis in isolation would have easily nominated this broad spectrum of functionally relevant targets and pathways.
Integrating the chemical and phosphoproteomics datasets, in-depth mining of the resulting midostaurin effector network and subsequent functional validation highlighted three kinases as relevant midostaurin targets in these NSCLC cells, namely AURKA, PDPK1, and TBK1. TBK1, a close homolog of IκB kinase ε (IKKε) that was originally identified to act in the innate immune response (48), was of particular interest in this context as it has recently been described to be synthetically lethal with mutant KRAS in several NSCLC cell lines (49). Notably, although this finding has been independently validated for several cell lines, later studies with broader NSCLC cell line panels reported a spectrum of sensitivity of KRAS-mutant NSCLC cell lines toward TBK1 silencing/inhibition (50–52). This is consistent with our observations that TBK1 inhibition causes only partial reduction of viability in these specific NSCLC cell lines. Midostaurin also showed activity in several KRAS-wt cells and conversely did not affect many other KRAS-mutant NSCLC cells, including some that are sensitive to TBK1 loss, such as H23 cells (49, 51). This further illustrates the importance of the multitargeted mechanism, because midostaurin's cellular activity in NSCLC cells was not simply reducible to TBK1 inhibition alone in KRAS-mutant cells. TBK1 has been shown to phosphorylate AKT at T308 and S473 (52), which is in agreement with our observations in A427 cells although TBK1 inhibition by BX795 had less prominent effects on T308. However, inhibition of PDPK1, another important midostaurin target, markedly reduced AKT phosphorylation also at T308, and further downstream at S473, as expected for this canonical activator of the PI3K-AKT signaling cascade. Consistently, midostaurin treatment elicited similar inhibition of T308 phosphorylation. However, midostaurin did not reduce S473 phosphorylation indicating either mTOR feedback signaling (53, 54) and/or inhibition of yet another target that rescues the effects at this stage. Also, we observed pronounced inhibition of CAMKK2-mediated phosphorylation of AMPK, a known tumor suppressor and negative regulator of mTOR signaling. Although AMPK regulates mTOR as part of the mTOR complex 1 (mTORC1) and AKT S473 is an mTORC2 target site, it has been recently shown that AMPK activation can also reduce mTORC2 activity and AKT S473 phosphorylation in NSCLC cells (55). Interestingly, recent studies found that inhibition of AURKA can overcome mTOR inhibitor resistance in gastrointestinal cancer cells and that AURKA can repress STK11(LKB1)/AMPK signaling in NSCLC cells (56, 57). These data agree with our findings that AURKA inhibition is a major component of midostaurin's MoA, maybe even more so in STK11-wild-type cells. In addition, we observed pronounced effects on other important AURKA downstream signals, namely phosphorylation of histone H3 S10 and PLK1 T210. Consistent with previous reports on PLK1 (51), these signals were furthermore reduced by TBK1 inhibition, albeit less strongly. Although PLK1 T210 has also been proposed to be a substrate of PDPK1 (58), we found no evidence for this in A427 cells suggesting that PDPK1 inhibition by midostaurin likely contributes to the overall MoA in these cells through other downstream targets. As reviewed in detail elsewhere, PLK1 and AURKA are well established to engage in multiple functional interactions during mitosis and cell division (59, 60). Furthermore, they play critical roles in controlling the G2/M cell cycle checkpoint and mitotic entry (61), which aligns well with the strong accumulation of cells in G2 upon midostaurin treatment. Interestingly, it has been recently reported that also TBK1 affects mitosis (62).
PLK1 as well as AURKA have been proposed as new markers for prognosis and cancer aggressiveness in addition to being functional targets for therapeutic intervention in several cancer types (59, 63). PLK1 is deregulated in many types of cancer and has been associated with oncogenesis. NSCLC patients with moderate PLK1 expression show 51.8% of 5-year survival rate in contrast to 24.2% for patients with elevated levels of PLK1 expression (63). Based on the known cancer relevance of PLK1 signaling and the only partial inhibition of PLK1 T210 phosphorylation by midostaurin, we evaluated the combination of midostaurin with the potent PLK1 inhibitor BI2536 and observed strong in vitro synergy at clinically relevant drug concentrations. Because midostaurin was recently FDA approved for AML and BI2536 is currently in phase I and II clinical trials, this combination might entail an attractive drug repurposing opportunity for midostaurin.
In summary, by using a combination of unbiased, layered functional proteomics and network-based data integration we were able to elucidate the mechanism of action of the multi-kinase inhibitor midostaurin in lung cancer cells, which was independent of its cognate target, but rather involved several noncanonical midostaurin targets. Analysis of the downstream signaling effects identified convergence on specific oncogenic pathways, which led to the rational design of a novel synergistic drug combination. The successful application of this integrated proteomics approach illustrates its potential to answer complex biological questions and reveal novel actionable cancer vulnerabilities that may not be apparent by genomics approaches.
DATA AVAILABILITY
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (http://www.ebi.ac.uk/pride) with the dataset identifiers PXD009041 (Phosphotyrosine Profiling), PXD010786 (IMAC Phosphoproteomics) and PXD010787 (Chemical Proteomics) (64).
CODE AVAILABILITY
Codes used in this manuscript will be shared upon request.
Supplementary Material
Acknowledgments
We thank all the authors for their input on the project design and their critical comments on the manuscript. We also would like to acknowledge the Moffitt Lung Cancer Center of Excellence and the Moffitt Chemical Biology (Chemistry Unit), Proteomics, Flow Cytometry and Analytical Microscopy Core Facilities.
Footnotes
* This work was supported by the NIH/NCI R01 CA181746 (to U.R.), the Austrian Marshall Plan Scholarships (to C.C.), the NIH/NCI F99/K00 Predoctoral to Postdoctoral Transition Award F99 CA212456 (to B.M.K.), the Moffitt NIH/NCI SPORE in Lung Cancer (Award No. P50 CA119997), and the H. Lee Moffitt Cancer Center and Research Institute. Moffitt Core Facilities are supported by the National Cancer Institute (Award No. P30-CA076292) as a Cancer Center Support Grant. Proteomics and Metabolomics is also supported by the Moffitt Foundation.
 This article contains supplemental Figures and Tables.
This article contains supplemental Figures and Tables.
1 The abbreviations used are:
- NSCLC
- non-small cell lung cancer
- AKT
- RAC-alpha serine/threonine-protein kinase
- AMPKα1
- 5′-AMP-activated protein kinase catalytic subunit alpha-1, PRKAA1
- AURKA
- Aurora kinase A
- AURKAi
- Aurora kinase A inhibitor
- CAMKK2
- calcium/calmodulin-dependent protein kinase kinase 2
- DAPI
- 4′, 6-diamidino-2-phenylindole
- DMF
- dimethylformamide
- EC50
- half maximal effective concentration
- EGFR
- epidermal growth factor receptor
- Emax
- maximal effective concentration
- ERK
- mitogen-activated protein kinase
- FDR
- false discovery rate
- HATU
- (1-[Bis(dimethylamino)methylene]1H-1,2,3-triazolo[4,5-b]pyridinum 3-oxid hexafluoro-phosphate
- HRP
- Horseradish peroxidase
- HSP90
- heat shock protein 90
- IRON
- iterative rank order normalization
- KEGG
- Kyoto encyclopedia of genes and genomes
- MoA
- mechanism of action
- NSAF
- normalized spectral abundance factor
- PARP1
- Poly [ADP-ribose] polymerase 1
- PDPK1
- 3-phosphoinositide-dependent protein kinase 1
- PDPK1i
- 3-phosphoinositide-dependent protein kinase 1 inhibitor
- PI3K
- phosphatidylinositol 3-kinase
- PKC
- protein kinase C
- PLK1
- Polo-like kinase 1
- pSTY
- global phosphoproteomics
- PTM
- post-translational modifications
- pY
- phosphotyrosine
- TBK1
- TANK-binding kinase 1
- TBK1i
- TANK-binding kinase 1 inhibitor
- TMT
- Tandem mass tag.
REFERENCES
- 1. Hyman D. M., Taylor B. S., and Baselga J. (2017) Implementing genome-driven oncology. Cell 168, 584–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Postow M. A., Callahan M. K., and Wolchok J. D. (2015) Immune checkpoint blockade in cancer therapy. J. Clin. Oncol. 33, 1974–1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lin J. J., and Shaw A. T. (2016) Resisting resistance: Targeted therapies in lung cancer. Trends Cancer 2, 350–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yaffe M. B. (2013) The scientific drunk and the lamppost: massive sequencing efforts in cancer discovery and treatment. Sci. Signal. 6, pe13. [DOI] [PubMed] [Google Scholar]
- 5. Conrads T. P., and Petricoin E. F. 3rd, (2016) The Obama Administration's Cancer Moonshot: A call for proteomics. Clin. Cancer Res. 22, 4556–4558 [DOI] [PubMed] [Google Scholar]
- 6. Zhang J., Yang P. L., and Gray N. S. (2009) Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 9, 28–39 [DOI] [PubMed] [Google Scholar]
- 7. Schenone M., Dancik V., Wagner B. K., and Clemons P. A. (2013) Target identification and mechanism of action in chemical biology and drug discovery. Nat. Chem. Biol. 9, 232–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ziegler S., Pries V., Hedberg C., and Waldmann H. (2013) Target identification for small bioactive molecules: finding the needle in the haystack. Angew Chem. Int. Ed Engl. 52, 2744–2792 [DOI] [PubMed] [Google Scholar]
- 9. Hopkins A. L. (2008) Network pharmacology: the next paradigm in drug discovery. Nat. Chem. Biol. 4, 682–690 [DOI] [PubMed] [Google Scholar]
- 10. Bodenmiller B., Wanka S., Kraft C., Urban J., Campbell D., Pedrioli P. G., Gerrits B., Picotti P., Lam H., Vitek O., Brusniak M. Y., Roschitzki B., Zhang C., Shokat K. M., Schlapbach R., Colman-Lerner A., Nolan G. P., Nesvizhskii A. I., Peter M., Loewith R., von Mering C., and Aebersold R. (2010) Phosphoproteomic analysis reveals interconnected system-wide responses to perturbations of kinases and phosphatases in yeast. Sci. Signal 3, rs4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pan C., Olsen J. V., Daub H., and Mann M. (2009) Global effects of kinase inhibitors on signaling networks revealed by quantitative phosphoproteomics. Mol. Cell. Proteomics 8, 2796–2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bantscheff M., and Drewes G. (2012) Chemoproteomic approaches to drug target identification and drug profiling. Bioorg. Med. Chem. 20, 1973–1978 [DOI] [PubMed] [Google Scholar]
- 13. Ong S. E., Schenone M., Margolin A. A., Li X., Do K., Doud M. K., Mani D. R., Kuai L., Wang X., Wood J. L., Tolliday N. J., Koehler A. N., Marcaurelle L. A., Golub T. R., Gould R. J., Schreiber S. L., and Carr S. A. (2009) Identifying the proteins to which small-molecule probes and drugs bind in cells. Proc. Natl. Acad. Sci. U.S.A. 106, 4617–4622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rix U., and Superti-Furga G. (2009) Target profiling of small molecules by chemical proteomics. Nat. Chem. Biol. 5, 616–624 [DOI] [PubMed] [Google Scholar]
- 15. Daub H. (2015) Quantitative proteomics of kinase inhibitor targets and mechanisms. ACS Chem. Biol. 10, 201–212 [DOI] [PubMed] [Google Scholar]
- 16. Fang B., Haura E. B., Smalley K. S., Eschrich S. A., and Koomen J. M. (2010) Methods for investigation of targeted kinase inhibitor therapy using chemical proteomics and phosphorylation profiling. Biochem. Pharmacol. 80, 739–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fabbro D., Ruetz S., Bodis S., Pruschy M., Csermak K., Man A., Campochiaro P., Wood J., O'Reilly T., and Meyer T. (2000) PKC412–a protein kinase inhibitor with a broad therapeutic potential. Anticancer Drug Des. 15, 17–28 [PubMed] [Google Scholar]
- 18. Kayser S., Levis M. J., and Schlenk R. F. (2017) Midostaurin treatment in FLT3-mutated acute myeloid leukemia and systemic mastocytosis. Expert Rev. Clin. Pharmacol. 10, 1177–1189 [DOI] [PubMed] [Google Scholar]
- 19. Lee H. J., Schaefer G., Heffron T. P., Shao L., Ye X., Sideris S., Malek S., Chan E., Merchant M., La H., Ubhayakar S., Yauch R. L., Pirazzoli V., Politi K., and Settleman J. (2013) Noncovalent wild-type-sparing inhibitors of EGFR T790M. Cancer Discov. 3, 168–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li J., Bennett K., Stukalov A., Fang B., Zhang G., Yoshida T., Okamoto I., Kim J. Y., Song L., Bai Y., Qian X., Rawal B., Schell M., Grebien F., Winter G., Rix U., Eschrich S., Colinge J., Koomen J., Superti-Furga G., and Haura E. B. (2013) Perturbation of the mutated EGFR interactome identifies vulnerabilities and resistance mechanisms. Mol. Syst. Biol. 9, 705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Borgdorff V., Rix U., Winter G. E., Gridling M., Muller A. C., Breitwieser F. P., Wagner C., Colinge J., Bennett K. L., Superti-Furga G., and Wagner S. N. (2014) A chemical biology approach identifies AMPK as a modulator of melanoma oncogene MITF. Oncogene 33, 2531–2539 [DOI] [PubMed] [Google Scholar]
- 22. Medard G., Pachl F., Ruprecht B., Klaeger S., Heinzlmeir S., Helm D., Qiao H., Ku X., Wilhelm M., Kuehne T., Wu Z., Dittmann A., Hopf C., Kramer K., and Kuster B. (2015) Optimized chemical proteomics assay for kinase inhibitor profiling. J. Proteome Res. 14, 1574–1586 [DOI] [PubMed] [Google Scholar]
- 23. Knezevic C. E., Wright G., Remsing Rix L. L., Kim W., Kuenzi B. M., Luo Y., Watters J. M., Koomen J. M., Haura E. B., Monteiro A. N., Radu C., Lawrence H. R., and Rix U. (2016) Proteome-wide Profiling of Clinical PARP Inhibitors Reveals Compound-Specific Secondary Targets. Cell Chem. Biol. 23, 1490–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sumi N. J., Kuenzi B. M., Knezevic C. E., Remsing Rix L. L., and Rix U. (2015) Chemoproteomics Reveals Novel Protein and Lipid Kinase Targets of Clinical CDK4/6 Inhibitors in Lung Cancer. ACS Chem. Biol. 10, 2680–2686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., and Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 26. Cox J., Hein M. Y., Luber C. A., Paron I., Nagaraj N., and Mann M. (2014) Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell Proteomics 13, 2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang F., Shen Y., Camp D. G. 2nd, and Smith R. D. (2012) High-pH reversed-phase chromatography with fraction concatenation for 2D proteomic analysis. Expert Rev. Proteomics 9, 129–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 29. Welsh E. A., Eschrich S. A., Berglund A. E., and Fenstermacher D. A. (2013) Iterative rank-order normalization of gene expression microarray data. BMC Bioinformatics 14, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., and MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J., Simonovic M., Roth A., Santos A., Tsafou K. P., Kuhn M., Bork P., Jensen L. J., and von Mering C. (2015) STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kuenzi B. M., Remsing Rix L. L., Stewart P. A., Fang B., Kinose F., Bryant A. T., Boyle T. A., Koomen J. M., Haura E. B., and Rix U. (2017) Polypharmacology-based ceritinib repurposing using integrated functional proteomics. Nat. Chem. Biol. 13, 1222–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Szklarczyk D., Morris J. H., Cook H., Kuhn M., Wyder S., Simonovic M., Santos A., Doncheva N. T., Roth A., Bork P., Jensen L. J., and von Mering C. (2017) The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 45, D362–D368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Blondel V. D., Guillaume J. L., Lambiotte R., and Lefebvre E. (2008) Fast unfolding of communities in large networks. J. Stat. Mech. 2008, P10008 [Google Scholar]
- 35. Huang da W, Sherman B. T., and Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 36. Bonacich P. (1987) Power and centrality: A family of measures. Am. J. Sociol. 92, 1170–1182 [Google Scholar]
- 37. Remsing Rix L. L., Kuenzi B. M., Luo Y., Remily-Wood E., Kinose F., Wright G., Li J., Koomen J. M., Haura E. B., Lawrence H. R., and Rix U. (2014) GSK3 alpha and beta are new functionally relevant targets of tivantinib in lung cancer cells. ACS Chem. Biol. 9, 353–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang W., Soares J., Greninger P., Edelman E. J., Lightfoot H., Forbes S., Bindal N., Beare D., Smith J. A., Thompson I. R., Ramaswamy S., Futreal P. A., Haber D. A., Stratton M. R., Benes C., McDermott U., and Garnett M. J. (2013) Genomics of drug sensitivity in cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 41, D955–D961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Davis M. I., Hunt J. P., Herrgard S., Ciceri P., Wodicka L. M., Pallares G., Hocker M., Treiber D. K., and Zarrinkar P. P. (2011) Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1046–1051 [DOI] [PubMed] [Google Scholar]
- 40. Klaeger S., Heinzlmeir S., Wilhelm M., Polzer H., Vick B., Koenig P. A., Reinecke M., Ruprecht B., Petzoldt S., Meng C., Zecha J., Reiter K., Qiao H., Helm D., Koch H., Schoof M., Canevari G., Casale E., Depaolini S. R., Feuchtinger A., Wu Z., Schmidt T., Rueckert L., Becker W., Huenges J., Garz A. K., Gohlke B. O., Zolg D. P., Kayser G., Vooder T., Preissner R., Hahne H., Tonisson N., Kramer K., Gotze K., Bassermann F., Schlegl J., Ehrlich H. C., Aiche S., Walch A., Greif P. A., Schneider S., Felder E. R., Ruland J., Medard G., Jeremias I., Spiekermann K., and Kuster B. (2017) The target landscape of clinical kinase drugs. Science 358, eaan4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Barabasi A. L., Gulbahce N., and Loscalzo J. (2011) Network medicine: a network-based approach to human disease. Nat. Rev. Genetics 12, 56–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brandes U. (2001) A faster algorithm for betweenness centrality. J. Math. Sociol. 25, 163–177 [Google Scholar]
- 43. Bain J., Plater L., Elliott M., Shpiro N., Hastie C. J., McLauchlan H., Klevernic I., Arthur J. S., Alessi D. R., and Cohen P. (2007) The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408, 297–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Najafov A., Sommer E. M., Axten J. M., Deyoung M. P., and Alessi D. R. (2011) Characterization of GSK2334470, a novel and highly specific inhibitor of PDK1. Biochem. J. 433, 357–369 [DOI] [PubMed] [Google Scholar]
- 45. Gorgun G., Calabrese E., Hideshima T., Ecsedy J., Perrone G., Mani M., Ikeda H., Bianchi G., Hu Y., Cirstea D., Santo L., Tai Y. T., Nahar S., Zheng M., Bandi M., Carrasco R. D., Raje N., Munshi N., Richardson P., and Anderson K. C. (2010) A novel Aurora-A kinase inhibitor MLN8237 induces cytotoxicity and cell-cycle arrest in multiple myeloma. Blood 115, 5202–5213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Frost A., Mross K., Steinbild S., Hedbom S., Unger C., Kaiser R., Trommeshauser D., and Munzert G. (2012) Phase i study of the Plk1 inhibitor BI 2536 administered intravenously on three consecutive days in advanced solid tumours. Curr. Oncol. 19, e28–e35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vose J. M., Friedberg J. W., Waller E. K., Cheson B. D., Juvvigunta V., Fritsch H., Petit C., Munzert G., and Younes A. (2013) The Plk1 inhibitor BI 2536 in patients with refractory or relapsed non-Hodgkin lymphoma: a phase I, open-label, single dose-escalation study. Leuk. Lymphoma 54, 708–713 [DOI] [PubMed] [Google Scholar]
- 48. Chien Y., Kim S., Bumeister R., Loo Y. M., Kwon S. W., Johnson C. L., Balakireva M. G., Romeo Y., Kopelovich L., Gale M. Jr., Yeaman C., Camonis J. H., Zhao Y., and White M. A. (2006) RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell 127, 157–170 [DOI] [PubMed] [Google Scholar]
- 49. Barbie D. A., Tamayo P., Boehm J. S., Kim S. Y., Moody S. E., Dunn I. F., Schinzel A. C., Sandy P., Meylan E., Scholl C., Frohling S., Chan E. M., Sos M. L., Michel K., Mermel C., Silver S. J., Weir B. A., Reiling J. H., Sheng Q., Gupta P. B., Wadlow R. C., Le H., Hoersch S., Wittner B. S., Ramaswamy S., Livingston D. M., Sabatini D. M., Meyerson M., Thomas R. K., Lander E. S., Mesirov J. P., Root D. E., Gilliland D. G., Jacks T., and Hahn W. C. (2009) Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462, 108–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cooper J. M., Ou Y. H., McMillan E. A., Vaden R. M., Zaman A., Bodemann B. O., Makkar G., Posner B. A., and White M. A. (2017) TBK1 provides context-selective support of the activated AKT/mTOR pathway in lung cancer. Cancer Res. 77, 5077–5094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kim J. Y., Welsh E. A., Oguz U., Fang B., Bai Y., Kinose F., Bronk C., Remsing Rix L. L., Beg A. A., Rix U., Eschrich S. A., Koomen J. M., and Haura E. B. (2013) Dissection of TBK1 signaling via phosphoproteomics in lung cancer cells. Proc. Natl. Acad. Sci. U.S.A. 110, 12414–12419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ou Y. H., Torres M., Ram R., Formstecher E., Roland C., Cheng T., Brekken R., Wurz R., Tasker A., Polverino T., Tan S. L., and White M. A. (2011) TBK1 directly engages Akt/PKB survival signaling to support oncogenic transformation. Mol. Cell 41, 458–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dibble C. C., Asara J. M., and Manning B. D. (2009) Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol. Cell Biol. 29, 5657–5670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. O'Reilly K. E., Rojo F., She Q. B., Solit D., Mills G. B., Smith D., Lane H., Hofmann F., Hicklin D. J., Ludwig D. L., Baselga J., and Rosen N. (2006) mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 66, 1500–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kang J. I., Hong J. Y., Lee H. J., Bae S. Y., Jung C., Park H. J., and Lee S. K. (2015) Anti-tumor activity of yuanhuacine by regulating AMPK/mTOR signaling pathway and actin cytoskeleton organization in non-small cell lung cancer cells. PloS one 10, e0144368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Katsha A., Wang L., Arras J., Omar O. M., Ecsedy J., Belkhiri A., and El-Rifai W. (2017) Activation of EIF4E by Aurora kinase A depicts a novel druggable axis in everolimus-resistant cancer cells. Clin. Cancer Res. 23, 3756–3768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zheng X., Chi J., Zhi J., Zhang H., Yue D., Zhao J., Li D., Li Y., Gao M., and Guo J. (2018) Aurora-A-mediated phosphorylation of LKB1 compromises LKB1/AMPK signaling axis to facilitate NSCLC growth and migration. Oncogene 37, 502–511 [DOI] [PubMed] [Google Scholar]
- 58. Tan J., Li Z., Lee P. L., Guan P., Aau M. Y., Lee S. T., Feng M., Lim C. Z., Lee E. Y., Wee Z. N., Lim Y. C., Karuturi R. K., and Yu Q. (2013) PDK1 signaling toward PLK1-MYC activation confers oncogenic transformation, tumor-initiating cell activation, and resistance to mTOR-targeted therapy. Cancer Discov. 3, 1156–1171 [DOI] [PubMed] [Google Scholar]
- 59. Lens S. M., Voest E. E., and Medema R. H. (2010) Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat. Rev. Cancer 10, 825–841 [DOI] [PubMed] [Google Scholar]
- 60. Zitouni S., Nabais C., Jana S. C., Guerrero A., and Bettencourt-Dias M. (2014) Polo-like kinases: structural variations lead to multiple functions. Nat. Rev. Mol. Cell Biol. 15, 433–452 [DOI] [PubMed] [Google Scholar]
- 61. Macurek L., Lindqvist A., Lim D., Lampson M. A., Klompmaker R., Freire R., Clouin C., Taylor S. S., Yaffe M. B., and Medema R. H. (2008) Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature 455, 119–123 [DOI] [PubMed] [Google Scholar]
- 62. Pillai S., Nguyen J., Johnson J., Haura E., Coppola D., and Chellappan S. (2015) Tank binding kinase 1 is a centrosome-associated kinase necessary for microtubule dynamics and mitosis. Nat. Commun. 6, 10072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Takai N., Hamanaka R., Yoshimatsu J., and Miyakawa I. (2005) Polo-like kinases (Plks) and cancer. Oncogene 24, 287–291 [DOI] [PubMed] [Google Scholar]
- 64. Vizcaino J. A., Csordas A., del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q. W., Wang R., and Hermjakob H. (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (http://www.ebi.ac.uk/pride) with the dataset identifiers PXD009041 (Phosphotyrosine Profiling), PXD010786 (IMAC Phosphoproteomics) and PXD010787 (Chemical Proteomics) (64).