


Abstract
Transcriptionally non-co-linear (NCL) transcripts can originate from trans-splicing (trans-spliced RNA; ‘tsRNA’) or cis-backsplicing (circular RNA; ‘circRNA’). While numerous circRNAs have been detected in various species, tsRNAs remain largely uninvestigated. Here, we utilize integrative transcriptome sequencing of poly(A)- and non-poly(A)-selected RNA-seq data from diverse human cell lines to distinguish between tsRNAs and circRNAs. We identified 24,498 NCL events and found that a considerable proportion (20–35%) of them arise from both tsRNAs and circRNAs, representing extensive alternative trans-splicing and cis-backsplicing in human cells. We show that sequence generalities of exon circularization are also observed in tsRNAs. Recapitulation of NCL RNAs further shows that inverted Alu repeats can simultaneously promote the formation of tsRNAs and circRNAs. However, tsRNAs and circRNAs exhibit quite different, or even opposite, expression patterns, in terms of correlation with the expression of their co-linear counterparts, expression breadth/abundance, transcript stability, and subcellular localization preference. These results indicate that tsRNAs and circRNAs may play different regulatory roles and analysis of NCL events should take the joint effects of different NCL-splicing types and joint effects of multiple NCL events into consideration. This study describes the first transcriptome-wide analysis of trans-splicing and cis-backsplicing, expanding our understanding of the complexity of the human transcriptome.
INTRODUCTION
Precursor mRNA (pre-mRNA) splicing can join exons in an order that is topologically inconsistent with the corresponding DNA template and generate non-co-linear (NCL) transcripts at the transcriptional level. NCL transcripts may originate from trans-splicing (i.e. trans-spliced RNA or ‘tsRNA’), which occurs between two or more separate pre-mRNAs derived from a single gene (intragenic tsRNA) or different genes (intergenic tsRNA) (1–3); or from cis-backsplicing (i.e. circular RNA or ‘circRNA’), which occurs within a single pre-mRNA (1,4). It was reported that circRNAs can act as microRNA sponges (5–10), regulate their parent genes (11–15) or play a regulatory role in development (16–18), the aging nervous system (19) and cancer growth/metastasis (10,20). circRNAs were also shown to be enriched in exosomes and suggested to be a promising biomarker for cancer diagnosis (21,22). As circRNAs are ubiquitous and have been widely detected in diverse species (16,23–28), tsRNAs remain largely unexplored in higher eukaryotes. However, accumulating evidence reveals the biological significance of tsRNAs. In humans, some tsRNAs were experimentally confirmed to be evolutionarily conserved in rhesus macaque/mouse (1). tsRNAs were also demonstrated to associate with anti-apoptotic function (29–31), prostate cancer (29,32) or pluripotency maintenance of human embryonic stem cells (hESCs) (33). Compared with co-linear mRNAs, most circRNAs are expressed at much lower levels (24,26,34) and tsRNA expression is lower still (35). Whether most NCL events are merely side-products of imperfect pre-mRNA splicing remains debatable (26,36). Actually, the biogenesis and functions of these two types of NCL events (tsRNAs and circRNAs) are still understudied.
In terms of biogenesis of NCL events, both trans- and cis-backsplicing events were reported to be produced by canonical splicing mechanisms (11,12,14,37–40) and regulated by cis- and trans-acting elements (11,12,14,17,30,39,41–45). It was observed that both tsRNA and circRNA junctions were predominantly located at canonical splice sites (e.g., GU-AG splice sites) (1,18,24,26,33,46–50). For circRNAs, several generalities of formation have been observed. For example, circRNAs exhibit a bias toward involving the middle exons of annotated genes (41,51). The majority of circRNAs are flanked by longer introns compared with their co-linear counterparts (24,41,52). In addition, the reverse complementary sequences residing in the introns flanking circularized exons, particularly inverted Alu repeats (IRAlus) in humans, are highly associated with the formation of circRNAs (7,24,41,52,53). Although the existence of complementary sequences in introns flanking trans-spliced exons was expected to promote the formation of paired duplex structures of transcripts allowing trans-splicing between different pre-mRNAs (38,54,55), no direct experimental evidence supports this model currently. Whether tsRNAs and circRNAs share similar generalities of formation also remains uninvestigated. Regarding the expression context, it was observed that the expression patterns of NCL events were not necessarily correlated with those of their corresponding co-linear mRNAs (16,17,26,34,53,56). A few circRNAs (7,24) and tsRNAs (33) are even more highly expressed than their co-linear counterparts, even though pre-mRNAs are regarded to be sources for both types of NCL RNAs. In addition, it was shown that multiple circRNAs can be generated from a single gene locus (7,11,23,24,41,51,57–59) and intragenic tsRNAs and circRNAs can share the same NCL junctions (1,35), further complicating the exploration of NCL expression. There thus remains a need for explication of expression patterns for different types of NCL events (particularly for the case that tsRNAs and circRNAs share the same NCL junctions).
To clarify different types of NCL events (tsRNA, circRNA or both sharing the same junction), we thus ask the following questions: (i) What is the transcriptome-wide distribution of different NCL-splicing types in human? (ii) Whether tsRNAs and circRNAs share similar generalities of formation? (iii) Can complementary sequences in flanking introns promote the formation of both tsRNA and circRNA isoforms simultaneously? and (iv) Whether tsRNAs and circRNAs exhibit similar expression patterns? Here we take advantage of polyadenylated and non-polyadenylated RNA-seq data from diverse human cell lines using NCLscan (35), which has been demonstrated to well control for alignment ambiguity and yield very high precision (>98%) without sacrificing sensitivity, to identify NCL events. On the basis of the concept that circRNAs are generally RNase R-resistant or non-polyadenylated but tsRNAs are not (1,5,14,23,26,41,57,60), we determined the presence of identified NCL events in poly(A)- and non-poly(A)-selected RNA-seq data to distinguish between tsRNAs and circRNAs and then systematically characterized different types of NCL events. We thus identified 24 498 NCL events in seven cell lines. By controlling for alignment ambiguity, read depth and experimental artifacts (RT-based artifacts), we showed that circRNA isoforms form the majority of intragenic NCL events and 20–35% of NCL junctions arise from both tsRNA and circRNA isoforms. We also utilized the Third Generation Sequencing platform, Oxford Nanopore Technologies (ONT) MinION sequencer (61), to generate long RNA-seq reads and thus distinguish tsRNAs from circRNAs and the corresponding co-linear mRNA background. We observed that some generalities of exon circularization were also present in tsRNAs. Importantly, we recapitulated the formation of a tsRNA-circRNA junction-sharing NCL event from a unique expression vector and experimentally confirmed that IRAlus can promote tsRNA and circRNA formation simultaneously, supporting the hypothesis that the formation of both types of NCL isoforms are associated with flanking complementary sequences. Furthermore, we showed that NCL-splicing types may vary among cell lines, and circRNAs and tsRNAs exhibit different expression patterns in terms of expression abundance/breadth, correlation with expression of their co-linear counterparts, transcript stability and subcellular localization preference. Our results thus indicate that tsRNA and circRNA isoforms may play different biological roles, shedding light on the fundamental biology of various types of non-canonical alternative splicing in the human transcriptome.
MATERIALS AND METHODS
Data retrieval
NCL candidates were identified by NCLscan 1.4 (35) on the basis of the human reference genome (GRCh37) and the GENCODE annotation (version 19), in which co-linear matches were eliminated by mapping RNA-seq reads against the reference genome, mitochondrial genome and GENCODE-annotated coding/non-coding transcripts. To evaluate the precision of NCLscan, we performed flux simulator (62) to generate paired-end RNA seq reads (a negative dataset) and showed that NCLscan did not identify any false positives. This result completely consists with two previous studies (35,63), which generated negative datasets using different RNA-seq read simulators (Mason (64) and ART (65)) and also reported that zero false call was identified by NCLscan on the corresponding negative datasets. In addition, on the basis of sequencing spike-ins (sequins) data (66), NCLscan also identified zero false call. Of note, the spike-ins data was downloaded from NCNI SAR at https://www.ncbi.nlm.nih.gov/sra/?term=SRR4011970 (accession number: SRR4011970), which was sequenced total RNA with 78 artificial genes (164 alternatively spliced isoforms) located in an ∼11 Mb artificial in silico chromosome. The spike-ins sequence must not contain any NCL events. GM12878 total RNA was spiked with RNA sequins Mix B prior to library preparation. These results thus support the good precision of NCLscan. The RNA-seq data used in this study are listed in Table 1. The expression level of NCL events in each sample was determined as the number of supported reads per million raw reads (RPM) (67). The quantitation of expression levels for co-linear transcripts was determined as fragments per kilobases of exon per million fragments mapped (FPKM) and evaluated with TopHat v2.0.9 (http://tophat.cbcb.umd.edu) and CuffLink v2.1.1 (Http://cufflinks.cbcb.umd.edu). The synonymous constraint elements (SCEs) based on 29 eutherian mammals were downloaded from the study of Lindblad-Toh et al. (68) at http://genomewiki.cse.ucsc.edu/index.php/29mammals. We defined the usage of a NCL donor (or acceptor) site as the count of all the distinct NCL event(s) with this donor (or acceptor) site. Distinct NCL events may share the same donor (or acceptor) sites. Alu elements were identified by RepeatMasker and were downloaded from the University of California Santa Cruz (UCSC) genome browser at https://genome.ucsc.edu/. Since the distance between potential IRAlu pairs may affect the pairing capacity of circRNA formation (41,67), we examined these generalities of IRAlus located within ≤5000 bp regions of flanking introns to donor/acceptor NCL splice sites for these three groups of NCL events. The find_circ 2 (5), CIRCexplorer (41) and CIRI (27) packages were downloaded at https://github.com/marvin-jens/find_circ, https://github.com/YangLab/ CIRCexplorer and https://sourceforge.net/projects/ciri/, respectively. The numbers of reads spanning the co-linearly spliced junctions at both NCL donor and acceptor sites were obtained from the read-to-genome STAR-alignments (STAR 2.5.2b, https://github.com/alexdobin/STAR) (69), with uniquely mapping reads crossing the junction. The A-to-I RNA editing sites were derived from the well-known public databases: DARNED (70), RADAR (71) and REDIportal (72). The editing level of a site was calculated by dividing the number of G by the total number of A and G. Only the editing sites with ≥10 mapped RNA-seq reads were considered.
Table 1. Summary of RNA-seq data used in this study.
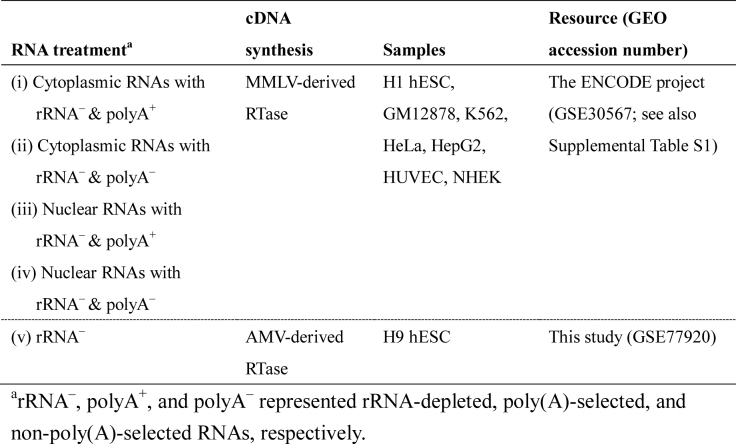
|
Data access
All the related data of this study was deposited in Supplementary Tables S1–9 and Figures S1–10. The catalog of the 24 498 NCL events identified by NCLscan in the seven cell lines and their corresponding expression levels (RPM) determined by different types of RNA-seq data are included in Supplementary Table S1. The catalog of the RT-independent NCL events, which are supported by both Avian Myeloblastosis Virus (AMV)- and Moloney Murine Leukemia Virus (MMLV)-derived RNA-seq reads from H9 hESCs, are included in Supplementary Table S4. Sequence generalities of formation for different types of NCL events are provided in Supplementary Table S5. The catalog of the intragenic NCL events identified by find_circ, CIRCexplorer and CIRI in the seven cell lines are provided in Supplementary Table S7. The simulated dataset (a negative dataset) generated by the flux simulator is publicly available at ftp://treeslab1.genomics.sinica.edu.tw/NCLscan/SimulationDatasets/fluxSimulato/. The generated AMV-based RNA-seq data from H9 hESCs and Oxford Nanopore RNA-seq data from HeLa cells were deposited in the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE77920.
Statistical analysis
All statistical analyses were performed using the R package (version 3.2.1). We used partial correlation analysis (73) to control for tsRNA or circRNA expression while evaluating the correlation between NCL event expression and the expression of their co-linear counterparts. All the used statistical tests, including the two-tailed Fisher’s exact test, two-tailed Wilcoxon rank-sum test, paired two-tailed Wilcoxon rank-sum test, two-tailed t-test and Spearman’s rank correlation, were stated in the corresponding figure legends.
AMV-based RNA-seq library construction and deep sequencing
To construct RNA-seq library for AMV-mediated reverse transcription (RT), Illumina TruSeq Stranded RNA-seq library prep kit was used with the following modifications. First of all, an input of 4 μg total RNA was applied to the Ribo-Zero- rRNA Removal Kits (Human-Mouse-Rat) (Epicentre), with final purification performed using Agencourt RNAClean SPRI beads (Beckman Coulter) instead of ethanol precipitation. Briefly, total RNA was heated at 68°C for 10 min and cooled to room temperature for 5 min with Ribo-Zero reaction buffer and Ribo-Zero rRNA removal solution. The heat-treated RNA sample was then mixed with pre-washed Ribo-Zero magnetic beads resuspended in Ribo-Zero magnetic bead suspension solution to which was added RiboGuard RNase inhibitor. The mixture was incubated at room temperature for 5 min and then shifted to 50°C for 5 min. The beads were then removed with Magnetic Particle Concentrator (MPC), the supernatant was further purified with two volumes of RNAClean SPRI beads, and the rRNA-depleted RNA was eluted in RNase-free water. The rRNA-depleted RNA was then added to Elution 2-Frag-Prime (Illumina), the mixture was incubated at 94°C for 7 min to fragment RNA and immediately chilled on ice. The fragmented RNA sample was equally divided into two aliquots for downstream differential processing. For first strand cDNA synthesis using AMV, 10 μl AMV Reaction Mix (2X) and 2 μl AMV enzyme (1U) (74) were added to each sample of fragmented, rRNA-depleted RNA. The 20 μl reaction was incubated at 25°C for 5 min and shifted to 42°C for 1 h, followed by inactivation at 80°C for 5 min. To synthesize second-strand cDNA, 16 μl Second Strand mix (Stranded RNA-seq library prep kit, Illumina) was added to each tube of the product from the first strand synthesis reaction, in which dUTP was used in place of dTTP. The reactions were incubated at 16°C for 1 h and then purified using the Agencourt AMPure XP kit (Beckman Coulter). The double stranded cDNA fragments were end-repaired and phosphorylated, followed by 3′-end adenylation and ligation to the TruSeq adaptors in the kit (Illumina). The ligation reactions were treated with 1 U USER Enzyme (Cat. #M5505S, NEB) (74M5505S) at 37°C for 30 min to remove uracils within the second strand of cDNA. The samples were subjected to 12 cycles of polymerase chain reaction (PCR) and purified using the Agencourt AMPure XP kit (Beckman Coulter). The purified libraries were profiled using Bioanalyzer 2100 system (Agilent) and quantified by Qubit dsDNA HS Assay Kit (Life Technologies) and the molar concentration was normalized using the NGS Library Quantification Kit (Kapa Biosystems). Paired-end (2 × 150 nt) sequencing was carried out using the rapid mode on a HiSeq2500 sequencer (Illumina).
Nanopore amplicon library preparation and nanopore data analysis
The polyadenylated RNA from HeLa cell was treated with DNase and reverse transcribed with the SuperScript III reverse transcriptase (Thermo Fisher Scientific). PCR were performed with the DreamTaq polymerase (Thermo Fisher Scientific), then purified the amplicons using KAPA pure beads (Kapa Biosystems), quantified by the Qubit fluorometer (Thermo Fisher Scientific). Library for nanopore sequencing was constructed using Ligation sequencing Kit 1D (SKQ-LSK108) and Native barcoding kit (EXP-NBD103) following Oxford Nanopore Technologies (ONT) protocol. Briefly, 1ug purified amplicons were end repaired and A tailed using NEBNext Ultra II End Repair/dA-Tailing Module (New England BioLabs). Individual barcode was added to the A-tailed amplicons using NEB Blunt / TA Ligase Master Mix (New England BioLabs). Purified barcoded amplicons were pooled at 700ng in 50ul and adaptor was added to using NEBNext Quick Ligation Module (New England BioLabs). Library was loaded into SpotON flowcell R9.5 (FLO-MIN107) and Sequencing script NC_48Hr_Sequencing_FLO-MIN107_SQK_LSK108 was executed on MinKNOW1.7.14. To detect tsRNA isoforms and the corresponding co-linear mRNA isoforms of FARSA, HIPK3, CAMSAP1, and POLR2A, seven barcoded amplicons were run onto one flow cell using the primer pairs listed in Supplementary Table S9. Similarly, for recapitulation of POLR2A NCL RNAs, seven barcoded amplicons (for T1-T7) were constructed with egfp primers (Supplementary Table S9) and were also run onto one flow cell. The raw signal data was processed with Albacore software (v2.0.2) for base calling. Reads containing sequences of adapters were trimmed by using fastx_clipper (http:// hannonlab.cshl.edu/fastx_toolkit/).
To discriminate between tsRNA isoforms and the corresponding co-linear mRNA isoforms, we made putative references of these two types of isoforms. For example, on the basis of the NCL junction of POLR2A exon10–exon9, we made two putative reference sequences for POLR2A tsRNA isoform (i.e. exon8–exon9–exon10–exon9–exon10 and exon9–exon10–exon9–exon10–exon11) and two putative reference sequences for its corresponding co-linear isoform (i.e. exon8–exon9–exon10 and exon9–exon10–exon11). We used BWA (version 0.7.16; for nanopore read-to-reference sequence alignment) to align nanopore long reads against the putative tsRNA and co-linear reference sequences with the parameters: bwa mem -x ont2d. Each mapped nanopore read should satisfy both of the two criteria: (i) the mapping quality of the mapped nanopore read must be ≥30; and (ii) mapped length of the nanopore read must be ≥90% of the length of the putative reference sequence.
Cell culture and transfection
K562 and HeLa cells were obtained from the American Type Culture Collection. The former were cultured in Iscove’s Modified Dulbecco’s Medium (IMDM) and the latter were in Dulbecco’s Modified Eagle Medium (DMEM). Both cell lines were cultured at 37°C with 5% CO2 in 10% fetal bovine serum (FBS) and 1% Antibiotic-Antimycotic solution (Thermo). The H9 hESCs were cultured on Mitomycin C-treated mouse embryonic fibroblast (MEF) feeders (2 × 104 cells/cm2) in DMEM/F12 medium with 20% Knockout Serum Replacement (Invitrogen) and 4 ng/ml bFGF (Sigma-Aldrich). The ESCs were transferred to new feeder medium every four days. All ESCs were maintained by MEF feeder cells. The MEFs used for ESCs were cultured in DMEM supplemented with 10% FBS (Level), 1 × non-essential amino acids (NEAA, Invitrogen), and 2 mM L-glutamine (Invitrogen) and treated with Mitomycin C. The protocol described by Dr Nicholas RF Hannan (75) was applied to in vitro differentiation of H9 ESCs into hepatic endoderm for 11 days. ESC lines were dispersed into small clumps using EDTA (Gibco; 0.5 mM for 2 min in 37°C, 5% CO2, 5% O2 incubator) and transferred onto VTN-N (Gibco) coated culture plates (Corning, NY, USA) and maintained for 48 h in Essential 8 media (Gibco). The initial stages of differentiation were carried out using CDM-PVA Medium consisting 0.5 g of PVA (Sigma) dissolved in 250 ml of IMDM/F-12, GlutaMAX (Invitrogen), 250 ml of IMDM (Invitrogen), 5 ml of chemically defined lipid concentrate (Invitrogen), 20 ml of thioglycerol 97% (Sigma), 350 ml of insulin (10 mg/ml; Roche), 250 ml of transferrin (30 mg/ml; Roche) and 5 ml of penicillin/streptomycin (10 000 U/ml; Invitrogen). Media was changed daily for all subsequent steps, and cells were differentiated at 37°C, 5% CO2, 5% O2. On days 2–3, cells were differentiated in CDM-PVA supplemented with Activin A (100 ng/ml; R&D), FGF2 (80 ng/ml; R&D), BMP4 (10 ng/ml; R&D), 10 mM LY-294002 (Promega) and 3 mM Stemolecule CHIR99021 (StemGent). On day 4, cells were differentiated in CDM-PVA supplemented with Activin A (100 ng/ml), FGF2 (80 ng/ml), BMP4 (10 ng/ml; R&D) and 10 mM LY-294002. On day 5, cells were differentiated in RPMI Medium (RPMI 1640 Medium, GlutaMAX (Invitrogen), 2% B-27 Serum-Free Supplement (50×) (Invitrogen), 1% MEM Non- Essential Amino Acids Solution (100×) (Invitrogen), 1% penicil- lin/streptomycin) supplemented with Activin A (100 ng/ml) and FGF2 (80 ng/ml). On day 6, cells were expanded in RPMI medium supplemented with Activin A (50 ng/ml). On day 7, cells were split using Cell Dissociation Buffer (Enzyme-free, Hank's; Invitrogen) and were plated in VTN-N coated culture plates at a density of 105 000 cells/cm2 in RPMI+Activin A (50 ng/ml)+Y-27632 2HCl (10 mM Selleck- chem). Cells were maintained in RPMI+Activin A (50 ng/ml) on days 8–11.
The plasmids of wild-type and a series of Alu deletion constructs for the POLR2A transcription segments (exons 9 and 10) cloned into pZW1 were directly obtained from Prof. Ling-Ling Chen (Chinese Academy of Sciences, China). Cells were transfected using TransIT®-LT1 Transfection Reagent (Mirus Bio) in accordance with the manufacturer’s instructions. Cells were harvested at 24 h after transfection. For the test of transcript stability, transcription was blocked by adding 2 μg ml−1 actinomycin D or Dimethyl Sulfoxide (DMSO; as a mock control) to the cell medium. Cell samples were collected at 0, 4, 8, 12 and 24 h after actinomycin D or DMSO treatment.
RNA extraction and RT-PCR
Total RNA was extracted using Trizol reagent (Invitrogen) according to the manufacturer’s protocol. After total RNA extraction, total RNA was treated with DNase I (Invitrogen) to eliminate genomic DNA contamination. The cDNA libraries prepared by MMLV-derived reverse transcriptase (Superscript III, Invitrogen) and AMV-derived reverse transcriptase (Promega) were primed with random hexamers and oligo(dT) primers. Reverse transcribed at 50°C for 2 h in mixed reagents provided by the manufacturers. All RT-PCR amplicons were amplified under 32 cycles using GoTag MasterMix (Promega) and quantitative RT-PCR (qRT-PCR) assays were performed using the SYBR 2× Master Mix (Thermo). All of the qRT-PCR reactions were performed three times per experiment. The RT-PCR/qRT-PCR primers used in this study are provided in Supplementary Table S9.
Purification of mRNAs from total RNA and RNase R treatment
The mRNAs were isolated using the Oligotex mRNA Min Kit (QIAGEN) according to the manufacturer’s protocol. After DNase I treatment, 20 μg of total RNA was put in a dry bath incubator at 70°C for 3 min to break up RNA secondary structure; the RNA was then incubated with Oligotex-particles solution at 30°C for 10 min. Oligotex-bound mRNAs were then centrifuged, collected and eluted with buffers provided in the kit. DNase-treated total RNA (2 μg) or purified mRNA (100 ng) were incubated for 1 h at 37°C with or without 3 U/μg RNase R (Epicentre). RNA was subsequently collected and condensed by phenol–chloroform extraction. For the validation of TS-circRNA events, we used GAPDH (which is poly(A)-tailed and must be degraded by RNase R treatment) and CDR1as (which is RNase R-resistant and must be non-polyadenylated) (1,5,6) as controls and performed qRT-PCR analyses to examine the expression fold changes for the selected TS-circRNA events before and after treatment.
Validation of the subcellular localization preference of tsRNA and circRNA isoforms for TS-circRNA events
To validate the subcellular localization preference of tsRNA and circRNA products, we selected eight TS-circRNA events that had been experimentally confirmed to exhibit both tsRNA and circRNA isoforms at the same NCL junctions (Figure 2F and G). We proceeded to separately isolate total nuclear and cytoplasmic RNA from diverse human cell lines (undifferentiated and differentiated H9 hESCs, HeLa and K562). The nuclear and cytoplasmic RNA were extracted using NE-PER nuclear and cytoplasmic extraction reagents (Thermo) and subsequently purified using the Trizol reagent method. We then separately purified mRNAs with poly(A) tails and treated total RNA with RNase R for both the isolated nuclear and cytoplasmic RNAs. Subsequently, qRT-PCR analyses were performed to examine the relative expression of the poly(A)-tailed RNA products (i.e. tsRNA isoforms) in the cytoplasm and nucleus and RNase R-treated RNA products (i.e. circRNA isoforms) in the cytoplasm and nucleus, respectively. We used GAPDH (which is known to be predominately cytoplasmic) and circEIF3J (which is an intron-retained circRNA and has been confirmed to be enriched in the nucleus (13)) as controls.
Figure 2.

Different types of NCL events. (A–C) Distribution of three types of NCL events: TS-only, circRNA-only and TS-circRNA events before (A) and after (B and C) controlling for read depth/RT-dependence. For (B and C), read depth was normalized by randomly selecting an equal number (60 million, left; 120 million, right) of reads from both poly(A)- and non-poly(A)-selected data for the examined cell lines (Supplementary Tables S2 and 3). For (C), the RT-independent NCL events, which were supported by both AMV- and MMLV-based reads, were considered only (see the text). (D) Comparisons of percentages of tsRNA-involved events (i.e. TS-only and TS-circRNA events) that were detected in both poly(A)-selected data from hESCs and non-poly(A)-selected data from non-ESC samples. (E) Comparison of percentages of circRNA-involved events (i.e. circRNA-only and TS-circRNA events) that were detected in both non-poly(A)-selected data from hESCs and poly(A)-selected data from non-ESC samples. (F) qRT-PCR analyses of the expression fold changes for the selected 42 TS-circRNA events and two controls, GAPDH (which is poly(A)-tailed and must be degraded by RNase R treatment) and CDR1as (which is RNase R-resistant and must be non-polyadenylated) (1,5,6), before and after RNase R treatment in H9 hESCs. (G) Comparisons of expression fold changes for the selected 40 TS-circRNA events in poly(A)-tailed RNAs (oligo-dT pull down) and poly(A)-tailed RNAs treated with RNase R in H9 hESCs. The green (F) and blue (G) asterisks represent statistical significances of expression fold changes between the selected events and the controls (GAPDH for (F), green dashed lines; CDR1as for (G), blue dashed line). For (G), the red asterisks represent statistical significances of expression fold changes between the poly(A)-tailed RNAs and the poly(A)-tailed RNAs treated with RNase R. qRT-PCR experiments were performed in triplicate and repeated twice. Error bars represent the mean values ± one standard deviation. The statistical significance was evaluated using the two-tailed Fisher’s exact test (D and E) and the two-tailed t-test (F and G), respectively. *P < 0.05, **P < 0.01 and ***P < 0.001. (H) Detection of expanded length of tsRNAs by nanopore long reads. Blue and orange arrows indicate two different pairs of primers of the three examined TS-circRNA events, FARSA, HIPK3 and CAMSAP1. Empty rectangles represent the exons located outside the predicted circles of exonic circRNAs (the blue solid rectangles). E, exon.
RNA in situ hybridization
HeLa cells were grown in 24-well overnight, and then fixed in 4% paraformaldehyde solution at room temperature for 10 min, rinsed with phosphate-buffered saline twice. Cells were incubated in hybridization buffer (2× Magnetic Particle Concentrator (SSC) , 10% dextran sulfate, 10% deionized formamide in nucleus free water) with Cy5-labeled probes antisense to the back-splice junction of target circRNAs at 40°C for 16 h. After hybridization, cells were washed with 2× SSC at 37°C for 30 min. Slides were mounted onto glass slides using DAPI-Fluoromount-G mounting medium (SouthernBiotech). The fluorescence signals were detected by confocal microscope system (Leica TCS-SP5-MP-SMD), and the signal intensities were quantified by the ImageJ software.
RNase protection assay (RPA)
The RPA approach (76) is not dependent on amplification or RT. In short, 10 μg sample RNAs were hybridized with the radiolabeled RNA (antisense) probe by RPA III kit (Ambion). The 32P-labeled RNA probe and control template were constructed using the MAXIscript SP6/T7 Transcription Kit (Ambion) according to manufacturer guidelines. After hybridization of RNA samples with isotope-labeled antisense probe, the mixture incubated with RNase A/T1 to digest unprotected single-stranded regions. The protected probe-RNA fragments were then separated on 6% acrylamide gel, then transfer the gel and exposed to X-ray film for 4 days at −80°C.
RESULTS
Identification and characterization of NCL transcripts
To conduct the genome-wide identification of NCL transcripts in the human transcriptome, we retrieved the RNA-seq data of seven human cell lines from the ENCODE project (77,78), each of which contained cytoplasmic/nuclear poly(A)- and non-poly(A)-selected RNA-seq data simultaneously (Table 1). These data allowed us to undertake follow-up analyses, including the discrimination between different types of intragenic NCL isoforms and the examination of subcellular localization preference for the identified NCL events. For each cell line, we first integrated all poly(A)- and non-poly(A)-selected RNA-seq data and then utilized NCLscan, which was demonstrated to be adept at effectively eliminating alignment artifacts with a good balance between sensitivity and precision while detecting NCL transcripts (35), to identify intragenic NCL transcript candidates. Of note, we only considered the NCL junctions located at well-annotated exon boundaries because NCL-splicing events were suggested to be produced by canonical spliceosomal mechanisms (11,12,14,37–40) and such non-co-linearly spliced junctions were suggested to be more reliable than those not matching exon boundaries (1,33,35,46,47). After that, 2000–9000 (24 498 in total) distinct intragenic NCL candidates were identified in the seven cell lines (Figure 1A, left and Supplementary Table S1). These candidates were found in 6731 genes, of which more than 97% (6542) were protein-coding genes. We normalized the numbers of identified NCL events and the expressed co-linear isoforms (considering the GENCODE-annotated isoforms with FPKM > 0.1) using the depth of RNA-seq reads. We found that the normalized number of the identified NCL events was 10–20 times smaller than those of the expressed co-linear isoforms, and the former was generally correlated with the latter (Pearson correlation coefficient r = 0.73) (Figure 1A). Of note, hESCs had the largest normalized number of expressed transcripts without respect to co-linear or NCL transcripts (Figure 1A), also reflecting a previous report that ESCs exhibit a very high level of transcriptome complexity (79).
Figure 1.

Properties of the identified NCL events in the examined cell lines. (A) Comparisons of the numbers of identified NCL events (left) and expressed co-linear isoforms (right). (B) Distribution of the number of NCL events produced from one gene. (C) Comparison of the percentages of NCL and non-NCL donor/acceptor splice sites located within SCEs in terms of the usage of NCL junction sites (top) and the average RPM of the NCL events detected in diverse cell lines (bottom). The control non-NCL donor and acceptor splice sites (10 000 donor and 10 000 acceptor sites) were randomly selected from the NCL-host genes. The red and blue dashed lines represent the percentages of control non-NCL donor and acceptor splice sites within SCEs, respectively. The statistical significance was evaluated using the two-tailed Fisher’s exact test. ***P < 0.001. (D) Schematic illustration of the methodology to estimate the no-co-linear ratio (RNCL) according to the number of reads spanning the NCL junction (NNCL) and that spanning the co-linearly spliced junctions (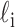 ) at both NCL donor and acceptor sites. (E) The cumulative distribution of NCL events plotted against RNCL. We only considered the NCL events that were located within non-single-exon genes and supported by NNCL≥3. The inset panel represented the number of highly expressed NCL events with RNCL>0.1. Of note, the NCL events were more highly expressed than their corresponding co-linear isoforms if RNCL>0.5. (F) Heatmap representation of expression patterns of highly abundant NCL events (with RNCL>0.1 in at least one cell line; 362 events). The numerical data represent log10-transformed RNCL.
) at both NCL donor and acceptor sites. (E) The cumulative distribution of NCL events plotted against RNCL. We only considered the NCL events that were located within non-single-exon genes and supported by NNCL≥3. The inset panel represented the number of highly expressed NCL events with RNCL>0.1. Of note, the NCL events were more highly expressed than their corresponding co-linear isoforms if RNCL>0.5. (F) Heatmap representation of expression patterns of highly abundant NCL events (with RNCL>0.1 in at least one cell line; 362 events). The numerical data represent log10-transformed RNCL.
We further found that 32–50% of the NCL-affected genes underwent multiple NCL events (Figure 1B). Of the 24 498 distinct NCL events, we found that 23.5% of the NCL donor sites (4354 out of 18 511 distinct sites) and 26.7% of the NCL acceptor sites (4621 out of 17 279 distinct sites) underwent multiple NCL events. We compared NCL junction sites with non-NCL sites in the NCL-affected genes, and found that NCL junction sites had a significantly higher percentage of junction sites located within SCEs (68) than non-NCL sites (all P-values < 10−7 by the two-tailed Fisher’s exact test; Figure 1C). Intriguingly, this percentage was higher in the NCL junction sites that underwent multiple NCL events (usage of NCL sites >1; see ‘Materials and Methods’ section) than in those that underwent a single NCL event (usage of NCL sites = 1) (Figure 1C, top) and was higher in highly expressed NCL events than in less expressed ones (Figure 1C, bottom). Of note, SCEs are coding sequences with extremely low synonymous mutation rates compared to the average rate of the whole coding sequences. Such synonymous constraints may be originated from the requirements of regulatory sites involved in translation initiation or transcript splicing, suggesting the potential role of SCEs in RNA secondary structures, RNA splicing, microRNA binding, transcription factor binding and nucleosome positioning (68,80). Our results thus suggested the regulatory importance of NCL events, especially for the NCL events with a high level of usage/expression.
To quantify the abundance of each NCL event as compared with that of its corresponding co-linear isoform(s), we estimated the NCL ratio (RNCL) according to the number of reads spanning the NCL junction (NNCL) and that spanning the co-linearly spliced junctions at both NCL donor and acceptor sites (Figure 1D). Although most NCL events were expressed at a very low level (RNCL < 0.01) as compared with their co-linear counterparts (Figure 1E), we observed that 362 NCL events were highly expressed (RNCL > 0.1 in at least one cell type) and 34 events were even more abundant than their co-linear counterparts (RNCL > 0.5 in at least one cell type) (Figure 1E, the inset panel). Regarding the 362 highly expressed NCL events, the heatmap analysis revealed that RNCL values varied considerably among cell lines (Figure 1F). Some distinct groups of NCL events were expressed predominately in one specific cell line. Several much larger groups were expressed mainly in H1 hESCs and HeLa cells (Figure 1F), also reflecting that there were more NCL events with RNCL > 0.1 in these two cell lines than in the other ones (Figure 1E, the inset panel). These results revealed that some NCL events were located within SCEs and were highly expressed in a specific cell line, suggesting their regulatory importance.
Numerous non-co-linearly spliced junctions arise from the products of both tsRNAs and circRNAs
Intragenic NCL events may arise from trans-splicing or cis-backsplicing (1,14,35). On the basis of the concept that circRNAs are generally non-polyadenylated but tsRNAs are not (1,5,14,23,26,41,57,60), we discriminated between tsRNAs and circRNAs according to the presence of the identified NCL events in poly(A)- and non-poly(A)-selected RNA-seq data from each cell line. Therefore, the NCL events were categorized into three groups:
Group I: TS-only events, which were detected in poly(A)-selected data only.
Group II: circRNA-only events, which were detected in non-poly(A)-selected data only.
Group III: TS-circRNA events, which were detected in both poly(A)- and non-poly(A)-selected data.
We found that circRNA-only events formed the majority (61–79%) of the NCL events and that >95% (circRNA-only and TS-circRNA events) exhibited circRNA products (Figure 2A). In particular, 20–35% of the NCL events (Figure 2A) were attributed to both tsRNA and circRNA products. Since the different read depth of the examined poly(A)- and non-poly(A)-selected RNA-seq data may bias the distribution of these three NCL groups, we normalized the RNA-seq data by randomly selecting an equal number (60 or 120 million) of reads from poly(A)- and non-poly(A)-selected data for each cell line (Supplementary Tables S2 and 3). After controlling for read depth of the examined RNA-seq data, circRNA products still comprised the majority of NCL events and a considerable percentage (18–32%) of the NCL events were TS-circRNA ones (Figure 2B).
It has been reported that an unexpectedly large number of in vitro artifacts arising from template switching during RT often masquerade as NCL events (1,33,81), and these RT-based artifacts cannot be easily eliminated by computational strategies or naive RT-PCR validation (1,14,33,81,82). We were therefore curious about whether such RT-based artifacts may bias the distribution of these three NCL groups. Since comparisons of different RTase products were shown to effectively detect RT-based artifacts (1,14,33,82,83) and such a strategy was demonstrated to act as effectively as RTase-free validation such as the RNase protection assay (RPA) (1,33), we performed paired-end deep sequencing of H9 hESCs prepared using AMV-derived RTases and performed NCLscan to identify NCL events based on the integration of the AMV-based reads and the H1 hESC reads (which were derived from MMLV-derived RTases; see Table 1). The NCL events supported by both AMV- and MMLV-based reads can be regarded as RT-independent events (Supplementary Table S4). After controlling for RT-dependence and read depth, the tendencies that circRNA-only events majorly contributed to NCL events and ∼30% of NCL events were TS-circRNA ones still observed (Figure 2C).
Previous studies reported that circRNAs may be detected in poly(A)-selected RNA-seq data because of residual circRNAs during poly(A) selection (25,27,35). We then examined whether incomplete depletion of poly(A)-tailed RNAs is the major cause of the detection of TS-circRNA events. To this end, considering the RT-independent NCL events of hESCs, we first examined hESC TS-only and TS-circRNA events that were also detected in non-ESC non-poly(A) samples. If incomplete depletion of poly(A)-tailed RNAs is the cause, the proportions of hESC TS-only and TS-circRNA events detected in the non-ESC non-poly(A) samples should be similar. However, for all six non-ESC samples examined, the percentages of hESC events detected in non-ESC non-poly(A)-selected samples were significantly higher in TS-circRNA events than in TS-only ones (all P-values < 0.01; Figure 2D). Similarly, we proceeded to examine hESC circRNA-only and TS-circRNA events that were also detected in non-ESC poly(A) samples, and found that the percentages of hESC events detected in non-ESC poly(A)-selected samples were significantly higher in TS-circRNA events than in circRNA-only ones, for all six non-ESC samples examined (all P-values < 0.001; Figure 2E). These results thus suggest that such a high proportion of TS-circRNA events in NCL products cannot be fully explained by incomplete depletion of poly(A)-tailed RNAs.
Moreover, we selected 42 TS-circRNA events and employed RT-PCR using AMV- and MMLV-derived RTase in parallel experiments in H9 hESCs, followed by sequencing the RT-PCR amplicons to validate their non-co-linearly spliced junction sites (see Supplementary Figure S1 and ‘Materials and Methods’ section). To validate the existence of circRNA isoforms, we treated total RNA from H9 with RNase R and confirmed that all 42 events existed as RNase R-resistant RNAs (Figure 2F). We proceeded to confirm the existence of tsRNA isoforms using purified mRNA with poly(A)-tails as template. The qRT-PCR analysis revealed that all the events existed as poly(A)-tailed RNAs (Figure 2G). To prevent potential contamination by circRNAs upon purification or potential poly(A) tracts located in the mature circRNA sequences, purified mRNAs were further treated with RNase R. Except for NCL event of TBC1D31, all the NCL events were degraded by the RNase R treatment, indicating the existence of tsRNA isoforms of these events (Figure 2G). These results thus showed that 98% (41 out of 42 events) of the selected events were derived from both trans-splicing and cis-backsplicing, in which both NCL-splicing types of isoforms share the same NCL junction sites.
To detect expanded length of tsRNAs, we used the Third Generation Sequencing platform, Oxford Nanopore Technologies (ONT) MinION sequencer (61), to generate extremely long RNA-seq reads from HeLa cells (which were prepared by oligo-dT selection; ‘Materials and Methods’ section) for the selected TS-circRNA events of FARSA, HIPK3 and CAMSAP1. Of note, all of these three TS-circRNA events had been previously confirmed to exhibit circRNA isoforms (7,39,41). Our results revealed that nanopore reads indeed spanned the NCL junctions of the three TS-circRNA events and mapped outside the predicted circles of the circRNA isoforms (Figure 2H). For example, for the case of FARSA (Figure 2H), we can find that nanopore reads spanned the NCL junction (i.e. exon7–exon5) and mapped outside the predicted exon5–exon6–exon7 circle of the circRNA isoform (i.e. exon4–exon5–exon6–exon7–exon5–exon6–exon7 and exon5–exon6–exon7–exon5–exon6–exon7–exon8). These results further distinguished tsRNAs from circRNAs and the corresponding co-linear mRNA background.
tsRNAs and circRNAs share similar sequence generalities of formation
Of the 8659 NCL events that exhibited tsRNA isoforms in at least one cell type (546 + 4357 + 3756; Figure 3A), a vast majority of events (93%; 8123 events) also exhibited circRNA isoforms and only 7% were TS-only events. These 8123 events were TS-circRNA events (4357 events) or exhibited dynamic NCL-splicing types (3756 events) across the examined cell lines. We thus asked whether generalities of exon circularization were also observed in tsRNA products. To this end, we examined some generalities of exon circularization in these three groups of events: TS-only events (546), circRNA-only events (15 839) and tsRNA or circRNA events (8123) (Figure 3A, see also Supplementary Table S5). First, we observed that the three groups all exhibited a bias toward involving the middle exons (25–50%) of annotated genes (Figure 3B), consistent with a previously reported tendency of circRNA biogenesis (41,51). Second, the median lengths of exonic NCL events (only known splice lengths without introns) were all ∼500 bp, and the lengths of most events were <1500 bp (Figure 3C), consistent with the previous report (7). Third, the tendency for circRNAs to possess longer flanking introns than expected (23,24,41,51,52) was also observed in both TS-only events and tsRNA or circRNA events (Figure 3D). Fourth, exon circularization was reported to be associated with the presence of pairs of IRAlus in the flanking introns (14,24,41,52,53,67). Circularization could be promoted by IRAlus and regulated by competition of IRAlus across flanking introns or within individual introns (designated ‘IRAluacross’ and ‘IRAluwithin’, respectively; Figure 3E) (41). Much like circRNA-only events, both TS-only events and tsRNA or circRNA events exhibited significantly higher percentages of flanking introns with IRAluacross>1 and (IRAluacross-IRAluwithin)≥1 than control introns (Figure 3F; all P-values < 0.001 by the two-tailed Fisher's exact test), suggesting that the sequence requirements in the flanking introns shown to promote circRNA biogenesis were also observed in tsRNAs. In addition, we examined potential composite motifs of NCL donor/acceptor sites and their flanking 10 nucleotides using Weblogo3 (84) and found no difference of sequence motif between these three groups (Supplementary Figure S2). We also found that all these three groups had a significantly higher percentage of junction sites located within SCEs than the control (Figure 3G; all P values < 0.05). These observations indicate that tsRNAs and circRNAs share similar sequence generalities.
Figure 3.

Sequence generalities of formation for different types of NCL events (see also Supplementary Table S5). (A) Distribution of TS-only events, circRNA-only events, TS-circRNA events and the events with dynamic NCL-splicing types across cell lines for the identified 24 899 NCL events. For simplicity, we integrated the last two groups of events for the analysis in (B–G). (B–D) Comparisons of distribution of genic position of exons (B), length of exonic NCL events (C), and length distribution of flanking introns at the donor (left) and acceptor sides (right) for the three types of NCL events (D). The control introns (10 000 introns, which do not overlap the flanking introns of the NCL events) were randomly selected from the NCL-host genes. The statistical significance in (D) was evaluated using the two-tailed Wilcoxon rank-sum test. ***P < 0.001. (E) Schematic illustration of a NCL event with five IRAluacross and four IRAluwithin pairs. (F) Comparisons of percentages of IRAluacross and (IRAluacross-IRAluwithin) ≥1 for the flanking introns of the three types of NCL events. (G) Comparisons of percentages of NCL junctions within SCEs for the three types of NCL events. The statistical significances in (F and G) were evaluated using the two-tailed Fisher’s exact test. *P < 0.05 and ***P < 0.001.
Inverted Alu repeats can simultaneously promote tsRNA and circRNA formation
We proceeded to examine whether deletions of IRAlu can simultaneously affect tsRNA and circRNA formation. We selected a TS-circRNA event (POLR2A TS-circRNA; Figure 4A) and cloned the exons located between the NCL donor and acceptor junctions along with their full-length flanking introns into the middle of the split egfp gene in pZW1 (41,85). Previous studies had validated the NCL junction of circPOLR2A using northern blots and RT-PCR (41,59). The circRNA isoform of POLR2A (termed circPOLR2A) had also been confirmed in hESCs (41). We found that the NCL junction of circPOLR2A can also arise from poly(A)-tailed RNA products (tsRNA isoforms) in all the examined cell lines except for GM12878 (Figure 4B). The RTase-based (i.e. qRT-PCR; Figure 4C) and non-RTase-based (i.e. RPA; Figure 4D) experiments also confirmed that this junction indeed existed as both poly(A)-tailed and RNase R-resistant RNAs, supporting the existence of both POLR2A tsRNA and circRNA isoforms. The nanopore long reads (this sequencing is of the endogenous locus), which spanned the NCL junction (i.e. exon10–exon9) and mapped outside the exon9–exon10 circle of circPOLR2A (i.e. exon8–exon9–exon10–exon9–exon10 and exon9–exon10–exon9–exon10–exon11; Figure 4E), further supported the existence of POLR2A tsRNA isoform. While the flanking IRAluacross pairs have been confirmed to promote the formation of circPOLR2A (41), it is unclear whether the deletion of such IRAluacross pairs may also prevent the expression of the POLR2A tsRNA isoform. To address this, we performed the similar experiments of recapitulation described by Zhang et al. (41) on HeLa cells (Figure 4F). We first designed divergent primers of POLR2A and performed qRT-PCR analyses of the expression fold changes before and after RNase R treatments (Figure 4G, left) and oligo-dT pull down (Figure 4G, right). Our results revealed that both recapitulated POLR2A circRNA and tsRNA isoforms can be generated when at least one IRAluacross pairs were formed (Figure 4G; T1, T3 and T4); in contrast, the expression of POLR2A tsRNA and circRNA isoforms was remarkably reduced when deletions eliminated IRAluacross pairing (Figure 4G; T2 and T5–T7). We also designed convergent PCR primers (Supplementary Figure S3A) to detect the POLR2A poly(A)-tailed RNA products contained the exon9–exon10 transcript fragment (including the co-linear transcript fragment of exon9–exon10 and tsRNA fragment of exon9–exon10–exon9–exon10) with oligo-dT pull down. Indeed, deletions of IRAluacross pairing can remarkably decrease the expression of the tsRNA isoform (i.e. the exon9–exon10–exon9–exon10 transcript fragment) (Supplementary Figure S3A). To prevent the possibility that the detected tsRNA fragment (i.e. exon9–exon10–exon9–exon10) was derived from the case of circRNAs with poly(A) tracts somewhere in the mature circRNA sequences, the purified mRNAs were further treated with RNase R. The exon9–exon10–exon9–exon10 tsRNA fragment was indeed degraded by the RNase R treatment (Supplementary Figure S3B). To further examine expression of the recapitulated POLR2A circRNA (exon skipping) and tsRNA (including the transcript fragment of exon9–exon10–exon9–exon10; see Figure 4H) isoforms, we designed primers in egfp and generated nanopore long reads from total RNA of the transfected cells. The similar trend that deletions of IRAluacross pairing remarkably decreased the expression of both circRNA and tsRNA isoforms was observed (Figure 4H). Our result also revealed that the tsRNA isoform had the lowest level of expression, followed by the circRNA isoform, and then by their corresponding co-linear isoform (Figure 4H). Taken together, these results show that IRAlu can simultaneously promote tsRNA and circRNA formation.
Figure 4.

Recapitulation of the formation of tsRNA and circRNA isoforms with IRAlus across their flanking introns. (A) Visualization of one identified TS-circRNA event in the human POLR2A locus from the UCSC genome browser. Green and red arrows indicate the direction of POLR2A transcription and polarity of the three Alu elements in the flanking introns, respectively. (B) The RPM values of POLR2A tsRNA and circRNA isoforms (measured by poly(A)- and non-poly(A)-selected data from the seven examined cell lines). (C) qRT-PCR analyses (similar to Figure 2F and G) of the expression fold changes for the POLR2A TS-circRNA event before and after RNase R treatments (top) and oligo-dT pull down and oligo-dT pull down with RNase R treatments (bottom). The statistical significance was evaluated using the two-tailed t-test. **P < 0.01 and ***P < 0.001. NS, no signal. (D) RTase-free validation of the tsRNA and circRNA isoforms for POLR2A TS-circRNA event by RPA. Total RNA from HeLa was treated by polyA pull-down (tsRNA isoform) and RNase R (circRNA isoform), respectively. RPA was performed by hybridizing 32P labeled RNA probe in excess to total RNA from HeLa or in vitro transcript containing chimeric junction (size standard). Negative control: probe only. Positive control: the probe hybridized with 100 ng synthesized complementary strand. The arrow indicates the size (295 bp) of the fully protected fragments. The lower band shown in the figure is partially protected probe. (E) Detection of expanded length of POLR2A tsRNA isoform by nanopore long reads. Of note, this sequencing is of the endogenous locus. Blue and orange arrows indicate two different pairs of primers of POLR2A TS-circRNA event. Empty rectangles (E8 and E11) represent the exons located outside the predicted circles of exonic circRNAs (the blue solid rectangles, i.e. E9 and E10). E, exon. (F) Schematic diagrams of egfp expression vectors with various genomic sequences for POLR2A NCL (T1–T7) (41). T1 represents the genomic region for POLR2A NCL RNA (i.e. exons 9 and 10) with its wild-type flanking introns; T2-T7 represent a series of Alu deletions (gray crosses) inserted into the pZW1 expression vector. The green solid rectangles indicate half egfp sequences from the expression vector backbone. EV, empty vector. (G) qRT-PCR analysis of expression fold changes relative to T1 circRNA expression (with RNase R treatment, left) and T1 tsRNA expression (with oligo-dT pull down, right). The expression levels of T1-T7 circRNAs (or tsRNAs) were normalized by egfp expression before RNase R treatments (or oligo-dT pull down). qRT-PCR experiments were performed in triplicate and repeated twice. Error bars represent the mean values ± one standard deviation. Black arrows indicate the PCR primers for spliced RNAs. (H) Analysis of the recapitulated circRNA (exon skipping; the far left panel), tsRNA (including the transcript fragment of E9-E10-E9-E10; the middle panel), and co-linear (including the transcript fragment of E9–E10; the far right panel) isoform expression by nanopore long reads. The numbers of mapped nanopore reads for T1–T7 are illustrated in the far right panel.
tsRNAs have a higher level of correlation with expression of the co-linear counterparts and a lower level of expression breadth, abundance and transcript stability than circRNAs
We next examined the expression profiles of tsRNAs (poly(A)-tailed RNAs) and circRNAs (non-polyadenylated RNAs). To examine expression profiles between samples, the expression levels of NCL events in each sample were determined as RPM values (67). We first found a weak correlation of expression between each other (all Spearman’s rank correlation coefficients P ≈ 0.2, Supplementary Table S6). We then compared the expression of these two types of NCL isoforms with that of their co-linear host genes (measured by FPKM), and asked which type of isoform was the major factor of NCL event expression. Since TS-circRNA events involved tsRNA and circRNA isoforms with the same NCL junctions, we can compare expression levels of these two types of isoforms for each NCL event. We then used partial correlation analysis (73) to evaluate the correlation of expression between NCL events and their corresponding co-linear mRNAs by controlling for tsRNA expression and circRNA expression, respectively. Of interest, the partial correlation decreased or even became insignificant after controlling for tsRNA expression; in contrast, such a partial correlation became stronger and more significant when circRNA expression was controlled for (Figure 5A). This result revealed that tsRNA and circRNA expression independently influenced the expression of their co-linear counterparts, and tsRNA expression was more strongly correlated with the expression of the corresponding co-linear mRNAs than circRNA expression.
Figure 5.

Comparing expression patterns of tsRNAs and circRNAs. (A) Spearman’s rank correlation coefficient (ρ) between NCL event expression (measured by RPM) and the expression of their corresponding co-linear host genes (measured by FPKM) before and after controlling for tsRNA or circRNA expression (measured by RPM). *P < 0.05, **P < 0.01 and ***P < 0.001. NS, not significant. The analyses were based on 1118, 1708, 1996, 1728, 2421, 1096 and 1087 TS-circRNA events in H1 hESC, GM12878, HeLa, HepG2, K562, HUVEC and NHEK cells, respectively. (B) Comparisons of expression breadth of the three groups of NCL events. The events with dynamic NCL-splicing types were not considered. (C) Comparisons of expression levels (measured by RPM; see Supplementary Table S1) for the three groups of NCL events. The statistical significance was evaluated using the two-tailed Wilcoxon rank-sum test. *P < 0.05, **P < 0.01 and ***P < 0.001. (D) Comparisons of expression levels for tsRNA and circRNA isoforms of TS-circRNA events. The statistical significance was evaluated using the paired two-tailed Wilcoxon rank-sum test. **P < 0.01 and ***P < 0.001. For each TS-circRNA event, the expression levels of tsRNA and circRNA isoforms were evaluated on the basis of poly(A)- and non-poly(A)-selected reads, respectively. (E) qRT-PCR for the abundance of two TS-circRNA events (HIPK3 and ANKRD17) and their corresponding co-linear mRNAs in K562 cells treated with Actinomycin D at five indicated time points. qRT-PCR experiments were performed in triplicate and repeated twice. Data are the means ± one standard deviation.
We then examined the cell-type specificity and expression level of tsRNAs and circRNAs. We found that most NCL events (16 197 out of 24 498 events; Supplementary Table S1) were present in only one cell type. Regarding the three groups of NCL events (546 TS-only, 4357 TS-circRNA and 15 839 circRNA-only events; see also Figure 3A), circRNA-involved events (i.e. TS-circRNA and circRNA-only events) tended to be more broadly expressed than TS-only events (Figure 5B). In view of the expression levels of NCL events, in general, circRNA-involved events were more abundant than TS-only ones (Figure 5C). Comparisons of tsRNA and circRNA isoforms for TS-circRNA events further revealed that circRNA isoforms were more highly expressed than tsRNA ones (Figure 5D), consistent with the result illustrated in Figure 4H. These results thus suggested that circRNAs had a higher level of expression breadth and abundance than tsRNAs.
It was reported that circRNAs were more stable than their corresponding co-linear mRNA isoforms (5,7,24,28). We then examined the stability of tsRNAs and circRNAs in K562 cells. We selected two TS-circRNA events (HIPK3 and ANKRD17) and treated total RNA of them with Actinomycin D, an inhibitor of transcription, at five time points (0, 4, 8, 12 and 24 h). tsRNA and circRNA were then separated by oligo-dT pull down and RNase R treatments (see ‘Materials and Methods’ section), respectively. Interestingly, as shown in Figure 5E, the transcript half-lives of circRNA isoforms were much longer (>24 h) than those of tsRNA isoforms and their co-linear mRNA counterparts (both <12 h). These results thus indicated that circRNA isoforms were more stable than tsRNA ones.
The tsRNAs and circRNAs exhibit different subcellular localization preferences
Last, we examined the subcellular localization preference of tsRNAs and circRNAs according to the poly(A)- and non-poly(A)-selected data from nuclear and cytoplasmic RNAs (Table 1). The heatmap analysis revealed that poly(A)-tailed RNA products tended to be more abundant in the cytoplasm than in the nucleus (Figure 6A), whereas the reverse was observed for non-polyadenylated RNAs (Figure 6B). This result suggested that tsRNAs were predominantly cytoplasmic, whereas circRNA ones were predominantly nuclear. Since intron-retained circRNAs (e.g. circEIF3J) were reported to be predominantly nuclear (13), we were curious whether the observations were biased by potential intron-retained circRNAs. Accordingly, we extracted the non-polyadenylated RNA products (including circRNA-only events and circRNA isoforms of TS-circRNA events) with the circles spanning only one exon, which must not retain intronic segments in circularized exons. The similar tendency that non-poly(A)-selected RNA products were predominant in the nucleus was observed (Figure 6C). To avoid the possibility that the observed trend was biased toward the NCL events identified from the used tool (i.e. NCLscan), we also performed three other well-known tools, find_circ (version 2) (5), CIRCexplorer (41) and CIRI (27), to detect NCL events (Supplementary Table S7) and found the similar results (Supplementary Figure S4). To control for read depth, we also normalized the RNA-seq data by randomly selecting an equal number (30 million) of reads from cytoplasmic poly(A) data, nuclear poly(A) data, cytoplasmic non-poly(A) and nuclear non-poly(A) data for each cell line and observed similar results (Supplementary Figure S5). The trend still held well after controlling for both RT-dependence and read depth (Supplementary Figure S6). Regarding TS-circRNA events, we further calculated the ratios of RPM values from cytoplasmic RNAs to those from nuclear RNAs for tsRNA (poly(A)-selected RNA products) and circRNA (non-poly(A)-selected RNA products) isoforms, respectively. Similarly, we found that the majority of tsRNA isoforms were predominantly cytoplasmic and the majority of circRNA ones were predominantly nuclear for each cell type (Supplementary Figure S7).
Figure 6.

Different subcellular localization preferences of tsRNA and circRNA products. (A and B) Heatmap representations of cytoplasmic and nuclear poly(A)-tailed RNA products (tsRNAs) (A) and cytoplasmic and nuclear non-polyadenylated RNA products (circRNAs) (B) from the seven human cell types, with rows representing NCL events and columns representing cell types. The numerical data represent the RPM values. The analyses were based on 7145 and 13 880 events for (A) and (B), respectively. (C) A similar analysis to that in (B) for cytoplasmic and nuclear non-polyadenylated RNA products with circles spanning only one exon. The analysis was based on 1097 events. (D) qRT-PCR analysis of the cytoplasmic to nuclear expression ratios with oligo-dT pull down (tsRNA isoforms, top) and RNase R treatments (circRNA isoforms, bottom) for the eight selected TS-circRNA events. GAPDH (which is known to be predominately cytoplasmic) and circEIF3J (which is an intron-retained circRNA confirmed to be enriched in the nucleus (13)) were examined as controls. qRT-PCR experiments were performed in triplicate and repeated twice. Error bars represent the mean values ± one standard deviation. (E) RNA fluorescence in situ hybridization for the TS-circRNA events of ANXA2 and CAMSAP1. The expression levels (RPM values) of tsRNA and circRNA isoforms for ANXA2 and CAMSAP1 were also provided (left).
To experimentally validate the subcellular localization preference of tsRNAs and circRNAs, we selected eight TS-circRNA events, which had been confirmed to exhibit both tsRNA and circRNA isoforms with the same NCL junction (see Figure 2F and G; Supplementary Figure S1), and performed qRT-PCR analysis of cytoplasmic and nuclear RNAs with oligo-dT pull down (i.e. tsRNA isoform) and with RNase R treatment (i.e. circRNA isoform) in diverse cell types (Figure 6D). In general, tsRNA isoforms were enriched in the cytoplasm, whereas circRNA isoforms tended to be enriched in the nucleus and exhibited dynamic subcellular localization among cell types (Figure 6D). Particularly, some cases of circRNA isoforms were much more nuclear in undifferentiated H9 hESCs compared to the other cell types (including differentiated H9 hESCs) (Figure 6D), suggesting that these circRNAs may be associated with the nuclear proteins involved in pluripotency maintenance of hESCs. In addition, except for TS-circRNA event of CAMSAP1, tsRNA and circRNA isoforms of the examined genes exhibited different patterns of subcellular localization preference during hESC in vitro differentiation, suggesting that these two types of NCL isoforms may play different roles in pluripotency-related regulation or pathways associated with early lineage differentiation. Furthermore, the RNA fluorescence in situ hybridization showed that the TS-circRNA event of ANXA2 predominated in the cytoplasm, in which the tsRNA isoform was more abundant than the circRNA isoform (Figure 6E, top). In contrast, the TS-circRNA event of CAMSAP1, which exhibited a higher expression level in the circRNA isoform than in the tsRNA isoform, was more abundant in the nucleus than in the cytoplasm (Figure 6E, bottom). These results thus showed that tsRNAs and circRNAs may exhibit different subcellular localization preference, even though both types of NCL isoform originated from the same sources.
DISCUSSION
In this study, we systematically investigated NCL events in the human transcriptome in diverse cell types. We found that the number of identified NCL events varied among the examined cell lines (2000–9000; 24 498 in total) and the normalized numbers of detected NCL events was positively correlated with those of the expressed co-linear isoforms (Figure 1A). This result suggests that NCL transcripts also provide a source of diversity for the transcriptome. Although most NCL were expressed at a much lower level compared with their co-linear counterparts (Figure 1E) and consequently were suspected to be side-products of imperfect pre-mRNA splicing (7,26,34), we found that some NCL events were abundant (Figure 1E). These highly abundant NCL events tended to locate within SCEs (Figure 1C) and to be cell type-specific (Figure 1F), suggesting that a number of NCL events are purposefully generated and play a specific role in different cell types.
Of the identified NCL events, we showed that NCL junctions can be derived from alternative NCL splicing types: trans-splicing, cis-backsplicing or both events sharing the same junction (Figure 2A–C). Although the majority (60–80%) of NCL events were derived from circRNA isoforms only, a considerable percentage (20–40%) of them were from tsRNA isoforms only or a mixture of tsRNA and circRNA isoforms (Figure 2A–C). Particularly, we validated that some NCL junctions, which have previously confirmed to be derived from circRNAs (e.g. circHIPK3 (7), circFARSA (39), CAMSAP1 (41) and circPOLR2A (41)), were also derived from tsRNA isoforms (Figures 2F–H and 4C–E). These results suggest that a considerable proportion of observed NCL junctions are derived from tsRNA and circRNA isoforms simultaneously, representing extensive alternative trans-splicing and cis-backsplicing in human cells. We consequently showed that some sequence generalities of exon circularization were also observed in tsRNAs (Figure 3B–G). Analysis of the recapitulated POLR2A NCL event further demonstrated that deletions eliminating IRAlus pairing across flanking introns can not only prevent the expression of the circRNA isoform but also that of the tsRNA isoform (Figure 4E). We thus conclude that both tsRNA and circRNA isoforms share similar sequence generalities of formation and that IRAlus can simultaneously promote the formation of both types of NCL isoforms.
However, tsRNAs and circRNAs exhibit different expression patterns in terms of correlation with the expression of their co-linear counterparts, expression breadth, abundance, transcript stability (Figure 5A–E) and subcellular localization preference (Figure 6). These results imply that although tsRNAs and circRNAs possess similar generalities of formation, they may play different roles in gene regulation. Particularly, compared with circRNA expression, tsRNA expression is more strongly correlated with the expression of their co-linear counterparts. This result reflects some recent observations that only a few circRNAs are co-regulated with their co-linear counterparts (16,56) and that circRNAs and their co-linear counterparts can even compete with each other for biogenesis during splicing (12,57). A possible explanation for this difference between tsRNAs and circRNAs is that tsRNAs are poly(A)-tailed but circRNAs are not. The decay rates of tsRNAs are similar to those of their corresponding co-linear mRNAs, whereas circRNAs are highly stable, with much longer transcript half-lives than their corresponding co-linear mRNAs (see also Figure 5E). Although tsRNAs exhibit a stronger correlation with expression of their co-linear counterparts compared with circRNAs, the Spearman’s rank coefficient of correlation is limited (all P ≤ 0.35, Figure 5A). Our previous study also showed that in some cases, tsRNAs and their co-linear counterparts exhibited very different expression patterns (33). In addition, it was reported that Alu repeats may promote the formation of both tsRNA and circRNA and often provides an ideal target for RNA-editing factor ADAR binding (86). We examined the editing level of the A-to-I RNA editing sites located in flanking Alu elements of circRNAs and tsRNAs (‘Materials and Methods’ section), and showed that the editing levels of the former were generally lower than those of the latter among the examined cell lines (Supplementary Figure S8). This result seems to reflect previous reports that ADAR1 expression was negatively correlated with circRNA biogenesis (16,52), although interactions between ADAR and these two types of NCL events await further investigation. These observations thus indicate that regulation and competition between canonical splicing (for co-linear mRNAs), cis-backsplicing and trans-splicing may be more complicated than we anticipated.
Of note, the subcellular localization preference of circRNAs observed in our large-scale analysis seemed to be different from previous reports that circRNAs tended to be predominately cytoplasmic (5,23,24,26). There are three possible explanations. First, only a few cases of circRNAs have been experimentally confirmed to be cytoplasmic. Although the fact that some circRNAs (e.g. CDR1as, circRNA of Sry, circHIPK3, circFOXO3, circ-TTBK2 and circCCDC66) (5–10) were confirmed to play a regulatory role in microRNA sponges suggests that circRNAs function in the cytoplasm, it was observed no significant excess of miRNA seed matches in the detected circRNAs, expecting that only few circRNAs can act as effectively as these circRNAs in this capacity (17,26,28,34,56,87). Second, a recent study has also demonstrated that some exonic circRNAs are predominantly nuclear, and one case exhibits dynamic subcellular localization during brain development (67). Indeed, we also showed that some circRNA isoforms exhibited dynamic subcellular localization among various cell types (Figure 6D). Finally, tsRNA isoforms considerably contribute to NCL events (Figures 2A–C and 3A) and tend to be predominately cytoplasmic (Figure 6). It may be perplexing for the investigation of subcellular localization preferences if circRNA isoforms cannot be well distinguished from tsRNA isoforms. Of note, a previous study quantified the circular fractions (defined as number of NCL junction reads/(number of total donor and acceptor reads+1)) of 514 circRNAs detected in the K562 RNA-seq data, which were also derived from the ENCODE project, and reported that these circRNAs were predominant in the cytoplasmic non-poly(A)-selected sample (26). We reasoned that such a different result may be due to the analysis focusing on a limited data set of circRNA candidates (514 events), as our study reveals a relatively large-scale profile of subcellular localization preferences for NCL events (see Supplementary Figure S9A). For comparison, we also quantified the circular fractions of the NCLscan-identified events (≥3118 events) in each of the subcellularly fractionated K562 sample, and found that non-poly(A)-tailed RNA products were generally more abundant in the nucleus than in the cytoplasm; such a trend was observed consistently, regardless of the used circRNA-detection tools (i.e. NCLscan, find-circ, CIRCexplorer and CIRI; Supplementary Figure S9B). Taken together, our observations suggest that circRNAs do not necessarily predominate in the cytoplasm, also reflecting a previous notion that exonic circRNAs were divided into cytoplasmic and nuclear circRNAs (88).
Also note that, although whether most tsRNAs are biologically significant remains understudied, our observations suggest that some tsRNAs may play a role of regulation. First, tsRNAs (including the TS-only group and the tsRNA or circRNA group) have a significantly higher percentage of donor/acceptor junction sites located within SCEs than the control (Figure 3G; all P-values < 0.05). Second, tsRNAs tend to have a high level of cell type specificity (Figure 5B), suggesting their specific role in different cell types. Third, some cases of tsRNA isoforms (e.g. tsRNA isoforms of PRKD3, ERBB2 and CAMSAP1; see Figure 6D) exhibited remarkably dynamic subcellular localization during hESC in vitro differentiation, suggesting the potential regulatory role of tsRNAs in pluripotency-related regulation or pathways associated with early lineage differentiation. Finally, although tsRNAs and circRNAs share some sequence generalities of formation (Figures 3 and 4), they exhibit quite different, or even opposite, expression patterns (Figures 5 and 6), suggesting that tsRNAs are not side-products produced from cis-backsplicing sites. These observations suggest that some tsRNAs are biologically important, although we cannot rule out the possibility that some of the observed tsRNAs could be due to genomic alterations in the specific cell lines.
Moreover, many RNA-seq-based methods have been developed to identify NCL events, and considerable effort has been made to remove false positives generated from sequencing or alignment errors. However, false positives from in vitro artifacts during RT (template switching events in particular) cannot be easily diagnosed by computational strategies or naive RT-PCR validation (1,14,33,81,82). To the best of our knowledge, only one systematic approach that can detect NCL RNAs and control for experimental artifacts has been proposed (81). Nevertheless, such an approach, which was based on Drosophila hybrid mRNAs and a mixed mRNA-negative control sample (81), is inapplicable to humans. On the basis of the previous observation that experiments with different RTase products can effectively minimize RT-based artifacts (1,14,33,82,83), here we integrated RNA-seq reads from both AMV- and MMLV-derived RTases to systematically identify RT-independent NCL events (Supplementary Table S4). We find that the RT-independent NCL events possess longer flanking introns and higher percentages of flanking introns with IRAluacross > 1 and (IRAluacross-IRAluwithin) ≥ 1 than the RT-dependent ones, regardless of NCL-splicing types (i.e. TS-only, circRNA-only and TS-circRNA), although such flanking intron lengths and percentages of both RT-independent and RT-dependent events are greater than expected (Supplementary Figure S10). Since the formation of NCL events is dependent on the pairing capacity of complementary sequences (41,67) (see also Figure 4F–H), this result appears to reflect the effectiveness of the strategy of integrative transcriptome sequencing. This study thus provides a feasible strategy that can systematically detect NCL events with control experiments in the human transcriptome. Also note that, we generated nanopore long reads to detect the expanded lengths of tsRNA isoforms. To the best of our knowledge, this is the first report that uses nanopore reads, which span the NCL junctions and map outside the circles of the predicted circRNA isoforms (Figures 2H and 4E), to distinguish tsRNAs from circRNAs and the corresponding co-linear mRNA background.
In summary, this study provides a portrayal of NCL events in diverse human cell types, expanding our understanding of the transcriptome complexity in humans. We highlight that an observed NCL junction may be derived from tsRNA and/or circRNA isoforms. We observed that tsRNA and circRNA isoforms share similar sequence generalities of formation. In particular, the processing of both types of NCL isoforms can be simultaneously facilitated by RNA paring of reversely complementary sequences across their flanking introns, leading to a phenomenon of alternative trans-splicing and cis-backsplicing in transcriptomes (Figure 7). However, we also showed that these two types of NCL isoforms exhibit different expression patterns. For TS-circRNA events, tsRNA and circRNA isoforms showed different patterns of subcellular localization preference across diverse cell types and during hESC differentiation, further suggesting their different regulatory roles. As ∼25% of NCL donor/acceptor junctions undergo multiple NCL events and 20–35% of NCL junctions are derived from both tsRNA and circRNA isoforms, we suggest that analysis of NCL events should take into consideration the effects of different NCL-splicing types and the joint effects of multiple NCL events. Our study thus opens up this important yet understudied class of transcripts for comprehensive characterization.
Figure 7.

A phenomenon of alternative cis-backsplicing and trans-splicing. Reversely complementary sequences across the NCL junctions (e.g. Alu1-Alu2 IRAluacross pair in Figure 4A) can promote both cis-backsplicing and trans-splicing efficiencies by taking the downstream splice donor and upstream acceptor sites close together.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Chan-Shuo Wu and Chi-Yu Weng for programming assistance, the High Throughput Genomics Core Facility of the Biodiversity Research Center in Academia Sinica, Taiwan for Illumina-based high-throughput transcriptome sequencing, and Han-Chieh Wu and Prof. Feng-Jui Chen for Oxford Nanopore sequencing. We also thank Yi-Fen Cheng, Chun-Ying Yu, Ching-Yu Chuang and Prof. Cheng-Fu Kao for providing samples and experimental assistance and Prof. Michael Hsiao and Prof. Che-Kun James Shen for providing experimental facilities. We extend our especial thanks to Prof. Ling-Ling Chen and her lab members for providing all the plasmids used in this study and Profs Junjie U Guo and David P Bartel for providing the information of their identified circRNAs detected in K562. Finally, we are grateful to Li-Yuan Hung and Prof. Pao-Yang Chen for their critical reading of the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Genomics Research Center, Academia Sinica, Taiwan (internal funding); Ministry of Science and Technology (MOST), Taiwan [MOST 103-2628-B-001-001-MY4]. Funding for open access charge: Genomics Research Center, Academia Sinica [internal funding], Taiwan; MOST, Taiwan [MOST 103-2628-B-001-001-MY4].
Conflict of interest statement. None declared.
REFERENCES
- 1. Yu C.Y., Liu H.J., Hung L.Y., Kuo H.C., Chuang T.J.. Is an observed non-co-linear RNA product spliced in trans, in cis or just in vitro. Nucleic Acids Res. 2014; 42:9410–9423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Konarska M.M., Padgett R.A., Sharp P.A.. Trans splicing of mRNA precursors in vitro. Cell. 1985; 42:165–171. [DOI] [PubMed] [Google Scholar]
- 3. Solnick D. Trans splicing of mRNA precursors. Cell. 1985; 42:157–164. [DOI] [PubMed] [Google Scholar]
- 4. Nigro J.M., Cho K.R., Fearon E.R., Kern S.E., Ruppert J.M., Oliner J.D., Kinzler K.W., Vogelstein B.. Scrambled exons. Cell. 1991; 64:607–613. [DOI] [PubMed] [Google Scholar]
- 5. Memczak S., Jens M., Elefsinioti A., Torti F., Krueger J., Rybak A., Maier L., Mackowiak S.D., Gregersen L.H., Munschauer M. et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013; 495:333–338. [DOI] [PubMed] [Google Scholar]
- 6. Hansen T.B., Jensen T.I., Clausen B.H., Bramsen J.B., Finsen B., Damgaard C.K., Kjems J.. Natural RNA circles function as efficient microRNA sponges. Nature. 2013; 495:384–388. [DOI] [PubMed] [Google Scholar]
- 7. Zheng Q., Bao C., Guo W., Li S., Chen J., Chen B., Luo Y., Lyu D., Li Y., Shi G. et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat. Commun. 2016; 7:11215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zheng J., Liu X., Xue Y., Gong W., Ma J., Xi Z., Que Z., Liu Y.. TTBK2 circular RNA promotes glioma malignancy by regulating miR-217/HNF1beta/Derlin-1 pathway. J. Hematol. Oncol. 2017; 10:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yang W., Du W.W., Li X., Yee A.J., Yang B.B.. Foxo3 activity promoted by non-coding effects of circular RNA and Foxo3 pseudogene in the inhibition of tumor growth and angiogenesis. Oncogene. 2016; 35:3919–3931. [DOI] [PubMed] [Google Scholar]
- 10. Hsiao K.Y., Lin Y.C., Gupta S.K., Chang N., Yen L., Sun H.S., Tsai S.J.. Non-coding effects of circular RNA CCDC66 promote colon cancer growth and metastasis. Cancer Res. 2017; 77:2339–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen L.L. The biogenesis and emerging roles of circular RNAs. Nat. Rev. Mol. Cell Biol. 2016; 17:205–217. [DOI] [PubMed] [Google Scholar]
- 12. Ashwal-Fluss R., Meyer M., Pamudurti N.R., Ivanov A., Bartok O., Hanan M., Evantal N., Memczak S., Rajewsky N., Kadener S.. circRNA biogenesis competes with pre-mRNA splicing. Mol. Cell. 2014; 56:55–66. [DOI] [PubMed] [Google Scholar]
- 13. Li Z., Huang C., Bao C., Chen L., Lin M., Wang X., Zhong G., Yu B., Hu W., Dai L. et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat. Struct. Mol. Biol. 2015; 22:256–264. [DOI] [PubMed] [Google Scholar]
- 14. Chen I., Chen C.Y., Chuang T.J.. Biogenesis, identification, and function of exonic circular RNAs. Wiley Interdiscip. Rev. RNA. 2015; 6:563–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang Y., Zhang X.O., Chen T., Xiang J.F., Yin Q.F., Xing Y.H., Zhu S., Yang L., Chen L.L.. Circular intronic long noncoding RNAs. Mol. Cell. 2013; 51:792–806. [DOI] [PubMed] [Google Scholar]
- 16. Rybak-Wolf A., Stottmeister C., Glazar P., Jens M., Pino N., Giusti S., Hanan M., Behm M., Bartok O., Ashwal-Fluss R. et al. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol. Cell. 2015; 58:870–885. [DOI] [PubMed] [Google Scholar]
- 17. Conn S.J., Pillman K.A., Toubia J., Conn V.M., Salmanidis M., Phillips C.A., Roslan S., Schreiber A.W., Gregory P.A., Goodall G.J.. The RNA binding protein quaking regulates formation of circRNAs. Cell. 2015; 160:1125–1134. [DOI] [PubMed] [Google Scholar]
- 18. Szabo L., Morey R., Palpant N.J., Wang P.L., Afari N., Jiang C., Parast M.M., Murry C.E., Laurent L.C., Salzman J.. Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development. Genome Biol. 2015; 16:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gruner H., Cortes-Lopez M., Cooper D.A., Bauer M., Miura P.. CircRNA accumulation in the aging mouse brain. Sci. Rep. 2016; 6:38907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhu M., Xu Y., Chen Y., Yan F.. Circular BANP, an upregulated circular RNA that modulates cell proliferation in colorectal cancer. Biomed. Pharmacother. 2017; 88:138–144. [DOI] [PubMed] [Google Scholar]
- 21. Li Y., Zheng Q., Bao C., Li S., Guo W., Zhao J., Chen D., Gu J., He X., Huang S.. Circular RNA is enriched and stable in exosomes: a promising biomarker for cancer diagnosis. Cell Res. 2015; 25:981–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lasda E., Parker R.. Circular RNAs co-precipitate with extracellular vesicles: a possible mechanism for circRNA clearance. PLoS One. 2016; 11:e0148407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Salzman J., Gawad C., Wang P.L., Lacayo N., Brown P.O.. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One. 2012; 7:e30733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jeck W.R., Sorrentino J.A., Wang K., Slevin M.K., Burd C.E., Liu J., Marzluff W.F., Sharpless N.E.. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013; 19:141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang P.L., Bao Y., Yee M.C., Barrett S.P., Hogan G.J., Olsen M.N., Dinneny J.R., Brown P.O., Salzman J.. Circular RNA is expressed across the eukaryotic tree of life. PLoS One. 2014; 9:e90859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guo J.U., Agarwal V., Guo H., Bartel D.P.. Expanded identification and characterization of mammalian circular RNAs. Genome Biol. 2014; 15:409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gao Y., Wang J., Zhao F.. CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015; 16:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Enuka Y., Lauriola M., Feldman M.E., Sas-Chen A., Ulitsky I., Yarden Y.. Circular RNAs are long-lived and display only minimal early alterations in response to a growth factor. Nucleic Acids Res. 2016; 44:1370–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gingeras T.R. Implications of chimaeric non-co-linear transcripts. Nature. 2009; 461:206–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li H., Wang J., Mor G., Sklar J.. A neoplastic gene fusion mimics trans-splicing of RNAs in normal human cells. Science. 2008; 321:1357–1361. [DOI] [PubMed] [Google Scholar]
- 31. Schoenfelder S., Clay I., Fraser P.. The transcriptional interactome: gene expression in 3D. Curr. Opin. Genet. Dev. 2010; 20:127–133. [DOI] [PubMed] [Google Scholar]
- 32. Rickman D.S., Pflueger D., Moss B., VanDoren V.E., Chen C.X., de la Taille A., Kuefer R., Tewari A.K., Setlur S.R., Demichelis F. et al. SLC45A3-ELK4 is a novel and frequent erythroblast transformation-specific fusion transcript in prostate cancer. Cancer Res. 2009; 69:2734–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu C.S., Yu C.Y., Chuang C.Y., Hsiao M., Kao C.F., Kuo H.C., Chuang T.J.. Integrative transcriptome sequencing identifies trans-splicing events with important roles in human embryonic stem cell pluripotency. Genome Res. 2014; 24:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Salzman J., Chen R.E., Olsen M.N., Wang P.L., Brown P.O.. Cell-type specific features of circular RNA expression. PLoS Genet. 2013; 9:e1003777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chuang T.J., Wu C.S., Chen C.Y., Hung L.Y., Chiang T.W., Yang M.Y.. NCLscan: accurate identification of non-co-linear transcripts (fusion, trans-splicing and circular RNA) with a good balance between sensitivity and precision. Nucleic Acids Res. 2016; 44:e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cocquerelle C., Mascrez B., Hetuin D., Bailleul B.. Mis-splicing yields circular RNA molecules. FASEB J. 1993; 7:155–160. [DOI] [PubMed] [Google Scholar]
- 37. Lei Q., Li C., Zuo Z., Huang C., Cheng H., Zhou R.. Evolutionary insights into RNA trans-splicing in vertebrates. Genome Biol. Evol. 2016; 8:562–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Berger A., Maire S., Gaillard M.C., Sahel J.A., Hantraye P., Bemelmans A.P.. mRNA trans-splicing in gene therapy for genetic diseases. Wiley Interdiscip. Rev. RNA. 2016; 7:487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Starke S., Jost I., Rossbach O., Schneider T., Schreiner S., Hung L.H., Bindereif A.. Exon circularization requires canonical splice signals. Cell Rep. 2015; 10:103–111. [DOI] [PubMed] [Google Scholar]
- 40. Wang Y., Wang Z.. Efficient backsplicing produces translatable circular mRNAs. RNA. 2015; 21:172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang X.O., Wang H.B., Zhang Y., Lu X., Chen L.L., Yang L.. Complementary sequence-mediated exon circularization. Cell. 2014; 159:134–147. [DOI] [PubMed] [Google Scholar]
- 42. Kramer M.C., Liang D., Tatomer D.C., Gold B., March Z.M., Cherry S., Wilusz J.E.. Combinatorial control of Drosophila circular RNA expression by intronic repeats, hnRNPs, and SR proteins. Genes Dev. 2015; 29:2168–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ma L., Yang S., Zhao W., Tang Z., Zhang T., Li K.. Identification and analysis of pig chimeric mRNAs using RNA sequencing data. BMC Genomics. 2012; 13:429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ling J.Q., Li T., Hu J.F., Vu T.H., Chen H.L., Qiu X.W., Cherry A.M., Hoffman A.R.. CTCF mediates interchromosomal colocalization between Igf2/H19 and Wsb1/Nf1. Science. 2006; 312:269–272. [DOI] [PubMed] [Google Scholar]
- 45. Williams A., Flavell R.A.. The role of CTCF in regulating nuclear organization. J. Exp. Med. 2008; 205:747–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kim P., Yoon S., Kim N., Lee S., Ko M., Lee H., Kang H., Kim J.. ChimerDB 2.0–a knowledgebase for fusion genes updated. Nucleic Acids Res. 2010; 38:D81–D85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Al-Balool H.H., Weber D., Liu Y., Wade M., Guleria K., Nam P.L., Clayton J., Rowe W., Coxhead J., Irving J. et al. Post-transcriptional exon shuffling events in humans can be evolutionarily conserved and abundant. Genome Res. 2011; 21:1788–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li H., Wang J., Ma X., Sklar J.. Gene fusions and RNA trans-splicing in normal and neoplastic human cells. Cell Cycle. 2009; 8:218–222. [DOI] [PubMed] [Google Scholar]
- 49. Robertson H.M., Navik J.A., Walden K.K., Honegger H.W.. The bursicon gene in mosquitoes: an unusual example of mRNA trans-splicing. Genetics. 2007; 176:1351–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fischer S.E., Butler M.D., Pan Q., Ruvkun G.. Trans-splicing in C. elegans generates the negative RNAi regulator ERI-6/7. Nature. 2008; 455:491–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Westholm J.O., Miura P., Olson S., Shenker S., Joseph B., Sanfilippo P., Celniker S.E., Graveley B.R., Lai E.C.. Genome-wide analysis of drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep. 2014; 9:1966–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ivanov A., Memczak S., Wyler E., Torti F., Porath H.T., Orejuela M.R., Piechotta M., Levanon E.Y., Landthaler M., Dieterich C. et al. Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep. 2015; 10:170–177. [DOI] [PubMed] [Google Scholar]
- 53. Liang D., Wilusz J.E.. Short intronic repeat sequences facilitate circular RNA production. Genes Dev. 2014; 28:2233–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rigatti R., Jia J.H., Samani N.J., Eperon I.C.. Exon repetition: a major pathway for processing mRNA of some genes is allele-specific. Nucleic Acids Res. 2004; 32:441–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dixon R.J., Eperon I.C., Samani N.J.. Complementary intron sequence motifs associated with human exon repetition: a role for intragenic, inter-transcript interactions in gene expression. Bioinformatics. 2007; 23:150–155. [DOI] [PubMed] [Google Scholar]
- 56. You X., Vlatkovic I., Babic A., Will T., Epstein I., Tushev G., Akbalik G., Wang M., Glock C., Quedenau C. et al. Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat. Neurosci. 2015; 18:603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chen L.L., Yang L.. Regulation of circRNA biogenesis. RNA Biol. 2015; 12:381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Salzman J. Circular RNA expression: its potential regulation and function. Trends Genet. 2016; 32:309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhang X.O., Dong R., Zhang Y., Zhang J.L., Luo Z., Zhang J., Chen L.L., Yang L.. Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome Res. 2016; 26:1277–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Suzuki H., Tsukahara T.. A view of pre-mRNA splicing from RNase R resistant RNAs. Int. J. Mol. Sci. 2014; 15:9331–9342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ip C.L.C., Loose M., Tyson J.R., de Cesare M., Brown B.L., Jain M., Leggett R.M., Eccles D.A., Zalunin V., Urban J.M. et al. MinION analysis and reference consortium: phase 1 data release and analysis. F1000Res. 2015; 4:1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Griebel T., Zacher B., Ribeca P., Raineri E., Lacroix V., Guigo R., Sammeth M.. Modelling and simulating generic RNA-Seq experiments with the flux simulator. Nucleic Acids Res. 2012; 40:10073–10083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zeng X., Lin W., Guo M., Zou Q.. A comprehensive overview and evaluation of circular RNA detection tools. PLoS Comput. Biol. 2017; 13:e1005420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Holtgrewe M., Emde A.K., Weese D., Reinert K.. A novel and well-defined benchmarking method for second generation read mapping. BMC Bioinformatics. 2011; 12:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Huang W., Li L., Myers J.R., Marth G.T.. ART: a next-generation sequencing read simulator. Bioinformatics. 2012; 28:593–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hardwick S.A., Chen W.Y., Wong T., Deveson I.W., Blackburn J., Andersen S.B., Nielsen L.K., Mattick J.S., Mercer T.R.. Spliced synthetic genes as internal controls in RNA sequencing experiments. Nat. Methods. 2016; 13:792–798. [DOI] [PubMed] [Google Scholar]
- 67. Veno M.T., Hansen T.B., Veno S.T., Clausen B.H., Grebing M., Finsen B., Holm I.E., Kjems J.. Spatio-temporal regulation of circular RNA expression during porcine embryonic brain development. Genome Biol. 2015; 16:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lindblad-Toh K., Garber M., Zuk O., Lin M.F., Parker B.J., Washietl S., Kheradpour P., Ernst J., Jordan G., Mauceli E. et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature. 2011; 478:476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kiran A., Baranov P.V.. DARNED: a DAtabase of RNa EDiting in humans. Bioinformatics. 2010; 26:1772–1776. [DOI] [PubMed] [Google Scholar]
- 71. Ramaswami G., Li J.B.. RADAR: a rigorously annotated database of A-to-I RNA editing. Nucleic Acids Res. 2014; 42:D109–D113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Picardi E., D’Erchia A.M., Lo Giudice C., Pesole G.. REDIportal: a comprehensive database of A-to-I RNA editing events in humans. Nucleic Acids Res. 2017; 45:D750–D757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kim S.H., Yi S.V.. Understanding relationship between sequence and functional evolution in yeast proteins. Genetica. 2007; 131:151–156. [DOI] [PubMed] [Google Scholar]
- 74. Gutierrez-Arcelus M., Ongen H., Lappalainen T., Montgomery S.B., Buil A., Yurovsky A., Bryois J., Padioleau I., Romano L., Planchon A. et al. Tissue-specific effects of genetic and epigenetic variation on gene regulation and splicing. PLoS Genet. 2015; 11:e1004958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hannan N.R., Fordham R.P., Syed Y.A., Moignard V., Berry A., Bautista R., Hanley N.A., Jensen K.B., Vallier L.. Generation of multipotent foregut stem cells from human pluripotent stem cells. Stem Cell Rep. 2013; 1:293–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Djebali S., Lagarde J., Kapranov P., Lacroix V., Borel C., Mudge J.M., Howald C., Foissac S., Ucla C., Chrast J. et al. Evidence for transcript networks composed of chimeric RNAs in human cells. PLoS One. 2012; 7:e28213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bernstein B.E., Birney E., Dunham I., Green E.D., Gunter C., Snyder M.. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Djebali S., Davis C.A., Merkel A., Dobin A., Lassmann T., Mortazavi A., Tanzer A., Lagarde J., Lin W., Schlesinger F. et al. Landscape of transcription in human cells. Nature. 2012; 489:101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wu J.Q., Habegger L., Noisa P., Szekely A., Qiu C., Hutchison S., Raha D., Egholm M., Lin H., Weissman S. et al. Dynamic transcriptomes during neural differentiation of human embryonic stem cells revealed by short, long, and paired-end sequencing. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:5254–5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lin M.F., Kheradpour P., Washietl S., Parker B.J., Pedersen J.S., Kellis M.. Locating protein-coding sequences under selection for additional, overlapping functions in 29 mammalian genomes. Genome Res. 2011; 21:1916–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. McManus C.J., Duff M.O., Eipper-Mains J., Graveley B.R.. Global analysis of trans-splicing in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:12975–12979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Houseley J., Tollervey D.. Apparent non-canonical trans-splicing is generated by reverse transcriptase in vitro. PLoS One. 2010; 5:e12271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kong Y., Zhou H., Yu Y., Chen L., Hao P., Li X.. The evolutionary landscape of intergenic trans-splicing events in insects. Nat. Commun. 2015; 6:8734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Crooks G.E., Hon G., Chandonia J.M., Brenner S.E.. WebLogo: a sequence logo generator. Genome Res. 2004; 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wang Z., Rolish M.E., Yeo G., Tung V., Mawson M., Burge C.B.. Systematic identification and analysis of exonic splicing silencers. Cell. 2004; 119:831–845. [DOI] [PubMed] [Google Scholar]
- 86. Levanon E.Y., Eisenberg E., Yelin R., Nemzer S., Hallegger M., Shemesh R., Fligelman Z.Y., Shoshan A., Pollock S.R., Sztybel D. et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat. Biotechnol. 2004; 22:1001–1005. [DOI] [PubMed] [Google Scholar]
- 87. Jeck W.R., Sharpless N.E.. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014; 32:453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Chen L., Huang C., Wang X., Shan G.. Circular RNAs in eukaryotic cells. Curr. Genomics. 2015; 16:312–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.