


Abstract
Embryonic stem cells (ESCs) and meiosis are featured by relatively higher frequent homologous recombination associated with DNA double strand breaks (DSB) repair. Here, we show that Pold3 plays important roles in DSB repair, telomere maintenance and genomic stability of both ESCs and spermatocytes in mice. By attempting to generate Pold3 deficient mice using CRISPR/Cas9 or transcription activator-like effector nucleases, we show that complete loss of Pold3 (Pold3−/−) resulted in early embryonic lethality at E6.5. Rapid DNA damage response and massive apoptosis occurred in both outgrowths of Pold3-null (Pold3−/−) blastocysts and Pold3 inducible knockout (iKO) ESCs. While Pold3−/− ESCs were not achievable, Pold3 iKO led to increased DNA damage response, telomere loss and chromosome breaks accompanied by extended S phase. Meanwhile, loss of Pold3 resulted in replicative stress, micronucleation and aneuploidy. Also, DNA repair was impaired in Pold3+/− or Pold3 knockdown ESCs. Moreover, Pold3 mediates DNA replication and repair by regulating 53BP1, RIF1, ATR and ATM pathways. Furthermore, spermatocytes of Pold3 haploinsufficient (Pold3+/−) mice with increasing age displayed impaired DSB repair, telomere shortening and loss, and chromosome breaks, like Pold3 iKO ESCs. These data suggest that Pold3 maintains telomere integrity and genomic stability of both ESCs and meiosis by suppressing replicative stress.
INTRODUCTION
Effective DNA repair, genomic stability and telomere maintenance are essential for unlimited self-renewal and clinical therapy of embryonic stem cells (ESCs) (1,2). During the rapid proliferation of ESCs in culture, DNA damage such as DSBs can occur in ESCs. Further understanding of how ESCs activate DNA damage response and repair and maintain genomic stability would benefit potential use of ESCs in stem cell therapy (3,4). Two major pathways exist for DNA DSB repair, homologous recombination (HR)-mediated repair (HRR) and non-homologous end-joining (NHEJ), the choice of which is tightly controlled (5). NHEJ occurs more commonly in the G1 and early S phases of the cell cycle. Mammalian somatic cells preferentially use NHEJ, whereas HRR is the predominant mechanism for DSB repair in the yeast Saccharomyces cerevisiae (6). ESCs differ from the differentiated somatic cells in terms of cell cycle progression. ESCs lack a G1 checkpoint and have very short cell cycle G1 and G2 phases, with about 75% of their cycle time in S-phase promoting the preferential use of HR (7,8), whereas HR is suppressed in G1 (9). Consistently, HR is a major DNA repair pathway in ESCs (1).
The structure and roles of Polδ were originally revealed in yeast. In fission yeast Schizosaccharomyces pombe, Polδ has four subunits, Pol3, Cdc1, Cdc27 and Cdm1 (10). In budding yeast S. cerevisiae, Polδ contains Pol3, Pol31 and Pol32, however, Pol32 is dispensable for growth, but a deletion mutant is sensitive to hydroxyurea and DNA damaging agents (11). It has been shown that mammalian homologue Pold3 is implicated in break-induced replication repair (BIR) and mitotic DNA synthesis in human cancers (12,13).
A high frequency of DNA DSBs and repair, which are required for spermatogenesis, is a unique feature of meiosis I prophase. Loss of function of genes involved in DNA damage repair leads to defective spermatogenesis (14–16). Pold3 is highly expressed in pachytene spermatocytes and round spermatids (17). Recently, Pold3 is essential for mouse development and also required for viability of adult animals (18). However, it remains elusive whether Pold3 has a specific role in spermatogenesis. We investigated potential functions and the underlying mechanisms of Pold3 in mouse ESC self-renewal and meiosis by various approaches, including knockout by CRISPR/Cas9, inducible knockout, or knockdown (KD) by RNA interference (shRNA).
MATERIALS AND METHODS
Vector construction
Transcription activator-like effector nucleases (TALEN) target site was designed using online website (https://tale-nt.cac.cornell.edu). TALEN expression vectors were gifts kindly provided by Bo Zhang (19). Asparagine-Isoleucine (NI), Asparagine-Glycine (NG), Histidine-Aspartic acid (HD), Asparagine-Asparagine (NN) were used as repeat-variable di-residue (RVD) to recognize nucleotides A, T, C and G, respectively. Transcription Activator-Like (TAL) effector repeats were constructed by serial cycles of digestion (NheI + HindIII or SpeI + HindIII) and ligation, then the TAL effector repeats were subcloned into the TALEN expression vectors containing the FokI cleavage domain and other necessary components. PX330-U6-Chimeric_BB-CBh-hSpCas9 (PX330) was obtained from Addgene (plasmid #42230) (20). sgRNAs were designed by online website (http://cripsr.mit.edu/). PX330 was digested with BbsI (Fermentas) and the linearized vector was gel purified. Pairs of oligos for target sites were annealed, and then ligated with linearized vector. The target vector was identified by U6 promoter and reverse oligo and verified by sequencing. Pold3 cDNA was cloned downstream of the tetO minimal promoter of the pBS31 vector using the EcoRI. Pold3 cDNA and 3xflag were ligated into pBS31 vector forming Pold3 3 × flag (Pold3 3F) constructor using EcoRI. All primers' sequences are provided in Supplementary Table S1.
In vitro transcription (IVT)
T7 promoter was added to Cas9 coding region and sgRNA template by polymerase chain reaction (PCR) amplification using IVT-cas9 F and R, T7-Pold3 F and R, respectively (21). T7-Cas9 PCR product was gel purified and used as the template for In Vitro Transcription (IVT) using mMESSAGE mMACHINE T7 ULTRA kit (Life Technologies). T7-Pold3 sgRNA PCR product was gel purified and used as the template for IVT using HiScribe™ T7 Quick High Yield RNA Synthesis Kit (NEB). Cas9 mRNA and Pold3 sgRNA were purified using MEGAclear kit (Life Technologies) and eluted in RNase-free water.
Micro-injection and generation of Pold3 knockout mice
B6D2F1 (C57BL/6 X DBA2) female mice and ICR (Institute for Cancer Research) albino mouse strains were used as embryo donors and foster mothers, respectively. Female B6D2F1 mice (6–8 weeks old) following superovulation stimulation were mated with B6D2F1 males, and fertilized eggs collected from oviducts. Cas9 mRNA and Pold3 sgRNA were injected into the cytoplasm of fertilized eggs with well-recognized pronuclei in HKSOM. The injected zygotes were cultured in KSOM with amino acids at 37°C under 5% CO2 in air to blastocyst stage by 3.5 days. Thereafter, 16–22 blastocysts were transferred into uterus of pseudopregnant ICR females at 2.5 dpc. All procedures in use of mice were performed according to the Animal Care Guidelines and approved by the Nankai University Animal Care and Use Committee.
Genotyping
One to two weeks old postnatal mice were genotyped using DNA extracted from their tails or ears. Genotyping of embryos was performed by PCR after isolation or outgrowth assays. Embryos or ESC clones were dissolved in DNA lysis buffer. PCR was carried out at 95°C for 5 min, followed by 35 cycles at 95°C for 30 s, 60°C for 30 s and 72°C for 40s. A total of 2 μl of Pold3 PCR products were digested with SacI or NcoI (TAKARA) at 37°C for more than 2 h. DNA fragments were visualized by agarose gel electrophoresis.
Collection of embryos at different developmental stage
Heterozygous male and female mutant mice were bred to obtain wild-type, heterozygous and homozygous mutant embryos. To generate embryos at different developmental stages, Pold3+/− females were injected intraperitoneally with pregnant mare's serum gonadotropin (5 IU per mouse; Sigma), followed by injection of human chorionic gonadotropin (5 IU per mouse; Sigma) 47 h later and mating overnight with Pold3+/− males. The next morning, males were removed and females were examined for the presence of a vaginal plug, designated as E0.5. Embryos were collected at various embryonic days after successful mating.
Blastocyst culture and ICM outgrowth
Embryos were collected at E3.5 or E4.5 by flushing the uterus of pregnant mice. Blastocysts were transferred on 0.2% gelatin-coated tissue culture plates with complete ESC medium (knock-out Dulbecco’s modified Eagle’s medium (Invitrogen) supplemented with 15% fetal bovine serum (FBS) (ES quality, Hyclone), 1000 U/ml mouse leukemia inhibitory factor (LIF; ESG1107; Millipore), 0.1 mM non-essential amino acids, 0.1 mM β-mercaptoethanol, 1 mM L-glutamine, penicillin (100 U/ml) and streptomycin (100 μg/ml). Embryos were cultured in the presence of 5% CO2 at 37°C and photographed daily using a microscope (Leica).
Generation of Pold3 knockdown (KD), Pold3 iKO (inducible knockout) and Pold3 3F (3 × flag) ESCs
N33 ESC line was derived from wild-type C57BL/6 mice (22). Control and shRNA sequences against Pold3 mRNA were used for KD experiments. Pairs of oligos were cloned into pSIREN-RetroQ (Clontech) and then the constructed vectors were introduced into Plat-E cells to package retrovirus. Cells were then infected with control and Pold3 KD retrovirus, and clones picked after about 5–7 days with 2 μg/ml puromycin.
The KH2 ESC line was developed by the Rudolf Jaenisch laboratory (23). pBS31-Pold3 or Pold3 3 × F plasmid and pCAGGS flpE plasmid were co-transfected into KH2 ESCs with lipofectamine 2000. iPold3 or Pold3 3 × F ESC clones were selected out with 140 μg/ml hygromycin and induced to express with Dox at concentration of 0.1 μg/ml. PX330-sgRNA-L and PX330-sgRNA-R were co-transfected into iPold3 KH2 ESCs, then Pold3 iKO clones were selected and genotyped. The cell lines were maintained by treatment with Dox. Pold3 can be induced to become KO by Dox withdrawal in Pold3 iKO KH2 ESCs. ESC lines were cultured in ESC medium on mitomycin C-inactivated mouse embryonic fibroblast (MEF) cells as feeder layers, prepared on 0.2% gelatin-treated culture dish.
Cell proliferation assay
1 × 105 ESCs were plated in 12-well plates. Cells were counted every 2 days and passaged into new 12-well plates with 1:8. Number of cells was counted 4 or 5 times (eight or ten days) for each cell line.
Cell cycle analysis
Cells were harvested and then fixed in freshly prepared pre-cooled 70% ethanol overnight at 4°C. Cells were centrifuged at 1000g for 5 min and stained with propidium iodide (PI, Beyotime) at 37°C for 30 min. Cell cycle phases were determined by FACS analysis and the data were processed using ModFit LT.
Embryoid body formation test
ESCs were removed off feeder cells based on their differences in the adherence to the bottom of dish. Cells were diluted to 4 × 104/ml. Every 30 μl was pipetted to form a hanging drop on the cover of a 100-mm dish. Embryoid bodies (EBs) formed on day 4 and then were transferred to six-well plates for adherent culture. Differentiated cells on day 15 were fixed for immunofluorescence staining using markers representative of three embryonic germ layers.
Immunofluorescence microscopy
Embryos at the indicated stage and mESCs were fixed in 4% paraformaldehyde in PBS at 4°C for 30 min, and then permeablilized with 0.2% Triton X-100 in PBS for 20 min at room temperature, followed by blocking in solution containing 3% goat serum, 0.1% BSA and 0.2% Triton X-100 for 2 h at room temperature. Samples were incubated with the primary antibodies overnight at 4°C. The primary antibodies included Oct4 (sc5279; Santa Cruz), Nanog (A300–397A; Bethyl), SSEA-1 (MAB4301; Millipore), β III-tubulin (CBL412; Chemicon), AFP (DAK-N1501; Dako), α -SMA (ab5694–100; Abcam), 53BP1 (ab36823; Abcam), TRF1 (TRF12-S; Alpha Diagnostic) TRF2 (05–521; Millipore), p-ATR (2853; CST), p-ATM (4526; CST), Rif1(sc515573; Santa Cruz), flag (F1804; Sigma) and γH2AX (05–636; Millipore). Embryos or mESCs were washed with PBS three times and incubated with appropriate fluorescein-labeled secondary antibodies at room temperature for 2 h. Goat Anti-Mouse IgG (H + L) FITC (115–095-003; Jackson), Goat Anti-Rabbit IgG (H + L) Alexa Fluor® 594 (111–585-003; Jackson) and Goat Anti-Mouse IgM Alexa Fluor® 488(A-21042; Invitrogen), diluted 1:200 with blocking solution, were used. Samples were washed and DNA was then stained for 10 min with 0.5 μg/ml Hoechst 33342 (H1398; MP), and placed in Vectashield mounting medium. Fluorescence was detected and imaged using a Zeiss Axio-Imager Z2 fluorescence microscope. Immunofluorescence (IF)-FISH was performed using TelC-Cy3 (F1002; Pangene) as described (24).
DNA synthesis
DNA synthesis was measured by BrdU incorporation. Embryos or ESCs were labeled with 10 μM BrdU in mouse ESC medium for 3 h, DNA was denatured after permeabilization with 0.25 N HCl and incubated with BrdU monoclonal antibody, Alexa Fluor® 594 for IF and IHC (A21304; Invitrogen).
DNA fiber assay
DNA fiber assay was performed as described (25,26), with slight modification. Cells were pulsed with 50 μM CldU (sc-221018; Santa Cruz), washed twice with 1 × PBS, and pulsed with 250 μM IdU (sc-205720; Santa Cruz). Cells were resuspended in PBS at 3 × 105/ml, and 2.5 μl cell suspension spotted on adhesion microscope slides (188105; CITOGLASS) and lysed with 7.5 μl lysis buffer (0.5% SDS, 200 mM Tris-HCl, pH 7.4, 50 mM EDTA) for 10 min. Slides were tilted 15° to stretch DNA fibers by gravitational flow. Fibers were fixed with methanol/ acetic acid (3:1) for 15 min at -20°C, denatured with 2.5 N HCl for 1 h, and blocked with 5% BSA plus 0.1% Tween for 30 min at RT. Rat anti-BrdU (1:200; Ab6326; Abcam) was used to detect CldU, and mouse anti-BrdU (1:50; Ab181664; Abcam) to detect IdU. Slides were incubated with the primary antibody overnight, and then washed once by stringency buffer (10 mM Tris-HCl, pH 7.4, 300 mM NaCl, 0.2% Tween and 0.2% NP40) for 5 min (stringency buffer reduces non-specific binding). Slides were washed twice with 1 × PBS, blocked again with 5% BSA for 30 min at RT, and then incubated with secondary antibodies for 2 h at RT, Alexa Fluor 488-conjugated donkey anti-Rat (A21208; Invitrogen) and Alexa Fluor 594-conjugated goat anti-mouse (A11032; Invitrogen). Coverslips were mounted using Vectashield (H-1000; Vector). Images are acquired using Zeiss Imager Z2 fluorescence microscope with a 100x objective lens and FITC and Rodamine filters. Fiber lengths in IdU tract were measured using ruler tool in Photoshop software.
TUNEL assay
Apoptosis assay for embryos, cultured cells and sections was performed by TUNEL staining following DeadEnd™ Fluorometric TUNEL System (G3250; Promega). Images were taken under a Leica confocal microscope.
Ionizing radiation and drug treatment
To induce DNA damage, ESCs were cultured under feeder-free condition and treated with a dose of 10 Gy irradiation by Gamma cell 40 Exactor (Best Theratronics) or 2.5 μM etoposide for 2 h. ESCs without treatment, or 2 h and 12 h later after treatment were collected for analysis. 1 × 105 ESCs were plated and treated with VE-821 (S8007; Selleck) in 12-well plates, and cells counted every two days or at day 3.
Western blot
Cells were lysed in cell lysis buffer for western blot on ice for 30 min and then sonicated for 1 min at 60 amplitude at 2 sec intervals. Samples were centrifuged at 10,000 g for 10 min at 4°C, then supernatants transferred into new tubes. The protein concentration was measured by bicinchoninic acid assay, and then the samples boiled with SDS buffer at 98°C for 8 min. 20 or 30 μg proteins of each sample were resolved by 8% or 10% Bis-Tris SDS-PAGE and transferred to polyvinylidene difluoride membranes (ISEQ00010; Millipore). Non-specific binding was blocked by incubation in 5% non-fat dry milk (9999S; CST) in TBS-T at room temperature for 2 h. Blots were then probed with primary antibodies, γH2AX (05–636; Millipore), p-CHK1 (2341S; CST), p-ATR (2853; CST), ATR (sc515173; Santa Cruz), p-ATM (4526; CST), RAD51 (Ab88572; Abcam), MLH1 (550838; BD Pharmingen), Oct4 (sc5279; Santa Cruz), Nanog (A300–397A; Bethyl) and β-actin (P30002; Abmart) by incubation overnight at 4°C in 5% skim milk in TBS-T. Immunoreactive bands were then probed for 2 h at room temperature with appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies, anti-Rabbit IgG-HRP (NA934V; GE Healthcare), or goat anti-Mouse IgG (H+L)/HRP (ZB-2305; ZSGB-BIO). Protein bands were detected by Chemiluminescent HRP substrate (WBKLS0500; Millipore). Custom-made polyclonal rabbit anti-Pold3 antibody was generated against the C-terminal region of Pold3 (C-KREPREERKGPKKG) (Abmart).
RNA extraction and real-time quantitative PCR
Total RNA was purified using a RNA mini kit (Qiagen), treated with DNase I (Qiagen), and the cDNA generated from 2 μg RNA using Oligo (dT) 18 primer (Takara) and M-MLV Reverse Transcriptase (Invitrogen). Real-time quantitative PCR reactions were set up in duplicate with the FS Universal SYBR Green Master (Roche) and carried out in an iCycler MyiQ2 Detection System (BIO-RAD). All reactions were carried out by amplifying target genes and internal control in the same plate. Each sample was repeated three times and normalized using GAPDH as the internal control. The amplification was performed for primary denaturation at 95°C for 10 min, then 40 cycles of denaturation at 95°C for 15 s, annealing and elongation at 58°C for 1 min, and the last cycle under 55–95°C for dissociation curve. Relative quantitative evaluation of target gene was determined by comparing the threshold cycles. Primers were confirmed for their specificity with dissociation curves.
Telomere quantitative fluorescence in situ hybridization (Q-FISH)
Telomere length and function (telomere integrity and chromosome stability) was estimated by Q-FISH (27,28). Cells were incubated with 0.3 μg/ml nocodazole for 3 h to enrich cells at metaphases. Chromosome spreads were made by a routine method. Metaphase-enriched cells were exposed to hypotonic treatment with 0.075 M KCl solution, fixed with methanol: glacial acetic acid (3:1) and spread onto clean and cold slides. Telomeres were denatured at 80°C for 3 min and hybridized with FITC-labeled (CCCTAA) peptide nucleic acid (PNA) probe (F1009; Panagene) at 0.5 μg/ml. Chromosomes were counter-stained with 0.5 μg/ml DAPI. Fluorescence from chromosomes and telomeres was digitally imaged on a Zeiss Imager Z2 microscope with FITC/DAPI filters, using AxioCam and AxioVision software 4.6. For quantitative measurement of telomere length, telomere fluorescence intensity was integrated using the TFL-TELO program (a gift kindly provided by P. Lansdorp, Terry Fox Laboratory).
TRF by Southern blot analysis
The average terminal restriction fragments (TRF) length was determined according to the commercial kit (TeloTAGGG Telomere Length Assay, 12209136001; Roche Life Science). Genomic DNA was extracted by traditional phenol:chloroform:isoamyl alcohol method. 3 μg DNA was digested with MboI (NEB) for 15 h and the DNA fragments separated by 1% agarose gel for 16 h at 6V/cm in 0.5 × TBE buffer using CHEF DR-III pulse-field system (Bio-Rad). Gels were denatured, neutralized, and transferred to nylon membrane (RPN2020B; GE Healthcare) for 48 h. The membrane was hybridized with digoxigenin (DIG)-labeled telomere probe at 42°C overnight and incubated with anti-DIG-alkaline phosphatase antibody. Telomere signal was detected by chemiluminescence after adding substrate solution on membrane.
Tissue collection and histology
Testis and caudal epididymis and other tissues were dissected immediately following euthanasia of mice. Tissues were then fixed in 10% formalin solution (HT501128; Sigma) overnight at 4°C, dehydrated in an ethanol series, and embedded in paraffin wax. 5-μm sections were cut with a microtome and then stained with hematoxylin and eosin for histological analysis.
Immunofluorescence of spread spermatocytes and sections
Spermatocytes spreading was performed using the drying-down technique previously described (29), and immunostaining of spermatocytes and sections performed as described previously (30). Briefly, dissected testis were placed in PBS, fixed in 1% paraformaldehyde, washed in PhotoFlo, air-dried, and stored at -20°C. Slides were washed in PBS-0.1% Triton X-100 for 10 min and blocked with blocking buffer (5% BSA, 1% goat serum and 0.1% Triton X-100 in PBS). The primary antibodies included anti-SYCP3 (NB 300–230, Novus Biologicals; Ab97672, Abcam), anti-SYCP1 (Ab15090; Abcam), anti-γH2AX (05–636; Millipore), anti-RAD51 (Ab88572; Abcam) and anti-MLH1 (550838; BD Pharmingen). Slides were washed 15 min for 3 times after incubation with primary antibody overnight at 4°C. Appropriate fluorescence-conjugated secondary antibodies were used to reveal specific proteins, and DAPI to reveal chromosomes/chromatin. For paraffin sections, sections were boiled for 3 min at 120°C in sodium citrate buffer for antigen retrieval. The remaining steps were described as above.
RNA-sequencing and analysis
Conventional RNA-Seq was performed for Pold3 iKO ESCs (P0, P1 and P2) by Novogene. The RNA-seq reads were aligned to the mouse reference genome mm9 using TopHat. Prior to differential gene expression analysis, the read counts were adjusted for each sequenced library by edgeR program package through one scaling normalized factor. Differential expression analysis of two conditions was performed using the DEGSeq R package (1.20.0). The P values were adjusted using the Benjamini & Hochberg method. All differentially expressed genes were determined by |log2(FoldChange)|> 1&q-value < 0.005.
Statistical analysis
Statistical significances were analyzed by ANOVA or t-tests using StatView software from SAS Institute. The data were considered significant when P < 0.05 (*), 0.01(**) or 0.001(***).
RESULTS
Targeted knockout of mouse Pold3 gene by CRISPR/Cas9 system
To explore the roles of Pold3 in mice, we first analyzed Pold3 mRNA and protein levels in several adult tissues by real-time quantitative PCR (RT-qPCR) and western blot respectively (Figure 1A and B). Pold3 was predominantly expressed in spleen, and especially in testis. To generate Pold3 knockout (KO) mice for investigation of its role in vivo, initially we attempted to establish Pold3−/− mouse ESCs which can be injected into blastocysts to produce chimeras from which Pold3−/− mice could be achieved in principle by cross mating. We originally designed a pair of TALEN targeting Exon 6 of Pold3 (Supplementary Figure S1A) and the indel (insert-deletion) efficiency was as high as 16% (Supplementary Figure S1B). Of 144 clones that were screened by PvuII digested, 7 clones were mutant in both alleles of Pold3 in mouse ESC cell line (N33, Supplementary Figure S1E). However, DNA sequencing results show that none of them were bi-allelic frame shift (Supplementary Figure S1C). Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/CRISPR-associated 9 (Cas9) becomes a common gene-editing tool which surpass Zinc-finger nucleases (ZFN) and TALEN because of its simplicity and high efficiency of gene editing in recent years (31). Two sgRNA, sgPold3–1 and sgPold3–2 were designed to target the same exon of Pold3 (Figure 1C). Much higher target efficiency was obtained using both sgRNAs by CRISPR/Cas9 than did TALEN (67% and 47% versus 16%) (Figure 1D). However, we still failed to achieve frame shift clones despite the high ratio of bi-allelic mutants in mouse ESCs when using CRISPR/sgPold3–1 (Supplementary Figure S1D and E). This suggests that Pold3−/− ESCs might be lethal and that Pold3 may play vital roles in mouse ESCs.
Figure 1.

Targeted disruption of Pold3 gene by CRISPR/Cas9 system in mice. (A) Pold3 mRNA levels in various mouse tissues by qPCR analysis. (B) Pold3 Protein levels in various mouse tissues by western blot. (C) Schematic of Cas9/sgRNA-targeting sites in Pold3. The designed sgRNAs (sgPold3–1 and sgPold3–2) target sequence are underlined, and the protospacer-adjacent motif sequence labeled in red. The restriction sites at or near target regions are labeled in purple. Sites of cleavage by Cas9 are indicated by upward or downward arrows. Forward and reverse primers are shown as rightward and leftward arrows. A 650 bp DNA fragment containing the Pold3 target site was amplified by PCR, and the fragments after SacI or NcoI digestion also shown. (D) Restriction assay of CRISPR-mediated cleavage by SacI or NcoI digestion at Pold3 loci in mouse ESCs. A successful digestion should give rise to two fragments. The uncut bands are resistant to enzyme cleavage due to indel mutation. Indel percentage (uncleaved band) was calculated by ImageJ software, % indel (uncleaved) = 100 × (1-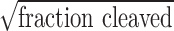 ). (E) Detection of Pold3 mutations in founder mice by NcoI or SacI digestion. Indel mutations were detected in #10, #11 and #12 three founder mice. (F) Sequencing results of Pold3 mutations induced by CRISPR/Cas9 in founder mice. Deletions are indicated by dashes, and insertion indicated by downward arrow. (G) Western blot analysis of Pold3 protein levels in testis and ovary from 2 to 3 month-old Pold3+/+ and Pold3+/− mice. (H) Embryos and progeny from Pold3+/− mouse intercross breeding and genotyping.
). (E) Detection of Pold3 mutations in founder mice by NcoI or SacI digestion. Indel mutations were detected in #10, #11 and #12 three founder mice. (F) Sequencing results of Pold3 mutations induced by CRISPR/Cas9 in founder mice. Deletions are indicated by dashes, and insertion indicated by downward arrow. (G) Western blot analysis of Pold3 protein levels in testis and ovary from 2 to 3 month-old Pold3+/+ and Pold3+/− mice. (H) Embryos and progeny from Pold3+/− mouse intercross breeding and genotyping.
We performed micro-injection of zygotes to generate Pold3 mutant mice as an alternative. Capped polyadenylated Cas9 mRNA with sgPold3–1 or sgPold3–2 were co-injected into mouse zygotes, and eGFP mRNA was injected as observation control. eGFP was expressed as expected (Supplementary Figure S1F and H). To evaluate the effectiveness of Cas9/sgPold3 in embryos, we assessed the target efficiency of altered alleles at the blastocyst stage, and up to 20% were modified at the target site in a collection of five blastocysts (Supplementary Figure S1G). A total of 60 B6D2F2 (B6D2F1 X B6D2F1) embryos injected with Cas9 mRNA and sgPold3 were transferred into three surrogate mothers (Supplementary Figure S1I). Three mutant founders generated by sgPold3 grew healthily (Figure 1E). DNA sequencing analysis revealed that #10 and #11 were mosaic mice with three different genetic modifications in the Pold3 locus, and #12 was mono-allelic mutant mouse. To generate bi-allelic frame-shift Pold3-deficient mice, #11 including 11 bp deletion was backcrossed with C57BL/6 mice, and then Pold3+/− male and female mice were bred to obtain Pold3−/− mice in principle. To confirm that the Pold3 allele was impaired in mutant mice, we analyzed Pold3 protein expression levels by western blot. Expectedly, Pold3 protein levels were reduced by about half in testis or ovary (Figure 1G). This demonstrated that Pold3 gene was disrupted successfully in vivo by CRISPR/Cas9 system and that only heterozygous (Pold3+/−) mice can be obtained.
Pold3 deficiency causes early embryonic lethality
Although no postnatal Pold3−/− pups were found among 179 live-born mice from the intercrosses, heterozygous Pold3+/− mice displayed no obvious abnormality in weight or fertility over 8 months (Figure 1H). These data suggest that one copy of functional Pold3 allele is sufficient for embryonic and postnatal development, whereas a homozygous deficiency of Pold3 leads to embryonic lethality. To determine the time of developmental failure of Pold3−/− embryos, embryos were collected from heterozygote intercross breeding at different times of gestation and individual embryos genotyped by PCR. As a result, no Pold3−/− embryos were found at E7.5 or after E7.5 (Figure 1H), suggesting that the time of embryonic lethality could occur prior to E7.5. Blastocysts at E3.5 or E4.5 were morphologically indistinguishable between wild-type and heterozygous or homozygous Pold3-deficient embryos (Supplementary Figure S2A).
To further assess why Pold3- homozygous deficient (Pold3−/−) embryos fail to develop normally beyond E4.5, we cultured in vitro for several days individually of E3.5 blastocysts derived from intercrosses of Pold3+/− mice. Trophoblasts of Pold3+/+ and Pold3+/− embryos spread over the culture dish after hatching and the robust inner cell mass (ICM) expanded as a round-shape aggregate on the trophoblast giant (TG) cells after 2 days. While all embryos were indistinguishable on day 0 and day 1 (Supplementary Figure S2A), the trophoblasts from Pold3−/− blastocysts attached and spread slowly over the culture dish to form a smaller sheet compared with outgrowths of WT blastocysts at day 2 and day 3 (Supplementary Figure S2A and B), and Pold3+/− outgrowths seemed normal. Meanwhile, Pold3−/− ICM cells failed to expand subsequently by day 2 and shrunk or diminished by day 3 in culture (Supplementary Figure S2A and C), suggesting that Pold3 may be more important for ICM than TG cells. These data indicate that Pold3 deficiency impairs outgrowth of ICM and TG cells.
To understand the underlying mechanisms associated with the failed outgrowth of Pold3−/− embryos, we carried out bromodeoxyuridine incorporation assay during blastocyst outgrowth. Pold3+/+ and Pold3+/− outgrowths displayed robust DNA synthesis by immunostaining with anti-BrdU antibody (Supplementary Figure S2D and E). However, both ICM and trophoblasts in Pold3−/− outgrowth ceased to proliferate. We also performed terminal deoxynucleotide end-labeling (TUNEL) assay on blastocyst outgrowths cultured for 3 days. Few TUNEL-positive cells were detected in Pold3+/+ and Pold3+/− outgrowths, in contrast to massive apoptosis found in Pold3−/− outgrowths (Supplementary Figure S2F and G). Meanwhile, γH2AX level was remarkably increased in Pold3−/− outgrowth (Supplementary Figure S2H and I), indicating that dramatic DNA damage response occurs without Pold3. Most of the TUNEL+ nuclei appeared to be fragmentized or broken, but most of the γH2AX+ nuclei remained intact. In vivo E6.5 embryos developed a cylinder-liker two-layered cellular structure, but Pold3-deficient embryos lacked such structures (Supplementary Figure S2J). These data demonstrate that Pold3 is essential for early embryonic development. Pold3 deficiency results in defective DNA replication and damage repair in the early embryos and ICM outgrowth.
Deletion of Pold3 leads to longer S phase and apoptosis in mouse ESCs
ESCs are derived from ICM outgrowth of blastocysts, and could be a good model to investigate the mechanism underlying early embryonic lethality. Interestingly, Pold3 was highly expressed in ESCs, but low in MEFs (Supplementary Figure S3A), implying that Pold3 may play important roles in ESCs. To examine the role of Pold3 in ESC self-renewal and pluripotency, we also generated Pold3 KD cell lines by RNA interference using two shRNA constructs (Supplementary Table S1) and assessed the capacity for proliferation, pluripotency, and differentiation of ESCs. The mRNA and protein levels of Pold3 were effectively reduced to about 20% of controls (Supplementary Figure S3B and C). Morphologically, Pold3 KD ESCs maintained characteristics of ESCs with compact clonal boundaries (Supplementary Figure S3D). Neither cell proliferation nor cell cycle progression was altered in Pold3 KD ESCs (Supplementary Figure S3E and F). Expression of pluripotency-associated genes was almost unchanged by immunofluorescence and qPCR analysis (Supplementary Figure S3G and H). Pold3 KD and control ESCs were differentiated in vitro by EB formation. Makers for three germ layers, βIII-tublin (ectoderm), alpha 1-fetoptrotein (AFP, endoderm) and alpha smooth muscle actin (α-SMA, mesoderm) were expressed similarly on day 15 (Supplementary Figure S3I). These data indicate that reduction of Pold3 by shRNA does not affect cell proliferation, pluripotency and differentiation of mouse ESCs, which is consistent with Pold3+/− mouse embryonic development.
Because Pold3−/− ESCs were not achievable by either TALEN or Cas9 methods in repeated experiments (Figure 1 and Supplementary Figure S1), we generated Pold3 inducible knockout (iKO) ESC line by flp/frt and CRISPR/Cas9 system (23) (Supplementary Figure S4A). An exogenous Pold3 gene was introduced into ESCs, and two sgRNAs that could not target the Pold3 cDNA were designed to delete the whole exon 6 of endogenous Pold3. Exogenous Pold3 can be induced to express with 0.1 μg/ml doxycycline (Dox) for 24 h (Supplementary Figure S4B). Pold3 iKO KH2 ESCs were generated and verified by western blot and sequencing (Supplementary Figure S4C–E). Morphologically, Pold3 iKO ESC clones became smaller and not compact with passage (Figure 2A). Culture Medium turned turbid after two passages without Dox (P2). Moreover, Pold3 iKO ESCs proliferated for no more than four passages and cell proliferation was severely compromised without Dox, but iPold3 (inducible Pold3 overexpression) ESCs did not show increased cell proliferation (Figure 2B). Analysis of cell cycle of Pold3 iKO ESCs with or withdrawal of doxycycline showed that ESCs without Pold3 displayed extended S phase and underwent apoptosis (Figure 2C and D). Consistent with deficient outgrowth, DNA replication was compromised shown by BrdU incorporation (Figure 2E) and cell apoptosis increased by TUNEL assay after loss of Pold3 (Figure 2F). However, these ESCs normally expressed core pluripotent genes including Nanog and Oct4 (32) (Figure 2G). Accompanied with decreasing protein level of Pold3, p-CHK1 protein level was elevated in Pold3 iKO ESCs, suggesting that loss of Pold3 leads to replicative stress.
Figure 2.

Cell cycle, apoptosis and RNA-seq analysis of Pold3 iKO ESCs. (A) Colony morphology of Pold3 iKO ESCs, cultured with Dox (P0), or without Dox for 48 h (P1) and for 96 h (P2). Scale bar, 100 μm. (B) Growth curve of WT, iPold3 (inducible exogenous Pold3 expression with endogenous Pold3) and iKO (inducible Pold3 knockout without endogenous Pold3) with or without Dox. (C and D) Cell cycle analysis of Pold3 iKO ESCs at P0, P1 and P2 by flow cytometry. Apo., apoptosis. Error bars indicate mean ± SEM (n = 3). *P < 0.05, **P < 0.01 and ***P < 0.001. (E) BrdU incorporation was detected in Pold3 iKO ESCs at P0, P1 and P2 by immunofluorescence. Scale bar, 20 μm. (F) TUNEL assay of apoptosis cells in Pold3 iKO ESCs at P0, P1 and P2. (G) Protein levels by western blot. (H) Heatmap cluster analysis of differentially regulated genes by RNA-seq analysis of Pold3 iKO ESCs. Pold3 iKO ESCs were cultured in the presence of Dox (P0) and 48 and 96 h after Dox withdrawal (P1 and P2, respectively). Differentially expressed genes were identified in these three comparisons with the criteria of more than 2-fold change and q-value < 0.005. (I) Venn diagram depicting the overlap of up- and downregulated genes among ESCs at P0, P1 and P2. (J) Scatterplot showing differentially expressed genes. Cell cycle, DNA repair and pluripotent associated genes are indicated.
These data indicate that complete loss of Pold3 leads to prolonged S phase, DNA damage and apoptosis. Prolonged S phase may imply stalled replication forks resulting from lack of Pold3.
RNA-seq reveals aberrant cell cycle and defective DNA repair induced by loss of Pold3
To investigate the molecular changes and mechanism during loss of Pold3, Pold3 iKO ESCs at different passages (P0, P1 and P2) were subjected to RNA-sequencing (RNA-Seq) analysis. Clustering analysis reveals that gene expression profile was similar in Pold3 iKO ESCs between P1 and P2, but genes were differentially expressed at P2 compared with P0 (Figure 2H). By comparison, 78 genes were upregulated and 13 downregulated in Pold3 iKO ESCs at P1 versus P0, and 178 upregulated genes and 50 downregulated genes identified in comparison of Pold3 iKO ESCs at P2 with P0. Venn diagrams also show that a large proportion of genes, 73 upregulated genes and 11 downregulated genes, are shared by P1 versus P0 and P2 versus P0 (Figure 2I). Fewer genes were differentially expressed between P1 and P0 and between P2 and P1. Most upregulated genes at P1 and P2 are enriched in p53 signaling pathway (Figure 2J and Supplementary Figure S5A). Core pluripotent genes did not change in their mRNA levels, consistent with the protein levels (Figure 2G), although expression of Lefty1, Lefty2 and Klf4 was decreased in Pold3 KO ESCs (Figure 2J and Supplementary Figure S5B). Trp53inp1 is associated with Trp73 to regulate cell cycle progression and apoptosis, regardless of p53 (33). 1700007K13Rik (RbEST47) involved in regulating S phase (34) was upregulated, consistent with the prolonged S phase. Downregulation of H1f0 may imply declined DNA synthesis (35). Increased Mdm2 expression leads to delayed DNA DSB repair and chromosome/chromatid breaks (36,37). These data further imply the replicative stress resulting from loss of Pold3.
Pold3 is required for telomere maintenance and chromosome stability of mouse ESCs
It is unclear whether Pold3 plays a role in telomere and genomic stability of mouse ESCs. Further, we performed telomere quantitative fluorescence in situ hybridization (Q-FISH) on metaphase spread to reveal relative telomere length as well as telomere integrity (27,28). Rapid telomere shortening was found in Pold3 iKO ESCs within only two passages after loss of Pold3 (Figure 3A, C and D), and this was further corroborated by Southern blot measurement of telomere TRF (Figure 3B). Loss of Pold3 also resulted in chromosomal abnormality and aneuploidy (Figure 3A). Moreover, loss of telomere was observed in Pold3 iKO ESCs at P2 in contrast to P0 (Figure 3C and E). Chromosome fusion slightly increased at P2 (Figure 3F). Notably, increased number of chromosome or chromatid breaks was found in Pold3 iKO ESCs (Figure 3G). In addition, DNA damage at telomeres, indicated by γH2AX foci co-localized at telomeres as telomere dysfunction-induced foci (TIF) (38), remarkably increased after loss of Pold3, in association with elevated γH2AX protein level (Figure 3H–J). Our data demonstrate that loss of Pold3 leads to telomere damage, shortening and loss, chromosome breakage and genomic instability.
Figure 3.

Deficiency of Pold3 leads to telomere loss and genomic instability in ESCs. (A) Representative telomere FISH images of Pold3 iKO ESCs. Green, telomeres, Blue, DAPI stained chromosome. Experiments were performed in Pold3 iKO ESCs with Dox (P0), or one passage (P1) or two passages (P2) after Dox withdrawal. Red arrowhead, chromosome break; White arrowhead, telomere loss. Right panel, Distribution in chromosome number of Pold3 iKO ESCs and ESCs at P0. About 20 chromosome spread was counted for each group. (B) Telomere length distribution shown as TRF by Southern blot analysis. (C) Enlarged telomere Q-FISH images, showing telomere loss (white arrowhead) and chromosome breakage (red arrowhead). Telomeres were labeled with telomere PNA probes (green), and chromosomes labeled with DAPI (blue). (D) Histogram showing distribution of relative telomere length as TFU by Q-FISH. Black bars on y axis indicate frequency of telomere signal-free ends. The medium telomere length (green bars) is shown as mean ± SD. About 20 chromosome spreads were quantified for each group. (E) Frequency of telomere loss per chromosome. (F) Frequency of chromosome fusion. (G) Frequency of chromosome breakage per chromosome. Bottom panel, Distribution of chromosome breaks in Pold3 iKO ESCs and ESCs at P0. About 20 chromosome spreads were counted for each group. (H) Immunofluorescence-FISH images of telomeres (red) and γH2AX (green). Co-localized foci are indicated by white arrows. Scale bar, 5 μm. (I) Statistical analysis of γH2AX foci per cell. Bottom panel, western blot analysis of γH2AX and H3 and β actin served as loading control. (J) Co-localized foci of telomeres and γH2AX per cell. 50 nuclei were randomly counted in (I) or (J). Data represent mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
Loss of Pold3 results in replicative stress
To further investigate the role of Pold3 at replicative stress, we analyzed DNA replication dynamics at the single-molecule level by DNA fiber assay. DNA fibers from Pold3 iKO (P0, P1 and P2) ESCs pulsed sequentially 30 min each with the nucleotide analogs, 5-chlorodeoxyuridine (CldU) and 5-iododeoxyuridine (IdU), were spread on glass slides. Replicating DNA incorporates CldU and then IdU during the sequential pulses, and is detected by immunofluorescence. Labeled DNA fibers were shorter in Pold3 iKO ESCs at P1 (8.96 μm) and further shortened at P2 (5.77 μm), compared to P0 (9.68 μm) ESCs by measuring the tract length of IdU (Figure 4A and B). Moreover, we found a higher frequency of stalled replication forks in Pold3 KO ESCs compared to controls (Figure 4C), indicative of the presence of serious replication stress in Pold3 KO ESCs. Replication stress can lead to micronucleus formation because of failed segregation of chromosome fragments broken at fragile site or unresolved replication intermediates (39,40). We also observed greatly increased micronucleated cells in cell populations at P1 and P2, compared to P0 (Figure 4D). These together might contribute to the chromosome breaks/deletions or aneuploidy (as shown in Figure 3A).
Figure 4.

Loss of Pold3 leads to DNA replicative stress and activates ATR and ATM signaling. (A) Representative immunofluorescence images of labeled DNA fibers are shown; Scale bar, 10 μm. Pold3 iKO ESCs at passage 1 (P1) and P2 were compared with control ESCs at P0. (B) Statistical analysis of tract length measured by IdU-labeled fiber, with 150 tracks in each assay. (C) DNA labeling showing ongoing fork (green CldU followed by red IdU track; green only tracks indicate stalled forks). % stalled fork = green only tracks/green tracks × 100%. mean ± SEM. (D) Representative images and statistical analysis of micronucleated cells in Pold3 iKO ESCs, compared with control ESCs at P0. Micronucleated cells were randomly counted within 10 fields and about 100 cells for each group. Scale bar, 5 μm. (E) Protein levels of p-ATR, ATR and p-ATM in Pold3 iKO ESCs by western blot. (F) Analysis of Pold3 (flag) localized with telomeres indicated by TRF1 in Pold3 3F ESCs with Dox. Twenty nuclei were randomly counted. (G) Immunofluorescence of TRF1 (red) and p-ATM (green). Co-localized foci are indicated by white arrows. Scale bar, 5 μm. Right panel, Statistical analysis of p-ATM foci per cell and co-localized foci of TRF1 and p-ATM per cell. Thirty nuclei were randomly counted. (H) Immunofluorescence of TRF2 (green) and p-ATR (red). Co-localized foci are indicated by white arrows. Scale bar, 5 μm. Right panel, Statistical analysis of p-ATR foci per cell and co-localized foci of TRF2 and p-ATR per cell. Thirty nuclei were randomly counted. Data represent mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
Given the increased γH2AX and p-CHK1 levels and delayed S-phase progression, we determined whether these linked to activation of the ATR or ATM kinases. Protein levels of p-ATR and ATR were increased following loss of Pold3 (Figure 4E). Moreover, p-ATM, activated after double strand breaks, was also increased. These data suggest that both ATR and ATM pathways are activated in Pold3 KO ESCs. By generation of Pold3 3 × flag cell line, we could see Pold3 localized with TRF1 at telomeres (Figure 4F). Furthermore, we tested whether ATR and ATM pathways are activated at telomeres. p-ATM showed only minimal expression at TRF1 at P0, but co-localized foci of p-ATM and TRF1 at telomeres dramatically increased after loss of Pold3 (Figure 4G). Likewise, p-ATR was found to increasingly co-localize with TRF2 in Pold3 KO ESCs (Figure 4H).
Hence, loss Pold3 induces replicative stress and DNA damage and this can be linked to telomere damage, shortening and loss, chromosome breaks, micronucleated cells and aneuploidy.
Reduction of Pold3 impairs DNA repair in mouse ESCs
Further, we tested whether expression levels of Pold3 influence DNA repair in mouse ESCs. DNA damage was induced by exposure to low dose of etoposide or γ-irradiation. Pold3 protein level was significantly increased 2 h after withdrawal of etoposide or γ-irradiation (Figure 5A), indicating that Pold3 may involve in DNA repair following DSB formation.
Figure 5.

Reduction of Pold3 impairs DNA repair in mouse ESCs. (A) Pold3 protein level increased following DSB induced by etoposide (2.5 μM) for 2 h or γ-irradiation (5 Gy). ESCs without treatment (−), or with treatment 2, 12 and 24 h after exposure to etoposide or γ-irradiation were harvested for western blot analysis. (B) Pold3 KD ESCs (N33) by exposure to 2.5 μM etoposide for 2 h express γH2AX protein at higher levels after 12 h recovery by western blot, while the protein level of Oct4 is not changed, like controls. (C) Pold3 KD ESCs (N33) by exposure to 2.5 μM etoposide for 2 h exhibit much more γH2AX foci after 12 h recovery compared with control by immunofluorescence. Scale bar, 5 μm. (D) Quantitative results of (C). Fifty cells were randomly counted in two repeated experiments. (E) Pold3 KD ESCs by exposure to 5 Gy γ-irradiation show more γH2AX foci after 12 h recovery compared with control by immunofluorescence. Scale bar, 5 μm. (F) Quantitative results of (E). n = 50 cells counted. (G) Pold3 KD ESCs by exposure to 5 Gy γ-irradiation display more 53BP1 foci after 12 h recovery compared with control by immunofluorescence. (H) Quantitative results of (G). n = 50 cells counted. (I) Genotyping of WT and Pold3+/− ESCs. (J) Reduced expression of Pold3 was confirmed by western blot in Pold3+/− ESCs, compared to WT ESCs. (K) Western blot analysis showing higher γH2AX protein level in Pold3+/− ESCs compared to control 12 h after exposure to etoposide for 2 h. Data represent mean ± SEM. **P < 0.01; ***P < 0.001.
To assess whether ESCs with reduced levels of Pold3 are more susceptible to DNA damage response, we performed immunofluorescence microscopy of γH2AX and 53BP1, commonly used DNA damage response markers (41), in Pold3 KD or Pold3+/− ESCs by exposure to low dose of either etoposide or γ-irradiation. After recovery for 12 h, higher γH2AX levels accumulated in Pold3 KD ESCs compared with control (Figure 5B). We also intuitively monitored γH2AX foci by immunofluorescence in etoposide-treated or γ-irradiated ESCs. Increased γH2AX foci appeared at 2 h, and the foci were significantly reduced likely indicative of DNA repair after 12 h recovery in ESC controls. However, more γH2AX foci persisted in Pold3 KD ESCs at 12 h (Figure 5C–F), consistent with western blot data (Figure 5B). Chemical proteomics shows that γH2AX-53BP1 interacts in the DNA damage response (42). Indeed, 53BP1 foci also persisted longer and showed higher incidence at 12 h in Pold3 KD ESCs than in the controls after exposure to γ-irradiation (Figure 5G and H). Also, higher γH2AX protein level was observed in Pold3+/− ESCs following exposure to etoposide for 2 h (Figure 5I–K).
It has been established that 53BP1 promotes NHEJ and inhibit homology-directed repair (review, (41)). Further, we showed that Pold3 co-localized with 53BP1 foci were visible 2 h after exposure to etoposide (Figure 6A). Rif1, a downstream factor of 53BP1 (5), was also co-localized with 53BP1 foci (Figure 6B). Rif1 foci still existed at higher incidence 12 h after exposure to etoposide in Pold3 KD ESCs than that of shControl (Figure 6C). Moreover, we used an ATR specific inhibitor VE-821 (43,44), which can induce predominantly replicative stress, to treat shPold3 ESCs compared with shControl. Cell number was markedly reduced in Pold3 KD ESCs compared with shControls by treatment with 8 μM VE-821 for 3 days or with 3 μM VE-821 for 6 days. Thus, Pold3 KD ESCs show increased sensitivity to ATR inhibition (Figure 6D), supporting the notion that DNA repair deficiency can be related to replication defects (13). These data further indicate that reduction of Pold3 increases replicative stress and impairs homology-directed DSB repair in mouse ESCs.
Figure 6.

Involvement of 53BP1 and Rif1 in DNA damage repair by Pold3. (A) Immunofluorescence and quantification of Pold3 (Flag) and 53BP1 foci after treatment with etoposide for 2 h. 20 nuclei were randomly counted. (B) Co-localization of Rif1 and 53BP1 by immunofluorescence after exposure to 2.5 μM etoposide for 2 h. Right panel bar figure, 20 cells were counted. (C) Following exposure to 2.5 μM etoposide for 2 h, Pold3 KD (shPold3) ESCs (N33) exhibit more Rif1 foci by 12 h compared with shControl. Scale bar, 5 μm. n = 50 cells counted. (D) Increased sensitivity to ATR inhibitor in Pold3 KD (shPold3) ESCs than in shControl ESCs. ESCs were plated in 12-plates, and various concentrations of VE-821 were added in the culture medium, and number of cells counted at day 3. Alternatively, ESCs were plated in 12-plates, and 3 μM VE-821 was added in the medium. Cells were counted and replated into new plates every two days. Data were analyzed using Two-way ANOWA. Data represent mean ± SEM. ***P < 0.001.
Pold3 heterozygosity results in germ cell loss with reproductive age
Recombination involves induction of massive DSB during meiosis, and failure to repair the DSB leads to defective meiosis and infertility (45,46). Interestingly, Pold3 was highly expressed in adult testis but minimal in fetal testis (Supplementary Figure S6A). Moreover, both Pold3 mRNA and protein expression levels in testis decreased with age and further in Pold3+/− testis (Supplementary Figure S6B and C). We hypothesized that reduced levels of Pold3 might accelerate reproductive aging. Old Pold3+/− male mice had smaller and lighter testis compared with old WT mice, but testis of the middle-aged mice by reproductive age showed no significant difference (Supplementary Figure S6D and E). A few abnormal, but no completely degenerated tubules were visible in middle-aged Pold3+/− mice. The phenotype became more pronounced with age, and old Pold3+/− mice manifested moderate to severe testicular degeneration characterized by loss of germ cells within the lumina of seminiferous tubules (Supplementary Figure S6F). An average of 1.8% and 27.5% of tubules contained vacuoles in middle-aged and old Pold3+/− mice, respectively, but the vacuoles were nearly undetectable in WT mice (Supplementary Figure S6G).
The observed phenotype is likely caused by impairment of spermatogenesis. More TUNEL positive cells were found in testis of old Pold3+/− males, and most apoptotic cells appeared to be spermatocytes by localization (Supplementary Figure S6H and I). It appeared that SYCP3 positive cells were reduced and a large number of γH2AX foci frequently observed in spermatocytes stained with SYCP3 in old Pold3+/− compared with age-matched WT mice (Supplementary Figure S6J). These data show that Pold3 haploinsufficiency increases apoptosis, accelerates germ cell loss and leads to reproductive premature aging.
Pold3 haploinsufficiency impairs meiosis progression and telomere integrity
To further characterize the defects of Pold3+/− testis, germ cell spreads were prepared and stained for key meiosis-associated proteins. SYCP1 is one of the key components of the synaptonemal complex for HR (47). Strikingly, breaks in chromosomes were observed in a few pachytene spermatocytes from middle-aged Pold3+/− males, and much more frequently in old Pold3+/− males, but rarely in age-matched WT males (Figure 7A, white arrow). SYCP1 distributed discontinuously in some of the abnormal spermatocytes, suggesting that meiotic synapsis was incomplete and partially impaired (Figure 7A, red arrow). The percentage of pachytene and diplotene spermatocytes was decreased, while the percentage of leptotene and zygotene increased in aged Pold3+/− mice (Figure 7B).
Figure 7.

Defective meiosis and telomeres in pachytene spermatocytes of Pold3 heterozygous (Pold3+/−) mice. (A) Meiotic chromosome spreads from Pold3+/+ (WT) and Pold3+/− males were stained with antibody to SYCP1 (red) and SYCP3 (green). (B) Frequency of meiotic prophase I stages. n = 3 mice for each group, and 100 spermatocytes for each mouse. (C) Staining of pachytene spermatocyte spreads by telomere-FISH (green) and immunofluorescence of SYCP3 (red). Chromosome breaks are indicated by white arrowheads. (D) Histogram showing distribution of relative telomere length as TFU by telomere Q-FISH. Black bars on y axis show frequency of telomere signal-free ends. The medium telomere length (green bars) is shown as mean ± SD. About 30 chromosome spreads were quantified for each group. (E) Statistical analysis of chromosome breaks in pachytene spermatocytes. (F) Representative image of telomere loss at the end of chromosome in pachytene spermatocyte, and image of individual chromosome at higher magnification. (G) Frequency of telomere loss per chromosome. Mean ± SEM. *P < 0.05; **P < 0.01.
To further validate the chromosome breakage, we performed telomere Q-FISH and immunofluorescence of SYCP1 on spermatocytes. Telomeres shortened with age, and shortened further in Pold3+/− pachytene spermatocytes, compared with those of WT mice (Figure 7C and D). Moreover, number of breaks in chromosomes increased statistically (Figure 7E), and telomere loss also occurred more frequently in old Pold3+/− pachytene spermatocytes compared with WT (Figure 7F and G).
Pold3 haploinsufficiency impairs meiotic DNA repair and recombination
HR is initiated by introducing DSB, and γH2AX is localized to the nucleoplasm from the leptotene to zygotene stage, but to the sex body which contains unpaired regions of the X and Y chromosomes at the pachytene stage (48). The pattern in distribution of γH2AX, a marker for DSBs, during meiosis is well documented (49,50). In leptotene and zygotene spermatocytes, it is usually interspersed throughout the nucleus and disappears by late zygotene, whereas in pachytene, strong γH2AX signal is solely restricted to the sex body. Staining of γH2AX in leptotene or zygotene spermatocytes did not differ between Pold3+/− and WT mice. However, abnormal distribution of γH2AX throughout the nuclei was frequently detected in Pold3+/− spermatocytes at pachytene and diplotene stages. γH2AX foci were increased in old Pold3+/− mice and number of γH2AX positive cells with more than 5 foci on the autosomes significantly rose in pachytene spermatocytes of middle-aged and old Pold3+/− mice (Figure 8A–C).
Figure 8.

Increased DSB and declined recombination in Pold3+/− spermatocytes with age. (A) Representative immunofluorescence images showing co-immunostaining of γH2AX and SYCP3 in Pold3+/+ and Pold3+/− spermatocytes, and accumulation of γH2AX in pachytene spermatocytes shown at higher magnification. Scale bar, 20 μm. (B and C) Statistical results of (A). n = 3 mice for each group, and 50 spermatocytes counted for each mouse. (D) Representative immunofluorescence images showing co-immunostaining of RAD51 and SYCP3. Scale bar, 20 μm. (E and F) Statistical results of (D). n = 3 mice for each group, and 50 spermatocytes counted for each mouse. (G) Representative immunofluorescence images showing co-immunostaining of MLH1 and SYCP3. Scale bar, 20 μm. (H and I) Statistical results of (G). Mean ± SEM. *P < 0.05; **P < 0.01.
RAD51, a DSB repair-associated recombinase needed for both mitotic and meiotic recombination events (16,51,52), maintained at higher levels in pachytene spermatocytes of old Pold3+/− mice (Figure 8D–F), consistent with the DSB repair defect. Both persistent γH2AX and remaining Rad51 foci on the pachytene and diplotene chromosomes demonstrate that DSB repair is impaired in old Pold3+/− testis, and this may be associated with defective HR. Indeed, the MLH1 foci, indicative of HR and crossover, declined and more cells with less than 17 MLH1 foci were found in spermatocytes of old Pold3+/− male, compared with those of age-matched WT mice (Figure 8G–I). Collectively, these results suggest that reduction of Pold3 impairs DSB repair ability and leads to genomic instability in meiosis.
DISCUSSION
We show that Pold3 is required for DSB repair in major developmental processes including early embryogenesis, ESC self-renewal and meiosis, all possessing robust HR mechanisms to repair DSB as well as telomere maintenance and integrity, important for genomic stability. Furthermore, we suggest that level of Pold3 affects aging, and show here that haploinsufficiency of Pold3 leads to meiosis defects with reproductive age.
Our data demonstrate that Pold3 is essential for early embryonic development. Some phenotypes of Pold3 knockout mice reported in our work using CRISPR/Cas9 gene editing (Figure 1H) are consistent with those of Murga’s (18), showing decreased mendelian ratios and reduced number of Pold3+/− mice born, and Pold3−/− embryonic lethality. Moreover, we could not obtain Pold3−/− ESC lines by either CRISPR/Cas9 or TALEN methods. Complete loss of POLD3 also was not achievable in U2OS cells using CRISPR/Cas9 (53). These data together further strengthen the notion that Pold3 is required for cell survival. Moreover, we observed that the Pold3−/− embryonic lethality occurs as early as E5.5-E6.5 due to DNA damage and apoptosis. Pold3+/− mice mostly survived to adulthood, but exhibited meiosis defects and reproductive premature aging, and this corroborates decreased lifespan reported by Murga et al. (18).
Robust DNA repair and telomere maintenance mechanisms ensure rapid proliferation and unlimited self-renewal capacity of ESCs (1,54). We show that Pold3 plays critical role in DSB repair and maintaining the telomere integrity and genomic stability of mouse ESCs. Earlier, POLD3/Pol32 is involved in break-induced telomere synthesis that underlies telomerase-independent telomere maintenance in budding yeast (55). Recently, the POLD3 subunit of Polδ is required for BIR and the conservative replication of telomeres in human alternative lengthening of telomeres (ALT) cell lines (12,53,56,57). ALT originally discovered in about 15% of human cancer cells utilizes homology-directed DNA repair to maintain telomeres (58). Interestingly, while decreased Pold3 causes telomere shortening in ALT cell lines, accompanied with accumulated increased numbers of TIFs, it does not change telomere length in telomerase-positive cells (53). These evidence demonstrate conserved role of Pold3 in telomerase-independent telomere maintenance in yeast and the ALT pathway in human cancers. Nevertheless, mouse ESCs utilize both telomerase and recombination based ALT-like pathways to robustly elongate telomeres ((59) review (54)). Our data extend these findings to demonstrate in mouse ESCs the existence of mechanistically multiple pathways involving Pold3-mediated DNA repair and telomere integrity.
Mechanistically, Pold3 is required for DNA replication and DNA repair by suppressing DNA replicative stress. Loss of Pold3 in ESCs results in increased replicative stress and impaired DNA damage repair. Also, replication stress can activate ALT mechanisms in primary and telomerase-positive cells (60). Mammalian telomeres resemble fragile sites and are prone to alterations or breakage under replication stress conditions (61,62). Consistently, telomere damage induced foci, telomere loss and chromosome fusion are increased in Pold3-deficient ESCs. Common fragile sites are prone to breaks or gaps in metaphase chromosomes when cells experience replicative stress (39,40), such that increased chromosome breakage and micronuclei are observed in Pold3-deficient ESCs.
Furthermore, we show that loss of Pold3 in ESCs activates ATM and ATR pathways. Moreover, ESCs with reduced Pold3 exhibit increased sensitivity to ATR inhibition. Activation of ATM kinase pathway elongates telomeres in telomerase positive mouse and human cells and parallels its role in DNA damage, and ATR may compensate for loss of ATM (44). ATM and ATR are required for telomere elongation by telomerase recruitment to telomeres in telomerase positive human cells (63). ATR kinase is crucial for DNA damage and replication stress responses and suppresses chromosome instability also by function at centromeres (64). However, ATR also has an important role in the suppression of telomere fragility and recombination (65). Similarly, ATM and ATR regulate damage signaling, telomere integrity, and survival in ALT cells, but may be not an essential component of the break-induced telomere synthesis (53). Although activated ATM and ATR pathways may extend telomeres by recruiting telomerase to telomeres in telomerase positive cells (44,63), their impact on rescuing DNA and telomere defects could be limited after loss of Pold3 in ESCs, as evidenced by chromosome breaks, telomere shortening and loss and aneuploidy. It is possible that Pold3 may mediate DNA replication and repair and maintain telomere integrity by suppressing ATR and ATM-activated DNA damage signaling.
53BP1 and RIF1 also may involve in Pold3-mediated DNA replication and telomere maintenance in mouse ESCs. Rif1 is an important contributor to the control of DSB repair by 53BP1, and RIF1 and 53BP1 cooperate to block DSB resection to promote NHEJ in G1, which is antagonized by BRCA1 in S phase to ensure a switch of DSB repair mode to HR (5,66). Reduction of Pold3 in ESCs causes elevated RIF1 and 53BP1 foci in response to DNA damage (Figure 6). Pold3 depletion results in delayed S phase, and shorter G1 in mouse ESCs, consistent with intra-S-phase checkpoint activation with increased levels of p-CHK1 (67), p-ATR and p-ATM, and DNA replication stress (68,69). Given that ESCs have relatively short G1 and even shorter G1 phase after loss of Pold3 (Figure 2D), the elevated Rif1 and 53BP1 may switch the DSB repair mode to HR in extended S phase. These data also are consistent with the concept that relatively long S phase and short G1 phase characteristic of ESCs are beneficial for HR-mediated DSB repair and telomere elongation and integrity. Telomere loss and dysfunction activates ATR-dependent DNA damage response and absence of 53BP1 can lead to amplification and persistent DNA damage signals (70). Replication stress elicits DNA damage response, activation of ATR and 53BP1 and DNA deletions and amplifications shown as micronuclei, and thus induces chromosomal instability (26). MUS81, BLM, BRCA and RAD52 also may involve in POLD3-induced DNA replication and HR (13,71). Together, these data suggest that Pold3 plays even more important roles in the genomic stability of ESCs than that of other cell types likely by regulating multiple pathways.
Pold3 also plays critical roles in DSB repair, HR and telomere maintenance in meiosis. BIR and telomeres are important for recombination in meiosis. Telomere clustering is often found in meiotic cells (72). Telomere damage or DSBs can induce telomere clustering and this results in HR-dependent repair and ALT mediated telomere maintenance (53,73,74). DSB response at ALT telomeres triggers long-range movement and clustering between chromosome termini, resulting in homology-directed telomere synthesis (75). A new study demonstrates that BLM and SLX4 become actively engaged at ALT telomeres to process HR-intermediate structures and that BLM requires both the Pol δ component POLD3 and RAD51 to initiate these events (56). Rad51 is required for the repair of telomeric DSBs (73). Rad51 recruitment and/or retention at DSBs facilitate repair by HR (76). We observed that Rad51 still accumulates in aged Pold3+/− spermatocytes, suggesting that Rad51 may remain to repair DSB. Moreover, telomeres gradually shorten and become lost and HR declines in pachytene spermatocytes of Pold3+/− mice with age. Short telomeres impair HR in meiosis as evidenced by reduced MLH1 foci on SYCP elements (30). Polδ is preferentially recruited during meiosis to promote heteroduplex DNA extension in yeast (77). Our data suggest that DNA damage repair defects resulting from reduced level of Pold3 can lead to telomere shortening and loss, and chromosome breaks, compromising meiosis.
Notably, haploinsufficiency of Pold3 leads to meiosis defects, increased DNA damage and apoptosis and declined germ cells with age. One of responses to DNA damage is to induce apoptotic cell death (78). Persistent γH2AX foci provides molecular marker of DNA damage and also can accelerate aging (79). Replicative stress in these cells due to reduced levels of Pold3 also may lead to aging (80). Moreover, impaired DSB repair and accumulation of DNA lesions contribute to age-associated rise of genomic instability and age-related diseases (81–85). Meanwhile, short telomeres also are sufficient to cause the degenerative defects associated with aging (86). Telomere damage can contribute to telomere dysfunction and genomic instability (87,88). Our data show that reduced level of Pold3 impairs DSB repair and HR, and induces telomere loss and these together can contribute to accelerated meiosis defects with age. Pold3 is involved in processing of meiotic DSB repair during meiotic recombination.
We conclude that Pold3 is essential for DSB repair and telomere maintenance, ensuring genome stability in ESCs and meiosis.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Bo Zhang for TALEN vectors, Deling Kong for providing instrument for ionizing radiation, Dongyi Xu for advising DNA fiber assay and Jian Ren for advising RNA-seq data analysis.
Author contributions: L.L. conceived the study, designed experiments and wrote the manuscript. Z.Z. designed and performed major experiments and data analysis, and prepared manuscript. L.W. performed micro-injection and embryo transfer experiments. F.G. helped with some vector construction. P.G. assisted IF-FISH experiment. H.W. advised RNA-seq analysis. F.W. helped DNA fiber assay. L.C. provided reagents and constructs.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
China Ministry of Science and Technology [2014DFA30450]; National Natural Science Foundation of China [31430052, 31571546]. Funding for open access charge: China Ministry of Science and Technology [2014DFA30450].
Conflict of interest statement. None declared.
REFERENCES
- 1. Tichy E.D., Stambrook P.J.. DNA repair in murine embryonic stem cells and differentiated cells. Exp. Cell Res. 2008; 314:1929–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Oliveira P.H., da Silva C.L., Cabral J.M.. Concise review: genomic instability in human stem cells: current status and future challenges. Stem Cells. 2014; 32:2824–2832. [DOI] [PubMed] [Google Scholar]
- 3. Lamm N., Ben-David U., Golan-Lev T., Storchova Z., Benvenisty N., Kerem B.. Genomic instability in human pluripotent stem cells arises from replicative stress and chromosome condensation defects. Cell Stem Cell. 2016; 18:253–261. [DOI] [PubMed] [Google Scholar]
- 4. Fu X., Cui K., Yi Q., Yu L., Xu Y.. DNA repair mechanisms in embryonic stem cells. Cell Mol. Life Sci. 2017; 74:487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zimmermann M., Lottersberger F., Buonomo S.B., Sfeir A., de Lange T.. 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science. 2013; 339:700–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang H., Zeng Z.C., Bui T.A., Sonoda E., Takata M., Takeda S., Iliakis G.. Efficient rejoining of radiation-induced DNA double-strand breaks in vertebrate cells deficient in genes of the RAD52 epistasis group. Oncogene. 2001; 20:2212–2224. [DOI] [PubMed] [Google Scholar]
- 7. Savatier P., Lapillonne H., Jirmanova L., Vitelli L., Samarut J.. Analysis of the cell cycle in mouse embryonic stem cells. Methods Mol. Biol. 2002; 185:27–33. [DOI] [PubMed] [Google Scholar]
- 8. van der Laan S., Tsanov N., Crozet C., Maiorano D.. High Dub3 expression in mouse ESCs couples the G1/S checkpoint to pluripotency. Mol. Cell. 2013; 52:366–379. [DOI] [PubMed] [Google Scholar]
- 9. Orthwein A., Noordermeer S.M., Wilson M.D., Landry S., Enchev R.I., Sherker A., Munro M., Pinder J., Salsman J., Dellaire G. et al. A mechanism for the suppression of homologous recombination in G1 cells. Nature. 2015; 528:422–426. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10. Zuo S., Gibbs E., Kelman Z., Wang T.S., O’Donnell M., MacNeill S.A., Hurwitz J.. DNA polymerase delta isolated from Schizosaccharomyces pombe contains five subunits. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:11244–11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gerik K.J., Li X., Pautz A., Burgers P.M.. Characterization of the two small subunits of Saccharomyces cerevisiae DNA polymerase delta. J. Biol. Chem. 1998; 273:19747–19755. [DOI] [PubMed] [Google Scholar]
- 12. Costantino L., Sotiriou S.K., Rantala J.K., Magin S., Mladenov E., Helleday T., Haber J.E., Iliakis G., Kallioniemi O.P., Halazonetis T.D.. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science. 2014; 343:88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Minocherhomji S., Ying S., Bjerregaard V.A., Bursomanno S., Aleliunaite A., Wu W., Mankouri H.W., Shen H., Liu Y., Hickson I.D.. Replication stress activates DNA repair synthesis in mitosis. Nature. 2015; 528:286–290. [DOI] [PubMed] [Google Scholar]
- 14. Cohen P.E., Pollard J.W.. Regulation of meiotic recombination and prophase I progression in mammals. Bioessays. 2001; 23:996–1009. [DOI] [PubMed] [Google Scholar]
- 15. Tarsounas M., Moens P.B.. Checkpoint and DNA-repair proteins are associated with the cores of mammalian meiotic chromosomes. Curr. Top. Dev. Biol. 2001; 51:109–134. [DOI] [PubMed] [Google Scholar]
- 16. Handel M.A., Schimenti J.C.. Genetics of mammalian meiosis: regulation, dynamics and impact on fertility. Nat. Rev. Genet. 2010; 11:124–136. [DOI] [PubMed] [Google Scholar]
- 17. Johnston D.S., Wright W.W., Dicandeloro P., Wilson E., Kopf G.S., Jelinsky S.A.. Stage-specific gene expression is a fundamental characteristic of rat spermatogenic cells and Sertoli cells. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:8315–8320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Murga M., Lecona E., Kamileri I., Diaz M., Lugli N., Sotiriou S.K., Anton M.E., Mendez J., Halazonetis T.D., Fernandez-Capetillo O.. POLD3 Is Haploinsufficient for DNA Replication in Mice. Mol. Cell. 2016; 63:877–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang P., Xiao A., Zhou M., Zhu Z., Lin S., Zhang B.. Heritable gene targeting in zebrafish using customized TALENs. Nat. Biotechnol. 2011; 29:699–700. [DOI] [PubMed] [Google Scholar]
- 20. Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A. et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013; 339:819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang H., Yang H., Shivalila C.S., Dawlaty M.M., Cheng A.W., Zhang F., Jaenisch R.. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013; 153:910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huang J., Wang F., Okuka M., Liu N., Ji G., Ye X., Zuo B., Li M., Liang P., Ge W.W. et al. Association of telomere length with authentic pluripotency of ES/iPS cells. Cell Res. 2011; 21:779–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Beard C., Hochedlinger K., Plath K., Wutz A., Jaenisch R.. Efficient method to generate single-copy transgenic mice by site-specific integration in embryonic stem cells. Genesis. 2006; 44:23–28. [DOI] [PubMed] [Google Scholar]
- 24. Dan J., Liu Y., Liu N., Chiourea M., Okuka M., Wu T., Ye X., Mou C., Wang L., Wang L. et al. Rif1 maintains telomere length homeostasis of ESCs by mediating heterochromatin silencing. Dev. Cell. 2014; 29:7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jackson D.A., Pombo A.. Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J. Cell Biol. 1998; 140:1285–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Saldivar J.C., Miuma S., Bene J., Hosseini S.A., Shibata H., Sun J., Wheeler L.J., Mathews C.K., Huebner K.. Initiation of genome instability and preneoplastic processes through loss of Fhit expression. PLoS Genet. 2012; 8:e1003077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zijlmans J.M., Martens U.M., Poon S.S., Raap A.K., Tanke H.J., Ward R.K., Lansdorp P.M.. Telomeres in the mouse have large inter-chromosomal variations in the number of T2AG3 repeats. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:7423–7428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Herrera E., Samper E., Martin-Caballero J., Flores J.M., Lee H.W., Blasco M.A.. Disease states associated with telomerase deficiency appear earlier in mice with short telomeres. EMBO J. 1999; 18:2950–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Peters A.H., Plug A.W., van Vugt M.J., de Boer P.. A drying-down technique for the spreading of mammalian meiocytes from the male and female germline. Chromosome Res. 1997; 5:66–68. [DOI] [PubMed] [Google Scholar]
- 30. Liu L., Franco S., Spyropoulos B., Moens P.B., Blasco M.A., Keefe D.L.. Irregular telomeres impair meiotic synapsis and recombination in mice. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:6496–6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hsu P.D., Lander E.S., Zhang F.. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014; 157:1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Loh Y.H., Wu Q., Chew J.L., Vega V.B., Zhang W., Chen X., Bourque G., George J., Leong B., Liu J. et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat. Genet. 2006; 38:431–440. [DOI] [PubMed] [Google Scholar]
- 33. Tomasini R., Seux M., Nowak J., Bontemps C., Carrier A., Dagorn J.C., Pebusque M.J., Iovanna J.L., Dusetti N.J.. TP53INP1 is a novel p73 target gene that induces cell cycle arrest and cell death by modulating p73 transcriptional activity. Oncogene. 2005; 24:8093–8104. [DOI] [PubMed] [Google Scholar]
- 34. Sung Y.H., Kim H.J., Lee H.W.. Identification of a novel Rb-regulated gene associated with the cell cycle. Mol. Cells. 2007; 24:409–415. [PubMed] [Google Scholar]
- 35. Meergans T., Albig W., Doenecke D.. Varied expression patterns of human H1 histone genes in different cell lines. DNA Cell Biol. 1997; 16:1041–1049. [DOI] [PubMed] [Google Scholar]
- 36. Alt J.R., Bouska A., Fernandez M.R., Cerny R.L., Xiao H., Eischen C.M.. Mdm2 binds to Nbs1 at sites of DNA damage and regulates double strand break repair. J. Biol. Chem. 2005; 280:18771–18781. [DOI] [PubMed] [Google Scholar]
- 37. Bouska A., Lushnikova T., Plaza S., Eischen C.M.. Mdm2 promotes genetic instability and transformation independent of p53. Mol. Cell. Biol. 2008; 28:4862–4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Takai H., Smogorzewska A., de Lange T.. DNA damage foci at dysfunctional telomeres. Curr. Biol. 2003; 13:1549–1556. [DOI] [PubMed] [Google Scholar]
- 39. Chan K.L., Palmai-Pallag T., Ying S., Hickson I.D.. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 2009; 11:753–760. [DOI] [PubMed] [Google Scholar]
- 40. Naim V., Rosselli F.. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat. Cell Biol. 2009; 11:761–768. [DOI] [PubMed] [Google Scholar]
- 41. Zimmermann M., de Lange T.. 53BP1: pro choice in DNA repair. Trends Cell Biol. 2014; 24:108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kleiner R.E., Verma P., Molloy K.R., Chait B.T., Kapoor T.M.. Chemical proteomics reveals a gammaH2AX-53BP1 interaction in the DNA damage response. Nat. Chem. Biol. 2015; 11:807–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reaper P.M., Griffiths M.R., Long J.M., Charrier J.D., Maccormick S., Charlton P.A., Golec J.M., Pollard J.R.. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 2011; 7:428–430. [DOI] [PubMed] [Google Scholar]
- 44. Lee S.S., Bohrson C., Pike A.M., Wheelan S.J., Greider C.W.. ATM kinase is required for telomere elongation in mouse and human cells. Cell Rep. 2015; 13:1623–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xu X., Aprelikova O., Moens P., Deng C.X., Furth P.A.. Impaired meiotic DNA-damage repair and lack of crossing-over during spermatogenesis in BRCA1 full-length isoform deficient mice. Development. 2003; 130:2001–2012. [DOI] [PubMed] [Google Scholar]
- 46. Yang F., Eckardt S., Leu N.A., McLaughlin K.J., Wang P.J.. Mouse TEX15 is essential for DNA double-strand break repair and chromosomal synapsis during male meiosis. J. Cell Biol. 2008; 180:673–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zickler D., Kleckner N.. Meiotic chromosomes: integrating structure and function. Annu. Rev. Genet. 1999; 33:603–754. [DOI] [PubMed] [Google Scholar]
- 48. Bolcun-Filas E., Schimenti J.C.. Genetics of meiosis and recombination in mice. Int. Rev. Cell Mol. Biol. 2012; 298:179–227. [DOI] [PubMed] [Google Scholar]
- 49. Mahadevaiah S.K., Turner J.M., Baudat F., Rogakou E.P., de Boer P., Blanco-Rodriguez J., Jasin M., Keeney S., Bonner W.M., Burgoyne P.S.. Recombinational DNA double-strand breaks in mice precede synapsis. Nat. Genet. 2001; 27:271–276. [DOI] [PubMed] [Google Scholar]
- 50. Vasileva A., Hopkins K.M., Wang X., Weisbach M.M., Friedman R.A., Wolgemuth D.J., Lieberman H.B.. The DNA damage checkpoint protein RAD9A is essential for male meiosis in the mouse. J. Cell Sci. 2013; 126:3927–3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Barlow C., Liyanage M., Moens P.B., Deng C.X., Ried T., Wynshaw-Boris A.. Partial rescue of the prophase I defects of Atm-deficient mice by p53 and p21 null alleles. Nat. Genet. 1997; 17:462–466. [DOI] [PubMed] [Google Scholar]
- 52. Sehorn M.G., Sigurdsson S., Bussen W., Unger V.M., Sung P.. Human meiotic recombinase Dmc1 promotes ATP-dependent homologous DNA strand exchange. Nature. 2004; 429:433–437. [DOI] [PubMed] [Google Scholar]
- 53. Dilley R.L., Verma P., Cho N.W., Winters H.D., Wondisford A.R., Greenberg R.A.. Break-induced telomere synthesis underlies alternative telomere maintenance. Nature. 2016; 539:54–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu L. Linking telomere regulation to stem cell pluripotency. Trends Genet. 2017; 33:16–33. [DOI] [PubMed] [Google Scholar]
- 55. Lydeard J.R., Jain S., Yamaguchi M., Haber J.E.. Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature. 2007; 448:820–823. [DOI] [PubMed] [Google Scholar]
- 56. Sobinoff A.P., Allen J.A., Neumann A.A., Yang S.F., Walsh M.E., Henson J.D., Reddel R.R., Pickett H.A.. BLM and SLX4 play opposing roles in recombination-dependent replication at human telomeres. EMBO J. 2017; 36:2907–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Roumelioti F.M., Sotiriou S.K., Katsini V., Chiourea M., Halazonetis T.D., Gagos S.. Alternative lengthening of human telomeres is a conservative DNA replication process with features of break-induced replication. EMBO Rep. 2016; 17:1731–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cesare A.J., Reddel R.R.. Alternative lengthening of telomeres: models, mechanisms and implications. Nat. Rev. Genet. 2010; 11:319–330. [DOI] [PubMed] [Google Scholar]
- 59. Zalzman M., Falco G., Sharova L.V., Nishiyama A., Thomas M., Lee S.L., Stagg C.A., Hoang H.G., Yang H.T., Indig F.E. et al. Zscan4 regulates telomere elongation and genomic stability in ES cells. Nature. 2010; 464:858–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. O'Sullivan R.J., Arnoult N., Lackner D.H., Oganesian L., Haggblom C., Corpet A., Almouzni G., Karlseder J.. Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nat. Struct. Mol. Biol. 2014; 21:167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sfeir A., Kosiyatrakul S.T., Hockemeyer D., MacRae S.L., Karlseder J., Schildkraut C.L., de Lange T.. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 2009; 138:90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Martinez P., Thanasoula M., Munoz P., Liao C., Tejera A., McNees C., Flores J.M., Fernandez-Capetillo O., Tarsounas M., Blasco M.A.. Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev. 2009; 23:2060–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tong A.S., Stern J.L., Sfeir A., Kartawinata M., de Lange T., Zhu X.D., Bryan T.M.. ATM and ATR signaling regulate the recruitment of human telomerase to telomeres. Cell Rep. 2015; 13:1633–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kabeche L., Nguyen H.D., Buisson R., Zou L.. A mitosis-specific and R loop-driven ATR pathway promotes faithful chromosome segregation. Science. 2018; 359:108–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. McNees C.J., Tejera A.M., Martinez P., Murga M., Mulero F., Fernandez-Capetillo O., Blasco M.A.. ATR suppresses telomere fragility and recombination but is dispensable for elongation of short telomeres by telomerase. J. Cell Biol. 2010; 188:639–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chapman J.R., Barral P., Vannier J.B., Borel V., Steger M., Tomas-Loba A., Sartori A.A., Adams I.R., Batista F.D., Boulton S.J.. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol. Cell. 2013; 49:858–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bartek J., Lukas C., Lukas J.. Checking on DNA damage in S phase. Nat. Rev. Mol. Cell Biol. 2004; 5:792–804. [DOI] [PubMed] [Google Scholar]
- 68. Ahuja A.K., Jodkowska K., Teloni F., Bizard A.H., Zellweger R., Herrador R., Ortega S., Hickson I.D., Altmeyer M., Mendez J. et al. A short G1 phase imposes constitutive replication stress and fork remodelling in mouse embryonic stem cells. Nat. Commun. 2016; 7:10660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zeman M.K., Cimprich K.A.. Causes and consequences of replication stress. Nat. Cell Biol. 2014; 16:2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Martinez P., Flores J.M., Blasco M.A.. 53BP1 deficiency combined with telomere dysfunction activates ATR-dependent DNA damage response. J. Cell Biol. 2012; 197:283–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bhowmick R., Minocherhomji S., Hickson I.D.. RAD52 facilitates mitotic DNA synthesis following replication stress. Mol. Cell. 2016; 64:1117–1126. [DOI] [PubMed] [Google Scholar]
- 72. Scherthan H., Jerratsch M., Dhar S., Wang Y.A., Goff S.P., Pandita T.K.. Meiotic telomere distribution and Sertoli cell nuclear architecture are altered in Atm- and Atm-p53-deficient mice. Mol. Cell. Biol. 2000; 20:7773–7783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mao P., Liu J., Zhang Z., Zhang H., Liu H., Gao S., Rong Y.S., Zhao Y.. Homologous recombination-dependent repair of telomeric DSBs in proliferating human cells. Nat. Commun. 2016; 7:12154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hu Y., Shi G., Zhang L., Li F., Jiang Y., Jiang S., Ma W., Zhao Y., Songyang Z., Huang J.. Switch telomerase to ALT mechanism by inducing telomeric DNA damages and dysfunction of ATRX and DAXX. Sci. Rep. 2016; 6:32280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cho N.W., Dilley R.L., Lampson M.A., Greenberg R.A.. Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell. 2014; 159:108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gao M., Wei W., Li M.M., Wu Y.S., Ba Z., Jin K.X., Li M.M., Liao Y.Q., Adhikari S., Chong Z. et al. Ago2 facilitates Rad51 recruitment and DNA double-strand break repair by homologous recombination. Cell Res. 2014; 24:532–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Maloisel L., Fabre F., Gangloff S.. DNA polymerase delta is preferentially recruited during homologous recombination to promote heteroduplex DNA extension. Mol. Cell. Biol. 2008; 28:1373–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Matt S., Hofmann T.G.. The DNA damage-induced cell death response: a roadmap to kill cancer cells. Cell Mol. Life Sci. 2016; 73:2829–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Siddiqui M.S., Francois M., Fenech M.F., Leifert W.R.. Persistent gammaH2AX: a promising molecular marker of DNA damage and aging. Mutat. Res. Rev. Mutat. Res. 2015; 766:1–19. [DOI] [PubMed] [Google Scholar]
- 80. Burhans W.C., Weinberger M.. DNA replication stress, genome instability and aging. Nucleic Acids Res. 2007; 35:7545–7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Li Z., Zhang W., Chen Y., Guo W., Zhang J., Tang H., Xu Z., Zhang H., Tao Y., Wang F. et al. Impaired DNA double-strand break repair contributes to the age-associated rise of genomic instability in humans. Cell Death Differ. 2016; 23:1765–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Vermeij W.P., Dolle M.E., Reiling E., Jaarsma D., Payan-Gomez C., Bombardieri C.R., Wu H., Roks A.J., Botter S.M., van der Eerden B.C. et al. Restricted diet delays accelerated ageing and genomic stress in DNA-repair-deficient mice. Nature. 2016; 537:427–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Vermeij W.P., Hoeijmakers J.H., Pothof J.. Genome integrity in aging: human syndromes, mouse models, and therapeutic options. Annu. Rev. Pharmacol. Toxicol. 2016; 56:427–445. [DOI] [PubMed] [Google Scholar]
- 84. de Boer J., Andressoo J.O., de Wit J., Huijmans J., Beems R.B., van Steeg H., Weeda G., van der Horst G.T., van Leeuwen W., Themmen A.P. et al. Premature aging in mice deficient in DNA repair and transcription. Science. 2002; 296:1276–1279. [DOI] [PubMed] [Google Scholar]
- 85. Lombard D.B., Chua K.F., Mostoslavsky R., Franco S., Gostissa M., Alt F.W.. DNA repair, genome stability, and aging. Cell. 2005; 120:497–512. [DOI] [PubMed] [Google Scholar]
- 86. Armanios M., Alder J.K., Parry E.M., Karim B., Strong M.A., Greider C.W.. Short telomeres are sufficient to cause the degenerative defects associated with aging. Am. J. Hum. Genet. 2009; 85:823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. McEachern M.J., Krauskopf A., Blackburn E.H.. Telomeres and their control. Annu. Rev. Genet. 2000; 34:331–358. [DOI] [PubMed] [Google Scholar]
- 88. Feldser D.M., Hackett J.A., Greider C.W.. Telomere dysfunction and the initiation of genome instability. Nat. Rev. Cancer. 2003; 3:623–627. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.