


Abstract
The integration of viral sequences into the host genome is an important driver of tumorigenesis in many viral mediated cancers, notably cervical cancer and hepatocellular carcinoma. We present ViFi, a computational method that combines phylogenetic methods with reference-based read mapping to detect viral integrations. In contrast with read-based reference mapping approaches, ViFi is faster, and shows high precision and sensitivity on both simulated and biological data, even when the integrated virus is a novel strain or highly mutated. We applied ViFi to matched genomic and mRNA data from 68 cervical cancer samples from TCGA and found high concordance between the two. Surprisingly, viral integration resulted in a dramatic transcriptional upregulation in all proximal elements, including LINEs and LTRs that are not normally transcribed. This upregulation is highly correlated with the presence of a viral gene fused with a downstream human element. Moreover, genomic rearrangements suggest the formation of apparent circular extrachromosomal (ecDNA) human-viral structures. Our results suggest the presence of apparent small circular fusion viral/human ecDNA, which correlates with indiscriminate and unregulated expression of proximal genomic elements, potentially contributing to the pathogenesis of HPV-associated cervical cancers. ViFi is available at https://github.com/namphuon/ViFi.
INTRODUCTION
Human tumor associated viruses are a major contributor to the global burden of cancer. Human papillomavirus (HPV) is detected in virtually all cervical cancers and nearly half of all infection-attributed cancers in women. Hepatitis B virus (HBV) and Hepatitis C virus (HCV) infection occur in 74% of all liver cancer cases worldwide (1).
Currently, the molecular mechanisms of viral carcinogenesis are incompletely understood. Human tumor associated viruses encode viral oncoproteins, such as HPV E6 and E7 (2,3), and HBx in HBV (4), that contribute to tumor formation by dysregulating the activity of cell cycle proteins in host cells (5–7). Human tumor associated viruses may also promote tumor formation via integration into the host genome. Although viral integration is seemingly random, a chance integration into a key genomic locus could provide a selective advantage for host cells if the virus integrates near a key growth controlling gene, effectively driving constitutive expression of a proliferative transcriptional program. Consistent with this model, in some viral-associated cancers, tumor cells from the same sample share a unique viral integration site near TERT, MYC or MLL4, suggesting that viral integration is an early and important driver of carcinogenesis (6). However, the majority of HPV-positive and HBV-positive tumors do not contain recurrent integration sites near known growth control genes. Thus, the impact of seemingly random viral integration on the development of viral-associated cancers is not well understood.
Next Generation Sequencing (NGS), including whole genome sequencing (WGS) and RNA-seq, permits detection of viral integration sites in human tumor tissue (8,9). Analytic pipelines have been developed for analysis of paired-end Illumina NGS data (VirusSeq (10), VirusFinder (11), ViralFusionSeq (12) in 2013; VERSE (13), Virus-Clip (14) and Vy-PER (15) in 2015). These pipelines vary in the methodologies used to infer viral integration from sequence data, but the overarching theme is similar: identify single end or paired-end reads that map to both the human and viral reference genome. However, bioinformatic inference of viral integration sites remains a challenge because shared repeat regions between human and viral genomes (15) are common, resulting in frequent false positives.
Phylogenetic methods could provide a powerful complementary strategy for more accurate and sensitive detection of viral integration sites in human cancer by using evolutionary relationships between known viral strains to identify novel or mutated integrated viral strains (16), thus yielding new insights into how random viral integration contributes to tumorigenesis. One commonly used evolutionary model for sequence identification is a profile Hidden Markov Model (HMM (17)). A profile HMM is statistical model for representing a multiple sequence alignment, and has been shown to outperform reference-based read mapping for the assignment of novel sequences to protein families (18). More recently, collections of HMMs known as ensemble of HMMs (eHMMs) have been shown to result in more accurate classification and identification of sequences compared to the use of a single HMM (19,20).
Here, we present Viral Integration and Fusion Identification (ViFi), a new tool for detecting viral integrations from WGS data and human-virus fusion mRNA from RNA-seq data (Figure 1). Unlike other viral integration detection pipelines that use reference-based alignment mapping for identifying viral reads, ViFi uses a combination of reference-based alignment mapping and eHMMs to represent the viral families of interest to identify viral reads. Previous methods using eHMMs modeled protein families or gene families and could only identify reads belonging to those protein or gene families (19,20); ViFi improves upon these previous techniques by constructing an eHMM on entire viral genomes, allowing the identification of viral reads from any region of the virus family of interest. In addition, ViFi incorporates mappability scores to reduce false positive detections. The end result is a tool which accurately detects viral integrations with high precision and recall, even when the viruses are highly mutated or are not found in the reference virus genomes.
Figure 1.

Overview of integration detection process. Integration detection is split into two phases. In the (A) pre-processing step, a BWA index is created from the human reference genome and input viral genomes (Hg19+viral). In addition, a multiple sequence alignment is estimated from the viral genomes, and a maximum likelihood tree is estimated from the alignment. The alignment is decomposed into an ensemble of profile Hidden Markov models. In the (B) viral detection step, the paired-end reads are mapped against the Hg19+viral index. Candidate paired-end reads are selected if, (i) one end of the read maps to the human genome and the other end maps to a viral genome, or (ii) one end of the read maps to the human genome and the other end scores high against the HMM ensemble. All other reads are discarded. The integration point is then inferred from the set of candidate reads.
We compared ViFi against competing tools, VERSE, Virus-Clip and ViralFusionSeq, on both simulated and biological datasets with experimentally verified integrations (Table 1). These datasets include simulated NGS of chromosome 1 containing integrated HPV (HPV-SIM), WGS from hepatocellular carcinoma (HCC) samples taken from patients with HBV integration (21) (HCC-WGS), RNA-seq datasets from HCC cell lines infected with HBV (22) (HCC-RNA). We also compared fusion mRNA sequences found by ViFi to results from other studies (23,24) on tumor cervical samples taken from the The Cancer Genome Atlas (TCGA-CESC; https://cancergenome.nih.gov/). In order to make the comparisons fair, all methods use the same set of reference genomes for each experiment (Materials and Methods).
Table 1. Overview of datasets. We provide an overview of the datasets used throughout this study.
Finally, we performed ViFi-based comprehensive analysis of matched paired WGS and RNA sequencing data from 68 cervical cancer samples profiled by The Cancer Genome Atlas (TCGA), which reveals unique genomic integration structures, including potentially circular amplicons containing fused human and viral DNA. A previous analysis of the cervical cancer samples from the TCGA revealed increased gene expression of genomic loci affected by integration (24). Our study refines this result and reveals that integration resulting in fusion viral/human sequences under the control of a viral regulatory element resulting in high-level indiscriminate proximal transcription of all nearby elements, including LINE/LTRs. These results demonstrate the ability of ViFi to provide unique structural information in viral associated tumors and to suggest new biological insights.
MATERIALS AND METHODS
ViFi
We present ViFi, a new computational method for the detection of integrated viruses from NGS data (Figure 1). Unlike other integration detection methods that use reference-based alignment mapping for identifying viral reads, ViFi uses a combination of reference-based alignment mapping and an ensemble of profile HMMs (20) (eHMM) to identify viral reads, and uses the mappability scores of the reads to reduce false positives. We outline our method below and provide a full description of the software, including the command line arguments and version numbers of external software used within ViFi, in Supplemental Section S8.
Pre-processing
ViFi begins with a pre-processing step (see Figure 1A) to build the reference database used to identify human and viral reads. The first step is to combine the human reference genome (Hg19; February 2009 release date) with the reference viral genomes of the viral family of interest into a single FASTA file and run BWA (25) to create a single BWA index (referred to as Hg19+viral index) on the combined set of human and viral genomes. The Hg19+viral index is used to rapidly identify reads as either human or viral by mapping the reads to the index.
In addition, ViFi also models the viral family of interest by using a collection of profile HMMs built from the viral reference genomes as follows. First, the FASTA file containing only the viral reference genomes are aligned into a multiple sequence alignment using PASTA (26), and a maximum likelihood tree is estimated from the multiple sequence alignment using RAxML (27). The input alignment and tree are then used to build the eHMM using Algorithm 1 (Figure 2 for graphical overview). Briefly, an HMM is computed from the alignment using HMMER’s hmmbuild (17) and is added as the first HMM in the set of HMMs. Next, if the input tree contains more than 10 leaves, the centroid edge (i.e. the edge that best separates the tree into two subtrees with roughly equal number of leaves) is removed to create two approximately equally-sized subtrees. The process then recurses on each of the subtrees and alignments induced by the leaf set of the subtrees, adding the profile HMMs computed on the induced alignments to the set of HMMs. This process repeats recursively on each subtree until there are at most 10 sequences in the subtree. This results in a collection of nested hierarchical profile HMMs which we call the ensemble of HMMs. This pre-processing step of building the ensemble of HMMs only needs to be run once for each viral family of interest.
Figure 2.

Algorithm for generating the ensemble of HMMs. The input is an initial multiple sequence alignment and a maximum likelihood tree that has been estimated from the multiple sequence alignment. The algorithm begins by adding the HMM built on the multiple sequence alignment to the ensemble. If the multiple sequence alignment has >10 sequences, the maximum likelihood tree is decomposed into two subtrees by deleting the centroid edge (i.e. the edge that produces a maximally balanced split of the sequence set into two sets). The subtrees are used to generate induced alignments. HMMs are built for each induced alignment and added to the ensemble. The process iterates on those subtrees that meet the criterion for decomposition (subset size >10).
Algorithm 1. Building eHMM from a multiple sequence alignment and maximum likelihood tree. The functions hmm build takes an alignment as input and returns a HMMER profile HMM computed the alignment, Number Of Leaves takes a tree as input and returns the number of leaves in the tree, bisectTree takes as input a tree and partitions the tree into two roughly equally sized subtrees by removing the centroid edge, and induced Alignment takes an alignment and tree as input and returns the induced alignment that contains only the sequences that are also in the tree.

Identification of candidate reads
One of the key steps in detecting integrations is to identify paired-end reads (called ‘candidate reads’) that map to both the human reference genome and to a viral reference genome. ViFi extends upon this approach by also attempting to identify viral reads that don’t map to any known viral reference genomes but do match to an evolutionary model representing the viral family of interest. We outline this approach below (Figure 1B).
ViFi begins by mapping all paired-end reads against the Hg19+viral index using BWA-mem. The paired-end reads are separated into four different groups: (i) reads in which both paired-end reads mapped Hg19 or both to the viral genomes, (ii) paired-end reads in which one end mapped to Hg19 and the other to a viral genome, (iii) paired-end reads in which one end mapped to Hg19 and the other is unmapped and (iv) all other reads. Typically, most existing viral integration detection tools focus on the set of reads in group (ii) and discard reads found in group iii). However, a read that is viral in origin might be unmapped because it is too evolutionarily divergent from the set of known viral genomes. ViFi attempts to rescue paired-end reads in this group by scoring the unmapped reads against the ensemble of HMMs created in the pre-processing step.
ViFi takes the set of paired-end reads in which one end maps to the human reference and the other is unmapped and creates a FASTA file containing the unmapped reads. ViFi then scores the FASTA file against each profile HMM in the ensemble of HMMs using HMMER’s nhmmer command. The scores represent how well the unmapped reads match to a model of the viral family of interest. If a read has a sufficient score to at least one of the profile HMMs in the eHMM (E-value below a threshold; default is 0.01), then the read is marked as a viral read and the paired-end read is also put into the candidate set. This step allows the detection of novel or evolutionarily divergent viral sequences belonging to the same family. Thus, ViFi’s set of candidate reads not only include paired-end reads that map to both human and viral genomes, but also include paired-end reads in which one end maps to the human genome and the other paired-end has sufficient score to profile HMMs of the viral family.
Identification of integration point
Once the candidate reads have been collected, the final step is to use the candidate reads to infer integration locations in the genome. The idea behind this procedure is to identify clusters of candidate reads that are sufficiently close together such that they might haven been generated from the same genomic integration, and then infer the possible range for the integration from the cluster of reads.
The first step is to remove poorly mapped reads that might result in false positive detection of integration sites. Read pairs with poor mappability scores, defined as an average Duke Uniqueness Score (28) (generated using sliding windows of 35-mers instead of the default window size of 20-mer) less than 0.33 or MAPQ score <10 are removed. These removed reads represent reads that might map to multiple locations. Next, the remaining read pairs grouped into clusters in order to identify potential integration points. A graph is created in which the read-pairs are vertices in the graph, and an edge is drawn between a pair of paired-end reads if their mapped human coordinates are sufficiently close (default threshold is within 300 bp of each other). The connected components of the graph define the read clusters, with each cluster representing a group of reads in which each read is at least within 300 bp of another read. Clusters with fewer than three reads are removed. Finally, we attempt to identify the location of the integration point from the clusters.
We report three different integration location ranges, going from most general to most specific. The first integration range, called the ‘relaxed’ range, is the most inclusive and is the range of all mapped positions in the read cluster (i.e. the most 5’ and 3’ positions in the set of reads). The second range, called the ‘stringent’ range, attempts to narrow the location of the integration using the mapped strand information. For all human-mapped reads in a cluster, we group the reads depending on whether or not they map to the forward strand (‘forward’ group) or the reverse strand (‘reverse’ group) of the human reference genome. For each group, we identity the coordinates of the most 3’ end position in both sets, and define the genomic integration as the range of these two numbers. For example, if the forward group reports that position chr19:30,303,492 is the most 3’ mapped position in the group, and the reverse group reports that position chr19:30,303,498 is the most 3’ mapped position in the group, the integration range would be reported as chr19:30,303,492–30,303,498.
The third range, called the ‘exact’ range, attempts to identity the exact integration location and can only be reported if there are split reads present in the read cluster. A split read is defined as a read that has a primary alignment to a human chromosome and a secondary alignment to a viral reference genome, or a read that has a secondary alignment to a human chromosome and a primary alignment to a viral reference genome. If a split read is detected, the primary and secondary alignments are combined into a single alignment by marking positions in the read that map to only to the human reference in the primary or secondary alignments with an ‘H’, marking positions in the read that map only to the viral reference genomes in the primary or secondary alignments with an ‘V’, marking positions that map to both human and viral genomes with an ‘M’, and marking all other positions with an ‘X’. Positions marked ‘M’ represent potential micro-homologies between the human and viral ends near the integration point. If a read contains at least five Hs and five Vs flanking 0 or more Ms (i.e. least 5 bp from both the human and viral genome flanking the integration), then the read covers the integration. The breakpoint (defined as the 3’ position closest to the boundary between the human and viral portion of the read) is reported. If multiple different breakpoints are reported due to multiple different split reads existing in a cluster, then all the breakpoints are reported. By reporting all breakpoints discovered from the split reads, it allows for the discovery of multiple integrations that might have only been classified as a single integration because they occurred within 300 bp of each other.
The output of ViFi is the list of read clusters discovered, and for each read cluster, the relaxed, stringent, and exact (if split reads are present) ranges are reported, as well as the read names of the reads in the cluster.
Viral reference genomes
We generated a PV reference genome set used on analyses on cervical cancer samples and a HBV reference genome set used for analyses on hepatocellular carcinoma samples. The PV reference genome was created by downloading all available reference PV genomes (337 total PV genomes) from PapillomaVirus Episteme website (PaVE (29); https://pave.niaid.nih.gov) on 15 August 2016. The HBV reference genome set was created by downloading the set of 73 reference HBV genomes used in (30), which includes genotypes A-I and a strain of Woolley Monkey HBV. The GI number for all the reference genomes and the map of GI numbers to sequence name is provided in Supplementary Table S1.
VERSE
We ran VERSE under its default setting (see Supplementary Section S5 for full details on running VERSE). We discovered that VERSE performed unnecessary I/O operations during its pipeline and improved upon its performance by reducing I/O operations through use of pipelines (see Supplementary Section S4).
ViralFusionSeq
We ran ViralFusionSeq under its default setting (see Supplementary Section S2 for full details on running ViralFusionSeq). We discovered several bugs while running ViralFusionSeq and had to fix the errors in the code in order to run the software (Supplementary Section S1). However, even with these corrections, we were unable to run the code to completion on any of the simulated datasets. In addition, even on a simple test case of 10 integrations on chr1 with 5x coverage, ViralFusionSeq required 93 hours on a dedicated node with 24 processors before failing with an error message. During this run, ViralFusionSeq produced more than 134GB of temporary files on an input dataset of <1GB. Due to the difficulty in running ViralFusionSeq, as well as the computational requirements of the software (Supplementary Section S1), we excluded it from our analyses.
Virus-Clip
We ran Virus-Clip under its default setting (see Supplementary Section S7 for full details on running Virus-Clip). We made minor modifications to the code in order to make the program more efficient and to take full advantage of the multiple processors on the node (see Supplementary Section S6).
Datasets
We use both simulated and biological datasets in our studies. We describe the datasets below.
HPV simulated NGS datasets (HPV-Sim)
We generated 16 model conditions with PV integrations (see Supplementary Section S9 for full description of simulation procedure). In order to examine the impact of viral sequence divergence on integration detection, we simulated different strains of HPV by evolving HPV16 down a phylogenetic tree with differing branch lengths under the Generalized Time Reversible (GTR) substitution model (31). We grouped the simulations into three categories, depending on the integrating virus strain’s similarity to the reference HPV16 strain: easy (99% similarity), medium (95% similarity), and hard (90% similarity). In addition, we generated one additional dataset with Brown Howler PV (AgPV1), a papillomavirus genome not included in the set of viral reference genomes to simulate detection of a novel HPV virus. AgPV1 is 44% similar to HPV16 and is 65% similar to the most closely related sequence in the set of reference genomes. For each simulated virus, we generated simulated viral integrations into the human chromosome 1, with the integration locations selected uniformly at random throughout the chromosome. Paired-end Illumina reads of 100 bp were generated from the simulated chr1 chromosome using ART Illumina Simulator (32). In order to study the impact of sequencing coverage and number of integrations on integration detection accuracy and running time, we generated a first set of model conditions in which we fix the coverage to be 25× and vary the number of integrations from 10, 25, 50 and 100, and a second set of model conditions in which we fix the number of integrations to be 10 and vary the coverage from 5×, 10× and 25×. In total, 96 datasets were generated for the simulation study.
HCC Sung 2012 WGS dataset (HCC-WGS)
Eighty eight HCC samples (81 HBV-positive and 7 HBV-negative; both tumor and adjacent tissue) were sequenced by Sung et al. (21) and deposited into the European Genome-phenome Archive (EGA) under the accession ERP001196. The authors identified recurrent genomic integrations (4 or more samples) in known oncogenes/oncogenic regions (FN1, TERT, MLL4, CCNE1, ROCK1, SENP5). A total of 399 genomic integrations were identified across the 88 samples. To confirm recurrent genomic integrations, the authors randomly selected 32 genomic integrations in six affected genes were able to successfully validate 22 of of the 32 integrations (72%) via PCR. Our study included the same 22 integrations as a comparison. As five samples contained pairs of integrations that were significantly closer than the insert size of the paired reads (less 20 bp from each other), we collapsed these integrations into a single integration, resulting in a final list of 17 verified integrations. The sample list used is provided in Supplementary Table S6.
HCC Lau 2014 cell line RNA-seq dataset (HCC-RNAseq)
Whole transcriptomes from six HCC cell lines were analyzed using RNA-seq by Lau et al. (22). 11 chimeric HBV-human fusion transcripts were detected in three of the cell lines and validated using Sanger sequencing. Our study included the original six cell line RNA-seq data and is available from the Sequence Read Archive under accession number SRP023539.
TCGA Cervical cancer datasets (TCGA-CESC)
We selected 68 patient cervical tumor samples in the TCGA database with matched WGS and RNA-seq tumor sequencing data for analyses. Of the 68 samples, 28 samples overlapped with the Tang et al. study (23) which examined the landscape of mRNA viral fusion events across the TCGA dataset, and 65 samples overlapped with The Cancer Genome Atlas Research Network study (24) which examined the genomic and molecular characteristics of cervical cancer. Our study includes a comparison of ViFi to results on the Tang and The Cancer Genome Atlas Research Network study study, as well as analyses on all 68 samples (Supplementary Table S5).
Scoring integration detection accuracy
As the exact breakpoint cannot be determined unless a split-end read overlaps the genomic integration location, we considered an integration is correctly detected if a method reports a detected integration within 300 bp (typical insert size of a paired-end read) of the true integration location. When a method reports a possible range for a genomic integration, we use the mean position as the estimated integration point and examine whether or not this mean position is within 300 bp of the true integration location to determine whether the range correctly includes the true integration. In order to make the comparisons fair across all methods, if a method reports multiple integrations within 300 bp of another, they are collapsed into a single cluster covering the integration point. This change mainly impacts Virus-Clip and reduces the number false positive integrations.
Annotation of genomic and mRNA transcripts proximal to integration sites
We annotated the genomic regions and transcripts that were proximal to integration sites using annotations from RefSeq genes (33) or RepeatMasker (34) annotations. For each integration, we took all positions within a 10 kb interval of the integration that were covered by at least three or more reads (WGS reads for genomic annotations, mRNA reads for transcript annotations), and then clustered the positions into segments. We merged any segments that were within 5 bp or any other segment into a single segment. We then reported the total number of unique annotations that intersected any of the segments within the interval. In addition, we also generated a distribution of expected annotations by selecting 1000 random intervals of the same genomic length from the sample containing the integration and recorded the total number of annotations that intersected the segments from the random intervals. We report the total annotations from the observed integrations compared to the distribution of total annotations from randomly selected intervals. To detect whether the total annotations from observed integrations was statistically significant, we modeled the distribution of annotations from the random intervals as a normal distribution and used it to computed a Z-score (one-tailed) for the total annotations from the observed integrations.
Quantifying transcription expression
In order to compare transcript expression across regions, we followed transcription analysis protocol outlined in (35). The RNA-seq datasets were aligned to the human reference using HISAT-2 (36). Next, StringTie (37) was used to perform annotation-free assembly of the transcripts for each sample. Afterward, all the gene and gene structures found in the individual samples were merged together using StringTie to create a consistent set of transcripts across all samples. Finally, the abundances of each transcript (reported as Fragments Per Kilobase of transcript per Million mapped reads (FPKM)) in each sample were computed using the merged transcript set.
Unlike other RNA-seq differential expression analyses where the number of conditions are typically more evenly distributed (i.e. half control and half treatment), our samples are split into two uneven groups: one sample with the integration at a particular loci, and all remaining samples without the integration at the same loci. Thus, we used a different process for performing differential expression analyses. In order to determine the impact of a specific integration on expression in integration region i, we compare the total FPKM of integration region of the sample containing the integration (FPKMi) against the distribution of the total FPKM of the same interval for all other samples not containing the integration (FPKMo = {FPKMj|j = 1..N, j ≠ i}), where N is the total number of samples. We compute the fold change in expression for integration region i as 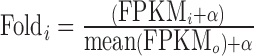 where α is a pseudocount value that we set to 0.01. We report the mean fold change as the geometric mean of the fold change in expression.
where α is a pseudocount value that we set to 0.01. We report the mean fold change as the geometric mean of the fold change in expression.
To determine whether or not the transcriptional activity of an integration region was significant within a sample, we compared the transcriptional activity of the integration region to the activity of all other transcripts within the same sample using the following protocol. For each sample, we first filtered out any transcript with low expression (defined as having an FPKM less than 0.01). Next, we computed the FPKMUQ value as the FPKM value of the 75th percentile of the filtered transcripts as a baseline of comparison (Supplementary Table S7). In other words, a transcript with expression exceeding FPKMUQ would be among the top 25% most expressed transcripts. We report both the number of times the FPKM of the integration region is greater than the FPKMUQ.
Correcting for copy number variation
In order to examine whether the increase in transcription expression was primarily caused by increased genomic amplification typically observed near viral integration regions, we attempted to correct for copy number variation as follows. For a particular integration region i for sample s, we compute the expression per copy number as 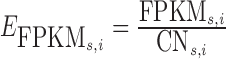 , where CNs, i is the average copy number of the region i for sample s. We define the average FPKM per copy number of a region without an integration as
, where CNs, i is the average copy number of the region i for sample s. We define the average FPKM per copy number of a region without an integration as 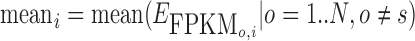 . To compute the fold-change from expected transcription for an integration region i, we compute
. To compute the fold-change from expected transcription for an integration region i, we compute 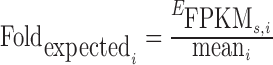 , which can be summarized as the FPKM of the region of the sample containing the integration divided by the average FPKM per copy number of the same region for all other samples not containing the integration multiplied by the copy number of the region of the sample containing the integration. We obtain the copy numbers of the region directly from the Masked Copy Number Segment files provided by the TCGA database.
, which can be summarized as the FPKM of the region of the sample containing the integration divided by the average FPKM per copy number of the same region for all other samples not containing the integration multiplied by the copy number of the region of the sample containing the integration. We obtain the copy numbers of the region directly from the Masked Copy Number Segment files provided by the TCGA database.
Statistical tests for differential expression
To compute a P-value for the statistical significance of an integration resulting in increased expression, we performed the following steps. For a particular integration region i in a sample si, we took the distribution FPKMo (i.e., normalized expression of the same region in of all other samples not containing that integration) and attempted to fit the distribution to a Normal, Log-Normal, Exponential, Gamma, and Weibull distribution (38) using the R package fitdistrplus (39). The model and parameters with the best fit (measured as lowest AIC score) were selected. Let M(p) be the model with the best fit and is parameterized by p. Let FPKMi be the observed normalized expression level in the integration region of the sample si. We then performed single-tailed parametric tests for the p-value of the FPKM from the region of the sample containing the integration to the best fit parameterized model, i.e. P-val=P(x ≥ FPKMi|M(p)). We corrected for multiple hypothesis testing by adjusting the P-value for significance using false discovery rate (40) (FDR) correction.
We performed a two-tailed paired Wilcoxon Signed-Rank Test to detect whether integration results in a statistically significant change in expression across all genomic segments. For each integration region i, let FPKMi be the normalized expression of the sample containing the integration region and mean(FPKMo) be the mean normalized expression of the same integration region in all other samples not containing an integration within that region, then the paired difference, Δi, for integration region i is defined as Δi = FPKMi − mean(FPKMo). We then found the p-value the two-tailed paired Wilcoxon Signed-Rank on the paired differences, computed using R’s stats library (41).
Classification of integration regions into simple, complex, fusionless
We define a simple integration as a single integration event with no other integrations within its integration region and shows concordant chimeric paired-end reads, allowing the identification of regions upstream and downstream of the viral gene. We define a complex integration as an integration that contains two or more integrations within its integration region and reveals multiple fusion mRNA sequences with discordant chimeric paired reads. Finally, regions without chimeric RNA are defined as ‘fusionless’. Supplementary Figure S8 outlines the classification of the integration regions into simple, complex, and fusionless integration regions.
AmpliconArchitect
In addition to running ViFi on each TCGA-CESC sample, we also ran AmpliconArchitect (AA; (42)), a directed assembly method for reconstructing complex genomic structures from WGS data. Briefly, AA uses discordant read-pair alignments and coverage information to connect genomic regions with high amplification. It then further breaks these regions into segments based upon coverage shift changes in the genomic regions. AA then builds a breakpoint graph by connecting segments using discordant read-pairs. From the breakpoint graph, AA reports possible cycles and paths in the graph that can be used to identify potential circular structures.
We leverage AA by using it to identify potential apparent extrachromosomal DNA (ecDNA). For each sample, we examine whether or not AA reported a cyclic human-viral structure with at least 2× amplification. We then take all discordant paired-end reads reported by ViFi or AA and filter out reads that might have multiple non-unique BLAT mapping to hg19 as follows. We define a BLAT score for a read aligned to a location on hg19 as the number of non-repeat base matches minus the number of mismatches and insertions into the query and template sequences. Thus, if the read aligns perfectly to a non-repeat location, its score would the length of the read. Next, we examine if the read has any other hits in which the BLAT score is within 90% of its best score; if so, then the read is not considered uniquely alignable and is discarded. Finally, we take the remaining reads and map it to cyclic structure to show the discordant and split reads that support the structure.
RESULTS
Comparison on simulated datasets
We simulated NGS datasets (Materials and Methods) with genomic integration of viral genomes, exploring the impact of viral strain diversity, coverage, and the number of integrations on detection accuracy and computational running time. The integrated viruses included three HPV16 strains simulated with either low, medium, or high rates of substitution mutation, and one papillomavirus (AgPV1) that was not included the reference PV database to simulate integration of a novel virus. Datasets containing the low, medium, or high mutation rates are referred to as ‘easy’, ‘medium’, ‘hard’, and datasets containing the AgPV1 virus are referred to as ‘novel’.
To better understand how the methods scale in accuracy and computational complexity with respect to coverage and the number of integrations, we generated datasets in which we fixed the coverage to be 25× and varied the number of integrations from 10, 25, 50 and 100, as well as datasets in which we fixed the number of integrations to be 10 and varied the coverage from 5×, 10× and 25×. For each model condition with a simulated integrated virus (parameterized by the mutation rate of the integrated virus, the coverage, and the number of integrations), we generated five replicate WGS datasets for a total of 16 model conditions and 96 datasets (∼320GB of sequencing data).
We ran ViFi, VERSE, Virus-Clip and ViralFusionSeq on the simulated HPV-SIM datasets (Figure 3A–C). All methods were run on a dedicated compute node with 24 cores and given 48 wall clock hours (1152 total core hours) to complete the analysis. Only ViFi and Virus-Clip were able to complete on all datasets. VERSE terminated prematurely on five of the medium model conditions and 18 of the hard model conditions, and failed to detect integrations four easy conditions, eight hard conditions, and all six novel datasets. ViralFusionSeq failed to complete any of the analyses within the allotted time (see Supplementary Section S1), and is excluded from the remainder of this study.
Figure 3.

ViFi performance on simulated datasets. Comparison of ViFi, Virus-Finder, and VERSE on simulated datasets where (A) the coverage is fixed to be 25x coverage and the number of integrations ranges from 10, 25, 50 and 100, and (B) the number of integrations is fixed to be 10 integrations and the coverage ranges from 5×, 10× and 25×. Each simulation has four model conditions. The first three model conditions (easy, medium, and hard) vary the percent similarity of simulated HPV16 genomes to the reference HPV16 genome, with five replicates per simulation. The last model condition uses Alouatta guariba papillomavirus 1 (AgPV1), a PV genome not included in the set of viral genomes to simulate integration of a novel HPV virus. AgPV1 is 44% similar to HPV16. Random noise (drawn from a uniform distribution between -0.01 and 0.01) was added to each point due to points often directly overlapping each other. VERSE is unable to detect integrations or terminates earlier on two easy cases, one medium case, 22 hard cases, and on all the AgPV1 datasets, and we exclude these results from the figure. (C) The mean wall clock running time (in hours) as a function of the number of integrations (top) and as a function of the coverage (bottom). All methods were run on a machine with 24 cores for a maximum of 48 wall clock hours (1152 total core hours). Only runs that report integrations were included.
On the easy model conditions at 25× coverage, all methods have high recall in detecting the integrated virus (Figure 3A). Both ViFi and VERSE are unaffected by the number of integrations and maintain high recall and precision (typically 90% or better) regardless of the number of integrated viruses. Virus-Clip, on the other hand, has lower precision (20–40%) and showed an odd behavior in which its precision decreased as the number of integrated viruses decreased. A closer inspection revealed several regions of chr1 result in false positive integration detection across independent runs of Virus-Clip. As the model conditions get more difficult (i.e. the integrated virus gets more divergent from those in the reference database), we begin to see more separation between ViFi and VERSE. ViFi continues to maintain high precision and recall, however VERSE’s recall drops as the viruses gets more evolutionarily divergence. Interestingly, Virus-Clip maintains nearly the same precision and recall under these difficult conditions. Performance differences between the methods are much more clear on the novel dataset with ViFi being the only method that can still accurately detect the integrated virus, while VERSE and Virus-Clip unable to detect any true positive integrations. These results demonstrate that incorporation of phylogenetic methods with read-based methods greatly enhanced the capability of ViFi to sensitively and precisely detect viral integrations that could not be detected by VERSE or Virus-Clip.
When we fix the number of integrations to be 10 and vary the coverage, we begin to see more performance differences between the methods (Figure 3B). In particular, all methods have lower recall as the coverage drops, though Virus-Clip is less impacted than ViFi or VERSE. We suspect this is because Virus-Clip requires only one split read to call an integration, where as both ViFi and VERSE require multiple supporting reads to call an integration. This design decision results in a tradeoff of lower recall on low coverage data, but allows the maintenance of high precision across all model conditions (ViFi mean precision of 99.8% and VERSE mean precision of 98.9%).
A comparison of the running times reveal that Virus-Clip is the most efficient method, and on average, was three times faster than ViFi (Figure 3C). However, it should be noted that we are using a version of Virus-Clip that we have optimized for this study (see Supplementary Section S6). ViFi is the next fastest method, and VERSE required the most running time. All methods had a linear increase in running time as the coverage increased. However, when the coverage is fixed and the number of integrations is increased, only VERSE was significantly impacted. As some cervical cancer samples can have up to 100 to 600 HPV integration events (43), it is vital for viral integration detection methods to run efficiently on samples with many integrations.
In summary, these results show that as long as there is sufficient coverage (10× or greater), ViFi had both high precision and recall (mean of 99.7% and 92.8% respectively) in detecting integrated viruses, even if the virus is highly mutated or a novel strain, and on low coverage data, ViFi maintains high precision. VERSE has high precision and recall under easy conditions in which the integrated virus is either in the reference database or similar enough to an existing virus in the reference database. However, VERSE’s recall drops considerably if the integrated virus is sufficiently different from those in the reference database. In addition VERSE’s computational requirements grows considerably as the number of integrations increase. Virus-Clip has high recall on all but the most difficult datasets at the cost of having lower precision. Due to Virus-Clip resulting in a large number of positives, we exclude it from the remainder of our analyses on the biological datasets.
Comparison on biological datasets with experimentally verified genomic integrations
Next, we compared the ViFi and VERSE on HCC-WGS and HCC-RNAseq datasets with experimentally verified HBV integrations. Of the 17 experimentally verified integration points in the HCC-WGS dataset, ViFi was able to detect 13 of the integration points, and VERSE was able to detect 12. In the HCC-RNAseq datasets, both ViFi and VERSE recovered 10 out of 11 verified fusion mRNA points (Supplementary Figure S1). Closer inspection of the integration points and fusion events missed by ViFi revealed that the number of reads supporting the integration was very low (less than three) or the integrations occurred near low complexity regions, making it difficult to recover the correction integration location.
Comparison of fusion mRNA detection on TCGA-CESC dataset
A 2013 study of RNA-seq datasets from the TCGA database by Tang et al. (23) explored the landscape of human-viral fusion gene expression. The authors observed that fusion mRNA transcripts are often found in HPV-related and HBV-related cancers. The authors verified the fusion transcripts found in a small subset of the datasets (8 out of the 178 datasets) by showing that the fusion transcripts were concordant with genomic integrations detected using matched WGS data. We expand upon this study by comparing the concordance of fusion mRNA transcripts and genomic integration events on 28 TCGA cervical cancer samples (TCGA-CESC) with matched WGS and RNA-seq data.
For each sample, we ran ViFi on the WGS data to detect genomic viral integrations. For each viral integration reported by ViFi, we report whether ViFi, VERSE, and the Tang et al. study also found one or more fusion mRNA events within a 100 kb interval around that integration point. A Venn diagram showing the overlap of fusion mRNA events found by the different methods show that for any case in which VERSE or Tang et al. found a fusion mRNA event, ViFi also detected the event (Figure 4A). In addition, ViFi detected four fusion mRNA events that were also supported by the WGS data that neither VERSE or Tang et al. identified, of which two of the fusion events were highly supported with uniquely mapped human reads (see Supplementary Figure S11). Thus, not only did ViFi detect more mRNA fusion transcripts than the Tang study or VERSE, these transcripts are highly likely to represent true fusion transcripts as the fusions are in concordance with genomic integration events.
Figure 4.

Comparison of ViFi on fusion event detection. (A) Venn diagram of the overlap of the WGS integration points with a matching mRNA event within 100 kb reported by ViFi, VERSE, and the Tang et al. (2013) study on the TCGA-CESC samples with both RNA-seq and WGS sequencing matched pair data. (B) Comparison of the fusion events detected by ViFi and The Cancer Genome Atlas Research Network 2017 study. Fusion are considered to have WGS support if ViFi detected a genomic integration within a 100 kb region of the fusion event.
A more recent study in 2017 by The Cancer Genome Atlas Research Network explored the genomic and molecular characteristics of cervical cancer on larger subset of the TCGA database (24). The group identified 220 unique fusion events using RNA-seq data from 228 cancer samples. We took the fusion events from the TCGA Research Network study that were on the same set of samples used in our study and collapsed any fusion events that fell within the same 100kb integration region into a single event, resulting in 78 total unique clusters, and perform the same step on the fusion events detected by ViFi, collapsing 212 fusion events into 125 clusters. A comparison of the clusters revealed that both methods had an overlap of 58 clusters, 67 were unique to ViFi, and 20 were unique to the TCGA Research Network study (Figure 4B). In order to explain this discrepancy, we compared the genomic integrations detected using ViFi from the matched WGS data with the fusion clusters reported by both methods. First, the fusion events that were detected by both ViFi and the TCGA Research Network study were strongly supported from the genomic data, with 52 out of 58 fusion clusters being proximal to a genomic integration. More importantly, 21 of the 67 fusion clusters detected uniquely by ViFi were proximal to a genomic integration, but no genomic integrations were detected proximal to any of the 20 unique fusion clusters reported by TCGA Research Network study.
Functional role of HPV integration in cervical cancer
Having demonstrated the ViFi can be applied to WGS and RNA-seq data to accurately detect viral integrations, we set out to gain deeper insight into how HPV integration may potentially promote cervical cancer by altering genome structure, gene transcription and possibly even activation of normally silent regions. We used ViFi to analyze matched WGS and RNA-seq data from 68 cervical cancer samples that were included in the TCGA analysis.
We detected a total of 226 HPV genomic integrations spread among 51 of the 68 cervical cancer samples (Supplementary Table S3). We also detected 376 fusion (viral-human) mRNA junctions, 87% of which were within 100kbp of a genomic integration (Figure 5A); 73% were within 50 kb, and 54% within 10 kb. A little over half of the genomic integrations 119 (52%) had at least one proximal fusion mRNA sequence within 10kb of the integration, suggesting that some of these fusion transcripts may be mediated by alternative splicing (44,45). These results confirm the robustness of automated ViFi analysis as well as the strong concordance between the genomic and RNA-seq data.
Figure 5.

Characterization of genomic integration sites and fusion mRNA. (A) Density plot of the distance of fusion mRNA junction to the nearest WGS integration breakpoint. (B) Number of annotated types covered by WGS or RNA-seq reads across all integration regions. The points give the number of specific functional annotations (e.g. LINE) across all 181 integration regions in the TCGA-CESC data set that are partially covered by at least three reads. Blue represents results from WGS data, and red represents results from RNA-seq data. The violin plot show the distribution of the total number of specific annotations across 1000 replicates that are partially covered by at least three reads, where each replicate is a collection of 181 randomly chosen intervals. The P-values of the observed annotation counts (Z-test) are all statistically significant for the RNA-seq data(P-value <10−20), but for the WGS data only the SINE elements (P-value <10−8) and genes (P-value <10−7) were enriched in a statistically significant manner.
The majority of fusion mRNA junctions were within 10 kb of a genomic integration. Therefore, we defined an ‘integration region’ as the 10 kb region flanking a genomic integration point in some sample; if a sample contains more than one integration within a region (i.e. multiple integrations within 10 kb of each other), the integration region is defined as the interval that includes all integrations within 10kb and their 10kb flanking region. Using this definition, we observed that the 226 integrations form 181 integration regions. We used integration regions to compare genomic features and transcription expression differences between samples with and without integrations.
Characterization of genomic integration location
Deeper sequence analysis (see Materials and Methods for details on annotation) revealed recurrent genomic integration sites in only 6 of the 51 cervical samples containing HPV genomic integrations; two in chr13:73,955,151–74,005,092 (two samples) and four within 1MB of chr8:128,747,810–128,889,296, which contains the MYC-PVT1 locus. Both of these hotspots were previously identified in Hu et al. (43). Overall, although 67% of the integration regions contained an annotated gene, indicating a modest but statistically significant enrichment for integration near coding genes (Z-test; P-value <10−7; Figure 5B), as has been previously reported (46), we detected no statistically significant enrichment for integrations near known oncogenes. Hu et al. reported an enrichment of integrations within 25 bps of genomic instability-related elements, including short tandem repeats (STR), short interspersed nuclear element (SINE/Alu), long terminal repeat/endogenous retroviruses (LTR/ERV1), and satellite DNA (43). Interestingly, when we examined whether there was an overall enrichment of genomic instability-related classes (SINE, LINE and LTR) within the integration regions, we found significant enrichment only for SINE elements (Z-test; P-value <10−8; Figure 5B) in the integration region. As the vast majority of integrations were not recurrent nor enriched for a specific annotation, the location of HPV genomic integration is unlikely to play a significant role in cervical cancer pathogenicity.
Impact of HPV integration on transcription
To gain further insight into potential mechanisms by which genomic integration of HPV may promote tumorigenesis, we examined the impact of HPV integration on transcription. For each integration region in a sample, the normalized transcriptional activity was compared to the mean normalized transcriptional activity of same region in all other samples lacking HPV genomic integration (Materials and Methods; Supplementary Table S4). A highly significant increase in transcription (4.09 × average increase; Wilcoxon signed rank test; P-value <10−10) was detected across all genomic segments containing HPV integrations (Supplementary Figure S2). In fact, we detected transcription of genomic regions that are normally silent. 39% of the integrations had no gene within its integration region, but transcripts from 49% of those regions were still detected. To understand the function of the transcripts, we marked all transcribed integration regions (Materials and Methods) and compared against RefSeq genes (33) or RepeatMasker (34) annotations. For each integration and a class of annotations (e.g. LINE), we counted the number of unique annotated elements that were transcribed, and compared that with the number of unique annotated elements of the same class in randomly selected segments (Figure 5B). We found a twelve fold increase in the number of transcribed LINEs, SINEs, LTRs in close proximity to an integration site (Z-test; P-value <10−20), and a 2-fold increase in the number of transcribed genes proximal to an integration. Moreover, we found a five to six-fold increase in expression of LINE and LTR elements in regions proximal to integration sites (Wilcoxon signed rank test; P-value <10−7, Supplementary Figure S2).
Thus, even though there was no significant enrichment in the number of LINE and LTR elements at the genomic level, there was a statistically significant enrichment of the transcription of LINE and LTR elements in the integration regions. These results suggests that random HPV integration into the genome result in transcription of normally silent regions of the genome, potentially promoting genomic instability and pathway disruption by de-repressing LINE and LTR elements (47–53).
We also noted that the increased transcriptional activity of these normally silent regions correlated with the presence of viral/human fusion transcripts (Figure 6A and Supplementary Figure S3). Considering only the genomic regions where fusion mRNA was found, we observed a 22.25× average increase in transcription when compared against samples without genomic integration (Wilcoxon signed rank test; P-value <10−13); whereas no significant change in transcriptional activity was detected when an integration region did not contain a fusion mRNA (0.82×; Wilcoxon signed rank test; P-value <0.17), and a 4.09 × average increase over all regions. Figure 6B provides the P-values per integration region (Also see Supplementary Figure S4 and Materials and Methods). Even when we correct for the copy number amplification typically found flanking the genomic integration regions (Materials and Methods), we still observe that there is increased transcriptional activity in regions with integrations compared to regions without integrations (Supplementary Figure S5).
Figure 6.

Impact of viral integration on proximal transcription. For each integration, we compare the expression change in the 10kb genomic interval around an integration in a sample to the mean expression change for the same 10 kb genomic interval for all other samples without the integration. (A) The distribution of log2-fold change in expression of human mRNA between segments with and without integrations, separated by whether the integration produces fusion mRNA or is a fusionless integration. The dashed line represents the geometric mean value of the distribution. (B) The –log(P-value) for expression change for integrations that produce fusion mRNA and fusionless integrations (see Materials and Methods for description of P-value computation). Each point on the x-axis corresponds to a distinct genomic fusion segment sorted by increasing p-value. The red dashed line denotes the threshold beyond which the samples do not show a significant change in expression (P-value >0.05 after FDR correction).
We observed that this increased transcriptional activity was not trivial. In genomic regions where fusion mRNA transcripts were found, 83% of those regions had transcriptional activity higher than 75% of the transcripts within that the same sample, and, on average, had 10.4-fold more transcriptional activity than the transcriptional activity of the median upper quantile transcript (Supplementary Figure S6). Thus, viral integration may cause dysregulation not only by expression of silenced or non-coding regions of the genome, but also by the large volume of transcripts being produced.
Regulation of transcription
To better understand why genomic integration and fusion transcripts are correlated with increased transcription, we characterized the orientation of the fusion transcripts. As the RNA-sequencing library preparation was not strand specific, we could not determine the active strand directly. However, because HPV genes are known to be transcriptionally active in cervical cancer cells (45), we can assume that the majority of viral transcripts were transcribed in the same direction as the viral gene. 82% of fusion mRNA sequences showed the human fragment to be downstream of the viral genes. (Supplementary Figure S7). Of the remaining 18% of the sequences where the human portion was upstream of the viral gene, 87% were within 10kb of an annotated gene. These observations suggest that transcription of fusion mRNA sequences are largely driven by the upstream regulatory elements within the viral genome. In the rare cases in which the viral gene is downstream of the human portion of the fusion transcript, human regulatory elements may drive expression.
To better characterize how HPV integration might dysregulate transcription of neighboring DNA sequences, we performed a more detailed analysis of the transcriptional activity near the site of HPV integration. We categorized each HPV integration as a simple, complex, or fusionless based on the number of integrations and concordant chimeric RNA reads within its integration region. Briefly, ‘simple’ integrations correspond to regions with a single genomic integration and concordant chimeric RNA reads, allowing the identification of regions upstream and downstream of the viral gene. Regions containing genomic integration but no chimeric RNA were defined as ‘fusionless’. All other regions with genomic integrations were classified as ‘complex’. (Figure 7; see Materials and Methods, Supplementary Figure S8 for details on classification). Using this characterization, we observed 68 simple, 51 complex, and 107 fusionless integrations in our dataset, enabling us to further examine the impact of viral integration-mediated fusions on transcription.
Figure 7.

Expression of human segments upstream and downstream of the integrated viral gene. (A) Expression fold-change within an integration region and (B) percent of samples in which the position in the integration region has a higher FPKM than its FPKMUQ. The blue line represents an integrated virus, with arrow representing the direction of transcription of the viral genome, and the red line represents the human genome. An integration is denoted as ‘fusionless’ when it does not contain a mapped chimeric (viral-human mRNA); otherwise, it is denoted as ‘simple’ when it is the only integration within a 10 kb window, and at least 75% of the chimeric paired-end reads supporting a fusion mRNA event are oriented in the same direction relative to the viral gene. All other regions are denoted ‘complex’. The position is reported relative to the integration point in the human genome, with negative position being upstream of the viral gene, and positive position being downstream of the viral gene. In total, there are 68 simple integrations, 51 complex integrations, and 107 integration events with no fusion mRNA sequences. We observe a high increase downstream of simple integrations, in the entire region of complex integrations, and no increase in expression in fusionless integrations.
Simple or complex integrations demonstrated a 5- to 17-fold increase in transcription proximal to the site of integration(Wilcoxon signed rank test; P-value <10−12) local to the integration point (Figure 7A), and the increased transcription was evident up to 100 kb around the integration point (Supplementary Figure S9). In cases with simple HPV integrations, we detected a sharp increase in expression downstream of the integrated viral sequence. Finally, fusionless integrations (genomic integration but no fusion transcript) showed a slight decrease in expression.
These results show that the transcriptional activity of the integration region is significantly increased compared to the same region across samples, however, it does not address whether the transcriptional activity is significant within a sample. To answer this question, we compared the transcriptional activity of each position in the integration region with its own FPKMUQ and reported the percentage of integration regions that had transcriptional activity greater than its FPKMUQ (Figure 7B). 60–80% of simple or complex integrations had higher proximal transcriptional activity than 75% of all other transcripts within the same sample proximal to the integration region. This activity was still notable up to a 100 kb region around the integration point (Supplementary Figure S10). Taken together, our results suggest that random HPV integration causes dysregulated expression of all proximal elements possibly driven by the viral regulatory elements.
Role of apparent hybrid circular extrachromosomal DNA (ecDNA)
We sought to identify a mechanistic structural basis for these findings by further integrating the RNA and WGS data, and closely inspecting several HPV integration sites. For each sample, we ran AmpliconArchitect (AA; (42)), a tool for reconstructing complex genomic structures from WGS data (Materials and Methods). AA revealed 34 out of the 68 samples contained genomic segments from both the human and viral genomes that could be arranged into a hybrid human-viral cyclic structure, of which, 14 of these structures had a copy count greater than four (Supplementary Figure S13–S26). For example, the sample TCGA-C5-A0TN had 235 chimeric paired-end reads between chr2:195,586,245 and HPV16:2,593, 149 discordant paired-end reads between chr2:195,603,512 and chr3:126,826,267, and 229 chimeric paired-end reads between chr3:126,849,186 and HPV:6,071 (Figure 8). Analyses of the reads the span these segments strongly support the fusion of these segments, with the human portion and viral portion of the split reads mapping back perfectly to their respective reference genome (Supplementary Figure S12). The AA reconstruction for this sample revealed a path containing gene-poor segments of chromosome 2 (chr2:195,586,245-195,603,512) and chromosome 3 (chr3:126,826,267-126,849,186) in a circular configuration with the partial HPV genome. Although it is initially surprising that these structures are composed of genomic material from multiple chromosomes, the identical elevated DNA copy number of each of the fragments, suggested that they were indeed a single structural unit. Even more compellingly, the viral/human fusion transcripts and the nearly uniformly elevated transcription of the normally silent genomic regions, is consistent with a circular structure that is highly reminiscent of circular extrachromosomal DNA (ecDNA), which has recently been shown to play a critical role in accelerated evolution in cancer (42). While we cannot rule out that this structure may be a result of tandem duplication, it would require a translocational insertion of one chromosomal segment into another chromosomal segment near the integration region, followed by tandem duplication events.
Figure 8.

Proposed apparent ecDNA structure for TCGA-C5-A0TN. Proposed apparent ecDNA structure for an integration from TCGA-C5-A0TN. The joined segments are chr2:195,586,245-195,603,512, chr3:126,826,267-126,849,186, and HPV16:0-7,905. There are 235 chimeric paired-end between chr2 and HPV16, 149 discordant paired-end reads between chr2 and chr3, and 229 chimeric paired-end reads between chr3 and HPV16. The genomic coverage fold amplification of the region relative to the average genomic coverage of the entire genome is shown in blue, and the mRNA coverage of the region is shown red. The FPKM fold change for the human mRNA in this region for this sample is 7200x. LINE and LTR elements are highlighted in teal and gold. The viral genes are highlighted in light green. The viral genome is is not complete and has a deletion of the E2 region. The assembled fusion transcript from this region is shown in the figure.
DISCUSSION
Human cancer-associated viruses most commonly integrate into the genome in seemingly random locations. Shared repeat regions between human and viral genomes arising from remnants of viral elements in the human genome, compromise the ability of current sequence-mapping approaches to accurately resolve viral integration sites in human cancers, limiting our ability to derive biological insights from the vast repertoire of cancer NGS data. Here we show that ViFi, a new method that integrates phylogenetic eHMMs to better detect evolutionarily divergent viruses with sequenced-based mappability scores, facilitates rapid, accurate, efficient and specific detection of viral integration sites, providing a powerful new way to obtain biological insights from the vast assembly of human cancer genome data.
The current version of ViFi includes eHMMs on the HPV and HBV viral families. However, additional viral families can be incorporated by downloading reference genomes from the viral family of interest and using the ViFi provided scripts to automatically build the eHMMs from those reference genomes. One potential weakness of this approach, however, is that the alignment method used internally within ViFi may have difficulty in aligning genomes with large amounts of genome rearrangement. To mitigate this problem, future versions of ViFi will also include eHMMs built from gene families, and thus would be unaffected by genome rearrangement.
In addition to providing a new computational resource tool to the community, our analyses have yielded a number of potentially important new biological insights about human cervical cancer that warrant further study. First, based on ViFi analyses of TCGA DNA and RNA NGS data from 68 matched human cervical cancer samples, we show that HPV integration plays a powerful role on local transcriptional activity, especially when fusion human-viral mRNA sequences are present. This includes a strong increase in transcription near the integration site, often times greater transcriptional activity than >75% of the other transcripts within the same sample, as well as transcription of elements that might normally not be expressed. Our results are consistent with the theory that integration results in the recruitment of transcription factors by HPV’s upstream regulatory region (URR) and subsequent transcription read-through to produce both viral-human mRNA transcripts as well as an uptick of expression downstream from the viral integration point.
This finding adds a new and unanticipated component to the dysregulated transcription that can be caused by viral integration, including by production of viral-human fusion transcripts. Viral-human fusion transcripts can alter functional pathways, as in the case of the viral-human fusion transcript HBx-LINE1 (22,54), which acts as a sponge for miRNA-122 and promotes hepatic cell epithelial-mesenchymal transition (EMT)-like changes and increases susceptibility to induced tumor formation (54). Recent studies for HPV-related cancers have noted differential expression profiles of miRNAs for HPV-positive and HPV-negative samples (55,56). One possibility is that fusion transcripts produced by integrated HPV might also act as a sponge for miRNA. Another possibility is that fusion transcripts might better disrupt host cellular pathways. Jeon and Lambert found that viral mRNA from integrated HPV are more stable due to the resulting disruption of the mRNA instability element in the viral genome after integration (57). Thus, fusion mRNA might also be more stable, especially in light of the observation that the human portion is typically downstream of the viral portion in fusion mRNA sequences.
Lastly, our analyses showing uniformly amplified regions of multiple chromosomes and HPV, with mapping reads suggesting that circular structure, coupled with the transcriptional patterns also suggesting circular structure, suggest a novel mechanism of small ecDNA formation that could contribute to viral carcinogenesis. Oncogene amplification on ecDNA has recently been shown to play a major role in accelerated evolution in cancer (42). The findings reported here raise the possibility that a different kind of apparent circular ecDNA, which is much smaller in size and lacks known human oncogenes, could provide a complementary mechanism of pathogenesis in some viral associated cancers through indiscriminate transcription of proximal genome elements on the circular structure.
Our findings show that an integrated approach using ViFi can reveal important new insights into the biological mechanisms that contribute to carcinogenesis through seemingly random viral integration into the genome. Analysis of genomic and transcriptomic profiles from cervical cancer samples suggests that recurrent integrations, oncogene expression, and/or viral gene expression may not be necessary for increased pathogenesis. Instead amplification and over-expression of proximal elements driven by viral gene chimerism and the possible role of viral integration in the production of a unique type of ecDNA formation may provide a novel and clearer explanation of the role of viral insertions in cancer pathogenesis. Future studies will be needed to confirm the presence of these apparent circular ecDNA structures and assess their presence in cervical cancer, assess their functional consequences, and examine their occurrence in other viral associated tumors.
DATA AVAILABILITY
ViFi is available on GitHub at https://github.com/namphuon/ViFi.
The majority of the data used in this paper are taken from publicly available sources cited within the paper, for which we have provided their accession numbers in the Methods section, and from the TCGA database, for which we have provided the sample identifiers in the Additional Supplemental File 2. Due to the large size of the simulated data (320 GB of total simulated data), the simulated data is available upon reasonable request. Scripts used to generate the simulated datasets are also available on the GitHub repository.
Supplementary Material
ACKNOWLEDGEMENTS
The results published here are in whole or part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/. Circos (58) was used to generate the Supplementary Figures S13–S26.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online.
FUNDING
National Science Foundation [DBI-1458557]; National Institutes of Health (NIH) [R01GM114362]; Extreme Science and Engineering Discovery Environment [TG-ASC160042]; Ludwig Institute for Cancer Research; National Institute for Neurological Diseases and Stroke [NS73831]; Defeat GBM Program of the National Brain Tumor Society; Ben and Catherine Ivy Foundation; Sharpe/National Brain Tumor Society Research Program; Ziering Family Foundation in memory of Sigi Ziering. Funding for open access charge: NIH [R01GM114362].
Conflict of interest statement. V.B. is a co-founder, has an equity interest and receives income from Digital Proteomics, LLC. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. D.P. was not involved in the research presented here.
REFERENCES
- 1. Plummer M., de Martel C., Vignat J., Ferlay J., Bray F., Franceschi S.. Global burden of cancers attributable to infections in 2012: a synthetic analysis. Lancet Glob. Health. 2016; 4:e609–e616. [DOI] [PubMed] [Google Scholar]
- 2. Duensing S., Münger K.. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res. 2002; 62:7075–7082. [PubMed] [Google Scholar]
- 3. Yim E.-K., Park J.-S.. The role of HPV E6 and E7 oncoproteins in HPV-associated cervical carcinogenesis. Cancer Res. Treat. 2005; 37:319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang T., Zhang J., You X., Liu Q., Du Y., Gao Y., Shan C., Kong G., Wang Y., Yang X. et al. Hepatitis B virus X protein (HBx) modulates oncogene YAP via CREB to promote growth of hepatoma cells. Hepatology. 2012; 56:2051–2059. [DOI] [PubMed] [Google Scholar]
- 5. Carrillo-Infante C., Abbadessa G., Bagella L., Giordano A.. Viral infections as a cause of cancer (review). Int. J. Oncol. 2007; 30:1521–1528. [DOI] [PubMed] [Google Scholar]
- 6. Moore P.S., Chang Y.. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer. 2010; 10:878–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mesri E.A., Feitelson M.A., Munger K.. Human viral oncogenesis: A cancer hallmarks analysis. Cell Host Microbe. 2014; 15:266–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Duncavage E.J., Magrini V., Becker N., Armstrong J.R., Demeter R.T., Wylie T., Abel H.J., Pfeifer J.D.. Hybrid capture and next-generation sequencing identify viral integration sites from formalin-fixed, paraffin-embedded tissue. J. Mol. Diagn. 2011; 13:325–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chandrani P., Kulkarni V., Iyer P., Upadhyay P., Chaubal R., Das P., Mulherkar R., Singh R., Dutt A.. NGS-based approach to determine the presence of HPV and their sites of integration in human cancer genome. Br. J. Cancer. 2015; 112:1958–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen Y., Yao H., Thompson E.J., Tannir N.M., Weinstein J.N., Su X.. VirusSeq: software to identify viruses and their integration sites using next-generation sequencing of human cancer tissue. Bioinformatics. 2013; 29:266–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang Q., Jia P., Zhao Z.. VirusFinder: Software for efficient and accurate detection of viruses and their integration sites in host genomes through next generation sequencing data. PLoS ONE. 2013; 8:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li J.W., Wan R., Yu C.S., Co N.N., Wong N., Chan T.F.. ViralFusionSeq: accurately discover viral integration events and reconstruct fusion transcripts at single-base resolution. Bioinformatics. 2013; 29:649–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang Q., Jia P., Zhao Z.. VERSE: a novel approach to detect virus integration in host genomes through reference genome customization. Genome Med. 2015; 7:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ho D.W., Sze K.M., Ng I.O.. Virus-Clip : a fast and memory-efficient viral integration site detection tool at single-base resolution with annotation capability. Oncotarget. 2015; 6:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Forster M., Szymczak S., Ellinghaus D., Hemmrich G., Rühlemann M., Kraemer L., Mucha S., Wienbrandt L., Stanulla M., Franke A.. Vy-PER: eliminating false positive detection of virus integration events in next generation sequencing data. Sci. Rep. 2015; 5:11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Matsen F.A., Kodner R.B., Armbrust E.V.. pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics. 2010; 11:538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eddy S.R. Profile hidden Markov models. Bioinformatics. 1998; 14:755–763. [DOI] [PubMed] [Google Scholar]
- 18. Skewes-Cox P., Sharpton T.J., Pollard K.S., DeRisi J.L.. Profile hidden markov models for the detection of viruses within metagenomic sequence data. PLoS ONE. 2014; 9:e105067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nguyen N.-p., Mirarab S., Liu B., Pop M., Warnow T.. TIPP: taxonomic identification and phylogenetic profiling. Bioinformatics. 2014; 30:3548–3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nguyen N.-p., Nute M., Mirarab S., Warnow T.. HIPPI: highly accurate protein family classification with ensembles of HMMs. BMC Genomics. 2016; 17:89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sung W.-K., Zheng H., Li S., Chen R., Liu X., Li Y., Lee N.P., Lee W.H., Ariyaratne P.N., Tennakoon C. et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012; 44:765–769. [DOI] [PubMed] [Google Scholar]
- 22. Lau C.C., Sun T., Ching A.K.K., He M., Li J.W., Wong A.M., Co N.N., Chan A.W.H., Li P.S., Lung R.W.M. et al. Viral-human chimeric transcript predisposes risk to liver cancer development and progression. Cancer Cell. 2014; 25:335–349. [DOI] [PubMed] [Google Scholar]
- 23. Tang K.-W., Alaei-Mahabadi B., Samuelsson T., Lindh M., Larsson E.. The landscape of viral expression and host gene fusion and adaptation in human cancer. Nat. Commun. 2013; 4:2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Burk R.D., Chen Z., Saller C., Tarvin K., Carvalho A.L., Scapulatempo-Neto C., Silveira H.C., Fregnani J.H., Creighton C.J., Anderson M.L. et al. Integrated genomic and molecular characterization of cervical cancer. Nature. 2017; 543:378–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li H., Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mirarab S., Nguyen N., Guo S., Wang L.-S., Kim J., Warnow T.. PASTA: ultra-large multiple sequence alignment for nucleotide and amino-acid sequences. J. Comp. Biol. 2014; 22:377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014; 30:1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rosenbloom K.R., Sloan C.A., Malladi V.S., Dreszer T.R., Learned K., Kirkup V.M., Wong M.C., Maddren M., Fang R., Heitner S.G. et al. ENCODE Data in the UCSC Genome Browser: Year 5 update. Nucleic Acids Res. 2013; 41:D56–D63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Van Doorslaer K., Li Z., Xirasagar S., Maes P., Kaminsky D., Liou D., Sun Q., Kaur R., Huyen Y., McBride A.A.. The Papillomavirus Episteme: a major update to the papillomavirus sequence database. Nucleic Acids Res. 2017; 45:D499–D506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jiang Z., Jhunjhunwala S., Liu J., Haverty P.M., Kennemer M.I., Guan Y., Lee W., Carnevali P., Stinson J., Johnson S. et al. The effects of hepatitis B virus integration into the genomes of hepatocellular carcinoma patients. Genome Res. 2012; 593–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tavaré S., Tavare S.. Some probabilistic and statistical problems in the analysis of DNA sequences. American Mathematical Society: Lectures on Mathematics in the Life Sciences. 1986; 17:American Mathematical Society; 57–86. [Google Scholar]
- 32. Huang W., Li L., Myers J.R., Marth G.T.. ART: A next-generation sequencing read simulator. Bioinformatics. 2012; 28:593–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. O’Leary N.A., Wright M.W., Brister J.R., Ciufo S., Haddad D., McVeigh R., Rajput B., Robbertse B., Smith-White B., Ako-Adjei D. et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016; 44:D733–D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tarailo-Graovac M., Chen N.. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics. 2009; 25:4.10.1–4.10.14. [DOI] [PubMed] [Google Scholar]
- 35. Pertea M., Kim D., Pertea G.M., Leek J.T., Salzberg S.L.. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016; 11:1650–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim D., Langmead B., Salzberg S.L.. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015; 12:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pertea M., Pertea G.M., Antonescu C.M., Chang T.-C., Mendell J.T., Salzberg S.L.. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015; 33:290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Weibull W. A statistical distribution function of wide applicability. J. Appl. Mech. 1951; 18:293–297. [Google Scholar]
- 39. Delignette-Muller M.L., Dutang C.. fitdistrplus : an R package for fitting distributions. J. Stat. Softw. 2015; 64:1–34. [Google Scholar]
- 40. Benjamini Y., Hochberg Y.. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995; 57:289–300. [Google Scholar]
- 41. R Core Team A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. 2015; Vienna. [Google Scholar]
- 42. Turner K.M., Deshpande V., Beyter D., Koga T., Rusert J., Lee C., Li B., Arden K., Ren B., Nathanson D.A. et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature. 2017; 543:122–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hu Z., Zhu D., Wang W., Li W., Jia W., Zeng X., Ding W., Yu L., Wang X., Wang L. et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat. Genet. 2015; 47:158–163. [DOI] [PubMed] [Google Scholar]
- 44. Ziegert C., Wentzensen N., Vinokurova S., Kisseljov F., Einenkel J., Hoeckel M., von Knebel Doeberitz M.. A comprehensive analysis of HPV integration loci in anogenital lesions combining transcript and genome-based amplification techniques. Oncogene. 2003; 22:3977–3984. [DOI] [PubMed] [Google Scholar]
- 45. Johansson C., Schwartz S.. Regulation of human papillomavirus gene expression by splicing and polyadenylation. Nat. Rev. Microbiol. 2013; 11:239–251. [DOI] [PubMed] [Google Scholar]
- 46. Akagi K., Li J., Broutian T.R., Padilla-Nash H., Xiao W., Jiang B., Rocco J.W., Teknos T.N., Kumar B., Wangsa D. et al. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res. 2014; 24:185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gasior S.L., Wakeman T.P., Xu B., Deininger P.L.. The human LINE-1 retrotransposon creates DNA double-strand breaks. J. Mol. Biol. 2006; 357:1383–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kinomoto M., Kanno T., Shimura M., Ishizaka Y., Kojima A., Kurata T., Sata T., Tokunaga K.. All APOBEC3 family proteins differentially inhibit LINE-1 retrotransposition. Nucleic Acids Res. 2007; 35:2955–2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Romanish M.T., Cohen C.J., Mager D.L.. Potential mechanisms of endogenous retroviral-mediated genomic instability in human cancer. Semin. Cancer Biol. 2010; 20:246–253. [DOI] [PubMed] [Google Scholar]
- 50. Sigurdsson M.I., Smith A.V., Bjornsson H.T., Jonsson J.J.. The distribution of a germline methylation marker suggests a regional mechanism of LINE-1 silencing by the piRNA-PIWI system. BMC Genet. 2012; 13:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yu H.-L., Zhao Z.-K., Zhu F.. The role of human endogenous retroviral long terminal repeat sequences in human cancer. Int. J. Mol. Med. 2013; 32:755–762. [DOI] [PubMed] [Google Scholar]
- 52. Rodić N., Burns K.H.. Long interspersed element-1 (LINE-1): passenger or driver in human neoplasms. PLOS Genet. 2013; 9:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Xiao-Jie L., Hui-Ying X., Qi X., Jiang X., Shi-Jie M.. LINE-1 in cancer: multifaceted functions and potential clinical implications. Genet Med. 2016; 18:431–439. [DOI] [PubMed] [Google Scholar]
- 54. Liang H.W., Wang N., Wang Y., Wang F., Fu Z., Yan X., Zhu H., Diao W., Ding Y., Chen X. et al. Hepatitis B virus-human chimeric transcript HBx-LINE1 promotes hepatic injury via sequestering cellular microRNA-122. J. Hepatol. 2016; 64:278–291. [DOI] [PubMed] [Google Scholar]
- 55. Lajer C.B., Garnæs E., Friis-Hansen L., Norrild B., Therkildsen M.H., Glud M., Rossing M., Lajer H., Svane D., Skotte L. et al. The role of miRNAs in human papilloma virus (HPV)-associated cancers: bridging between HPV-related head and neck cancer and cervical cancer. Br. J. Cancer. 2012; 106:1526–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gao D., Zhang Y., Zhu M., Liu S., Wang X.. MiRNA expression profiles of HPV-infected patients with cervical cancer in the uyghur population in China. PLoS ONE. 2016; 11:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jeon S., Lambert P.F.. Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of E6 and E7 mRNAs: implications for cervical carcinogenesis. Proc. Natl. Acad. Sci. U.S.A. 1995; 92:1654–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Krzywinski M., Schein J., Birol I., Connors J., Gascoyne R., Horsman D., Jones S.J., Marra M.A.. Circos: an information aesthetic for comparative genomics. Genome Res. 2009; 19:1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ViFi is available on GitHub at https://github.com/namphuon/ViFi.
The majority of the data used in this paper are taken from publicly available sources cited within the paper, for which we have provided their accession numbers in the Methods section, and from the TCGA database, for which we have provided the sample identifiers in the Additional Supplemental File 2. Due to the large size of the simulated data (320 GB of total simulated data), the simulated data is available upon reasonable request. Scripts used to generate the simulated datasets are also available on the GitHub repository.