

Abstract
Objective
To evaluate the role of the copy number loss in SFMBT1 in a Caucasian population.
Methods
Five hundred sixty-seven Finnish and 377 Norwegian patients with idiopathic normal pressure hydrocephalus (iNPH) were genotyped and compared with 508 Finnish elderly, neurologically healthy controls. The copy number loss in intron 2 of SFMBT1 was determined using quantitative PCR.
Results
The copy number loss in intron 2 of SFMBT1 was detected in 10% of Finnish (odds ratio [OR] = 1.9, p = 0.0078) and in 21% of Norwegian (OR = 4.7, p < 0.0001) patients with iNPH compared with 5.4% in Finnish controls. No copy number gains in SFMBT1 were detected in patients with iNPH or healthy controls. The carrier status did not provide any prognostic value for the effect of shunt surgery in either population. Moreover, no difference was detected in the prevalence of hypertension or T2DM between SFMBT1 copy number loss carriers and noncarriers.
Conclusions
This is the largest and the first multinational study reporting the increased prevalence of the copy number loss in intron 2 of SFMBT1 among patients with iNPH, providing further evidence of its role in iNPH. The pathogenic role still remains unclear, requiring further study.
Idiopathic normal pressure hydrocephalus (iNPH) is a late-onset progressive neurologic disease presenting typically with gait difficulties together with enlarged ventricles and tightened parasagittal cortical sulci, whereas other symptoms such as cognitive impairment and urinary incontinence are often present.1,2 Recently, the 2 available diagnostic guidelines1,2 have been criticized, and urgent revision and/or unification of the diagnostic manual have been requested.3–5
Patients with iNPH are characterized by abnormal CSF circulation and evidence of delayed cerebral clearance, as recently shown in an MRI CSF tracer study.6 Although there are no biomarkers to aid in the diagnostics, and the etiology of iNPH remains unclear, there is an increasing amount of proof indicating a potential genetic component in iNPH.7–12 The prevalence of familial iNPH, i.e., at least 2 patients with iNPH, in the first-degree relatives has been reported to range from 4.8% to 7%.10,11 Data also contain a pair of identical twins having iNPH11 and a family in which an autosomal dominant inheritance pattern is observed.9
A segmental copy number loss in the SFMBT1 gene was reported in Japan to be present in 50% of patients, who present concomitantly with clinical features of iNPH and enlarged ventricles,13 and in 26% of patients with iNPH, who experienced a positive shunt response.14 These results have previously not been confirmed outside of Japan and with sufficiently large cohorts. Our aim was to evaluate the prevalence of the copy number loss of SFMBT1 in patients with iNPH of Caucasian origin.
Methods
Standard protocol approvals, registrations, and patient consents
This study was conducted in the Department of Neurosurgery in Kuopio University Hospital, the Brain Research Unit of the University of Eastern Finland, and the Department of Neurosurgery, Oslo University Hospital, Rikshospitalet, Oslo, Norway, according to the Declaration of Helsinki. Kuopio University Hospital Research Ethics Board approved the study (5/2008 and 276/2016). In Norway, the study was approved by the Regional Committee for Medical and Health Research Ethics (REK) of Health Region South-East, Norway (2015/1313), and the Institutional Review Board of Oslo University Hospital (2015/8128).
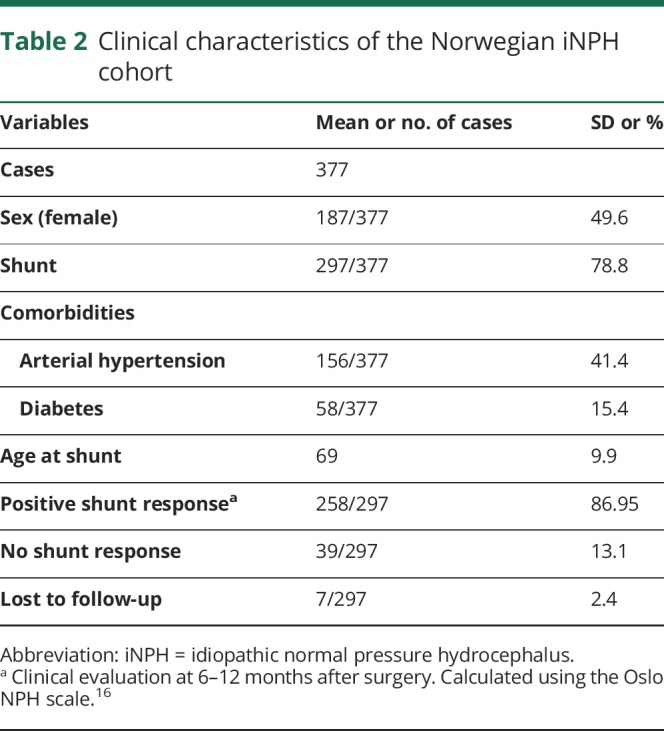
The study consisted of 567 Finnish (mean age 70 years; SD 8.3; men: n = 254; table 1) and 377 Norwegian (mean age 68 years; SD 11; men: n = 190 (50%); table 2) patients with possible iNPH. All patients and controls provided their written informed consent.
Table 1.
Clinical characteristics of the Finnish iNPH cohort
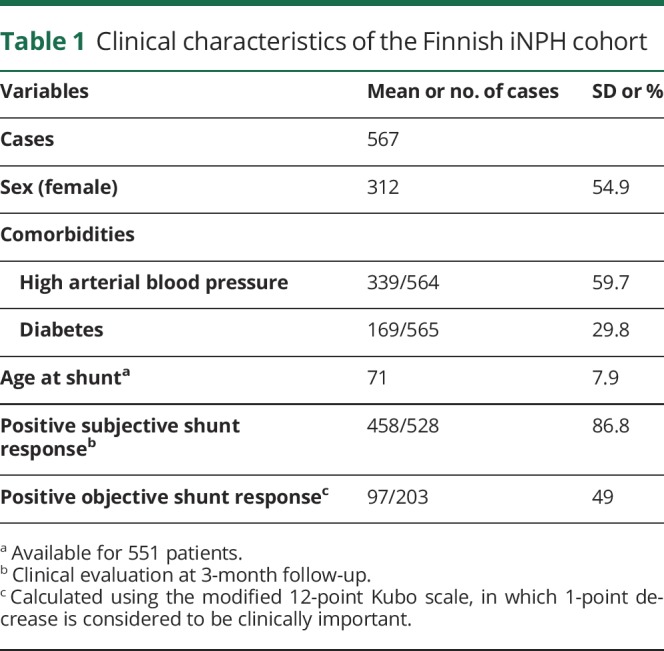
Table 2.
Clinical characteristics of the Norwegian iNPH cohort
All Finnish and Norwegian patients suspected of having iNPH have been clinically evaluated by a neurologist and a neurosurgeon. The patients were referred to a neurosurgeon by a neurologist if the patients were observed to have at least one of the core symptoms associated with iNPH, which include gait difficulties, cognitive impairment, and urinary incontinence together with enlarged ventricles (Evans Index >0.30)15 disproportionate to the size of the sulci of the cerebral convexities in MRI or CT imaging scans. In addition, most Finnish and Norwegian patients have undergone, as a prognostic test for shunt benefit, a 24-hour intraventricular pressure monitoring, spinal tap, extended lumbar drainage, or lumbar infusion test.
The Norwegian patients were diagnosed, and the decision to perform the shunt surgery was done following a previously published protocol,16 which included a clinical judgment of the severity of iNPH symptoms using a Oslo iNPH Grading Scale (ranging from 3 to 15),16 MRI or CT imaging scans for the evaluation of the ventriculomegaly, evaluation of comorbidities, and finally a diagnostic overnight ICP monitoring.
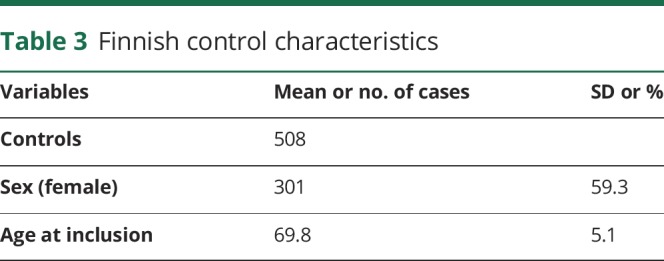
All the Finnish and Norwegian patients fulfilled the clinical diagnostic criteria for possible iNPH.1,2 The control group consisted of 508 (mean age 70 years; SD 5.1; male: n = 207) Finnish subjects acquired for neurogenetic studies. All subjects have undergone clinical and neuropsychological testing lead by an experienced neurologist specialized in neurodegenerative conditions to exclude any signs of cognitive impairment (table 3).
Table 3.
Finnish control characteristics
Genomic DNA was extracted from venous blood samples using QIAamp DNA blood mini extraction kit (QIAGEN). The copy number loss/gain in the intron 2 region of SFMBT1 was detected using quantitative PCR (qPCR) and the delta-delta method as previously described.14 Our collaborator (T. Kato) provided a positive control sample, and a commercial negative control was used.
All statistical analyses were performed using SPSS statistic version 23 (IBM corp. USA). The χ2 test was used for categorical dichotomous variables for groups >2. The Fisher exact test was used for pairwise comparison. p < 0.05 was considered statistically significant.
Power calculations were completed before the execution of the research and were based on the results of the study by Sato et al.14 in which the expected prevalence was divided by a factor of 2 (26/2% = 13%). With a power of 0.85, p = 0.05, the sample size was determined to be n ≥ 362. Both the Finnish and Norwegian iNPH cohorts and Finnish controls fulfill this requirement.
Data availability statement
According to Finnish law, the full individual clinical data set cannot be shared publicly. However, the data set can/will be shared by academic collaboration agreement upon request.
Results
The copy number loss in SFMBT1 was detected in 9.9% of the Finnish patients with iNPH, in 21% of the Norwegian patients with iNPH, and in 5.4% of the Finnish controls (figures 1 and 2). Statistically significant difference was detected between Finnish and Norwegian patients with iNPH (p < 0.0001). No copy number gains in SFMBT1 were detected in Finnish or Norwegian patients with iNPH or healthy controls. In the Finnish iNPH cohort, 9/71 suspected familial patients, who are carriers for the copy number loss, were detected, and therefore, no aggregation of the copy number loss variant was observed in familial iNPH (12.1% vs 10%, p = 0.5). There was no significant difference in age at onset, sex, shunting prevalence, or shunt response between Finnish and Norwegian patients. In addition, no difference was detected in the prevalence of hypertension or T2DM between SFMBT1 copy number loss carriers and noncarriers (table 4) or in the frequency of the genetic variant between shunt-responsive and nonresponsive patients in either Finnish (odds ratio [OR] = 1.0, confidence interval [CI] 95% 0.44–2.4, p = 0.93) or Norwegian (OR = 0.68, CI 95% 0.31–1.5, p = 0.33) cohort.
Figure 1. Flowchart of the study cohort.

Flowchart showing the Finnish and Norwegian cohorts. No difference was detected between the frequency of the genetic variant between shunt-responsive and nonresponsive patients in either Finnish (OR = 1.0, CI 95% 0.44–2.4, p = 0.93) or Norwegian (OR = 0.68, CI 95% 0.31–1.5, p = 0.33) cohort. aLost to follow-up (n = 7). bLost to follow-up response (n = 63). CI = confidence interval; OR = odds ratio.
Figure 2. The prevalence of the copy number loss in the SFMBT1 among Finnish and Norwegian patients with iNPH.
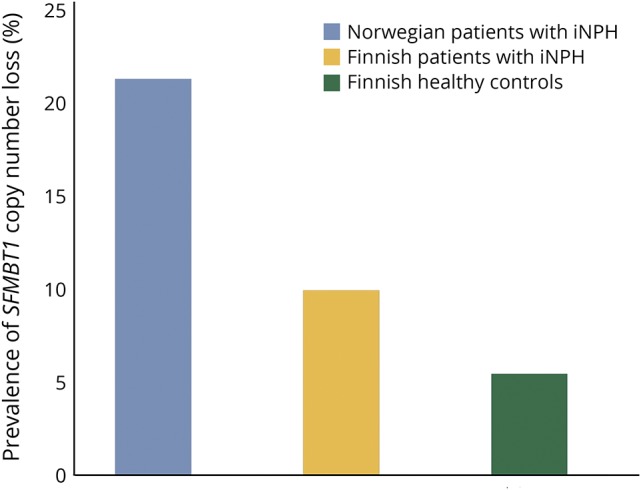
The prevalence of the copy number loss in SFMBT1 among Norwegian iNPH patients is 21% (n = 79/377), 9.9% (n = 56/567) among Finnish iNPH patients, and 5.4% (n = 27/508) among Finnish controls. Statistical significance was observed; Norwegian iNPH patients vs Finnish controls (OR = 4.7, CI 95% 3.0–7.5, p < 0.001), Finnish iNPH patients vs Finnish controls (OR = 1.9, CI 95% 1.2–3.1, p = 0.0078), and Norwegian iNPH patients vs Finnish iNPH patients (OR = 2.5, CI 95% 1.7–3.6, p < 0.001). p < 0.05 was considered statistically significant. The Fisher exact test was used for pairwise comparisons. CI = confidence interval; iNPH = idiopathic normal pressure hydrocephalus; OR = odds ratio.
Table 4.
Prevalence of hypertension and diabetes

Discussion
This is the first study on the prevalence of the copy number loss in SFMBT1 among patients with iNPH of non-Asian origin. Although there are studies describing familial aggregation11 and even in some rare cases autosomal dominant inheritance pattern,9 SFMBT1 is the first gene to be associated with iNPH. Our findings are principally in line with the Japanese results providing compelling further evidence on the role of the copy number loss in SFMBT1 in iNPH.
The copy number loss in SFMBT1 is detected only in 10%–20% of the patients with iNPH, which would suggest that iNPH has both a polygenetic and multifactorial origin. Ethnicity seems to modify the prevalence of this genetic variant between Finnish and Norwegian iNPH patients but surprisingly is similar between selected Norwegian and Japanese populations. In addition, a small percentage of cognitively intact controls carry the genetic variant providing further evidence on the copy number loss in SFMBT1 of being only a possible risk-increasing genotype. In the Japanese study, a small percentage of patients and controls were found to carry a copy number gain variant, but no statistically significant difference between patients with iNPH and controls was detected.14 Of interest, in the Nordic cohorts, no copy number gain variants were detected in either patients with iNPH or healthy controls. Therefore, it appears that the copy number loss in SFMBT1 is associated with iNPH, and the results are not influenced by genomic instability in the SFMBT1 intron region.
Hypertension, T2DM, schizophrenia, and Alzheimer disease are common among patients with iNPH.17–22 The present results could reflect a higher occurrence of hypertension and T2DM in iNPH, as previously demonstrated. However, in this study cohort, we found no statistically significant difference in the prevalence of hypertension or T2DM between SFMBT1 copy number loss carriers and noncarriers, further validating its independent association with iNPH.
In the present study, both shunt-responsive and nonresponsive patients were included, and no difference was observed between these groups regarding the carrier status of the copy number loss in SFMBT1. The original discovery was done in subjects with enlarged brain ventricles,13 and the Japanese study on copy number loss in SFMBT114 included only shunt-responsive iNPH patients, and therefore, the prevalence of the copy number loss in SFMBT1 among patients who are nonresponsive to shunt surgery is unknown in the Japanese population.14 Because of the similar prevalence between both shunt-responsive and nonresponsive patient groups, SFMBT1 seems to be linked with enlargement of brain ventricles but not with shunt response.
The diagnosis of iNPH is presently based on typical clinical characteristics and radiologic presentation. In addition, different prognostic tests are used to evaluate the potential shunt benefit, but no biomarkers have been found to help in the differential diagnostics. The current vague diagnostic criteria of iNPH1,2 should be noted in the interpretation of the current results and further genetic studies on iNPH. These include the definition of the diagnosis, heterogeneity of the disease course and variable radiologic features, and common comorbid neurodegenerative diseases.4 This poses a challenge for the differential diagnostics of iNPH, and both false and missed diagnoses are likely to be common. Potentially, the genetic information could, in the future, be included in different risk calculators (e.g., reference 23) to provide clinician tools that help decide referrals concerning these types of uncommon diseases.
The SFMBT1 locus has been previously identified to be involved in elevated serum urate levels, fasting glucose, and high blood pressure.24–26 The physiologic role of the SFMBT1 protein is poorly understood but relates to histone binding and is involved in different transcription corepressor activities.27,28 However, the SFMBT1 protein seems to be present in anatomical structures involved with CSF development and circulation such as the choroid plexus, the ependymal cell lining of the ventricles, and the smooth muscle and endothelial cells of the arteries.13 A CSF tracer study has revealed disturbed CSF circulation and delayed CSF tracer clearance within the brains of patients with iNPH.6 An increasing body of evidence links the pathophysiology behind iNPH to processes taking place at the glia-vascular interface. It has been shown that patients with iNPH showed evidence of alterations in the brain capillary ultrastructure, including alterations in pericytes and endothelial cells.29 Furthermore, the perivascular expression of the water channel aquaporin-4 (AQP4) was reduced in iNPH.30
Our study has considerable strengths including a well-characterized large multinational cohort consisting of both shunted and nonshunted patients. The most substantial limitation in the current study is that no healthy Norwegian control individuals were available and that no internal validation was used. The prevalence of the copy number variant could be different among neurologically healthy population. In addition, in the current study, asymptomatic ventriculomegaly was not routinely evaluated from the healthy Finnish controls. Therefore, it is possible that the some of the healthy controls that are carriers for the copy number loss variant in SFMBT1 have enlarged ventricles but are asymptomatic. This might skew the current result, and the association between iNPH and the copy number loss in SFMBT1 could be even stronger than reported. Although a strong association between the copy number loss in SFMBT1 and iNPH was found in both the Japanese and the Caucasian cohorts, there are still limitations to the studies concentrating on the copy number loss in SFMBT1 and iNPH. First, no nationwide data exist on the prevalence of the copy number loss in SFMBT1, and therefore, the controls used provide only a rough estimation of the prevalence in the normal population. Second, the role of the SFMBT1 gene in the development of cardiovascular diseases needs to be investigated in depth. In addition, the role of SFMBT1 should be studied in other neurodegenerative diseases to find whether the genetic variation in SFMBT1 is unique to iNPH. Third, the present results need still to be confirmed in larger multinational cohorts both in cross-sectional and prospective study settings.
Normal pressure hydrocephalus is divided into 3 different groups: idiopathic, where the etiology is unclear, secondary when there is a predisposing factor to be found, and most recently detected familial type, where iNPH is seen also in the first-degree relatives.9,11 So far, no difference in the phenotype has been observed between iNPH and familial NPH. Because copy number variation of SFMBT1 was not enriched in familial iNPH, it does not seem to explain familial aggregation of iNPH, and therefore, other genetic variations are expected to associate with iNPH. The effect of the copy number loss in SFMBT1 to the clinical phenotype should be described. This might shed light into the different forms of iNPH with so far undistinguishable phenotypes.
It has been reported that some of the asymptomatic people with ventriculomegaly are carriers for the copy number loss in SFMBT1.13 Ventriculomegaly is a key radiologic finding in iNPH, and Evans Index >0.3 is included as a requirement in the diagnostic criteria of iNPH,1,2 although it has been recently suggested that the cutoff value for ventricular enlargement should be age and sex dependent and the pathologic lower limit of Evan's Index increased to >0.32.31 It has been suggested that people who are asymptomatic with enlarged ventricles are at an increased risk of developing iNPH, and it has been reported that in the Japanese population, 25% of people with asymptomatic ventriculomegaly will eventually develop iNPH.32 However, the cohort sizes have been limited in the studies in which the connection between AVIM and iNPH has been proposed. Therefore, no universal consensus on this matter exists.
The prevalence of the copy number loss in SFMBT1 could be increased among the families in which iNPH is frequently observed, and therefore, first-degree relatives of patients with iNPH should be included in future genetic studies. It might be that the copy number loss in SFMBT1 is a risk gene for iNPH but requires other unknown triggering risk factors to result in clinical disease.
Future studies are still urgently needed to elucidate the genetics of iNPH. Understanding even some of the pathologic processes causing iNPH provides possibilities to develop targeted or even preventive therapies in the future. This study encourages functional studies on SFMBT1 to clarify the role in the disease process of iNPH.
Acknowledgment
The authors acknowledge Marjo Laitinen for her help in setting up the qPCR method and RN Marita Parviainen for managing the KUH iNPH register.
Glossary
- CI
confidence interval
- iNPH
idiopathic normal pressure hydrocephalus
- OR
odds ratio
Author contributions
V.E. Korhonen: study concept and design, data acquisition, data analysis and interpretation, and drafting of the manuscript. S. Helisalmi: study concept and design, data acquisition, and critical revision of the manuscript for important intellectual content. A. Jokinen, I. Jokinen, J.-M. Lehtola, M. Oinas, K. Lönnrot, C. Avellan, A. Kotkansalo, J. Frantzen, J. Rinne, A. Ronkainen, M. Kauppinen, and A. Junkkari: data acquisition and critical revision of the manuscript for important intellectual content. M. Hiltunen and H. Soininen: critical revision of the manuscript for important intellectual content. M. Kurki: critical revision of the manuscript for important intellectual content (statistics). J.E. Jääskeläinen, A.M. Koivisto, H. Sato H, and T. Kato: critical revision of the manuscript for important intellectual content. A.M. Remes: study concept and design and critical revision of the manuscript for important intellectual content. P.K. Eide and V. Leinonen: study concept and design, data acquisition, critical revision of the manuscript for important intellectual content, and study supervision.
Study funding
This work was supported by the Academy of Finland (no 307866), the Sigrid Juselius Foundation, the Kuopio University Hospital Research Fund, the Kuopio University Hospital VTR fund, the Emil Aaltonen Foundation, the Cultural Foundation of Finland, the North Savo Regional Fund, the Olvi Foundation, and the Finnish Medical Association. In Norway, the study was supported by grants from Health South-East, Norway (grant 2011067).
Disclosure
V.E. Korhonen has received research support from the Kuopio University Hospital Research Fund, the Maire Taponen Foundation, the University of Eastern Finland, the Olvi Foundation, the Emil Aaltonen Foundation, the Finnish Cultural Foundation, the North Savo Regional fund, and the Finnish Medical Association. S. Helisalmi, A. Jokinen, I. Jokinen, J.-M. Lehtola, M. Oinas, and K. Lönnrot report no disclosures. C. Avellan has received research support from the Maire Taponen Foundation, Lastentautien tutkimussäätiö, Stiftelsen Dorothea Olivia, Karl Walter och Jarl Walter Perkléns minne, and Svenska kulturfonden. A.E. Kotkansalo reports no disclosures. J. Frantzen serves/has served on the scientific advisory board of and as a consultant to Bonalive Biomaterials Ltd. J. Rinne, A. Ronkainen, and M. Kauppinen report no disclosures. A. Junkkari has received research support from the state research fund (VTR), Kuopio University Hospital (KUH), the University of Eastern Finland (UEF), the Finnish Cultural Foundation, and the Maire Taponen Foundation. M. Hiltunen serves/has served on the editorial boards of the Journal of Alzheimer's Disease, Journal of Alzheimer's Disease & Parkinsonism, and The Scientific World Journal. H. Soininen serves/has served on the scientific advisory board of AC Immune and serves/has served on the editorial board of the Journal of Alzheimer's Disease. M. Kurki reports no disclosures. J.E. Jääskeläinen serves/has served on the editorial board of Acta Neurochirurgica; has received research support from the Finnish Academy of Sciences, the Juho Vainio Foundation Päivikki, the Sakari Sohlberg Foundation, and Kuopio University Hospital. A.M. Koivisto, H. Sato, T. Kato, A.M. Remes, and P.K. Eide report no disclosures. V. Leinonen has received funding for travel and/or speaker honoraria from B. Braun Aesculap; serves/has served on the editorial board of the Journal of Alzheimer's Disease; and has received research support from NeuroVision Imaging, LLC, and Kuopio University Hospital. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/NG.
References
- 1.Relkin N, Marmarou A, Klinge P, Bergsneider M, Black PM. INPH guidelines, part II: diagnosing idiopathic normal-pressure hydrocephalus. Neurosurgery 2005;57:4–16. [DOI] [PubMed] [Google Scholar]
- 2.Mori E, Ishikawa M, Kato T, et al. Guidelines for management of idiopathic normal pressure hydrocephalus: second edition. Neurol Med Chir (Tokyo) 2012;52:775–809. [DOI] [PubMed] [Google Scholar]
- 3.Williams MA, Relkin NR. Diagnosis and management of idiopathic normal-pressure hydrocephalus. Neurol Clin Pract 2013;3:375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Espay AJ, Da Prat GA, Dwivedi AK, et al. Deconstructing normal pressure hydrocephalus: ventriculomegaly as early sign of neurodegeneration. Ann Neurol 2017:503–513. [DOI] [PubMed] [Google Scholar]
- 5.Andersson J, Rosell M, Kockum K, Söderström L, Laurell K. Challenges in diagnosing normal pressure hydrocephalus: evaluation of the diagnostic guidelines. eNeurologicalSci 2017;7:27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ringstad G, Vatnehol SAS, Eide PK. Glymphatic MRI in idiopathic normal pressure hydrocephalus. Brain 2017;140:2691–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Portenoy RK, Berger A, Gross E. Familial occurrence of idiopathic normal-pressure hydrocephalus. Arch Neurol 1984;41:335–337. [DOI] [PubMed] [Google Scholar]
- 8.Cusimano MD, Rewilak D, Stuss DT, Barrera-Martinez JC, Salehi F, Freedman M. Normal-pressure hydrocephalus: is there a genetic predisposition? Can J Neurol Sci 2011;38:274–281. [DOI] [PubMed] [Google Scholar]
- 9.Takahashi Y, Kawanami T, Nagasawa H, Iseki C, Hanyu H, Kato T. Familial normal pressure hydrocephalus (NPH) with an autosomal-dominant inheritance: a novel subgroup of NPH. J Neurol Sci 2011;308:149–151. [DOI] [PubMed] [Google Scholar]
- 10.McGirr A, Cusimano MD. Familial aggregation of idiopathic normal pressure hydrocephalus: novel familial case and a family study of the NPH triad in an iNPH patient cohort. J Neurol Sci 2012;321:82–88. [DOI] [PubMed] [Google Scholar]
- 11.Huovinen J, Kastinen S, Komulainen S, et al. Familial idiopathic normal pressure hydrocephalus. J Neurol Sci 2016;368:11–18. [DOI] [PubMed] [Google Scholar]
- 12.Korhonen V, Solje E, Suhonen NM, et al. Frontotemporal dementia as a comorbidity to idiopathic normal pressure hydrocephalus (iNPH): a short review of literature and an unusual case. Fluids Barriers CNS 2017;14:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kato T, Sato H, Emi M, et al. Segmental copy number loss of SFMBT1 gene in elderly individuals with ventriculomegaly: a community-based study. Intern Med 2011;50:297–303. [DOI] [PubMed] [Google Scholar]
- 14.Sato H, Takahashi Y, Kimihira L, et al. A segmental copy number loss of the SFMBT1 gene is a genetic risk for shunt-responsive, idiopathic normal pressure hydrocephalus (iNPH): a case-control study. PLoS One 2016;11:e0166615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shprecher D, Schwalb J, Kurlan R. Normal pressure hydrocephalus: diagnosis and treatment. Curr Neurol Neurosci Rep 2008;8:371–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eide PK, Sorteberg W. Diagnostic intracranial pressure monitoring and surgical management in idiopathic normal pressure hydrocephalus: a 6-year review of 214 patients. Neurosurgery 2010;66:80–90. [DOI] [PubMed] [Google Scholar]
- 17.Leinonen V, Koivisto AM, Savolainen S, et al. Post-mortem findings in 10 patients with presumed normal-pressure hydrocephalus and review of the literature. Neuropathol Appl Neurobiol 2012;38:72–86. [DOI] [PubMed] [Google Scholar]
- 18.Leinonen V, Koivisto AM, Alafuzoff I, et al. Cortical brain biopsy in long-term prognostication of 468 patients with possible normal pressure hydrocephalus. Neurodegener Dis 2012;10:166–169. [DOI] [PubMed] [Google Scholar]
- 19.Malm J, Graff-Radford NR, Ishikawa M, et al. Influence of comorbidities in idiopathic normal pressure hydrocephalus—research and clinical care. A report of the ISHCSF task force on comorbidities in INPH. Fluids Barriers CNS 2013;10:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Israelsson H, Carlberg B, Wikkelsö C, et al. Vascular risk factors in INPH. Neurology 2017;88:577–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eide P, Pripp A. Increased prevalence of cardiovascular disease in idiopathic normal pressure hydrocephalus patients compared to a population-based cohort from the HUNT3 survey. Fluids Barriers CNS 2014;11:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vanhala V, Junkkari A, Korhonen VE, et al. Prevalence of Schizophrenia in idiopathic normal pressure hydrocephalus. Neurosurgery Epub 2018 May 8. [DOI] [PMC free article] [PubMed]
- 23.Luikku AJ, Hall A, Nerg O, et al. Multimodal analysis to predict shunt surgery outcome of 284 patients with suspected idiopathic normal pressure hydrocephalus. Acta Neurochir (Wien) 2016;158:2311–2319. [DOI] [PubMed] [Google Scholar]
- 24.Yang HC, Liang YJ, Chen JW, et al. Identification of igf1, slc4a4, wwox, and sfmbt1 as hypertension susceptibility genes in han Chinese with a genome-wide gene-based association study. PLoS One 2012;7:e32907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Köttgen A, Albrecht E, Teumer A, et al. Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat Genet 2012;45:145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chung RH, Chiu YF, Hung YJ, et al. Genome-wide copy number variation analysis identified deletions in SFMBT1 associated with fasting plasma glucose in a Han Chinese population. BMC Genomics 2017;18:591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu S, Trievel RC, Rice JC. Human SFMBT1 is a transcriptional repressor protein that selectively binds the N-terminal tail of histone H3. FEBS Lett 2007;581:3289–3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang J, Bonasio R, Strino F, et al. SFMBT1 functions with LSD1 to regulate expression of canonical histone genes and chromatin-related factors. Genes Dev 2013;27:749–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eidsvaag VA, Hansson HA, Heuser K, Nagelhus EA, Eide PK. Brain capillary ultrastructure in idiopathic normal pressure hydrocephalus: relationship with static and pulsatile intracranial pressure. J Neuropathol Exp Neurol 2017;76:1034–1045. [DOI] [PubMed] [Google Scholar]
- 30.Eide PK, Hansson HA. Astrogliosis and impaired aquaporin-4 and dystrophin systems in idiopathic normal pressure hydrocephalus. Neuropathol Appl Neurobiol 2018;44:474–490. [DOI] [PubMed] [Google Scholar]
- 31.Brix MK, Westman E, Simmons A, et al. The Evans’ Index revisited: new cut-off levels for use in radiological assessment of ventricular enlargement in the elderly. Eur J Radiol 2017;95:28–32. [DOI] [PubMed] [Google Scholar]
- 32.Iseki C, Kawanami T, Nagasawa H, et al. Asymptomatic ventriculomegaly with features of idiopathic normal pressure hydrocephalus on MRI (AVIM) in the elderly: a prospective study in a Japanese population. J Neurol Sci 2009;277:54–57. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
According to Finnish law, the full individual clinical data set cannot be shared publicly. However, the data set can/will be shared by academic collaboration agreement upon request.