

Abstract
Objective
We report 3 siblings with the characteristic features of ataxia-telangiectasia-like disorder associated with a homozygous MRE11 synonymous variant causing nonsense-mediated mRNA decay (NMD) and MRE11A deficiency.
Methods
Clinical assessments, next-generation sequencing, transcript and immunohistochemistry analyses were performed.
Results
The patients presented with poor balance, developmental delay during the first year of age, and suffered from intellectual disability from early childhood. They showed oculomotor apraxia, slurred and explosive speech, limb and gait ataxia, exaggerated deep tendon reflex, dystonic posture, and mirror movement in their hands. They developed mild cognitive abilities. Brain MRI in the index case revealed cerebellar atrophy. Next-generation sequencing revealed a homozygous synonymous variant in MRE11 (c.657C>T, p.Asn219=) that we show affects splicing. A complete absence of MRE11 transcripts in the index case suggested NMD and immunohistochemistry confirmed the absence of a stable protein.
Conclusions
Despite the critical role of MRE11A in double-strand break repair and its contribution to the Mre11/Rad50/Nbs1 complex, the absence of MRE11A is compatible with life.
Neurological defects, including microcephaly, ataxia or neurodegeneration, are a hallmark for autosomal recessive mutations in individual genes encoding components of the Mre11/Rad50/Nbs1 (MRN) complex. Mutations in the NBS1 encoding gene, NBN, are associated with Nijmegen breakage syndrome (NBS) (OMIM# 251260) characterized by microcephaly, immunodeficiency, growth and intellectual disability, radiosensitivity, and cancer predisposition.1–3 Mutations in RAD50 gene RAD50 are related to NBS-like disorder (NBSLD) (OMIM# 613078).4 The clinical features of patients with NBSLD are very similar to those with NBN, including microcephaly and intellectual disability but not infections, immunodeficiency, or cancer predisposition. Mutations in MRE11A homolog, double-strand break repair nuclease gene MRE11 are associated with a very rare chromosomal breakage syndrome (OMIM# 604391). The clinical features are characterized by progressive cerebellar degeneration and ionizing radiation hypersensitivity, similar to the ataxia telangiectasia (OMIM# 208900) caused by mutations in ATM, encoding ataxia telangiectasia mutated (ATM).5,6 The neurologic features have a later onset, slower progression and it is referred to as ataxia-telangiectasia-like disorder (ATLD) (OMIM# 604391).5,7,8 Unlike patients with ataxia telangiectasia, however, patients with ATLD show no telangiectasia or obvious immunodeficiency.7–9 In 1999, first association of ATLD with mutations in MRE11 was reported,5 followed by additional families with clinical features of ATLD9–16 or NBSLD.17
Here, we report a family with characteristic features of ATLD associated with a novel homozygous apparently synonymous variant in exon 7 of MRE11 that ablates normal splicing, induces nonsense-mediated mRNA decay (NMD), and deficiency of MRE11A protein.
Methods
Standard protocol approvals, registrations, and patient consents
The study was approved by the ethical standards of the relevant institutional review board, the Ethics Review Committee in the Gothenburg Region (Dn1: 842-14), and the Human Research Ethics Committee of the University of Western Australia. Informed consent was obtained from the parents included in this study after appropriate genetic counseling. Blood samples were obtained from patients and their parents.
Clinical evaluation
Medical history was obtained and physical examination was performed as part of routine clinical workup.
Genetic analysis
Next-generation sequencing (NGS) (a targeted neuromuscular sub-exomic sequencing [NSES] panel and/or whole exome sequencing [WES]) was performed on patients' DNA. Confirmatory bidirectional Sanger sequencing was performed in the patients and all available unaffected family members (e-Methods, links.lww.com/NXG/A128).
Analyses of muscle biopsy
A muscle biopsy was obtained in the index patient (V:2). Morphologic and histochemical analyses of paraffin-embedded muscle tissue were performed according to standard protocols. Sections of skeletal muscle tissue were processed for transcript, histologic and immunohistochemical assessments (e-Methods, links.lww.com/NXG/A128).
Transcript analysis
To analyze the impact of the c.657C>T variant in splicing efficiency of MRE11 exon 7 in the index case (V:2), polymerase chain reaction (PCR) was performed on complementary DNA with primer pairs covering exon 1 through 8 (e-Methods, links.lww.com/NXG/A128).
Chromosomal assay
Peripheral blood samples were obtained from 9 individuals of the family (III:1, IV:1, IV:2, IV:8, IV:9, V:1, V:2, V:3, and V:4). Chromosomal breakage tests using mitomycin C (MMC) induction in cultures was carried out according to standard protocols. Metaphases were stained and scored for spontaneous chromosomal anomalies. Twenty metaphase spreads were studied from routine culture, 100 spreads from culture were prepared with the addition of 2 concentrations of MMC. These were compared with 100 spreads from age-matched normal controls. Unfortunately, no additional tissue was available for assessment of the effect of MRE11 c.657C>T variant on sensitivity to ionizing radiation and the impact on NBS1 and RAD50 expression levels or the entire MRN complex.
Data availability
Data not published within the article are available online in supplemental material (links.lww.com/NXG/A128).
Results
Clinical characteristics of patients
Three affected siblings were born to an apparently healthy consanguineous couple (figure 1). Their clinical presentations were consistent with the characteristic features of ATLD. The affected individuals presented with developmental delay during the 1st year of age. They developed poor balance and suffered from mild intellectual disability from early childhood. The symptoms progressed gradually. Chromosomal study revealed a normal 46, XY (V:2 and V:4) or 46, XX (V:3) pattern. There was no history of severe or recurrent infections in the index case or in his 2 affected siblings. Follow-up at 23 (V:2), 20 (V:3), and 18 (V:4) years of age revealed oculomotor apraxia, slurred and explosive speech, limb and gait ataxia, exaggerated deep tendon reflexes (+3), dystonic posture, mirror movement in their hands, and poor balance (video 1). They had no sensory deficits. IQ was between 50 and 69 in all affected siblings. There was no facial dysmorphism (video 1); however, measurements including height, weight, and head circumference were below the 3rd percentile in all 3 affected siblings. Systemic examinations of the affected siblings were normal without any evidence of skeletal deformity or skin lesions. The laboratory evaluation, including thyroid function, liver function test, and alpha-fetoprotein, revealed levels in the reference range. Echocardiography of the youngest sibling (V:4) at 16 years of age was unremarkable. Nerve conduction velocity and EMG recording in the index case (V:2) did not show any neuropathic or myopathic features. Brain MRI in the index case (V:2) at 21 years of age revealed cerebellar atrophy (figure 2). The cousin of the affected siblings (V:5) (figure 1) was a boy with Down syndrome, who died at 6 months of age because of tetralogy of Fallot. However, he was not clinically evaluated for ATLD. There is no family history of cancer.
Figure 1. Pedigree of the family.

Pedigree and recessive inheritance of MRE11. In the pedigree, squares represent males; circles, females; open symbols, unaffected family members; and slash, deceased. The affected individuals are represented with shaded symbols. Arrow indicates the proband in the family (V:2). +/− indicates heterozygous presence of the variant; +/+ indicates homozygous appearance of the variant.
Figure 2. Sagittal T1-weighted MRI of brain.
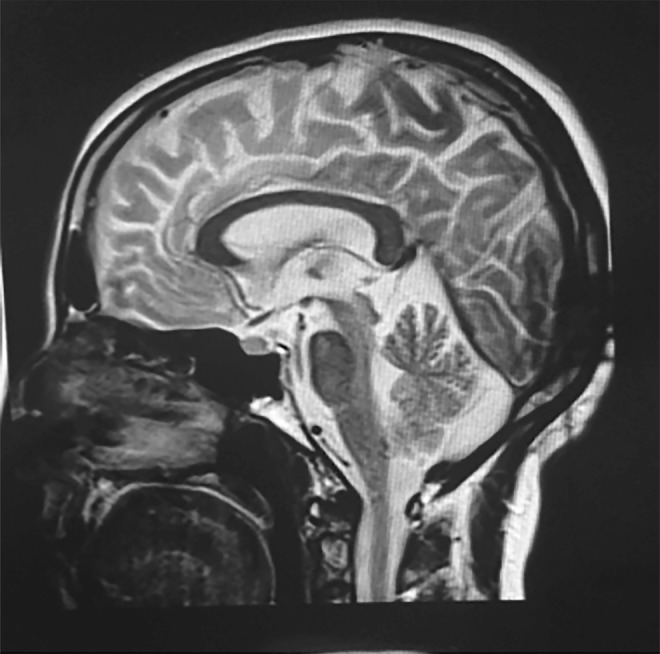
Brain MRI from the index case (V:2) shows significant atrophy of the cerebellar vermis. The brainstem is relatively preserved.
The proband (V:2) shows no dysmorphic facial features, however poor balance is evident.Download Supplementary Video 1 (36.4MB, mp4) via http://dx.doi.org/10.1212/000295_Video_1
Genetic findings
Data from NGS of DNA from 2 affected (V:2 and V:3) and 1 unaffected family members (V:1) were analyzed. Targeted sequencing of 336 known neurogenic disease genes, including 32 ataxia-associated genes in DNA from the index case V:2 identified a MRE11 variant changing the nucleotide 3 bases from the 3′ end of exon 7 (c.657C>T, rs775017362) in the homozygous state. The variant did not alter the coded amino acid (AAC>AAT, p.Asn219=). No rare, likely pathogenic heterozygous or homozygous variants were identified in other neurogenetic- or ataxia-associated genes included in the targeted panel, including ATM, APTX, or SETX. Simultaneous WES on DNA samples from individuals V:1, V:2, and V:3 to look for variants in novel disease genes was performed. The filtering strategy of initially concentrating on homozygous coding variants in known neurogenetic disease genes, selected based on variant databases Human Genome Mutation Database and ClinVar, and most recent literature allowed the identification of only the same homozygous c.657C>T variant in MRE11 as identified by NSES analysis. The MRE11 variant was identified in the heterozygous state in individual V:1 (figure 3A). The c.657C>T substitution was present at very low frequency in the genome Aggregation Databases (1/245,540 alleles). In silico analysis predicted the MRE11 variant to be possibly disease causing, presumably identified as potentially affecting splicing (MutationTaster, mutationtaster.org/). In silico prediction with the SpliceAid2 (introni.it/spliceaid.html) indicated that the variant was located in the splicing regulatory sequences, suggesting altered splicing factors binding sites. In addition, in silico prediction with the Human Splicing Finder version 3.0 and MaxEntScan suggested that the variant has a deleterious effect on the gene and creates an exonic splice site loss, leading to a mRNA frameshift and subsequently to a premature termination codon.
Figure 3. Genetic findings and expression analysis of MRE11A at transcript and protein levels.

(A) Sanger sequence analysis demonstrates the segregation of the MRE11 c.657C>T variant in the family. The unaffected parents (III:1 and (IV:2) and additional family members (IV:1, IV:8, IV:9, and V:1) are heterozygous for the MRE11 c.657C>T variant. The affected individuals for whom DNA was available (V:2, V:3, and V:4) are homozygous for the variant. The variant does not lead to alteration of asparagine amino acid (AAC>AAT, p.Asn219=). The variant is located in splice region (indicated by a box) changing the nucleotide of the 3′ end of exon 7 (ENST00000323929.7). The 5′ end of intron 7 is shown by an arrow. (B) reverse-transcriptase polymerase chain reaction analysis of RNA from skeletal muscle tissues of the index case (V:2) and a control. Expression analysis of fragment covering exon 1 through 8 coding regions of MRE11 (1168 bp) at the transcript level in the patient and a control indicated the absence of MRE11 transcript in the patient. Amplification of ACTB served as quality control of complementary DNA. (C) Immunostaining of MRE11A in skeletal muscle biopsy specimen from the index patient (V:2) indicates no nucleoplasm expression, whereas the control muscle specimen shows positive immunostaining of MRE11A localized to nucleoplasm. Scale bar, 50 µm.
The appearance of the MRE11 variant was examined in all available family members by sequencing analysis (figure 3A). Sanger sequencing confirmed segregation of the variant with the disease phenotype. The 3 affected individuals (V:2, V:3, and V:4) were homozygous for the c.657C>T variant; the unaffected parents (III:1 and IV:2) and 1 unaffected sibling (V:1) were heterozygous carriers of the MRE11 variant. In addition, the unaffected individuals IV:1, IV:8, and IV:9 were heterozygous for the MRE11 variant (figure 3A).
Transcript analysis
Muscle biopsy from the index case (V:2) was available for testing the effects of the homozygous c.657C>T MRE11 exon 7 variant on transcript and protein levels. In silico prediction suggested splicing defect with a likely consequence of a premature termination codon. Accordingly, the reverse-transcriptase PCR analysis indicated no detectable expression levels of MRE11 transcript in the index case (figure 3B), suggesting that this variant destabilizes the transcript by NMD.
Histological and immunohistochemical analysis of skeletal muscle
Immunohistochemical analysis on the muscle biopsy from the index case (V:2) was performed to assess the nuclear expression of MRE11A protein. Skeletal muscle biopsies from 2 individuals without neurodegenerative disorders were used as controls. In contrast to control skeletal muscle biopsies showing detectable expression levels of MRE11A protein in the nucleoplasm, MRE11A immunohistochemistry using a polyclonal antibody showed no staining in the skeletal muscle biopsy from the index case (figure 3C), indicating lack of a stable truncated protein.
Chromosomal aberration assay
Chromosomal breakage more than or equal to tenfold of control is clinically significant. Chromosomal breakage tests carried out for 9 individuals of the family (III:1, IV:1, IV:2, IV:8, IV:9, V:1, V:2, V:3, and V:4) indicated no significant structural alterations, such as chromosomal breaks, chromosomal translocations or gaps, when compared with the controls at 450–550 band resolution.
Discussion
In this study, we describe a family with 3 affected siblings, at 23, 20, and 18 years of age, diagnosed with ATLD. Consistent with this diagnosis, the siblings developed progressive cerebellar ataxia, developmental delay, and mild intellectual disability but with absence of telangiectasia or facial dysmorphism and no history of severe infections, immunodeficiency, or cancer.
Next-generation sequencing revealed homozygosity in the affected individuals for the MRE11 (c.657C>T, p.Asn219=) rs775017362 variant, which is present in population databases at frequencies compatible with recessive inheritance. The variant does not change the amino acid, but in silico analysis predicted that the variant would affect splicing efficiency, most likely resulting in exon 7 skipping, leading to a frameshift and a premature termination codon (p.Ser183Valfs*31). The homozygous variant could only be detected from the sequencing of genomic DNA. No MRE11 alleles were revealed by transcript assessment, making it likely that this variant destabilizes the transcript by NMD. This correlated with immunolabeling findings, demonstrating no MRE11A staining in the skeletal muscle biopsy from the index case, suggesting the absence of a stable protein.
So far, several families with MRE11 variants have been reported.5,9–13,17 A majority of reported variants in MRE11, homozygous or compound heterozygous splicing, nonsense or missense mutations, have been associated with a spectrum of clinical severity of ATLD5,9–13 (table). However, MRE11 variants have been described in 2 unrelated Japanese patients with characteristic features of NBSLD,17 which is otherwise associated with mutations in RAD50.4 In both Japanese patients, variants of close-by nucleotides to the mutated nucleotide in our patients were found. One patient was compound heterozygote for c.658A>C and c.659+1G>A and the other patient was compound heterozygote c.658A>C and c.338A>G. The MRE11 c.658A>C variant does not lead to alteration of amino acid (p.Arg220=) and the results from reverse-transcriptase PCR analysis in these patients carrying the transcript of the c.658A>C allele indicated that this variant leads to exon 7 skipping, but that some small amount of RNA was correctly spliced.17 Correspondingly, a reduced amount of wild-type–sized MRE11A protein was detected by immunoblot analysis.17 This indicates that c.657C>T alteration in our patients has a greater impact on splicing efficiency than the c.658A>C variant, leading to complete destabilization of the transcript by NMD and subsequent depletion of MRE11A.
Table.
Clinical findings of cases with recessive MRE11 variants


The MRN complex is involved in sensing of DNA double-strand breaks, DNA recombination, and multiple cell-cycle checkpoints.18,19 Cooperation between the ATM and MRN complexes is essential in the DNA damage response, which is not fully determined.20 MRE11A, a member of the MRN complex, is involved in homologous and mitotic and meiotic recombination, telomere length maintenance, and DNA double-strand break repair.21 MRE11A possesses DNA exonuclease and endonuclease activities that are highly conserved during evolution.22 The relatively mild impact of MRE11A deficiency, which permits viability in our patients, is intriguing. In addition, it is in sharp contrast to the early embryonic lethality of nuclease-deficient and null alleles of murine Mre11, associated with marked genome instability.21,23 However, despite the contribution of MRE11A in the MRN complex and the cooperation between the MRN complex and ATM in the DNA double-strand break repair pathways to maintain genomic integrity,20 loss of MRE11A only modestly impaired double-strand break repair in the chicken DT40 and human TK6 cell lines.24 Furthermore, animal models of NBS1 or MRE11A do not completely recapitulate phenotypes observed in NBS or ATLD patients, including the neurologic aspects.25–27
Notably, complete absence of ATM kinase, in patients with ataxia telangiectasia, a cancer-prone neurodegenerative disease, is not lethal.28 The majority of the variants in ATM in patients with classic ataxia telangiectasia are biallelic truncating mutations that result in a total loss of destabilized ATM protein.28 Given the vital role of ATM in the DNA damage response for DNA repair, cell cycle checkpoint activation, and apoptosis,29 the viability associated with loss of ATM in ataxia telangiectasia patients is intriguing. Mouse models deficient in ATM recapitulate accurate ataxia telangiectasia disease phenotypes,30–32 but loss of ATM kinase activity causes early embryonic lethality in mice, indicating that inhibition of ATM kinase activity does not equate to loss of the ATM protein.33 It was speculated that embryonic lethality in mice with ATM kinase inactivity was the result of ATM kinase recruitment at DNA breaks, which may impair the function of other proteins by blocking their access to DNA damage.33
Although MRE11A, NBS1, and RAD50 are components of the MRN complex, mutations in MRE11, NBN, and RAD50 are associated with different clinical phenotypes, suggesting that the components have distinct functions and roles independent of the MRN complex. Patients with NBS and ataxia telangiectasia have a predisposition to cancer, particularly an increased risk of developing lymphoid tumors,20 which may reflect the involvement of ATM and MRN complex in DNA damage response. Nevertheless, development of cancer in ATLD patients associated with MRE11 has not been a frequent finding in reported cases, with only 2 patients so far,10 and it is not a clinical feature in the family reported here. It thus remains unknown whether patients with ATLD have a predisposition to cancer, given the few patients with MRE11 mutation that have been described so far.
Patients with ataxia telangiectasia, NBS, and ATLD usually show an increased level of chromosomal translocation in the peripheral blood involving chromosome 7 and 14.20 Homozygous MRE11 variants often alter not only the levels of MRE11A but also the levels of 2 other components of the MRN complex, NBS1 and RAD50, leading to inactivation of the entire MRN protein complex.5,9,11,17 Although, the effect of MRE11 c.657C>T variant on sensitivity to ionizing radiation and the impact on NBS1 and RAD50 expression levels or the entire MRN complex in our patients remain unknown, none of the homozygous or heterozygous carriers of the MRE11 variant in our family show any chromosomal abnormalities. This is presumably because of retained contribution of damage sensor or mediators involved in the MRN complex and sustained ATM kinase activity in maintaining chromosomal integrity, as observed in other ATLD patients associated with MRE11 mutation.34 However, absence of chromosomal alterations in this family is intriguing and requires further investigations.
We describe patients with characteristic features of ATLD, associated with a homozygous MRE11 splicing variant leading to RNA decay and deficiency of MRE11A protein.
Acknowledgment
The authors thank the family members who provided samples and clinical information for this study. The authors thank Dr. Mansoor Salehi for providing us with the DNA samples and clinical data of patients.
Glossary
- ATLD
ataxia-telangiectasia-like disorder
- ATM
ataxia telangiectasia mutated
- MMC
mitomycin C
- NBS
Nijmegen breakage syndrome
- NBSLD
Nijmegen breakage syndrome-like disorder
- NGS
Next-generation sequencing
- NMD
nonsense-mediated mRNA decay
- NSES
neuromuscular sub-exomic sequencing
- WES
whole exome sequencing
Appendix 1. Author contributions

Study funding
The study was supported by grants from the European Union's Seventh Framework Programme for research, technological development and demonstration under grant agreement no. 608473 (H.T.) and the Swedish Research Council (H.T.). N. Laing is supported by Australian National Health and Medical Research Council Principal Research Fellowship (APP1117510), N. Laing and H. Goullée by NHMRC EU Collaborative Grant APP1055295. The funders had no role in the design of the study and collection, analysis, decision to publish, interpretation of data or preparation of the manuscript.
Disclosures
The authors report no disclosures relevant to the manuscript. M. Sedghi, M. Salari, A.-R. Moslemi, A. Kariminejad, M. Davis, H. Goulée, and B. Olsson report no disclosures. N. Laing has received travel funding from the World Muscle Society, the Asian Oceanian Myology Centre, Sanofi, and the Ottawa Neurology Meeting; has served on the editorial board for Neuromuscular Disorders Executive Associate Editor and Neuromuscular Disorders; has received publishing royalties for The Sarcomere and Skeletal muscle disease; has received research support from the governmental entity, the Australian National Health, and Medical Research Council; and has received research and support (from foundations and societies) from the U.S. Muscular Dystrophy Association, the Association Francaise contre les Myopathies, the Foundation Building Strength for Nemaline Myopathy, the Motor Neuron Disease Research Institute of Australia, the Western Australian Health and Medical Research Infrastructure Fund, and the Perpetual Foundation. H. Tajsharghi reports no disclosures. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/NG.
References
- 1.Matsuura S, Tauchi H, Nakamura A, et al. Positional cloning of the gene for Nijmegen breakage syndrome. Nat Genet 1998;19:179–181. [DOI] [PubMed] [Google Scholar]
- 2.Carney JP, Maser RS, Olivares H, et al. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell 1998;93:477–486. [DOI] [PubMed] [Google Scholar]
- 3.Varon R, Vissinga C, Platzer M, et al. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell 1998;93:467–476. [DOI] [PubMed] [Google Scholar]
- 4.Waltes R, Kalb R, Gatei M, et al. Human RAD50 deficiency in a Nijmegen breakage syndrome-like disorder. Am J Hum Genet 2009;84:605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stewart GS, Maser RS, Stankovic T, et al. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell 1999;99:577–587. [DOI] [PubMed] [Google Scholar]
- 6.Gatti RA, Boder E, Vinters HV, Sparkes RS, Norman A, Lange K. Ataxia-telangiectasia: an interdisciplinary approach to pathogenesis. Medicine (Baltimore) 1991;70:99–117. [PubMed] [Google Scholar]
- 7.Hernandez D, McConville CM, Stacey M, et al. A family showing no evidence of linkage between the ataxia telangiectasia gene and chromosome 11q22-23. J Med Genet 1993;30:135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klein C, Wenning GK, Quinn NP, Marsden CD. Ataxia without telangiectasia masquerading as benign hereditary chorea. Mov Disord 1996;11:217–220. [DOI] [PubMed] [Google Scholar]
- 9.Pitts SA, Kullar HS, Stankovic T, et al. hMRE11: genomic structure and a null mutation identified in a transcript protected from nonsense-mediated mRNA decay. Hum Mol Genet 2001;10:1155–1162. [DOI] [PubMed] [Google Scholar]
- 10.Uchisaka N, Takahashi N, Sato M, et al. Two brothers with ataxia-telangiectasia-like disorder with lung adenocarcinoma. J Pediatr 2009;155:435–438. [DOI] [PubMed] [Google Scholar]
- 11.Delia D, Piane M, Buscemi G, et al. MRE11 mutations and impaired ATM-dependent responses in an Italian family with ataxia-telangiectasia-like disorder. Hum Mol Genet 2004;13:2155–2163. [DOI] [PubMed] [Google Scholar]
- 12.Fernet M, Gribaa M, Salih MAM, Seidahmed MZ, Hall J, Koenig M. Identification and functional consequences of a novel MRE11 mutation affecting 10 Saudi Arabian patients with the ataxia telangiectasia-like disorder. Hum Mol Genet 2005;14:307–318. [DOI] [PubMed] [Google Scholar]
- 13.Bohlega SA, Shinwari JM, Al Sharif LJ, Khalil DS, Alkhairallah TS, Al Tassan NA. Clinical and molecular characterization of ataxia with oculomotor apraxia patients in Saudi Arabia. BMC Med Genet 2011;12:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chaki M, Airik R, Ghosh AK, et al. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell 2012;150:533–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nemeth AH, Kwasniewska AC, Lise S, et al. Next generation sequencing for molecular diagnosis of neurological disorders using ataxias as a model. Brain 2013;136:3106–3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miyamoto R, Morino H, Yoshizawa A, et al. Exome sequencing reveals a novel MRE11 mutation in a patient with progressive myoclonic ataxia. J Neurol Sci 2014;337:219–223. [DOI] [PubMed] [Google Scholar]
- 17.Matsumoto Y, Miyamoto T, Sakamoto H, et al. Two unrelated patients with MRE11A mutations and Nijmegen breakage syndrome-like severe microcephaly. DNA Repair (Amst) 2011;10:314–321. [DOI] [PubMed] [Google Scholar]
- 18.D'Amours D, Jackson SP. The Mre11 complex: at the crossroads of dna repair and checkpoint signalling. Nat Rev Mol Cell Biol 2002;3:317–327. [DOI] [PubMed] [Google Scholar]
- 19.Petrini JH, Stracker TH. The cellular response to DNA double-strand breaks: defining the sensors and mediators. Trends Cell Biol 2003;13:458–462. [DOI] [PubMed] [Google Scholar]
- 20.Taylor AM, Groom A, Byrd PJ. Ataxia-telangiectasia-like disorder (ATLD)-its clinical presentation and molecular basis. DNA Repair (Amst) 2004;3:1219–1225. [DOI] [PubMed] [Google Scholar]
- 21.Buis J, Wu Y, Deng Y, et al. Mre11 nuclease activity has essential roles in DNA repair and genomic stability distinct from ATM activation. Cell 2008;135:85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furuse M, Nagase Y, Tsubouchi H, Murakami-Murofushi K, Shibata T, Ohta K. Distinct roles of two separable in vitro activities of yeast Mre11 in mitotic and meiotic recombination. EMBO J 1998;17:6412–6425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiao Y, Weaver DT. Conditional gene targeted deletion by Cre recombinase demonstrates the requirement for the double-strand break repair Mre11 protein in murine embryonic stem cells. Nucleic Acids Res 1997;25:2985–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoa NN, Akagawa R, Yamasaki T, et al. Relative contribution of four nucleases, CtIP, Dna2, Exo1 and Mre11, to the initial step of DNA double-strand break repair by homologous recombination in both the chicken DT40 and human TK6 cell lines. Genes Cells 2015;20:1059–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Theunissen JW, Kaplan MI, Hunt PA, et al. Checkpoint failure and chromosomal instability without lymphomagenesis in Mre11(ATLD1/ATLD1) mice. Mol Cell 2003;12:1511–1523. [DOI] [PubMed] [Google Scholar]
- 26.Williams BR, Mirzoeva OK, Morgan WF, Lin J, Dunnick W, Petrini JH. A murine model of Nijmegen breakage syndrome. Curr Biol 2002;12:648–653. [DOI] [PubMed] [Google Scholar]
- 27.Shull ER, Lee Y, Nakane H, et al. Differential DNA damage signaling accounts for distinct neural apoptotic responses in ATLD and NBS. Genes Dev 2009;23:171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gilad S, Khosravi R, Shkedy D, et al. Predominance of null mutations in ataxia-telangiectasia. Hum Mol Genet 1996;5:433–439. [DOI] [PubMed] [Google Scholar]
- 29.Banin S, Moyal L, Shieh S, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 1998;281:1674–1677. [DOI] [PubMed] [Google Scholar]
- 30.Barlow C, Hirotsune S, Paylor R, et al. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell 1996;86:159–171. [DOI] [PubMed] [Google Scholar]
- 31.Elson A, Wang Y, Daugherty CJ, et al. Pleiotropic defects in ataxia-telangiectasia protein-deficient mice. Proc Natl Acad Sci U S A 1996;93:13084–13089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu Y, Ashley T, Brainerd EE, Bronson RT, Meyn MS, Baltimore D. Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev 1996;10:2411–2422. [DOI] [PubMed] [Google Scholar]
- 33.Daniel JA, Pellegrini M, Lee BS, et al. Loss of ATM kinase activity leads to embryonic lethality in mice. J Cell Biol 2012;198:295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Regal JA, Festerling TA, Buis JM, Ferguson DO. Disease-associated MRE11 mutants impact ATM/ATR DNA damage signaling by distinct mechanisms. Hum Mol Genet 2013;22:5146–5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The proband (V:2) shows no dysmorphic facial features, however poor balance is evident.Download Supplementary Video 1 (36.4MB, mp4) via http://dx.doi.org/10.1212/000295_Video_1
Data Availability Statement
Data not published within the article are available online in supplemental material (links.lww.com/NXG/A128).