

Abstract
In Alzheimer’s disease (AD), the brain’s primary immune cells microglia become activated and are found in close apposition to amyloid beta (Aβ) protein plaques and neurofibrillary tangles (NFT). The present study evaluated microglia density and morphology in a large group of aged chimpanzees (n = 20, ages 37-62 years) with varying degrees of AD-like pathology. Using immunohistochemical and stereological techniques, we quantified the density of activated microglia and morphological variants (ramified, intermediate, and amoeboid) in postmortem chimpanzee brain samples from prefrontal cortex, middle temporal gyrus, and hippocampus, areas that show a high degree of AD pathology in humans. Microglia measurements were compared to pathological markers of AD in these cases. Activated microglia densities were consistently present across brain areas. In the hippocampus, CA3 displayed a higher density than CA1. Aβ42 plaque volume was positively correlated with higher microglial activation and with an intermediate morphology in the hippocampus. Aβ42-positive vessel volume was associated with increased hippocampal microglial activation. Activated microglia density and morphology were not associated with age, sex, pretangle density, NFT density, or tau neuritic cluster density. Aged chimpanzees displayed comparable patterns of activated microglia phenotypes as well as an association of increased microglial activation and morphological changes similar to AD patients. In contrast to human AD brains, activated microglia density was not significantly correlated with tau lesions. This evidence suggests that the chimpanzee brain may be relatively preserved during normal aging processes but not entirely protected from neurodegeneration as previously assumed.
Keywords: microglia, Alzheimer’s disease, chimpanzee, neuroinflammation, amyloid beta protein, neurofibrillary tangle, RRID: AB_2313952, RRID: AB_2313890, RRID: AB_223647, RRID: AB_2315150, RRID: AB_839504
Graphical abstract
In Alzheimer’s disease, microglia activation occurs near amyloid beta (Aβ) protein plaques and neurofibrillary tangles. Analyses of microglia densities in aged chimpanzee brains revealed increased microglial activation with greater Aβ42 plaque and vessel volumes, but not tau lesions, in the hippocampus, indicating chimpanzees are not entirely protected from neurodegeneration.
1. INTRODUCTION
Microglia are the primary immune cells in the brain and are widely dispersed across neocortical layers but show higher concentrations in the hippocampus (Graeber & Streit, 1990; Kongsui, Beynon, Johnson, & Walker, 2014; Lawson, Perry, Dri, & Gordon, 1990; Ransohoff & Perry, 2009). In the healthy brain, microglia use highly motile ramified processes to survey the cellular environment (Nimmerjahn, Kirchhoff, & Helmchen, 2005). When infection, trauma, or neurodegeneration occur, microglia undergo rapid changes in cell morphology, gene expression, and function, a process known as microglial activation (Finsen, Lehrmann, Castellano, Kiefer, & Zimmer, 1996; Gehrmann et al., 1992; Jørgensen, 1993; Kofler et al., 2012; Lehrmann, Christensen, Zimmer, Diemer, & Finsen, 1997; Morioka, Kalehua, & Streit, 1992, 1993). Phenotypically, microglial activation results in a graded response of decreased arborization, enlarged cell soma, and shortened or total loss of cellular processes. Activated microglia also migrate to lesion or infection sites and can increase in density through mitotic proliferation to provide additional defense and restoration of tissue homeostasis (Kettenmann, Hanisch, Noda, & Verkhratsky, 2011).
Microglia are macrophages implicated in the inflammatory response to the formation of amyloid beta protein (Aβ) plaques and neurofibrillary tangles (NFT) in Alzheimer’s disease (AD) (Akiyama et al., 2000; Cunningham, 2013; Gandy & Heppner, 2013; Heneka, Kummer, & Latz, 2014; Hickman & El Khoury, 2014; Krstic & Knuesel, 2013; V Hugh Perry & Holmes, 2014; Prokop, Miller, & Heppner, 2013; Sudduth, Schmitt, Nelson, & Wilcock, 2013). It has been proposed that activated microglia stimulate neurons to overproduce Aβ, which results in the formation of extracellular plaques and hyperphosphorylation of the microtubule-stabilizing protein tau (Meraz Rios, Toral-Rios, Franco-Bocanegra, Villeda-Hernández, & Campos-Peña, 2013; Wyss-Coray & Rogers, 2012). This process promotes increased microglial activation creating a positive feedback loop that plays a role in the pathological cascade that drives the development of AD.
In vitro work in human neuronal cell lines has demonstrated that inflammatory factors released from stimulated microglia upregulated mRNA and protein expression of all six tau isoforms and the production of amyloid precursor protein (APP), which is cleaved into Aβ peptides in AD (Lee, McGeer, & McGeer, 2015). Activated microglia migrate towards Aβ plaques and NFT, participate in the clearance of Aβ, and proliferate at sites of Aβ deposition in the hippocampus (Cagnin et al., 2001 ; Hickman, Allison, & El Khoury, 2008; Higuchi, 2009; Maier et al., 2008; Maphis et al., 2015; Marlatt et al., 2014; Mattiace, Davies, & Dickson, 1990; Lynette G. Sheffield, Marquis, & Berman, 2000; Tahara et al., 2006; Von Gunten et al., 2005). Studies have shown colocalization of NFT, neuropil threads, and neuritic plaques displaying dystrophic (fragmented) microglial cells and an increase in microglia density in AD brains with high densities of NFT (Ekonomou et al., 2015; Streit, Braak, Xue, & Bechmann, 2009). Furthermore, the pharmacological inhibition of colony-stimulating factor 1 receptor, a necessary component for microglial signaling and survival, reduced 80% of total microglia and rescued dendritic loss, prevented neuronal loss, and improved contextual memory despite unaltered Aβ plaque loads (Spangenberg et al., 2016).
Nonhuman primates also display microglial activation in response to Aβ deposits. Microinjection of insoluble fibrillary Aβ (fAβ) in the cerebral cortex of aged rhesus monkeys (Macaca mulatta) resulted in profound neuron loss, tau phosphorylation, and microglial proliferation (Geula et al., 1998). Moreover, inhibition of microglial activation with a macrophage/microglia inhibitory factor eliminated fAβ toxicity in elderly macaque monkeys (Leung et al., 2011). Yet, until recently, evidence of the spontaneous co-occurrence of both Aβ and tau pathological markers of AD had not been found in species other than humans with the exception of a single aged chimpanzee and aged gorillas (Perez et al., 2013, 2016; Rosen et al., 2008). Our recent work in aged chimpanzees (Pan troglodytes) demonstrated both Aβ and tau lesions, suggesting the pathologic hallmarks of AD are not limited to the human brain (Edler et al., 2017). This study builds upon that foundation by evaluating activated microglia density and morphological changes of microglia in association with AD pathology in the same group of aged chimpanzees.
2. MATERIALS AND METHODS
2.1. Specimens and Sample Processing
Postmortem brain samples from 20 aged chimpanzees (Pan troglodytes; Table 1) were acquired from American Zoo and Aquarium-accredited zoos, American Association for Accreditation of Laboratory Animal Care-accredited research institutions, and the National Chimpanzee Brain Resource. All animals were maintained in accordance with each institution’s animal care and use guidelines. Ninety-five percent of the founder chimpanzee population in the United States is Pan troglodytes, and all chimpanzees in this study were classified as this subspecies (Ely et al., 2005). Chimpanzees in this study did not participate in formal behavioral or cognitive testing. Available health information for these apes was previously reported in supplemental table (S1) in Edler et al., 2017. Sex and age was balanced as equally as possible. Samples from four brain regions, including prefrontal cortex (PFC, Brodmann’s areas 9 and 10), middle temporal gyrus (MTG, Brodmann’s area 21), and hippocampal subregions CA1 and CA3, were obtained from 8 male (ages 39-62 years) and 12 female (ages 37-58 years) chimpanzees.
Table 1.
Subject demographics
| Subject | Age | Sex | Pathology Score | Aβ Plaque (Thal) | Aβ Vessel (CAA) | Pretangle | NFT (Braak) | Tau Neuritic Cluster (CERAD) |
|---|---|---|---|---|---|---|---|---|
| 1 | 37 | F | 4 | Phase 1 | Minimal | + | − | Sparse |
| 2 | 39 | F | 0 | − | Minimal | + | − | − |
| 3 | 40 | F | 2 | Phase 1 | Minimal | + | − | − |
| 4 | 40 | F | 0 | − | Minimal | + | − | − |
| 5 | 41 | F | 0 | − | Minimal | + | − | − |
| 6 | 43 | F | 7 | Phase 2 | Mild | + | − | Sparse |
| 7 | 44 | F | 0 | − | Minimal | + | − | − |
| 8 | 45 | F | 5 | Phase 1 | Mild | + | Stage II | Sparse |
| 9 | 49 | F | 3 | Phase 1 | Minimal | + | Stage I | − |
| 10 | 51 | F | 4 | Phase 1 | Minimal | + | − | Sparse |
| 11 | 58 | F | 11 | Phase 3 | Moderate | + | − | Moderate |
| 12 | 58 | F | 9 | Phase 1 | Severe | + | Stage I | Sparse |
| 13 | 39 | M | 9 | − | Minimal | + | +* | Moderate |
| 14 | 40 | M | 1 | − | Minimal | + | − | Sparse |
| 15 | 41 | M | 4 | Phase 1 | Minimal | + | − | Sparse |
| 16 | 41 | M | 4 | − | Mild | + | − | Sparse |
| 17 | 41 | M | 0 | − | Minimal | + | − | − |
| 18 | 46 | M | 3 | Phase 2 | Minimal | + | − | Sparse |
| 19 | 57 | M | 19 | Phase 4 | Severe | + | Stage V | Moderate |
| 20 | 62 | M | 10 | Phase 4 | Severe | + | − | − |
Aβ (amyloid-beta protein) includes APP/Aβ and Aβ42, CAA (cerebral amyloid angiopathy), CERAD (Consortium to Establish a Registry for AD), F (female), M (male), NFT (neurofibrillary tangle), Thal (clinical phases for accumulation of Aβ plaques), Braak (clinical staging system used to classify tangle distribution). Pathology score refers to a pathology scoring system adapted from staging guidelines for Aβ and NFT deposition in AD and CAA with higher numbers corresponding to more severe pathology (Edler et al., 2017).
+ positive for pathology,
does not follow staging pattern.
Depending on availability, samples were taken from the right or left hemispheres. Brains were collected postmortem (postmortem interval < 20 h) and immersion fixed in 10% buffered formalin solution for at least 10 days. Specimens were transferred to a 0.1 M buffered saline solution containing 0.1% sodium azide at 4°C for storage prior to sectioning. Samples were cryoprotected in a graded series of 10, 20, and 30% sucrose solutions, and cut frozen into 40 μm-thick sections perpendicular to the main axis of the gyrus contained in each block using a Leica SM2000R freezing sliding microtome (Buffalo Grove, IL). Sections were placed into individual centrifuge tubes containing a cyroprotection solution (30% dH2O, 30% ethylene glycol, 30% glycerol, 10% 0.244 M phosphate-buffered saline [PBS]), numbered sequentially, and stored at −20°C until immunohistochemical processing. Every tenth section was stained for Nissl substance with a 0.5% cresyl violet solution to reveal cell somata and to define cytoarchitectural boundaries.
2.2. Identification and Regional Sampling
All regions were identified using Nissl-stained sections. Brain areas and layers were selected based on NFT staging and Aβ deposition phases as reported in humans with AD and in aged chimpanzees (Braak & Braak, 1991; Edler et al., 2017; M Gearing, Rebeck, Hyman, Tigges, & Mirra, 1994; Marla Gearing, Tigges, Mori, & Mirra, 1996; Montine et al., 2012; Rosen et al., 2008). Sampled regions included layer III in areas 9 and 10 of the dorsolateral PFC, layer III in area 21 of the MTG, and the stratum pyramidale in the hippocampal subfields CA1 and CA3. In AD, pyramidal neurons in layers III and V of the neocortex and stratum pyramidale in the CA1 field display extensive neuron and synapse loss, and the distribution of neuritic Aβ plaques and NFT is most prevalent in these cortical layers (Akram et al., 2008; Bussière et al., 2003; P R Hof, Cox, & Morrison, 1990; West, Coleman, Flood, & Troncoso, 1994).
2.3. Immunohistochemistry and Antibody Characterization
Table 2 describes the antibodies used in the present study including: 6E10, a mouse IgG1 antibody raised against amino acid residue 1-16 (EFRHDS) of Aβ, with an epitope at residues 3-8, which recognizes Aβ isoforms and its precursor APP protein (1:7,500, Covance SIG-39320 [Biolegend], San Diego, CA); Aβ42, a rabbit IgG monoclonal antibody raised against amino acids 707-713 of the C-terminus of human Aβ A4 protein that does not cross react with Aβ40 (1:2,500, Invitrogen 700254, Grand Island, NY); AT8, a mouse IgG1 monoclonal antibody raised against partially purified human PHF-tau, with an epitope at residues phosphoserine202/phosphothreonine205 that recognizes a tau phosphorylated isoform (1:2,500, ThermoFisher MN1020, Waltham, MA); PHF-1, a mouse IgG1 monoclonal antibody for tau with epitopes near phosphoserines 396 and 404 (1:10,000, Peter Davies, New York, NY); and Iba1 (1:10,000, Wako 019-19741), a rabbit IgG polyclonal antibody raised to synthetic peptide corresponding to amino acids 81-93 of human Allograft Inflammatory Factor-1 (AIF-1, TGPPAKKAISELP). Antibody specificity was determined by western blot 6E10 (Covance), Aβ42 (Invitrogen), AT8 (Goedert, Jakes, & Vanmechelen, 1995), PHF-1 (Otvos et al., 1994), and Iba1 (Wako).
Table 2.
Summary of antibodies
| Antigen | Antibody | Pathology/Protein | Dilution | Company/Catalog # |
|---|---|---|---|---|
| APP/Aβ (6E10) | mouse IgG1 monoclonal to Aβ residues 1-16 (EFRHDS) | APP/Aβ40/Aβ42 plaques and vessels | 1:7,500 | Covance (Biolegend), SIG-39320, RRID: AB_2313952 |
| Aβ42 | rabbit IgG monoclonal raised against C-terminus of human Aβ A4 protein | Aβ42 plaques and vessels | 1:2,500 | Invitrogen, 700254, RRID: AB_2313890 |
| Tau (AT8) | mouse IgG-ι monoclonal to a partially purified human PHF-tau with epitope at residues pSer202/pThr205 | early, middle, and late tau | 1:2.500 | ThermoFisher, MN1020, RRID: AB_223647 |
| Tau (PHF-1) | mouse IgG1 monoclonal to epitopes near phosphorylated Ser396/404 | middle and late tau | 1:10,000 | Gift from Peter Davies, RRID: AB_2315150 |
| Iba1 | rabbit IgG polyclonal raised against the C-terminus of amino acids 81-93 of human AIF-1 | activated microglia | 1:10,000 | Wako, 019-19741, RRID: AB_839504 |
Every 20th section was immunohistochemically processed using a rabbit anti-ionized calcium-binding adapter molecule 1 (Iba 1) polyclonal antibody recognizing its C-terminus (1:10,000 dilution, Wako, 019-19741) following established protocols utilizing the avidin-biotin-peroxidase method (Raghanti et al., 2008, 2009). Iba1 is specifically expressed in macrophage/microglia and detects activated microglia. Additional sections were double immunostained for Iba1 and tau, either using antibodies PHF-1 (1:10,000 dilution, gift from Dr. Peter Davies) or AT8 (1:2,500, ThermoFisher, MN1020), to determine the co-localization of microglia and NFT. Free-floating sections were pretreated for antigen retrieval by incubation in 0.05% citraconic acid (pH 7.4) at 86°C for 30 min. Endogenous peroxidase was quenched in a hydrogen peroxide (2.5%) and methanol (75%) solution for 20 min at room temperature (RT), and sections were pre-blocked for 1 h in a solution of 0.1 M phosphate-buffered saline (PBS, pH 7.4), 0.6% Triton X, 4% normal serum, and 5% bovine serum albumin at RT. Sections then were placed in primary antibody diluted in PBS for 48 h at 4°C. Next, sections were incubated in a biotinylated secondary antibody (1:200 dilution) in a solution of PBS and 2% normal serum (1 h, RT), followed by an avidin-peroxidase complex (1 h, RT, PK6100, Vector Laboratories, Burlingame, CA) and either 3,3′-diaminobenzidine with nickel enhancement or Vector NovaRED (SK-4100/SK-4800, Vector Laboratories). For immunofluorescent staining, free-floating sections were rinsed in PBS (5 min × 6) and pre-blocked (1 h, RT) in a 10% BSA working stock, 1% Triton X, and PBS. After rinsing, sections then were placed in a primary antibody cocktail of AT8 (1:2,500) and Iba1 (1:2,000; Table 2) diluted in PBS and 10% BSA working stock for 48 h at 4°C. Sections then were rinsed and incubated in a secondary antibody cocktail (1:200) of goat anti-mouse Alexa Fluor 488 (Abcam), goat anti-rabbit Alexa Fluor 594 (Thermo Fisher), PBS, and 10% BSA working stock (1 h, RT). After a final rinse, sections were mounted on slides, dried, coverslipped using Vector Hard Set Mounting Media, and stored at 4°C. Photomicrographs of the immunofluorescent staining were taken on a Leica DMi8 confocal microscope at 63x (N.A. 1.30 oil).
2.4. Morphology Identification
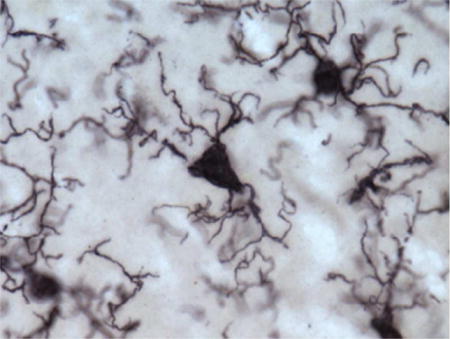
To measure potential phagocytic activity in microglia, we quantified densities for three activated morphological subtypes—ramified, intermediate, and amoeboid—as previously defined (Kettenmann et al., 2011). Briefly, ramified morphology included long, highly arborized processes and a small cell soma (Fig. 1a). The intermediate stage of activation was noted by shorter, thicker prolongations, less arborization, and an enlarged cell body (Fig. 1 b, d). An amoeboid shape with a round or enlarged cell soma and loss of processes identified the final phase (Fig. 1c).
Figure 1.
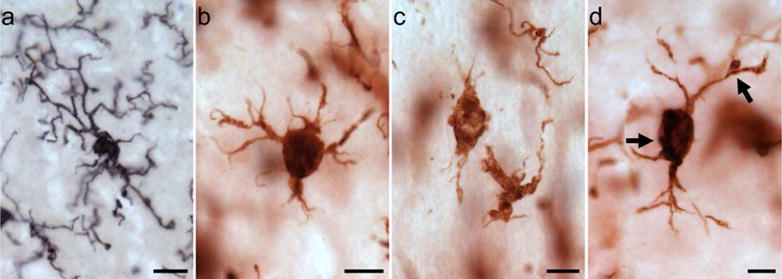
Photomicrographs of activated microglia (Iba1-ir) morphologies in the MTG (a) and PFC (b–d) of a 39-year-old female (a; subject 2) and a 40-year-old female (b–d; subject 3) chimpanzee: (a) ramified morphology with small cell soma and fine processes, (b) intermediate morphology with enlarged cell soma and thickened, shorter processes, (c) amoeboid morphology with loss of nearly all processes, and (d) PHF-1/Iba1 expressing microglia with intermediate morphology (black arrows denote PHF-1 staining). Scale bar for each panel = 250 μm.
2.5. Data Acquisition
Quantitative analyses were performed using computer-assisted stereology with an Olympus BX-51 photomicroscope equipped with a digital camera and Stereoinvestigator software version 11 (MBF Bioscience, Williston, VT) by a single observer blinded to age and sex of study subjects. Initial subsampling techniques were performed for each probe to determine appropriate sampling parameters (Slomianka & West, 2005). Densities for Iba1-immunoreactive (ir) activated microglia, activated microglia displaying ramified, intermediate, and amoeboid morphologies, and microglia expressing PHF-1/Iba1 immunoreactivity were obtained using the optical fractionator probe at 40x (N.A. 0.75) under Köhler illumination. Grid size was set at 250 × 250 μm with a disector height of 8 μm and an upper and lower guard zone of 2%. Beginning at a random starting point, three equidistant sections (every 20th section) per region of interest and individual were selected for analysis. Mounted section thickness was measured every fifth sampling site. A different marker for each morphology type was placed when encountered within the optical disector frame. Occasionally morphology could not be distinguished, and in those instances, a morphology marker was not placed; undetermined morphology accounted for approximately 4% of activated microglia. An additional marker was placed when microglia were immunoreactive for both PHF-1 and Iba1. Microglia densities (per mm3) for each region were calculated as the population estimate divided by planimetric volume (Sherwood, Raghanti, & Wenstrup, 2005). To correct for tissue shrinkage in the z- axis, the height of the disector was multiplied by the ratio of section thickness to the actual weighted mean thickness after mounting and dehydration. No correction was necessary for the x and y dimensions because shrinkage in section surface area is minimal (Dorph-Petersen, Nyengaard, & Gundersen, 2001). For each individual, densities for PFC and MTG were averaged to calculate neocortical (NC) density. The same process was executed for CA1 and CA3 to compute average hippocampal (HC) density. Densities for all four regions were averaged for total activated, ramified, intermediate, amoeboid, and PHF-1/Iba1-ir microglia densities. The ratio of each morphology type to total activated microglia density was calculated for NC and HC, by dividing the region’s ramified, intermediate, and amoeboid density by the region’s activated microglia density (e.g., NC ramified density/NC activated microglia density). Penetration of antibodies was determined by examining each section through the z-axis. The mean number of sampling sites for each area per individual was 48 ± 10 and mean number of markers for each area per individual was 246 ± 82.
2.6. Statistical Analyses
We previously collected data for APP/Aβ and Aβ42 plaque and vessel volume as well as AT8-ir pretangle, NFT, and tau neuritic cluster densities for each case included in the present study (Edler et al., 2017). All densities and volumes were checked for linearity, and because skewness from means was close to zero, densities and volumes were transformed using the formula: arcsin [sqrt (density/1,000)]. To evaluate neuropathologic changes for each individual, a value was computed utilizing a pathology scoring system adapted from staging guidelines for Aβ and NFT deposition in AD and CAA (Edler et al., 2017). Principal component analysis (PCA) was performed to reduce the number of pathological variables to the most relevant factors, and regression factors (PCA-generated brain pathology score) from this prior analysis were employed for further regression analyses with microglia densities in this study. Regression analyses were utilized to determine relationships between NC, HC, and total activated microglia density and morphological densities and ratios with chronological age, PCA-generated pathology score, sex, APP/Aβ and Aβ42 plaque and vessel volumes (%), and pretangle, NFT, and tau neuritic cluster densities (mm3). Two-way ANOVAs with Bonferroni post hoc tests were used to examine sex and brain region differences in activated microglia and morphology densities. Statistical analyses were conducted using IBM SPSS Statistics, Version 22 (Armonk, NY). The level of significance (α) was set at 0.05.
3. RESULTS
3.1. Regional Densities of Activated Microglia
Activated microglia morphologies subtypes were observed throughout the neocortex and hippocampus (Figs. 1,2). Dystrophic microglia were found mainly in neocortical layers I and II (Fig. 2e). Iba1-ir ramified microglia displayed a spherical, triangular, or elongated shape and numerous fine fibers (Fig. 2f–h). Intermediate and amoeboid morphology often exhibited an increased intensity of staining and were observed in close proximity to blood vessels and tau pathology (Fig. 2i–l). Microglia were occasionally taupositive (PHF-1; Fig. 2c–d), and territories of intermediate and amoeboid-shaped microglia sometimes overlapped compared to the typical pattern of extracellular space seen between ramified microglia.
Figure 2.
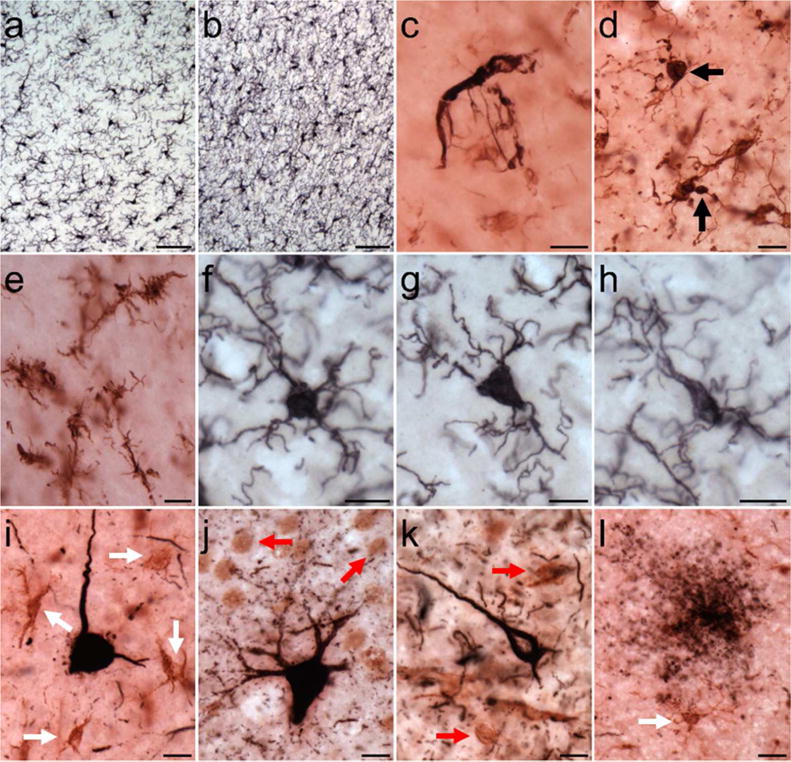
Photomicrographs of Iba1 (a,b, and f–h), PHF-1/Iba1 (c–e), and AT8/Iba1 (i–l) immunostaining in aged chimpanzees (subject 20: a–b; subject 13: c,i,k; subject 4: d; subject 8: e; subject 2: f–h; subject 19: j,l): (a) microglia in neocortex, (b) microglia in hippocampus, (c,d) PHF-1-ir microglia (black arrows), (e) dystrophic microglia in neocortex, (f–h) ramified microglia with spherical (f), triangular (g), and elongated (h) cell somas in neocortex, (i–j) AT8-ir pretangles surrounded by intermediate microglia (i, white arrows) in the neocortex and amoeboid microglia (j, red arrows) in the hippocampus, (k) AT8-ir NFT adjacent to amoeboid microglia in neocortex, and (l) tau neuritic cluster next to intermediate microglia. Scale bars = 250 μm (a–b) or 25 μm (c–l).
Activated microglia density was collected in all 20 chimpanzees, and the average density across the four brain regions examined was 5,209/mm3. Two-way ANOVA revealed a significant main effect of region (F372 = 4.79, p < 0.01) but not sex (F172 = 0.11, p = 0.74) with no interaction (F372 = 0.19, p = 0.90). A Bonferroni post hoc test did not detect differences between the NC (5,187/mm3) and HC (5,231/mm3) or within the neocortex between PFC (5,702/mm3) and MTG (4,672/mm3) (all p values ≥ 0.07). However, activated microglia density was significantly greater in CA3 (6,384/mm3) than CA1 (4,078/mm3) in the hippocampus (p ≤ 0.01; Fig. 3).
Figure 3.
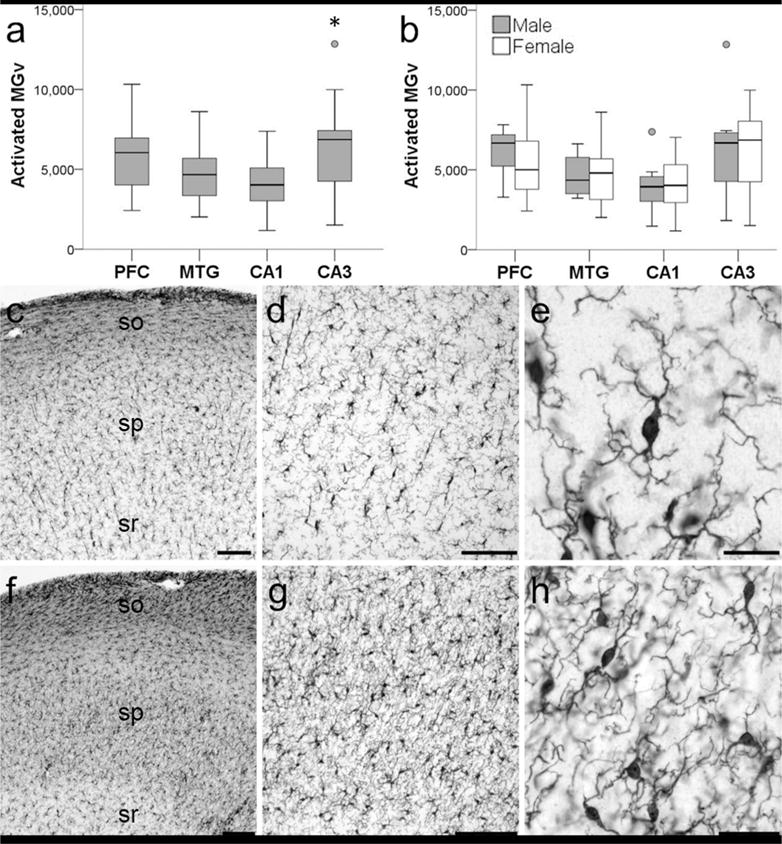
Photomicrographs showing activated Iba 1-ir microglia in the hippocampus of a 62-year old male chimpanzee (subject 20). Activated Iba1-ir microglia density (MGv, mm3) was significantly higher in CA3 compared to CA1 in the hippocampus of aged chimpanzees (a; * represents a significant difference, p = 0.01). Sex differences were not observed in activated Ibal-ir microglia density (b; p = 0.68). Whiskers represent 1 SD. Small circles represent outliers (1.5 × interquartile range). Hippocampal subfield CA1 (c–e) has significantly decreased microglial activation than subfield CA3 (f–h). Stratum oriens (so), stratum pyramidale (sp), stratum radiatum (sr). Scale bars = 250 μm (c–d, f–g) or 25 μm (e,h).
Microglia densities were compiled in 18 subjects for each activation state based on morphology, since morphology could not be consistently identified in all regions for two individuals. Of the total activated microglia, ramified morphology accounted for 9%, intermediate for 79%, and amoeboid for 8%. Morphological composition was similar in the NC (ramified, 11%; intermediate, 78%; and amoeboid, 9%) and HC (ramified, 8%; intermediate, 75%; and amoeboid, 6%; Fig. 4). Average ramified microglia density across brain areas was 428/mm3. Ramified microglia densities were comparable between NC (PFC = 551/mm3, MTG = 368/mm3) and HC (CA1 = 237/mm3, CA3 = 653/mm3), and analyses did not find significant effects for region (F347 = 1.62, p = 0.20), sex (F147 = 0.80, p = 0.38), and interaction between region and sex (F347 = 0.20, p = 0.90; Fig. 5a,d). Average intermediate microglia density was 4,195/mm3 among all regions, and intermediate microglia density in NC was 4,110/mm3 and in HC was 4,689/mm3. Intermediate microglia density in PFC (4,274/mm3) was greater than MTG (3,775/mm3), and CA3 density (5,691/mm3) was higher than CA1 (3,686/mm3). ANOVA yielded a significant effect for region (F349 = 2.91, p = 0.04), but not for sex (F149 = 0.69, p = 0.41) and regional interaction (F349 = 0.65, p = 0.58; Fig. 5b,e). However, post hoc analyses found no significant difference among regions in intermediate microglia density (all p values ≥ 0.06). For all four regions, average amoeboid microglia density was 423/mm3. Amoeboid microglia density was consistent in the NC (PFC = 527/mm3, MTG = 377/mm3) and HC (CA1 = 236/mm3, CA3 = 495/mm3). Again, analyses for amoeboid microglia density did not find significant effects for region (F349 = 0.69, p = 0.56), sex (F149 = 0.61, p = 0.44), and interaction between region and sex (F349 = 0.55, p = 0.65; Fig. 5c,f).
Figure 4.
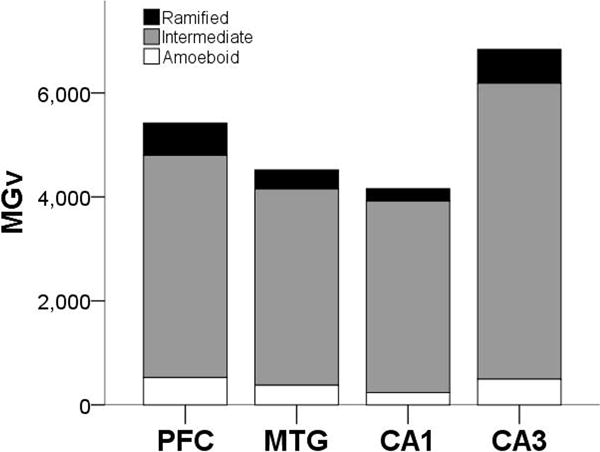
Proportion of Iba1-ir activated microglia (MGv, mm3) by morphology and region.
Figure 5.
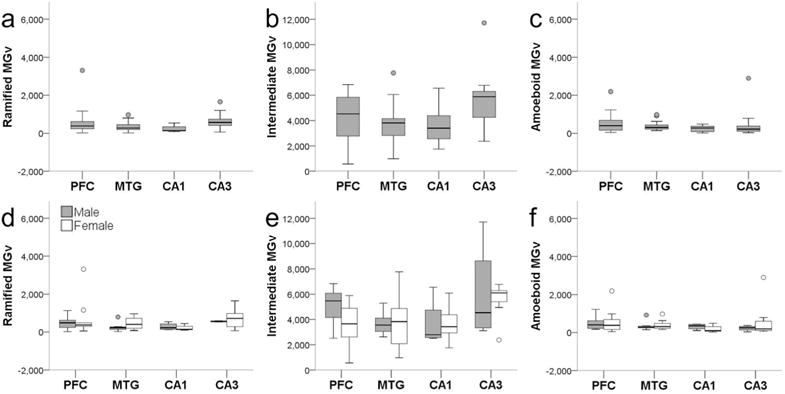
Iba1-ir ramified (a,d), intermediate (b,e), and amoeboid (c,f) microglia densities (MGv, mm3) did not differ by region (a–c; all p values ≥ 0.07) or sex (d–f; all p values ≥ 0.34). Whiskers represent 1 SD. Small circles represent outliers (1.5 × interquartile range).
3.2. Regional PHF-1/Iba1-ir Microglia Density
Microglia that showed colocalization of PHF-1 and Iba1 immunoreactivity exhibited tau deposition most frequently in the cell soma and occasionally in processes, while PHF-1 immunoreactivity was observed mainly in intermediate and amoeboid morphologies (Figs. 1d and 2c-e, Fig. 6). PHF-1/Iba1-ir microglia were identified in all subjects with an average density across all brain areas of 960/mm3. ANOVA revealed a significant main effect of region (F372 = 4.67, p ≤ 0.01), but no effect of sex (F172 = 1.89, p = 0.17) or interaction (F372 = 0.10, p = 0.96; Fig. 7). Post hoc analyses detected a higher PHF-1/Iba1-ir microglia density in PFC (1,728/mm3) than CA1 (317/mm3) and CA3 densities (483/mm3; all p values ≤ 0.02). PHF-1/Iba1-ir microglia density in MTG (1,136/mm3) did not differ significantly from other regions (all p values ≥ 0.27), nor did CA1 vary from CA3 density (p = 1.00). Regression analyses confirmed that increases in PHF-1/Iba1-ir density was associated with an increase in the intermediate microglia morphology in the NC (p = 0.03; Fig. 8a).
Figure 6.
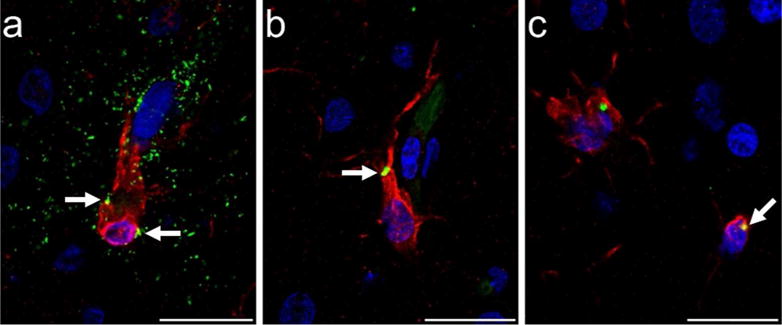
Photomicrographs of tau (AT8, green) immunoreactivity in microglia (Iba1, red) in PFC of a 39-year-old male chimpanzee (subject 13): (a) microglial cell surrounded by a tau-ir (AT8, green) neuritic cluster with minimal tau localization intracellularly (white arrows, yellow), and (b,c) intracellular tau deposition in microglia (white arrows, yellow). Scale bars = 25 μm.
Figure 7.
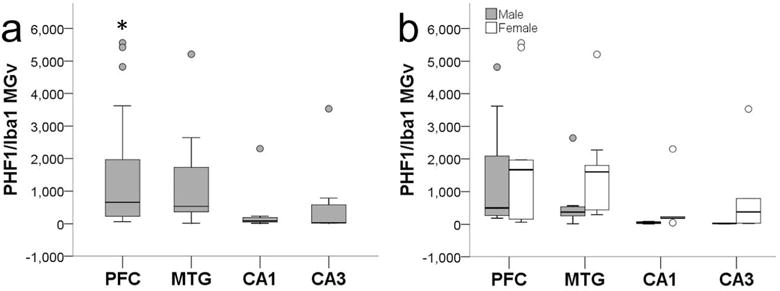
PHF-1/Iba1-ir microglia density (MGv, mm3) was significantly higher in PFC compared to CA1 and CA3 (a; all p values ≤ 0.04). Sex differences were not observed in aged chimpanzees (b; p = 0.10). Whiskers represent 1 SD. Small circles represent outliers (1.5 × interquartile range).
Figure 8.
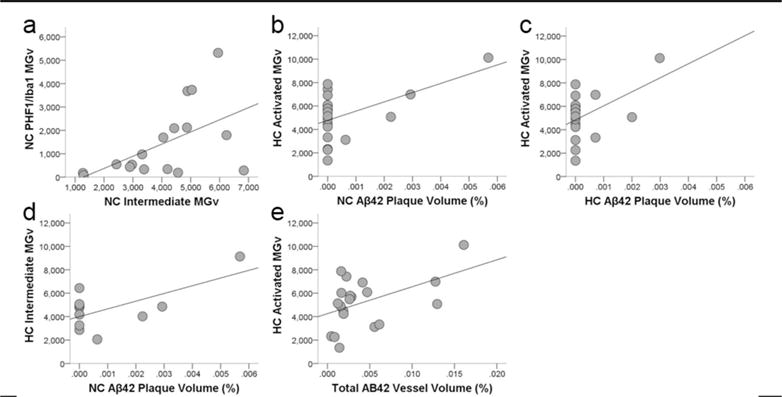
Scatter plots showing that colocalization of tau in activated microglia (PHF-1/Iba1-ir microglia density, MGv, mm3) was associated with increased intermediate morphology in the neocortex (a; R2 = 0.24, p = 0.03). Aβ42 plaque volume (%) in the neocortex (b; R2 = 0.25, p = 0.02) and hippocampus (c; R2 = 0.18, p = 0.04) correlated with increased activation of microglia (Iba1-ir microglia density, mm3) and intermediate morphology (d; R2 = 0.35, p = 0.03) in the hippocampus. Aβ42 vessel volume also was associated with greater activated microglial density in the hippocampus (e; R2 = 0.19, p = 0.03).
3.3. Age and Pathology Correlations
Linear regression analyses did not reveal a correlation between chronological age and activated microglial density (p ≥ 0.45; Table 3). Densities of the different microglia morphologies, ratios of microglia subtype densities/activated microglia densities, and PHF-1/Iba1-ir microglia density also were not associated with chronological age (all p values ≥ 0.25; Table 3). The PCA-generated pathology score (Edler et al., 2017), an assessment for overall Aβ and tau pathology in individuals, was not linked with activated microglia density, morphology, ratios, or PHF-1/Iba1-ir microglia density (all p values ≥ 0.08; Table 3).
Table 3.
Correlation coefficients for activated, morphological, and PHF-1/Iba1-expressing microglia densities (MGv) versus chronological age and PCA-generated pathology score
| Chronological Age | PCA-generated Pathology Score | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Adj R2 | t | P | Adj R2 | t | p | |
| NC Activated MGv | −0.04 | 0.50 | 0.62 | −0.04 | 0.03 | 0.97 |
| HC Activated MGv | −0.03 | 0.73 | 0.47 | −0.05 | 0.17 | 0.87 |
| Total Activated MGv | −0.02 | 0.75 | 0.46 | −0.06 | 0.12 | 0.91 |
|
| ||||||
| NC Ramified MGv | −0.05 | −0.23 | 0.82 | −0.02 | −0.77 | 0.45 |
| HC Ramified MGv | −0.05 | 0.17 | 0.87 | −0.04 | 0.56 | 0.58 |
| Total Ramified MGv | −0.03 | −0.68 | 0.51 | −0.03 | −0.61 | 0.55 |
|
| ||||||
| NC Intermediate MGv | −0.04 | −0.44 | 0.67 | −0.04 | 0.47 | 0.64 |
| HC Intermediate MGv | −0.01 | 0.90 | 0.38 | −0.01 | 0.89 | 0.38 |
| Total Intermediate MGv | −0.05 | −0.30 | 0.77 | −0.05 | 0.31 | 0.76 |
|
| ||||||
| NC Amoeboid MGv | −0.04 | −0.44 | 0.67 | −0.04 | 0.44 | 0.67 |
| HC Amoeboid MGv | −0.03 | 1.29 | 0.21 | −0.11 | 1.83 | 0.08 |
| Total Amoeboid MGv | −0.06 | 0.09 | 0.93 | −0.00 | 0.97 | 0.35 |
|
| ||||||
| NC PHF-1/Iba1 MGv | −0.05 | −0.43 | 0.67 | −0.03 | 0.63 | 0.54 |
| HC PHF-1/Iba1 MGv | −0.03 | −0.71 | 0.49 | −0.05 | −0.32 | 0.76 |
| Total PHF-1/Iba1 MGv | −0.03 | −0.67 | 0.51 | −0.05 | 0.26 | 0.80 |
Regression analyses determined APP/Aβ plaque and vessel volumes were not correlated with activated, morphologic, or PHF-1/Iba1 microglia densities (all p values ≥ 0.08, data not shown). Conversely, higher levels of Aβ42 plaque volume were associated with increased microglial activation and intermediate morphology in the hippocampus (all p values ≤ 0.04; Fig. 8b–d). Activated microglia density in the hippocampus was linked to increased Aβ42 vessel volume (p = 0.03; Fig. 8e). No relationship was found between activated microglia density or morphology and pretangle, NFT, or tau neuritic cluster densities (all p values ≥ 0.09).
4. DISCUSSION
Research examining neuroinflammation in nonhuman primates is scarce. Here, we present a quantitative study of activated microglia and morphological densities in association with AD-like lesions in aged chimpanzees.
Regional differences in activated microglia were noted in the hippocampus of aged chimpanzees with higher density in CA3 than CA1, including those apes displaying both Aβ and tau pathology (Fig. 3). A control group in a human AD study exhibited comparable, though nonsignificant, results with greater Iba1-ir microglia density in CA3 (~160/mm2) than CAI (~115-120/mm2) (Marlatt et al., 2014). However, in the same study, the AD brains showed an increase in activated microglia density in CA1 but not CA3 compared to controls. In AD, the CA1 subfield of the hippocampus typically is one of the first areas affected by NFT and also associated with the greatest neuron loss, while CA3 remains relatively unaffected by tau lesions and neuron numbers are preserved (Braak & Braak, 1991 ; Mueller et al., 2010; Rössler, Zarski, Bohl, & Ohm, 2002). Similar to humans, aged chimpanzees with AD-like pathology displayed higher pretangle, NFT, and tau neuritic cluster loads in CA1, although volume occupied by Aβ-positive plaques and blood vessels was greater in CA3 (Edler et al., 2017). Additionally, Aβ42 immunoreactive plaque and vessel volumes were correlated with higher activated microglia densities in the hippocampus of these chimpanzees, indicating that increased microglial activation in CA3 compared to CA1 may be related to Aβ and not tau pathology (Miller et al., 1993). This concept is supported by the lack of association of pretangle, NFT, and tau neuritic cluster densities with microglial activation in chimpanzees. In addition, a recent study of APP-overexpressing mice (hAPP-J20) demonstrated a correlation of greater total Aβ and microglial activation with neuronal loss in CA1, suggesting activation of microglia is closely associated with Aβ expression and neuron loss (Wright et al., 2013). Taken together, these results demonstrate some important differences between humans and chimpanzees with AD pathology (Table 4). Both tau and Aβ pathology are associated with increased microglial activation in human AD, while only Aβ lesions appear related to greater microglial activation in the hippocampus of aged chimpanzees. Also, CA3 may be more susceptible to pathological changes than CA1 in the chimpanzee, which contrasts to human AD where CA1 is one of the most severely affected areas. Future work will investigate whether increased microglial activation and Aβ deposition in CA3 of aged chimpanzees is correlated with neuron loss.
Table 4.
AD-related pathologic and inflammatory traits observed in chimpanzees and humans.
| DIVERGENT TRAITS | SHARED TRAITS | |
|---|---|---|
|
| ||
| Chimpanzee | Human | Chimpanzee + Human |
|
| ||
| Deposition of Aβ is primarily in the brain’s vessels and occurs prior to formation of plaques. | Deposition of Aβ is primarily in the form of plaques, although CAA occurs in most AD patients. | Aβ pathology increases with age. |
| Aβ42 is the predominant peptide in severe CAA. | Aβ40 is the primary protein in humans with severe CAA. | Aβ pathology is associated with increased tau pathology. |
| Tau neuritic clusters lack an Aβ core. | Neuritic plaques contain an Aβ core. | Tau deposition occurs in microglia. |
| Pretangles in the neocortex increase with age. | NFT increase with age in the hippocampus. | NFT density is higher in hippocampal subfield CA1 compared to CA3. |
| Activated microglia density is higher in CA3 compared to CA1. | AD patients had higher microglial activation in CA1, despite control groups presenting with greater microglia density in the CA3. | Severe CAA is associated with increased tau pathology (NFT in humans; pretangles and tau neuritic clusters in chimpanzees). |
| Microglial activation is correlated with Aβ but not NFT lesions. | Microglial activation is associated with Aβ and NFT pathology. | Aβ42 is correlated with increased microglial activation (plaques in humans, vessels in chimpanzees). |
| Neuron loss in association with AD pathology has not yet been quantified in chimpanzees. | Selective neuronal loss occurs in the prefrontal cortex and hippocampus. | |
| Antemortem cognitive testing is rare in aged apes. Mild cognitive deficits in short-term and spatial memory, attention, and executive function have been noted. | Severe memory, cognitive, and behavioral deficits are observed. | |
The predominant activated microglia phenotype was intermediate (79%) compared to ramified (9%) or amoeboid (8%) subtypes. Normal adult human brains demonstrated an analogous pattern in the dorsal anterior cingulate cortex with 66% of Iba1-ir microglia displaying an intermediate morphology, 16% a ramified appearance, and 18% an amoeboid shape (Torres-Platas et al., 2014). Morphological microglia densities were consistent across the neocortex and hippocampus in male and female chimpanzees. While microglial morphology did not vary by sex in chimpanzees or humans, a study in 60-day old rats found that females had significantly more Iba1-ir microglia with thicker and longer processes (i.e., intermediate) than males in the CA1, CA3, dentate gyrus, and amygdala (Marlatt et al., 2014; Schwarz, Sholar, & Bilbo, 2012). In addition, young, middle-aged, and old female B6 mice had 25-40% more microglia in the dentate gyrus and CA1 than age-matched male C57Bl/6J mice (Mouton et al., 2002). Whether these regional or sex variances are species-specific requires further investigation of microglia in rodents, primates, and humans.
Among elderly chimpanzees, age was not associated with changes in activated microglia density or morphology. These data are congruent with previous findings in older nonhuman primates and rodents. Activated microglia density in the visual cortex, substantia nigra, and ventral tegmental area of rhesus macaques did not show a significant increase with age (Kanaan, Kordower, & Collier, 2010; Peters & Sethares, 2004). Similarly, changes in activated microglia density in the substantia nigra of young and middle-aged rats were not detected (Ogura, Ogawa, & Yoshida, 1994). In contrast, aged rats demonstrated a mild-to-moderate increase in microglia activation in CA1 and CA3 hippocampal subfields (VanGuilder et al., 2011). Moreover, some studies in humans demonstrate increased microglial activation, particularly in white matter tracts, with age (T. E. Morgan et al., 1999; Ogura et al., 1994; V H Perry & Gordon, 1991; L G Sheffield & Berman, 1998; Sloane, Hollander, Moss, Rosene, & Abraham, 1999). Though difficult to compare accurately due to variances in data collection methods and antigens, prior reports suggest that humans may have greater microglial densities in the dorsolateral PFC and temporal cortex (Table 5) (Bachstetter et al., 2015; Davies, Ma, Jegathees, & Goldsbury, 2016; DiPatre & Gelman, 1997; Doorn et al., 2014; Fabricius, Jacobsen, & Pakkenberg, 2013; Lyck et al., 2009; Maltseva, Volchegorskii, & Shemyakov, 2017; J. T. Morgan et al., 2010; Pelvig, Pakkenberg, Stark, & Pakkenberg, 2008; Radewicz, Garey, Gentleman, & Reynolds, 2000; Jin G. Sheng, Mrak, & Griffin, 1997; Steiner et al., 2008; Tetreault et al., 2012). Activated interleukin-1 alpha (IL-1α) microglia density is greater with age in humans (J G Sheng, Mrak, & Griffin, 1998). IL-1α is a protein produced by activated macrophages and responsible for the production of inflammation. Additionally, age-related morphological changes in IL-1α microglia density were identified; intermediate and amoeboid morphologies, but not ramified, were more prevalent with age in humans independent of postmortem interval and sex (J G Sheng et al., 1998). The number of amoeboid microglia was significantly increased in the human substantia nigra despite activated microglia densities not differing by age (Jyothi et al., 2015). As the aim of the current study was to identify AD pathology- associated microglial activation, we collected data in the oldest available individuals, and therefore, it is likely that the present sample did not include individuals young enough to detect age-related differences in microglia densities. Future examination of activated microglia density and morphology in young chimpanzees as well as young, middle-aged, and elderly humans is needed to address age- and species- related differences in microglial activation.
Table 5.
Microglia densities in brain regions of human (control only) reported in the literature.
| Reference | Age (years) | Sex (n) | Region | Microglia Density | Antigen | Method | Major Results |
|---|---|---|---|---|---|---|---|
| DiPatre Gelman, 1997 | 38-73 | NR (17) | EC (II-V) Subiculum CA1 CA2 CA3 DG |
~35-50/mm2 ~25-50/mm2 ~55-80/mm2 ~25-55/mm2 ~30-55/mm2 ~25-45/mm2 |
Ferritin | 2D model-based stereology | Increase with age in all hippocampal subfields |
| Sheng, Mrak, Griffin, 1998 | 2-93 | M (12) F (10) |
Mesial temporal | ~32-50/mm2 | IL-1α | 2D model-based stereology | Increase with age in females and males |
| Radewicz et al., 2000 | 55-90 | NR (11) | DLPFC STG ACC |
89/mm2 88/mm2 ~135/mm2 |
HLA-DR | 2D model-based stereology | Minor increase with age in STG but not DLPFC or ACC |
| Pelvig et al., 2008 | 18-93 | M (13) F (18) |
Neocortex | 2 × 109 (M) 1.8 × 109 (F) |
Modified Giesma | 3D design-based stereology | Increase with age in females but not males |
| Steiner et al., 2008 | 47-63 | M (5) F (5) |
DLPFC ACC Mediodorsal thalamus HC |
5-10/mm2 12.5-15/mm2 18-22.5/mm2 15-18/mm2 |
HLA-DR | 2D model-based stereology | No change with age |
| Lyck et al., 2009 | 59-88 | M (2) F (1) |
FC TC PC OC |
0.86-1.17 × 109 0.61-1.12 × 109 0.74-1.44 × 109 0.34-1.01 × 109 |
CD45 | 3D design-based stereology | Not analyzed |
| Morgan et al., 2010 | 1-44 | M (9) | DLPFC (gray) DLPFC (white) |
~28,000-40,000/mm3 ~28,000-50,000/mm3 |
Iba1 | 3D design-based stereology | Decrease with age (nonsignificant trend) |
| Tetreault et al., 2012 | 2-23 | M (11) F (1) |
Fronto-insular | ~4,000-11,000/mm3 | Iba1 | 3D design-based stereology | No change with age |
| PV1 | ~4,500-7,800/mm3 | ||||||
| Fabricius, Jacobsen, Pakkenberg, 2013 | 65-105 | F (23) | Neocortex | 2.7 × 109 (65-75 y) 3.0 × 109 (76-85 y) 2.3 × 109 (94-105 y) |
Modified Giesma | 3D design-based stereology | No change with age |
| Doorn et al., 2014 | 66-93 | NR (11) | AON | 6,650/mm3 (amoeboid) 28,790/mm3 (ramified) |
CD68 | 3D design-based stereology | No change with age |
| Bachstetter et al., 2015 | 81-93 | M (6) F (3) |
Subiculum CA1 CA2/3 DG HC (avg) |
13.9/mm2 (CD68) 66.5/mm2 (Iba1) 16.0/mm2 (CD68) 60.8/mm2 (Iba1) 17.0/mm2 (CD68) 74.0/mm2 (Iba1) 10.7/mm2 (CD68) 80.5/mm2 (Iba1) 22.1/mm2 (CD68) 77.7/mm2 (Iba1) |
CD68, Iba1 | Nuclear algorithm | Not analyzed |
| Davies et al., 2016 | 68-89 | M (2) F (3) |
ACC (M-IM) ITG (M-IM) |
~97-140/mm2 ~100-165/mm2 |
Iba1 | 2D model-based stereology | No change with age |
| Maltseva et al., 2017 | 0-21 | M, F (121) |
PMC (area 6, V) PV1 (area 17, IV) Caudate nucleus |
20,168/mm3 22,657/mm3 30,559/mm3 |
Silver | 2D model-based stereology | Increase with age until adolescence or early adulthood |
NR (not reported), M (male), F (female), EC (entorhinal cortex), DG (dentate gyrus), DLPFC (dorsolateral prefrontal cortex), STG (superior temporal gyrus), ACC (anterior cingulate cortex), HC (hippocampus), FC (frontal cortex), TC (temporal cortex), PC (parietal cortex), OC (occipital cortex), AON (anterior olfactory nucleus), ITG (inferior temporal gyrus), PMC (premotor cortex), PV1 (primary visual cortex), IL-1α (interleukin one-alpha), HLA-DR (human leukocyte antigen-D related), CD45 (cluster of differentiation 45), Iba1 (ionized-binding calcium adapter 1), CD68 (cluster of differentiation 68)
Similar to findings in humans, nonhuman primates, and rodent models of AD, aged chimpanzees exhibited greater levels of microglial activation and an increase in intermediate-shaped microglia morphologies associated with Aβ42 plaque and vessel volume. Several studies demonstrate the presence of a robust immune response in AD, including production of inflammatory cytokines, genomic associations, and microglial activation (Carpenter, Carpenter, & Markesbery, 1993; Forny-Germano et al., 2014; Griciuc et al., 2013; Guerreiro et al., 2013; Jonsson et al., 2013; Ledo et al., 2013; Njie et al., 2012). Cultures of microglia cells isolated from aged mice displayed elevated production of proinflammatory molecules IL-6 and TNF-α, and microglia from these animals display a decreased ability to internalize Aβ (Njie et al., 2012). Recent molecular studies highlight the expression of polymorphisms in immune-related genes, such as TREM2 (triggering receptor expressed on myeloid cells 2), CD33 (sialic acid binding Ig-like lectin 3), and HLA-DR (human leukocyte antigen-D related) in association with AD (Griciuc et al., 2013; Guerreiro et al., 2013; Jonsson et al., 2013). HLA-DR-ir microglia density was significantly higher in the MTG of AD patients than in controls, and CD33-ir microglia density was positively correlated with insoluble Aβ42 levels and plaque loads in AD brains (Carpenter et al., 1993; Griciuc et al., 2013). Aβ oligomers also trigger astrocyte and microglial activation in mice and long-tailed macaque monkeys (Forny-Germano et al., 2014; Ledo et al., 2013). Additionally, evidence indicates cerebrovascular Aβ deposition promotes neuroinflammation in AD and cerebral amyloid angiopathy (CAA) disorders. In sporadic and familial CAA, leptomeningeal and cortical vessels were associated with an increased activation of monocyte/macrophage lineage cells (Yamada et al., 1996). A transgenic mouse model (Tg-SwDI) of CAA showed abundant reactive astrocytes and activated microglia strongly associated with the cerebral microvascular fibrillar Aβ deposits (Miao et al., 2005).
In addition to Aβ-positive plaques and vasculature, tau pathology correlates with neuroinflammation. Tau deposition was significantly increased in activated, intermediate microglia as measured by PHF-1/Iba1 immunoreactivity in aged chimpanzees (Fig. 8a). Furthermore, while tau lesions were not significantly associated with increased microglial activation, intermediate and amoeboid morphologies were noted adjacent to pretangles, NFT, and tau neuritic clusters (Fig. 2i–l). Microglial activation has been implicated in driving tau hyperphosphorylation, aggregation, and neurodegeneration in human models of tauopathies (Bellucci, Bugiani, Ghetti, & Spillantini, 2011; Gebicke-Haerter, 2001; Gerhard et al., 2006; Ishizawa & Dickson, 2001). Activated microglia are adjacent to tau-positive neurons in the brains of patients with progressive supranuclear palsy and corticobasal degeneration (Gerhard et al., 2006; Ishizawa & Dickson, 2001). Microglial activation also has been demonstrated to precede tau pathology in the P301S mouse model of tauopathy, and chemically or genetically enhanced microglial activation significantly accelerated tau pathology in the hTau mice (Bhaskar et al., 2010; Yoshiyama et al., 2007). Recent work also showed that transfer of purified microglia derived from hTau mice induced tau hyperphosphorylation in the non-transgenic mouse brain, and inclusion of an IL-1 receptor antagonist significantly reduced microglia-induced tau pathology. These results suggest that reactive microglia may drive tau pathology in humans and rodents but not in chimpanzees (Maphis et al., 2015). Rather, the combination of Aβ40 and Aβ42 is correlated to increased tau lesions in aged chimpanzees, whereas Aβ42 alone appears to activate microglia, potentially resulting in greater uptake of tau.
4.1 Conclusions
Elderly chimpanzees exhibit changes in activated microglia density and morphology related to AD pathology. The chimpanzee hippocampus, particularly the CA3 subfield, seems most susceptible to neuroinflammation with increased microglial activation as well as a greater number of microglia with intermediate morphology in relation to Aβ deposition but not tau lesions. This outcome diverges from humans with AD who display greater microglial activation and morphological changes in response to both tau and Aβ pathology, especially in the CA1 field. Such evidence supports prior work demonstrating the chimpanzee brain may be relatively preserved during normal aging processes compared to the human brain (Allen, Bruss, & Damasio, 2005; Bishop, Lu, & Yankner, 2010; Erwin, Nimchinsky, Gannon, Perl, & Hof, 2001; Erwin, Perl, Nimchinsky, & Hof, 1999; Finch, 2003; Jernigan et al., 2001; Sherwood et al., 2011). Conversely, the chimpanzee brain may not be entirely protected from neurodegeneration as previously assumed and supported by the correlation of Aβ pathology and increased neuroinflammation. Growing evidence suggests the detrimental cognitive effects of AD and other neurodegenerative diseases in humans may be the result of an increased lifespan and the further exaggeration of normal aging processes rather than evolutionary modifications in cerebral structure and function between chimpanzees and humans (Hof, Gilissen, & Sherwood, 2002; Rapoport & Nelson, 2011; Sherwood et al., 2011; Walker & Cork, 1999). Future investigations will clarify the relationships by studying the effect of normal aging on microglia density and morphology as well as neuroinflammation in relation to α-synuclein pathology (e.g., Lewy bodies) in elderly chimpanzees.
Acknowledgments
We express our gratitude to Cheryl Stimpson, Bridget Wicinski, and Emily Munger for their expert technical assistance and to Dr. Jason R. Richardson. Dr. Peter Davies generously provided the PHF-1 antibody.
Grant sponsors: National Science Foundation; Grant number: BCS-1316829 (M.A.R.). Grant Sponsor: National Institutes of Health; Grant numbers: NS042867, NS073134, and the National Chimpanzee Brain Resource NS092988 (W.D.H., C.C.S.), AG017802 (J.J.E.), AG014308 (J.M.E.), AG005138 (P.R.H.), and AG014449 and AG043375 (E.J.M.). Grant sponsor: James S. McDonnell Foundation; Grant number: 220020293 (C.C.S.). Grant sponsor: Sigma Xi (M.K.E.). Grant sponsor: Kent State University Research Council (M.A.R.). Grant Sponsor: Kent State University Graduate Student Senate (M.K.E.).
References
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Wyss-Coray T. Inflammation and Alzheimer’s disease. Neurobiology of Aging. 2000;21(3):383–421. doi: 10.1016/s0197-4580(00)00124-x. http://doi.org/10.1016/S0197-4580(00)00124-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akram A, Christoffel D, Rocher AB, Bouras C, Kovari E, Perl DP, Hof PR. Stereologic estimates of total spinophilin-immunoreactive spine number in area 9 and the CA1 field: relationship with the progression of Alzheimer’s disease. Neurobiology of Aging. 2008;29(9):1296–1307. doi: 10.1016/j.neurobiolaging.2007.03.007. http://doi.org/10.1016/j.neurobiolaging.2007.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen JS, Bruss J, Damasio H. The aging brain: the cognitive reserve hypothesis and hominid evolution. American Journal of Human Biology: The Official Journal of the Human Biology Council. 2005;17(6):673–89. doi: 10.1002/ajhb.20439. http://doi.org/10.1002/ajhb.20439 [DOI] [PubMed] [Google Scholar]
- Bachstetter AD, Van Eldik LJ, Schmitt FA, Neltner JH, Ighodaro ET, Webster SJ, Nelson PT. Disease-related microglia heterogeneity in the hippocampus of Alzheimer’s disease, dementia with Lewy bodies, and hippocampal sclerosis of aging. Acta Neuropathologica Communications. 2015;3(1):32. doi: 10.1186/s40478-015-0209-z. http://doi.org/10.1186/s40478-015-0209-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellucci A, Bugiani O, Ghetti B, Spillantini MG. Presence of reactive microglia and neuroinflammatory mediators in a case of frontotemporal dementia with P301S mutation. NeuroDegenerative Diseases. 2011;8(4):221–229. doi: 10.1159/000322228. http://doi.org/10.1159/000322228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010;68(1):19–31. doi: 10.1016/j.neuron.2010.08.023. http://doi.org/10.1016/j.neuron.2010.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop NA, Lu T, Yankner BA. Neural mechanisms of ageing and cognitive decline. Nature. 2010;464(7288):529–535. doi: 10.1038/nature08983. http://doi.org/10.1038/nature08983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathologica. 1991;82(4):239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Bussière T, Gold G, Kövari E, Giannakopoulos P, Bouras C, Perl DP, Hof PR. Stereologic analysis of neurofibrillary tangle formation in prefrontal cortex area 9 in aging and Alzheimer’s disease. Neuroscience. 2003;117(3):577–592. doi: 10.1016/s0306-4522(02)00942-9. [DOI] [PubMed] [Google Scholar]
- Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE, Banati RB. In-vivo measurement of activated microglia in dementia. Lancet. 2001;358(9280):461–467. doi: 10.1016/S0140-6736(01)05625-2. http://doi.org/10.1016/S0140-6736(01 )05625-2. [DOI] [PubMed] [Google Scholar]
- Carpenter AF, Carpenter PW, Markesbery WR. Morphometric analysis of microglia in Alzheimer’s disease. Journal of Neuropathology and Experimental Neurology. 1993;52(6):601–608. doi: 10.1097/00005072-199311000-00007. [DOI] [PubMed] [Google Scholar]
- Cunningham C. Microglia and neurodegeneration: the role of systemic inflammation. Glia. 2013;61(1):71–90. doi: 10.1002/glia.22350. http://doi.org/10.1002/glia.22350 [DOI] [PubMed] [Google Scholar]
- Davies DS, Ma J, Jegathees T, Goldsbury C. Microglia show altered morphology and reduced arborization in human brain during aging and Alzheimer’s disease. Brain Pathology. 2016;(16) doi: 10.1111/bpa.12456. http://doi.org/10.1111/bpa.12456 [DOI] [PMC free article] [PubMed]
- DiPatre PL, Gelman BB. Microglial cell activation in aging and Alzheimer disease: partial linkage with neurofibrillary tangle burden in the hippocampus. Journal of Neuropathology and Experimental Neurology. 1997;56(2):143–149. doi: 10.1097/00005072-199702000-00004. [DOI] [PubMed] [Google Scholar]
- Doorn KJ, Goudriaan A, Blits-Huizinga C, Bol JGJM, Rozemuller AJ, Hoogland PVJM, Van Dam AM. Increased amoeboid microglial density in the Olfactory Bulb of Parkinson’s and Alzheimer’s Patients. Brain Pathology. 2014;24(2):152–165. doi: 10.1111/bpa.12088. http://doi.org/10.1111/bpa.12088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorph-Petersen KA, Nyengaard JR, Gundersen HJ. Tissue shrinkage and unbiased stereological estimation of particle number and size. Journal of Microscopy. 2001;204(Pt 3):232–246. doi: 10.1046/j.1365-2818.2001.00958.x. [DOI] [PubMed] [Google Scholar]
- Edler MK, Sherwood CC, Meindl RS, Hopkins WD, Ely JJ, Erwin JM, Raghanti MA. Aged chimpanzees exhibit pathologic hallmarks of Alzheimer’s disease Neurobiology of Aging. Elsevier Inc; 2017. http://doi.org/10.1016/j.neurobiolaging.2017.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekonomou A, Savva GM, Brayne C, Forster G, Francis PT, Johnson M, Ballard CG. Stage-specific changes in neurogenic and glial markers in Alzheimer’s disease. Biological Psychiatry. 2015;77(8):711–719. doi: 10.1016/j.biopsych.2014.05.021. http://doi.org/10.1016/j.biopsych.2014.05.021 [DOI] [PubMed] [Google Scholar]
- Ely JJ, Dye B, Frels WI, Fritz J, Gagneux P, Khun HH, Lee DR. Subspecies composition and founder contribution of the captive U.S. chimpanzee (Pan troglodytes) population. American Journal of Primatology. 2005;67(2):223–241. doi: 10.1002/ajp.20179. http://doi.org/10.1002/ajp.20179 [DOI] [PubMed] [Google Scholar]
- Erwin JM, Nimchinsky EA, Gannon PJ, Perl DP, Hof PR. Functional Neurobiology of Aging. San Diego: Academic Press; 2001. The study of brain aging in great apes; pp. 447–455.http://doi.org/https://doi.org/10.1016/B978-012351830-9/50031-7 [Google Scholar]
- Erwin JM, Perl DP, Nimchinsky EA, Hof PR. Stereologic analyses of neuron number and volume in the entorhinal cortex of aged chimpanzees. Society of Neuroscience Abstract. 1999:25–105. [Google Scholar]
- Fabricius K, Jacobsen JS, Pakkenberg B. Effect of age on neocortical brain cells in 90+ year old human females-a cell counting study. Neurobiology of Aging. 2013;34(1):91–99. doi: 10.1016/j.neurobiolaging.2012.06.009. http://doi.org/10.1016/j.neurobiolaging.2012.06.009 [DOI] [PubMed] [Google Scholar]
- Finch CE. Neurons, glia, and plasticity in normal brain aging. Neurobiology of Aging. 2003;24(Suppl 1):S123–7. doi: 10.1016/s0197-4580(03)00051-4. discussion S131. [DOI] [PubMed] [Google Scholar]
- Finsen B, Lehrmann E, Castellano B, Kiefer R, Zimmer J. The role of microglial cells and brain macrophages in transient global cerebral ischemia. In: Ling E, Tan C, Tan C, editors. Topical Issues in Microglia Research. Singapore: Singapore Neuroscience Association; 1996. pp. 297–318. [Google Scholar]
- Forny-Germano L, e Silva NML, Batista AF, Brito-Moreira J, Gralle M, Boehnke SE, De Felice FG. Alzheimer’s disease-like pathology induced by Aβ oligomers in non- human primates. Journal of Neuroscience. 2014;34(41):13629–13643. doi: 10.1523/JNEUROSCI.1353-14.2014. http://doi.org/10.1523/JNEUROSCI.1353-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandy S, Heppner FL. Microglia as dynamic and essential components of the amyloid hypothesis. Neuron. 2013;78(4):575–577. doi: 10.1016/j.neuron.2013.05.007. http://doi.org/10.1016/j.neuron.2013.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gearing M, Rebeck GW, Hyman BT, Tigges J, Mirra SS. Neuropathology and apolipoprotein E profile of aged chimpanzees: implications for Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(20):9382–9386. doi: 10.1073/pnas.91.20.9382. https://doi.org/10.1073/pnas.91.20.9382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gearing M, Tigges J, Mori H, Mirra SS. Aβ40 is a major form of β-amyloid in nonhuman primates. Neurobiology of Aging. 1996;17(6):903–908. doi: 10.1016/s0197-4580(96)00164-9. http://doi.org/10.1016/S0197-4580(96)00164-9 [DOI] [PubMed] [Google Scholar]
- Gebicke-Haerter PJ. Microglia in neurodegeneration: molecular aspects. Microscopy Research and Technique. 2001;54(1):47–58. doi: 10.1002/jemt.1120. http://doi.org/10.1002/jemt.1120 [DOI] [PubMed] [Google Scholar]
- Gehrmann J, Bonnekoh P, Miyazawa T, Oschlies U, Dux E, Hossmann KA, Kreutzberg GW. The microglial reaction in the rat hippocampus following global ischemia: immuno-electron microscopy. Acta Neuropathologica. 1992;84(6):588–595. doi: 10.1007/BF00227735. [DOI] [PubMed] [Google Scholar]
- Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, Brooks DJ. In vivo imaging of microglial activation with [11 C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiology of Disease. 2006;21(2):404–412. doi: 10.1016/j.nbd.2005.08.002. http://doi.org/10.1016/j.nbd.2005.08.002 [DOI] [PubMed] [Google Scholar]
- Geula C, Wu CK, Saroff D, Lorenzo A, Yuan M, Yankner BA. Aging renders the brain vulnerable to amyloid beta-protein neurotoxicity. Nature Medicine. 1998;4(7):827–831. doi: 10.1038/nm0798-827. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Vanmechelen E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neuroscience Letters. 1995;189(3):167–169. doi: 10.1016/0304-3940(95)11484-e. [DOI] [PubMed] [Google Scholar]
- Graeber MB, Streit WJ. Microglia: immune network in the CNS. Brain Pathol. 1990;1:2–5. doi: 10.1111/j.1750-3639.1990.tb00630.x. [DOI] [PubMed] [Google Scholar]
- Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, Tanzi RE. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013;78(4):631–43. doi: 10.1016/j.neuron.2013.04.014. http://doi.org/10.1016/j.neuron.2013.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Hardy J. TREM2 variants in Alzheimer’s disease. The New England Journal of Medicine. 2013;368(2):117–127. doi: 10.1056/NEJMoa1211851. http://doi.org/10.1056/NEJMoa1211851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nature Reviews Immunology. 2014;14(7):463–477. doi: 10.1038/nri3705. http://doi.org/10.1038/nri3705 [DOI] [PubMed] [Google Scholar]
- Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. Journal of Neuroscience. 2008;28(33):8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008. http://doi.org/10.1523/JNEUROSCI.0616-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SE, El Khoury J. TREM2 and the neuroimmunology of Alzheimer’s disease. Biochemical Pharmacology. 2014;88(4):495–498. doi: 10.1016/j.bcp.2013.11.021. http://doi.org/10.1016/j.bcp.2013.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi M. Visualization of brain amyloid and microglial activation in mouse models of Alzheimer’s disease. Current Alzheimer Research. 2009;6(2):137–143. doi: 10.2174/156720509787602906. [DOI] [PubMed] [Google Scholar]
- Hof PR, Cox K, Morrison JH. Quantitative analysis of a vulnerable subset of pyramidal neurons in Alzheimer’s disease: I. Superior frontal and inferior temporal cortex. The Journal of Comparative Neurology. 1990;301(1):44–54. doi: 10.1002/cne.903010105. http://doi.org/10.1002/cne.903010105 [DOI] [PubMed] [Google Scholar]
- Hof PR, Gilissen EP, Sherwood CC. Comparative Neuropathology of Brain Aging in Primates. In: Erwin JM, Hof PR, editors. Aging in nonhuman primates. Vol. 31. Karger, Basel; 2002. pp. 130–154. [Google Scholar]
- Ishizawa K, Dickson DW. Microglial activation parallels system degeneration in progressive supranuclear palsy and corticobasal degeneration. Journal of Neuropathology and Experimental Neurology. 2001;60(6):647–657. doi: 10.1093/jnen/60.6.647. [DOI] [PubMed] [Google Scholar]
- Jernigan TL, Archibald SL, Fennema-Notestine C, Gamst AC, Stout JC, Bonner J, Hesselink JR. Effects of age on tissues and regions of the cerebrum and cerebellum. Neurobiology of Aging. 2001;22(4):581–594. doi: 10.1016/s0197-4580(01)00217-2. http://doi.org/10.1016/S0197-4580(01)00217-2 [DOI] [PubMed] [Google Scholar]
- Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Stefansson K. Variant of TREM2 associated with the risk of Alzheimer’s disease. The New England Journal of Medicine. 2013;368(2):107–116. doi: 10.1056/NEJMoa1211103. http://doi.org/10.1056/NEJMoa1211103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen M. Microglial and Astroglial Reactions to Ischemic and Kainic Acid-Induced Lesions of the Adult Rat Hippocampus. Experimental Neurology. 1993;120(1):70–88. doi: 10.1006/exnr.1993.1041. http://doi.org/10.1006/exnr.1993.1041 [DOI] [PubMed] [Google Scholar]
- Jyothi HJ, Vidyadhara DJ, Mahadevan A, Philip M, Parmar SK, Manohari SG, Alladi PA. Aging causes morphological alterations in astrocytes and microglia in human substantia nigra pars compacta. Neurobiology of Aging. 2015;36(12):3321–3333. doi: 10.1016/j.neurobiolaging.2015.08.024. http://doi.org/10.1016/j.neurobiolaging.2015.08.024 [DOI] [PubMed] [Google Scholar]
- Kanaan NM, Kordower JH, Collier TJ. Age-related changes in glial cells of dopamine midbrain subregions in rhesus monkeys. Neurobiology of Aging. 2010;31(6):937–952. doi: 10.1016/j.neurobiolaging.2008.07.006. http://doi.org/10.1016/j.neurobiolaging.2008.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiological Reviews. 2011;91(2):461–553. doi: 10.1152/physrev.00011.2010. http://doi.org/10.1152/physrev.00011.2010 [DOI] [PubMed] [Google Scholar]
- Kofler J, Lopresti B, Janssen C, Trichel AM, Masliah E, Finn OJ, Wiley Ca. Preventive immunization of aged and juvenile non-human primates to beta-amyloid. Journal of Neuroinflammation. 2012;9(1):84. doi: 10.1186/1742-2094-9-84. http://doi.org/10.1186/1742-2094-9-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kongsui R, Beynon SB, Johnson SJ, Walker F. Quantitative assessment of microglial morphology and density reveals remarkable consistency in the distribution and morphology of cells within the healthy prefrontal cortex of the rat. Journal of Neuroinflammation. 2014;11(1):182. doi: 10.1186/s12974-014-0182-7. http://doi.org/10.1186/s12974-014-0182-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krstic D, Knuesel I. Deciphering the mechanism underlying late-onset Alzheimer disease. Nature Reviews Neurology. 2013;9(1):25–34. doi: 10.1038/nrneurol.2012.236. http://doi.org/10.1038/nrneurol.2012.236 [DOI] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39(1):151–170. doi: 10.1016/0306-4522(90)90229-w. http://doi.org/10.1016/0306-4522(90)90229-W [DOI] [PubMed] [Google Scholar]
- Ledo JH, Azevedo EP, Clarke JR, Ribeiro FC, Figueiredo CP, Foguel D, Ferreira ST. Amyloid-beta oligomers link depressive-like behavior and cognitive deficits in mice. Molecular Psychiatry. 2013 Oct;18(10):1053–1054. doi: 10.1038/mp.2012.168. http://doi.org/10.1038/mp.2012.168 England. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, McGeer E, McGeer PL. Activated human microglia stimulate neuroblastoma cells to upregulate production of beta amyloid protein and tau: implications for Alzheimer’s disease pathogenesis. Neurobiol Aging. 2015;36(1):42–52. doi: 10.1016/j.neurobiolaging.2014.07.024. http://doi.org/10.1016/j.neurobiolaging.2014.07.024 [DOI] [PubMed] [Google Scholar]
- Lehrmann E, Christensen T, Zimmer J, Diemer NH, Finsen B. Microglial and macrophage reactions mark progressive changes and define the penumbra in the rat neocortex and striatum after transient middle cerebral artery occlusion. Journal of Comparative Neurology. 1997;386(3):461–476. http://doi.org/10.1002/(SICI)1096-9861(19970929)386:3<461::AID-CNE9>3.0.CO;2-# [PubMed] [Google Scholar]
- Leung E, Guo L, Bu J, Maloof M, El J, Geula C, Geula C. Microglia activation mediates fibrillar amyloid-β toxicity in the aged primate cortex. Neurobiology of Aging. 2011;32(3):387–397. doi: 10.1016/j.neurobiolaging.2009.02.025. http://doi.org/10.1016/j.neurobiolaging.2009.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyck L, Santamaria ID, Pakkenberg B, Chemnitz J, Schrøder HD, Finsen B, Gundersen HJG. An empirical analysis of the precision of estimating the numbers of neurons and glia in human neocortex using a fractionator-design with sub-sampling. Journal of Neuroscience Methods. 2009;182(2):143–156. doi: 10.1016/j.jneumeth.2009.06.003. http://doi.org/10.1016/jJneumeth.2009.06.003 [DOI] [PubMed] [Google Scholar]
- Maier M, Peng Y, Jiang L, Seabrook TJ, Carroll MC, Lemere CA. Complement C3 deficiency leads to accelerated amyloid beta plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. Journal of Neuroscience. 2008;28(25):6333–6341. doi: 10.1523/JNEUROSCI.0829-08.2008. http://doi.org/10.1523/JNEUROSCI.0829-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltseva NV, Volchegorskii IA, Shemyakov SE. Age-related changes in the number of microglial cells, lipid peroxidation, and oxidative protein-modification products in the human cerebrum at early stages of ontogenesis. Neurochemical Journal. 2017;1l(1):50–56. http://doi.org/10.1134/S181971241701007X [Google Scholar]
- Maphis N, Xu G, Kokiko-Cochran ON, Jiang S, Cardona A, Ransohoff RM, Bhaskar K. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain. 2015;138(6):1738–1755. doi: 10.1093/brain/awv081. http://doi.org/10.1093/brain/awv081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlatt MW, Bauer J, Aronica E, van Haastert ES, Hoozemans JJM, Joels M, Lucassen PJ. Proliferation in the Alzheimer hippocampus is due to microglia, not astroglia, and occurs at sites of amyloid deposition. Neural Plasticity. 2014;2014:1–12. doi: 10.1155/2014/693851. http://doi.org/10.1155/2014/693851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiace La, Davies P, Dickson DW. Detection of HLA-DR on microglia in the human brain is a function of both clinical and technical factors. The American Journal of Pathology. 1990;136(5):1101–1114. [PMC free article] [PubMed] [Google Scholar]
- Meraz Rios MA, Toral-Rios D, Franco-Bocanegra D, Villeda-Hernández J, Campos-Peña V. Inflammatory process in Alzheimer’s disease. Frontiers in Integrative Neuroscience. 2013 doi: 10.3389/fnint.2013.00059. Retrieved from http://journal.frontiersin.org/article/10.3389/fnint.2013.00059. [DOI] [PMC free article] [PubMed]
- Miao J, Xu F, Davis J, Otte-Holler I, Verbeek MM, Van Nostrand WE. Cerebral microvascular amyloid beta protein deposition induces vascular degeneration and neuroinflammation in transgenic mice expressing human vasculotropic mutant amyloid beta precursor protein. The American Journal of Pathology. 2005;167(2):505–515. doi: 10.1016/s0002-9440(10)62993-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DL, Papayannopoulos IA, Styles J, Bobin SA, Lin YY, Biemann K, Iqbal K. Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer’s disease. Archives of Biochemistry and Biophysics. 1993;301(1):41–52. doi: 10.1006/abbi.1993.1112. http://doi.org/10.1006/abbi.1993.1112 [DOI] [PubMed] [Google Scholar]
- Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Hyman BT. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathologica. 2012;123(1):1–11. doi: 10.1007/s00401-011-0910-3. http://doi.org/10.1007/s00401-011-0910-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JT, Chana G, Pardo CA, Achim C, Semendeferi K, Buckwalter J, Everall IP. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biological Psychiatry. 2010;68(4):368–376. doi: 10.1016/j.biopsych.2010.05.024. http://doi.org/10.1016/j.biopsych.2010.05.024 [DOI] [PubMed] [Google Scholar]
- Morgan TE, Xie Z, Goldsmith S, Yoshida T, Lanzrein AS, Stone D, Finch CE. The mosaic of brain glial hyperactivity during normal ageing and its attenuation by food restriction. Neuroscience. 1999;89(3):687–699. doi: 10.1016/s0306-4522(98)00334-0. http://doi.org/10.1016/S0306-4522(98)00334-0 [DOI] [PubMed] [Google Scholar]
- Morioka T, Kalehua AN, Streit WJ. Progressive expression of immunomolecules on microglial cells in rat dorsal hippocampus following transient forebrain ischemia. Acta Neuropathologica. 1992;83(2):149–157. doi: 10.1007/BF00308474. [DOI] [PubMed] [Google Scholar]
- Morioka T, Kalehua AN, Streit WJ. Characterization of microglial reaction after middle cerebral artery occlusion in rat brain. Journal of Comparative Neurology. 1993;327(1):123–132. doi: 10.1002/cne.903270110. http://doi.org/10.1002/cne.903270110 [DOI] [PubMed] [Google Scholar]
- Mouton PR, Long JM, Lei DL, Howard V, Jucker M, Calhoun ME, Ingram DK. Age and gender effects on microglia and astrocyte numbers in brains of mice. Brain Research. 2002;956(1):30–35. doi: 10.1016/s0006-8993(02)03475-3. http://doi.org/10.1016/S0006-8993(02)03475-3 [DOI] [PubMed] [Google Scholar]
- Mueller SG, Schuff N, Yaffe K, Madison C, Miller B, Weiner MW. Hippocampal atrophy patterns in mild cognitive impairment and Alzheimer’s disease. Human Brain Mapping. 2010;31(9):1339–1347. doi: 10.1002/hbm.20934. http://doi.org/10.1002/hbm.20934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314 LP–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Njie EMG, Boelen E, Stassen FR, Steinbusch HWM, Borchelt DR, Streit WJ. Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function. Neurobiology of Aging. 2012;33(1):195.e1–195.e12. doi: 10.1016/j.neurobiolaging.2010.05.008. http://doi.Org/10.1016/j.neurobiolaging.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura K, Ogawa M, Yoshida M. Effects of ageing on microglia in the normal rat brain: immunohistochemical observations. Neuroreport. 1994;5(10):1224–1226. doi: 10.1097/00001756-199406020-00016. [DOI] [PubMed] [Google Scholar]
- Otvos LJ, Feiner L, Lang E, Szendrei GI, Goedert M, Lee VM. Monoclonal antibody PHF-1 recognizes tau protein phosphorylated at serine residues 396 and 404. Journal of Neuroscience Research. 1994;39(6):669–673. doi: 10.1002/jnr.490390607. http://doi.org/10.1002/jnr.490390607 [DOI] [PubMed] [Google Scholar]
- Pelvig DP, Pakkenberg H, Stark AK, Pakkenberg B. Neocortical glial cell numbers in human brains. Neurobiology of Aging. 2008;29(11):1754–1762. doi: 10.1016/j.neurobiolaging.2007.04.013. http://doi.org/10.1016/j.neurobiolaging.2007.04.013 [DOI] [PubMed] [Google Scholar]
- Perez SE, Raghanti MA, Hof PR, Kramer L, Ikonomovic MD, Lacor PN, Mufson EJ. Alzheimer’s disease pathology in the neocortex and hippocampus of the western lowland gorilla (Gorilla gorilla gorilla) Journal of Comparative Neurology. 2013;521(18):4318–38. doi: 10.1002/cne.23428. http://doi.org/10.1002/cne.23428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez SE, Sherwood CC, Cranfield MR, Erwin JM, Mudakikwa A, Hof PR, Mufson EJ. Early Alzheimer’s disease-type pathology in the frontal cortex of wild mountain gorillas (Gorilla beringei beringei) Neurobiology of Aging. 2016;39:195–201. doi: 10.1016/j.neurobiolaging.2015.12.017. http://doi.org/10.1016/j.neurobiolaging.2015.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry VH, Gordon S. Macrophages and the nervous system. International Review of Cytology. 1991;125:203–244. doi: 10.1016/s0074-7696(08)61220-6. [DOI] [PubMed] [Google Scholar]
- Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nature Reviews Neurology. 2014;10(4):217–224. doi: 10.1038/nrneurol.2014.38. http://doi.org/10.1038/nrneurol.2014.38 [DOI] [PubMed] [Google Scholar]
- Peters A, Sethares C. Oligodendrocytes, their progenitors and other neuroglial cells in the aging primate cerebral cortex. Cerebral Cortex. 2004;14(9):995–1007. doi: 10.1093/cercor/bhh060. http://doi.org/10.1093/cercor/bhh060 [DOI] [PubMed] [Google Scholar]
- Prokop S, Miller KR, Heppner FL. Microglia actions in Alzheimer’s disease. Acta Neuropathologica. 2013;126(4):461–477. doi: 10.1007/s00401-013-1182-x. [DOI] [PubMed] [Google Scholar]
- Radewicz K, Garey LJ, Gentleman SM, Reynolds R. Increase in HLA-DR immunoreactive microglia in frontal and temporal cortex of chronic schizophrenics. Journal of Neuropathology and Experimental Neurology. 2000;59(2):137–150. doi: 10.1093/jnen/59.2.137. http://doi.org/10.1093/jnen/59.2.137 [DOI] [PubMed] [Google Scholar]
- Raghanti MA, Spocter MA, Stimpson CD, Erwin JM, Bonar CJ, Allman JM, Sherwood CC. Species-specific distributions of tyrosine hydroxylase-immunoreactive neurons in the prefrontal cortex of anthropoid primates. Neuroscience. 2009;158(4):1551–1559. doi: 10.1016/j.neuroscience.2008.10.058. http://doi.org/10.1016/j.neuroscience.2008.10.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghanti MA, Stimpson CD, Marcinkiewicz JL, Erwin JM, Hof PR, Sherwood CC. Cortical dopaminergic innervation among humans, chimpanzees, and macaque monkeys: a comparative study. Neuroscience. 2008;155(1):203–220. doi: 10.1016/j.neuroscience.2008.05.008. http://doi.org/10.1016/j.neuroscience.2008.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annual Review of Immunology. 2009;27:119–45. doi: 10.1146/annurev.immunol.021908.132528. http://doi.org/10.1146/annurev.immunol.021908.132528 [DOI] [PubMed] [Google Scholar]
- Rapoport SI, Nelson PT. Biomarkers and evolution in Alzheimer disease. Progress in Neurobiology. 2011;95(4):510–3. doi: 10.1016/j.pneurobio.2011.07.006. http://doi.org/10.1016/j.pneurobio.2011.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen RF, Farberg AS, Gearing M, Dooyema J, Long PM, Anderson DC, Walker LC. Tauopathy with paired helical filaments in an aged chimpanzee. The Journal of Comparative Neurology. 2008;509(3):259–270. doi: 10.1002/cne.21744. http://doi.org/10.1002/cne.21744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rössler M, Zarski R, Bohl J, Ohm TG. Stage-dependent and sector-specific neuronal loss in hippocampus during Alzheimer’s disease. Acta Neuropathologica. 2002;103(4):363–369. doi: 10.1007/s00401-001-0475-7. http://doi.org/10.1007/s00401-001-0475-7 [DOI] [PubMed] [Google Scholar]
- Schwarz JM, Sholar PW, Bilbo SD. Sex differences in microglial colonization of the developing rat brain. Journal of Neurochemistry. 2012;120(6):948–963. doi: 10.1111/j.1471-4159.2011.07630.x. http://doi.org/10.1111/j.1471-4159.2011.07630.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield LG, Berman NE. Microglial expression of MHC class II increases in normal aging of nonhuman primates. Neurobiology of Aging. 1998;19(1):47–55. doi: 10.1016/s0197-4580(97)00168-1. [DOI] [PubMed] [Google Scholar]
- Sheffield LG, Marquis JG, Berman NEJ. Regional distribution of cortical microglia parallels that of neurofibrillary tangles in Alzheimer’s disease. Neuroscience Letters. 2000;285(3):165168. doi: 10.1016/s0304-3940(00)01037-5. http://doi.org/10.1016/S0304-3940(00)01037-5 [DOI] [PubMed] [Google Scholar]
- Sheng JG, Mrak RE, Griffin WS. Enlarged and phagocytic, but not primed, interleukin-1 alpha-immunoreactive microglia increase with age in normal human brain. Acta Neuropathologica. 1998;95(3):229–234. doi: 10.1007/s004010050792. [DOI] [PubMed] [Google Scholar]
- Sheng JG, Mrak RE, Griffin WST. Neuritic plaque evolution in Alzheimer’s disease is accompanied by transition of activated microglia from primed to enlarged to phagocytic forms. Acta Neuropathologica. 1997;94(1):1–5. doi: 10.1007/s004010050664. http://doi.org/10.1007/s004010050664 [DOI] [PubMed] [Google Scholar]
- Sherwood CC, Gordon AD, Allen JS, Phillips KA, Erwin JM, Hof PR, Hopkins WD. Aging of the cerebral cortex differs between humans and chimpanzees. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(32):13029–34. doi: 10.1073/pnas.1016709108. http://doi.org/10.1073/pnas.1016709108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood CC, Raghanti MA, Wenstrup JJ. Is humanlike cytoarchitectural asymmetry present in another species with complex social vocalization? A stereologic analysis of mustached bat auditory cortex. Brain Research. 2005;1045(1–2):164–174. doi: 10.1016/j.brainres.2005.03.023. http://doi.org/10.1016/j.brainres.2005.03.023 [DOI] [PubMed] [Google Scholar]
- Sloane JA, Hollander W, Moss MB, Rosene DL, Abraham CR. Increased microglial activation and protein nitration in white matter of the aging monkey. Neurobiology of Aging. 1999;20(4):395–405. doi: 10.1016/s0197-4580(99)00066-4. http://doi.org/10.1016/S0197-4580(99)00066-4 [DOI] [PubMed] [Google Scholar]
- Slomianka L, West MJ. Estimators of the precision of stereological estimates: An example based on the CA1 pyramidal cell layer of rats. Neuroscience. 2005;136(3):757–767. doi: 10.1016/j.neuroscience.2005.06.086. http://doi.org/10.1016/j.neuroscience.2005.06.086 [DOI] [PubMed] [Google Scholar]
- Spangenberg EE, Lee RJ, Najafi AR, Rice RA, Elmore MRP, Blurton-jones M, Green KN. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid- b pathology. 2016:1–17. doi: 10.1093/brain/aww016. http://doi.org/10.1093/brain/aww016 [DOI] [PMC free article] [PubMed]
- Steiner J, Bielau H, Brisch R, Danos P, Ullrich O, Mawrin C, Bogerts B. Immunological aspects in the neurobiology of suicide: Elevated microglial density in schizophrenia and depression is associated with suicide. Journal of Psychiatric Research. 2008;42(2):151–157. doi: 10.1016/j.jpsychires.2006.10.013. http://doi.org/10.1016/jjpsychires.2006.10.013 [DOI] [PubMed] [Google Scholar]
- Streit WJ, Braak H, Xue QS, Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathologica. 2009;118(4):475–485. doi: 10.1007/s00401-009-0556-6. http://doi.org/10.1007/s00401-009-0556-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudduth TL, Schmitt FA, Nelson PT, Wilcock DM. Neuroinflammatory phenotype in early Alzheimer’s disease. Neurobiology of Aging. 2013;34(4):1051–1059. doi: 10.1016/j.neurobiolaging.2012.09.012. http://doi.org/10.1016/j.neurobiolaging.2012.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahara K, Kim HD, Jin JJ, Maxwell JA, Li L, Fukuchi K. Role of toll-like receptor signalling in Abeta uptake and clearance. Brain: A Journal of Neurology. 2006;129(Pt 11):3006–3019. doi: 10.1093/brain/awl249. http://doi.org/10.1093/brain/awl249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetreault NA, Hakeem AY, Jiang S, Williams BA, Allman E, Wold BJ, Allman JM. Microglia in the cerebral cortex in autism. Journal of Autism and Developmental Disorders. 2012;42(12):2569–2584. doi: 10.1007/s10803-012-1513-0. http://doi.org/10.1007/s10803-012-1513-0 [DOI] [PubMed] [Google Scholar]
- Torres-Platas SG, Comeau S, Rachalski A, Dal Bo G, Cruceanu C, Turecki G, Mechawar N. Morphometric characterization of microglial phenotypes in human cerebral cortex. Journal of Neuroinflammation. 2014;11(1):1–13. doi: 10.1186/1742-2094-11-12. http://doi.org/10.1186/1742-2094-11-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanGuilder HD, Bixler GV, Brucklacher RM, Farley JA, Yan H, Warrington JP, Freeman WM. Concurrent hippocampal induction of MHC II pathway components and glial activation with advanced aging is not correlated with cognitive impairment. Journal of Neuroinflammation. 2011;8(1):138. doi: 10.1186/1742-2094-8-138. http://doi.org/10.1186/1742-2094-8-138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Gunten A, Kövari E, Rivara CB, Bouras C, Hof PR, Giannakopoulos P. Stereologic analysis of hippocampal Alzheimer’s disease pathology in the oldest-old: Evidence for sparing of the entorhinal cortex and CA1 field. Experimental Neurology. 2005;193(1):198–206. doi: 10.1016/j.expneurol.2004.12.005. http://doi.org/10.1016/j.expneurol.2004.12.005 [DOI] [PubMed] [Google Scholar]
- Walker L, Cork L. The neurobiology of aging in nonhuman primates. In: Terry R, Katzman R, Bick K, Sisodia S, editors. Alzheimer Disease. Philadelphia: Lippincott, Williams and Wilkins; 1999. pp. 233–243. [Google Scholar]
- West MJ, Coleman PD, Flood DG, Troncoso JC. Differences in the Pattern of Hippocampal Neuronal Loss in Normal Aging and Alzheimers-Disease. Lance. 1994;344:769–772. doi: 10.1016/s0140-6736(94)92338-8. http://doi.org/10.1016/S0140-6736(94)92338-8 [DOI] [PubMed] [Google Scholar]
- Wright AL, Zinn R, Hohensinn B, Konen LM, Beynon SB, Tan RP, Vissel B. Neuroinflammation and Neuronal Loss Precede Aβ Plaque Deposition in the hAPP-J20 Mouse Model of Alzheimer’s Disease. PLoS ONE. 2013;8(4) doi: 10.1371/journal.pone.0059586. http://doi.org/10.1371/journal.pone.0059586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Rogers J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harbor Perspectives in Medicine. 2012;2(1):a006346. doi: 10.1101/cshperspect.a006346. http://doi.org/10.1101/cshperspect.a006346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Itoh Y, Shintaku M, Kawamura J, Jensson O, Thorsteinsson L, Otomo E. Immune reactions associated with cerebral amyloid angiopathy. Stroke. 1996;27(7):1155–1162. doi: 10.1161/01.str.27.7.1155. [DOI] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Lee VMY. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53(3):337–351. doi: 10.1016/j.neuron.2007.01.010. http://doi.org/10.1016/j.neuron.2007.01.010 [DOI] [PubMed] [Google Scholar]