

Abstract
Irisin, a muscle-origin protein derived from the extracellular domain of the fibronectin domain-containing 5 protein (FNDC5), has been shown to modulate mitochondria welfare through paracrine action. Here we test the hypothesis that irisin contributes to cardioprotection after myocardial infarction by preserving mitochondrial function in cardiomyocytes. Animal model studies show that intravenous administration of exogenous irisin produces dose-dependent protection against ischemia/reperfusion (I/R)-induced injury to the heart as reflected by the improvement of left-ventricular ejection fraction and the reduction in serum level of cTnI (n=15, P<0.05). I/R-induced apoptosis of cardiomyocytes is reduced after irisin treatment. The irisin-mediated protection has, at least in part, an effect on mitochondrial function because administration of irisin increases irisin staining in the mitochondria of the infarct area. Irisin also reduces I/R-induced oxidative stress as determined by mitochondrial membrane potential evaluation and superoxide FLASH event recording (n=4, P<0.05). The interaction between irisin and SOD2 plays a key role in the protective process because irisin treatment increases SOD activity (n=10, P<0.05) and restores the mitochondria-localization of SOD2 in cardiomyocytes (n=5, P<0.05). These results demonstrate that irisin plays a protective role against I/R injury to the heart. Targeting the action of irisin in mitochondria presents a novel therapeutic intervention for myocardial infarction.
Keywords: irisin, heart, ischemia-reperfusion injury, SOD2
Introduction
Ischemic injury contributes to deleterious outcomes in multiple vital organs such as heart, lungs, brain and kidneys1–4. Restoration of ischemic blood flow is required to prevent tissue infarction, while reperfusion often causes exacerbation of tissue injuries. Blood reflow and reoxygenation is frequently associated with an activation of profound inflammatory response and a rise of reactive oxygen species (ROS)2, 5–7as an intermediate stage in this pathogenesis. This leads to mitochondrial dysfunction and plasma membrane disintegration8–10, activating the mitochondrial pathway to cell death11, 12.
Irisin is a muscle-origin protein derived from the extracellular domain of the fibronectin domain-containing 5 protein (FNDC5)13, 14. Boström et al showed that exercise-induced release of irisin had beneficial effects on adipose tissue in part by increasing the amount of mitochondrial uncoupling protein-1 in brown adipocytes15. While the lipogenic effect of irisin may correlate with the chronic action of irisin15–19, the acute action of irisin in non-adipose tissues has only recently been explored. Here we test the hypothesis that irisin released from skeletal muscle protects the heart from ischemia-reperfusion (I/R) injury. Our present study provides evidence that irisin protects against I/R-induced mitochondrial dysfunction through modulation of superoxide dismutase function. We also find that intravenous administration of exogenous irisin can ameliorate I/R-induced myocardial injury in rodent models.
Methods
Myocardial ischemia-reperfusion model
Sprague-Dawley rats (250 to 260 g) were purchased from the Animal Center of Daping Hospital, the Third Military Medical University (Chongqing, China). The rats were randomized into three groups: the sham-operated group, myocardial ischemia/reperfusion (I/R) group, and irisin treatment I/R group (recombinant irisin, Phoenix Biotech, Burlingame, CA). Irisin was administered prior to reperfusion through intravenous injection after dissolving in saline solution. After anesthetization with an intraperitoneal injection of sodium pentobarbital (50 mg/kg), rat myocardial I/R model was performed as described below20. Briefly, after a thoracotomy, the left anterior descending coronary artery (LAD) of the rats was ligated for 30 mins, and then the ligature around the LAD was released for 24-hour reperfusion. The blood was collected and centrifuged at 3000 g for 10 minutes to separate serum. After euthanasia by an overdose of sodium pentobarbital (200 mg/kg) with intraperitoneal injection, the heart tissues were obtained immediately for following analysis.
The experimental protocol was approved by the Institutional Animal Care and Use Committee at Third Military Medical University as well as approved by the IACUC of Kansas City University of Medicine and Biosciences and The Ohio State University. All surgeries were performed under appropriate anesthesia, and all efforts were made to minimize animal suffering. All experiments conformed to the guidelines of the American Association for the Accreditation of Laboratory Animal Care.
Infarct size measurement
The infarct size was defined by phosphate-buffered 1% TTC staining.22 Briefly, the heart was snap-frozen and cut into six 1-mm-thick transverse slices from the apex to the base, parallel to the atrioventricular groove. Each slice was incubated in a 1% solution of phosphate-buffered TTC at 37°C for 20 minutes to differentiate infarct area (pale) from viable (brick red) myocardial area. The ratio of the area of the infarct band to the total area of the LV was calculated and presented as a percentage. Slices were then fixed in 10% formalin, and the infarct area from each section were measured using ImageJ software (National Institutes of Health, Bethesda, MD), and the values obtained were averaged. Individuals conducting the experiment were blinded to the experimental groups.
Echocardiography
After 24-hour reperfusion, the rats were again anesthetized with intraperitoneal injection of 40 mg/kg sodium pentobarbital for echocardiography.22 Echocardiographic parameters [LVEF according to Simpson's rule and LV internal diameter at diastole (LVIDd)] were determined by 2-dimensional (2-D) and M-mode echocardiography at the mid-papillary muscle level. Left ventricular ejection fraction (LVEF) was measured by 2-D tracing using the formula; LVEF (%) = [(LV diastolic area-LV systolic area) / LV diastolic area] ×100, and automatically calculated by the echocardiography software. The individuals conducting the experiment and the reading of the echocardiographic images were blinded to the animal treatments.
Histopathological examination and TUNEL assay
Formalin fixed hearts were processed, embedded in paraffin and cut as 5 μm thick sections. Nonspecific binding of the secondary antibody was blocked using 10% goat serum in PBS for 20 min. To determine the distribution of irisin in hearts after I/R injury, sections were incubated with the primary antibody (rabbit anti-irisin antibody, 1: 50, Phoenix Biotech, Burlingame, CA) at 4°C overnight in a humidity chamber. Specific binding of primary antibody was detected with the respective HRP Detection kit (ZSBG). Nuclei were counterstained with hematoxylin staining solution. Slides were covered using neutral balsam and appropriate cover-slips. Negative controls were incubated either omitting the primary antibody or blocking the primary antibody with the respective immunizing peptide at 4°C overnight. No unspecific binding of the secondary antibody and only minimal unspecific binding of the primary antibody was detected.
To detect the cardiomyocyte apoptosis, an in-situ apoptotic cell death detection kit based on the TUNEL assay was used. The percentage of apoptotic nuclei per section was calculated by counting the number of TUNEL-staining cardiomyocyte nuclei divided by the total number of DAPI-positive nuclei. Cardiomyocyte cytoplasm was identified by sarcomeric tropomyosin antibody. The number of apoptotic nuclei were counted in ten randomly high-power fields (HPFs) per section of the peri-infarct zone, and a total of five sections per animal were analyzed for each rat to quantify levels of apoptosis. All of the sections came from six rats in every group. Images were taken at a magnification of 630× with a LSM 780 NLO & DuoScan System (Carl Zeiss, Oberkochen, Germany). All of the manual counts were performed in a blinded fashion.
Cell preparation and culture
H9c2 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and cultured in DMEM-F12, supplemented with 10% FBS, at 37°C and 5% CO2 in the CO2 incubator. Cells were passed regularly and sub-cultured to about 80-90% of confluence and rendered quiescent in serum-free DMEM for 24 hrs prior to experimentation. H9c2 cells were exposed to an anoxic chamber with 5% CO2 and 95% N2 at 37°C for 16 hrs followed by reoxygenation for up to 3 hrs. Exogenous irisin was conjugated with FITC via a dye labeling kit (G-Biosciences, St Louis, MO), and used to treat cells immediately after anoxia. After reoxygenation, the cells were stained with mito-tracker green or indicated antibodies.
Immunoblotting and immunoprecipitation
After washing twice by ice-cold PBS, cells and heart tissues were lysed in ice-cold lysis buffer (20 mM Tris-HCl, pH 7.4; 2 mM EDTA, pH 8.0; 2 mM EGTA; 100 mM NaCl; 10 μg/ml leupeptin; 10 μg/ml aprotinin; 2 mM phenylmethylsulfonyl fluoride; 1% NP-40). Protein concentration was determined by BCA assay (Beyotime, Shanghai, China). For co-immunoprecipitation experiments, 500μg of lysate was subjected to co-immunoprecipitation using agarose beads (Santa Cruz Biotechnology, Paso Robles, CA) and anti-SOD1 or anti-SOD2 antibodies at 4°C overnight. After immunoaffinity isolation of target proteins, the beads of immunoprecipitates were suspended in sample buffer, washed with PBS for three times, and boiled in 2X SDS loading buffer for 5 minutes at 95°C. The rabbit anti-irisin antibody (1:1000, Phoenix Biotech) was used for immunoblotting.
For immunoblotting, protein (50 μg) was separated by 15% SDS-PAGE gel and transferred electrophoretically to 0.45 μm nitrocellulose membrane (Pall, Port Washington, NY). Membranes were blocked in 0.5% casein solution in PBS at room temperature for two hrs. The membranes were then probed with rabbit anti-irisin antibody (1:1000, Phoenix Biotech), goat anti-SOD1, rabbit anti-SOD2 antibody (1:300, Santa Cruz Biotechnology), rabbit anti-cytochrome c oxidase polypeptide IV (COX IV) antibody, rabbit anti-total caspase-3, rabbit anti-cleaved caspase-3 (1:400, Abcam Technology, Cambrige, UK), or rabbit anti-cytochrome c antibody (1:500, Protein Technology, Chicago, IL), respectively, at 4°C overnight. The primary antibodies were then identified by relative IRDye conjugated secondary antibodies diluted 1:15000 (LI-COR Biosciences) for two hours at room temperature. Finally, the membrane signals were imaged with an Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, Nebraska) at scan intensity of 6 to 7 and scan resolution of 84 μm. The band density was analyzed using Quantity One software. The densitometric value of each protein was normalized with relative loading control protein signal.
Mitochondrial membrane potential assay
Mitochondrial membrane potential was assessed using a JC-1 (5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine iodide) kit (Beyotime, Shanghai, China)23. Briefly, cells cultured in 24-well plates after indicated treatments were incubated with an equal volume of JC-1 staining solution (10 μg/ml) for 20 minutes at 37°C in the dark and rinsed twice with buffer (provided as part of the kit and pre-cooled at 4°C). An uncoupling agent, carbonyl cyanide phenylhydrazones (CCCP), was used as a positive control. JC-1 fluorescence was measured by using a confocal microscopy at 10× magnification (Confocal Fluorescence Imaging Microscope, Leica TCS-SP5, Bensheim, Germany) under single excitation (Argon-ion 488 nm) and dual emission (shift from green 530 nm to red 590 nm). To quantify the fluorescence intensity, 10 different images taken from each group were randomly selected and analyzed by Image Pro Plus 6.0 software (Media Cybernetics; Silver Spring, MD). The loss of mitochondrial membrane potential (MMP) induced the depolymerization of JC-1, resulting in less J-aggregates (red) and more JC-1 monomer (green). Therefore, MMP was quantified as the ratio of red and green fluorescence of JC-1.
Cell viability assay
A Cell Counting Kit-8 (CCK-8, Dojindo, Kumamoto, Japan) was employed in the experiment to evaluate cell viability.22 Briefly, H9c2 cells were seeded at 3×104 per well in 100 μl culture medium into 96-well plate and incubated with CCK-8 for 2 hour at 37°C. The absorbance was subsequently measured on a microplate spectrophotometer Model 680 (Bio-Rad, Hercules, CA) at an absorbance of 450 nm. Three independent experiments were performed.
Mitochondrial isolation
According to previously published procedures,24 H9c2 cells were washed with ice-cold PBS and then pelleted by centrifugation at 1,000 g for 10 min at 4°C in PBS. The resulting pellet was then resuspended in 1 ml of mitochondrial isolation buffer (in mM: NaCl 10; MgCl2 2.5; Tris 10; pH 7.5 adjusted by HCl). After incubated in an ice bath for 10 minutes, the cell suspension was transferred to a 1-ml glass homogenizer and added 0.7 ml 2.5x mitochondrial suspension (in mM: mannitol 525; sucrose 175; Tris 12.5; EDTA•Na2 1; pH 7.5 adjusted by HCl). The supernatant was purified by centrifugation at 12,000 g for 5 min at 4°C for two times and then was centrifuged at 6,000 g for 20 min at 4°C to pellet the mitochondria. The pellet was resuspended in mitochondrial suspension. Protein concentrations were determined using a commercial BCA protein assay kit (Beyotime).
Assay for serum LDH and cTnI
Before harvesting the heart, about 6 ml arterial blood was taken from abdorminal aorta for measurement of LDH, cTnI, and Irisin concentrations. Serum was separated by centrifugation at 3000 g for 10 minutes. The supernatant was stored at -80°C until used. Lactate dehydrogenase (LDH) and cardiac troponin I (cTnI) were assayed by automatic biochemical detecting system DXC800 (Beckman Coulter, Brea, CA) and respectively expressed as IU/L, U/L and mg/L.
Caspase-3 activity assay
As a marker of irreversible end point of apoptotic pathway,25 caspase-3 activity was determined using a commercial kit (Beyotime), which is based on the ability of caspase-3 to change acetyl-Asp-Glu-Val-Asp p-nitroanilide into the yellow formazan product, p-nitroaniline. According to the manufacturer’s protocol, after incubating the mixture composed of 10 μl of tissue lysate, 80 μl of reaction buffer and 10 μl of 2 mM caspase-3 substrate in 96-well microtiter plates at 37 °C for 4 hrs, caspase-3 activity was quantified in the samples with a microplate spectrophotometer Model 680 (Bio-Rad, Hercules, CA) at an absorbance of 405 nm.
Measurement of SOD activity
SOD activity measurement was performed by nitroblue tetrazolium (NBT) method to detect oxidative stress with assay kit (Beyotime).26, 27 Briefly, activity was analyzed during the ethanol phase of the lysate after 1.0 ml ethanol/chloroform mixture (5/3, v/v) was mixed with the same volume of sample and centrifuged. The SOD activity in tissue homogenates was detected by the inhibition of NBT reduction caused by the xanthine-XO system as the superoxide generator, and the absorbance was finally determined at 450 nm using a microplate reader Model 680. One unit of SOD means the enzyme amount causing 50% inhibition in the NBT reduction rate. The results were expressed in U/min/g tissue.
ROS generation assay
For intracellular ROS measurement, about 3 × 105 cells were incubated for 15 min at 37°C with the oxidation-sensitive fluorescent probe DCFH-DA (5 μM).28 Then fluorescence intensity was measured using a confocal microscopy at 10× magnification (Confocal Fluorescence Imaging Microscope, Leica TCS-SP5, Bensheim, Germany) coupled with ImagePro Plus software for the quantification of fluorescent changes. DCFH-DA is a fat-soluble probe, easily penetrates through cell membrane, and is hydrolyzed into DCFH, which can be oxidized into bright DCF. The intracellular probe oxidation-induced changes of immunofluorescence was measured as ROS generation.
For tissue ROS measurement, the fresh frozen heart sections from each rat of each respective group were collected. Cardiac slides were incubated with another oxidation-sensitive fluorescent probe DHE (10 μM) for 20 minutes at 37°C. The fluorescence intensity recording and data analysis was carried out as mentioned above. Like DCFH-DA, DHE are also widely used to monitor changes in myocardial O2− levels in response to oxidative injury.
Confocal microscopy of immunostaining
For colocalization analysis of mitochondria and irisin/SOD2, H9c2 cells were grown on coverslips that were fixed with 4% paraformaldehyde (30 minutes). Exogenous FITC-labeled irisin was given to cells immediately after anoxia. When reoxygenation ended, the cells were stained with mito-tracker green (50nM, 30minutes). Then, the cells were treated with indicated antibodies overnight at 4°C. To identify the primary antibodies, relative fluorescent probes conjugated secondary antibodies at room temperature for 1 ~ 2 hrs. Immunofluorescence images were acquired with a LSM 780 NLO&DuoScan System (Carl Zeiss, Germany).
Mitochondrial FLASH event recording and analysis
A circularly permuted yellow fluorescent protein (cpYFP) transgenic mouse colony was generated using vector (mt-cpYFP/pUCCAGGS)21. Cardiomyocytes were isolated from cpYFP transgenic mice in the age range of 8 ~12 weeks29. The animal was injected with 70 μl heparin (70 USP units/mouse) i.p. 10 minutes prior to anesthesia. The heart was removed from a mouse under anesthesia using isoflurane. After the aorta of the heart was cannulated by a 22 gauge feeding needle (Fine Science Tools, Item No. 18061-22), the heart was perfused via the left ventricle for 4 minutes with a perfusion buffer (in mM: NaCl, 120; KCl, 5.4; MgSO4, 1.6; NaH2PO4, 1.2; glucose, 5.6; NaHCO3, 20; taurine, 5; 2,3-butanedione monoxime (BDM), 10; aerated with 5 % CO2/95 % O2; pH 7.2) using gravity from 70 cm height through the Langendorff system. The heart was perfused for 4 minutes with isolation solution (perfusion buffer containing 1 mg/ml collagenase Type 2 (Worthington), 0.02 mg/ml protease Type XIV (Sigma-Aldrich), and then perfused for 8~12 min with 50 μM CaCl2 contained isolation solution. Afterwards, the heart was transferred to a 35 mm dish with 2.5 ml of 50 μM CaCl2 isolation solution and the aorta, atria, and great vessels were trimmed away. The remaining ventricle tissue was gently teased into 10-12 small pieces. The heart pieces were carefully transferred to a 15-ml polypropylene conical tube and 7.5 ml of 50 μM CaCl2 isolation solution was added. Cells were aspired up and down around 10 times with a 7-ml transfer pipette. After removing debris by 250 μm nylon mesh, cardiomyocytes were centrifuged for 1 min at 45 x g, followed by removal of the supernatant and re-suspended in 10 ml calcium solution containing 100 μM CaCl2 and 2.5% FBS. After cells were allowed to settle down for 10 min at room temperature, the supernatant was removed and cells were re-suspended in the same solution to stop enzyme activity completely. The process of settling down and re-suspending by using 250 μM, 500 μM, 1.2 mM calcium solutions for calcium tolerance was repeated. At this point, cardiomyocytes were used in this study. Cells were cultured in N2 gas for 3 hours for anoxia treatment, and then the cells were returned to normal condition for reoxygenation. Live cell imaging was conducted to record FLASH events in cardiomyocytes using a Zeiss LSM 510 confocal microscope equipped with a 40x, 1.2W (water immersion) objective and a sampling rate of 1s/frame. The cpYFP imaging was achieved by excitation at 488 nm and collecting emission at >505 nm. Imaging experiments were done at room temperature (23°C). IDL 7.0 (IDL, ITT Visual Information Solutions) was used for image processing. Sigmaplot 11.0 and Microsoft Excel were used for FLASH signal data analysis.
Statistical analysis
Data was reported as means±SEM. Statistical analyses were performed by one-way ANOVA followed by Holm-Sidak's post hoc multiple comparison test (for comparison of more than two groups) or Student's t-test (for comparison of two groups). Data are represented as mean ± S.E.M. A value of P<0.05 was considered significant.
Results
Exogenous irisin protects against I/R-injured myocardium
We performed experiments with the rat model of heart I/R to test if systemic application of irisin could protect against I/R induced tissue injury. Studies from other investigators have reported the basal serum levels of irisin to be in the range of 6-20 ng/ml in humans and rodents30. We thus treated rats with intravenous (i.v.) administration of 1 μg/kg irisin. Assuming a 15 ml blood volume with a rat of 250 g body weight, i.v. administration of 1 μg/kg should produce elevation of ~17 ng/ml of irisin in the blood circulation, and ~2-3 fold increase over the basal level.
Results found that infarct areas of the heart were significantly reduced with i.v. administration of 1.0 μg/kg irisin (Fig. 1a and 1b). Specificity of irisin effects were tested using denatured protein that was boiled for 30 minutes at 100°C. In the presence of heat-denatured irisin, the protective effects on cardiac I/R injury were lost. Myocardial injury was further determined by measurement of circulating level of troponin I (cTnI) as a biomarker for heart injury, and echocardiography for assessment of heart function. We found that irisin could ameliorate I/R-induced injury, as reflected by the improvement of left-ventricular ejection fraction (LVEF, Fig. 1c) and the reduction in serum level of cTnI (Fig. 1d). Moreover, the cardioprotection of irisin was shown in a dose-dependent manner, determined by measurement of cTnI concentrations (Fig. 1e).
Figure 1. Irisin treatment protects I/R-induced injury to the rat heart.
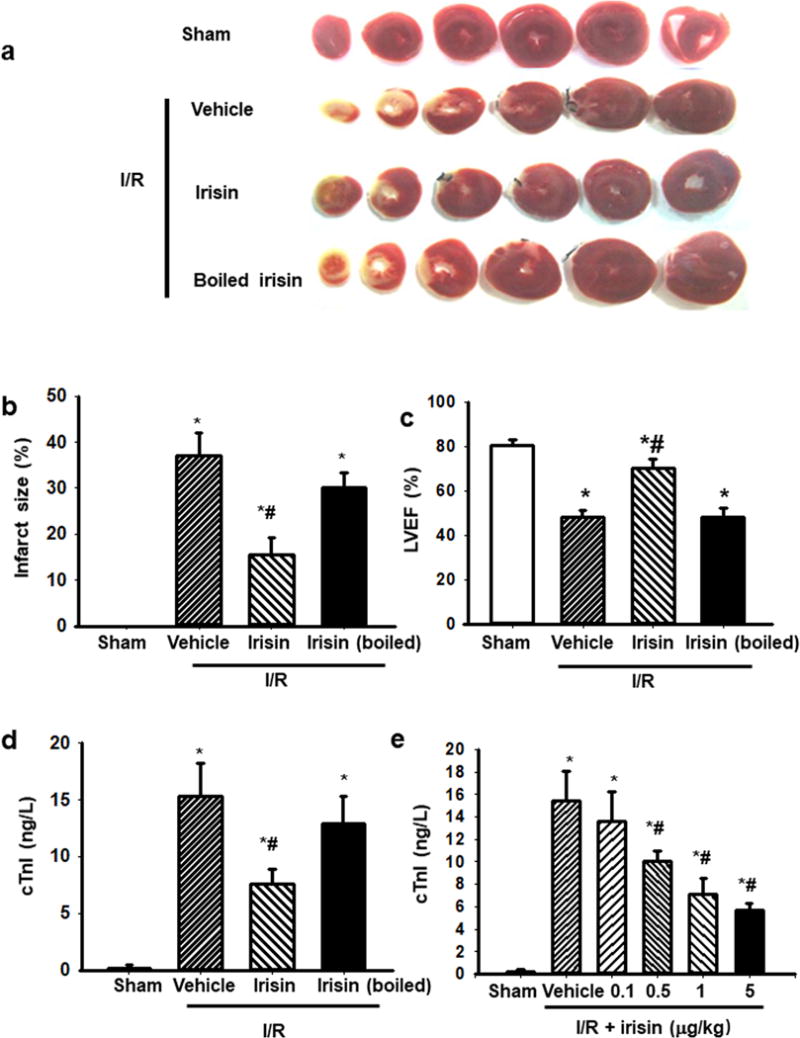
(a) Myocardial infarction after I/R injury was shown by TTC staining of viable cardiac tissues. Irisin treatment reduced infarct area in hearts subjected to I/R as compared to vehicle or denatured irisin treatment. Sham and boiled irisin serve as controls. (b) The infarct areas from multiple experiments were quantified and represented as mean ± SEM (n=12 per group, * P<0.05 vs. sham control, #: P<0.05 vs. I/R vehicle). (c) Echocardiogram showed that i.v. administration of irisin improved left ventricle ejection fraction (LVEF) after I/R injury as compared to controls. (n=12 per group, * P<0.05 vs. sham control, #: P<0.05 vs. I/R vehicle). (d) Rats receiving irisin showed reduced serum troponin I (cTnI) level after I/R (n=15, * P<0.05 vs. sham control, #: P<0.05 vs. I/R vehicle). (e) The cardio-protective effect of exogenous irisin (e.g. reduction of cTnI) was dose-dependent (n=6, * P<0.05 vs. sham control, #: P<0.05 vs. I/R vehicle).
As apoptosis is one contributing factor to the pathogenesis of I/R injury, the effect of irisin on apoptosis was analyzed by TUNEL staining. TUNEL assay revealed that I/R-induced apoptosis of cardiomyocytes was protected by irisin (Fig. 2a), and I/R-induced release of cytochrome c from mitochondria was reduced by administration of irisin to the rat model of MI (Fig. 2b). Moreover, Fig. 2c showed that caspase-3 was activated in an I/R-treated heart, and administration of irisin to I/R injured heart led to significant reduction of caspase-3 activity. These results suggest that irisin-mediated cardio-protection involves mitochondria-dependent apoptosis.
Figure 2. Irisin protects against I/R-induced apoptosis in cardiomyocytes.
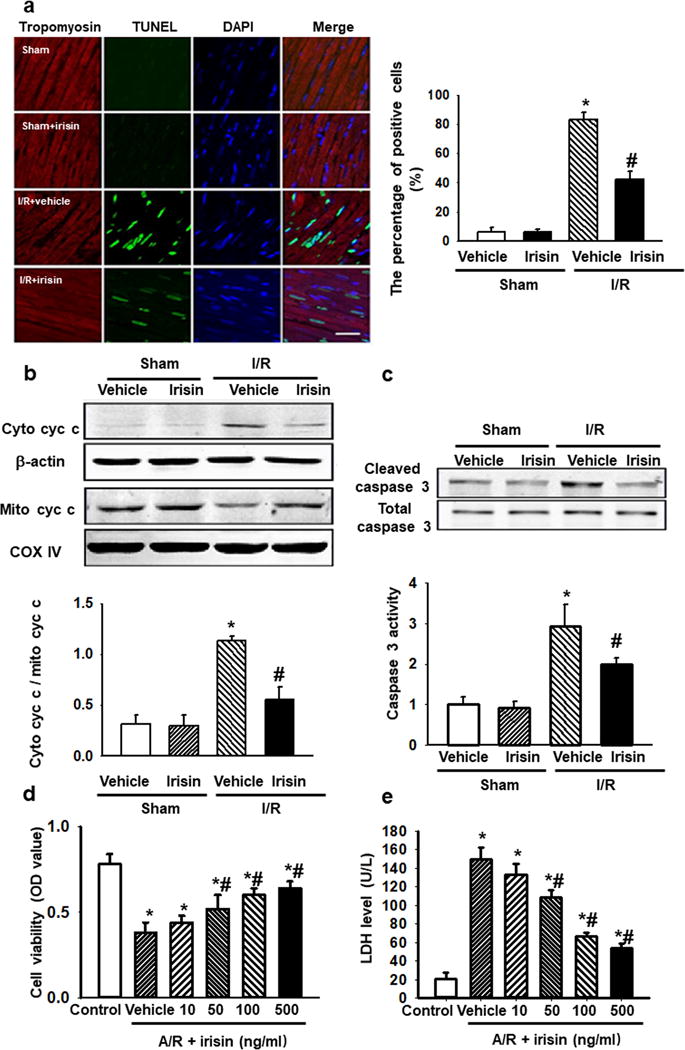
(a) Representative TUNEL staining images showed less TUNEL positive nuclei in irisin treated rat cardiomyocytes after I/R injury. (Bar = 50 μm). The percentage of apoptotic nuclei per section was quantified. n=6 rats in each group with 10 fields averaged from each animal (* P<0.05 vs. sham, # P<0.05 vs. I/R vehicle). (b and c) Western blot showed irisin treatment reduced both cytochrome c release from mitochondria (b), and caspase 3 activation (c) in I/R injured rat hearts. The total caspase-3, COX-IV and β-actin were used as internal control. (n=9, * P<0.05 vs. sham control, # P<0.05 vs. I/Rvehicle). (d and e) H9c2 cells were subjected to A/R with or without irisin treatment (10-500ng/ml). Cell viability (d) and LDH concentration in the culture medium (e) were determined. Irisin treatment prevented A/R induced cell death and preserves cell integrity in a dose-dependent manner (n=6, *P<0.01 vs. control, #P<0.01 vs. A/R vehicle).
Consistent with the in-vivo study, the protection on cardiomyocyte was confirmed in H9c2 cells. Treatment of H9c2 cells with anoxia/reoxygenation (A/R) led to an elevation of lactate dehydrogenase (LDH) activity (Fig. 2d) in the culture medium as indication of injury to H9c2 cells that was accompanied by reduced cell viability. Irisin applied to the culture medium produced dose-dependent protection against A/R-induced injury to H9c2 cells (Fig. 2e).
Irisin targets mitochondria to ameliorate I/R-induced myocardium injury
To further elucidate the mechanism of irisin in myocardium protection, the localization of irisin in myocardium was determined by immunostaining. Immunohistochemical (IHC) staining was used to examine the concentration of irisin in the injured hearts. As shown in Fig. 3a, there was basal irisin staining in the heart from the sham controls. I/R induced mild elevation of endogenous irisin in heart tissue in the infarcted area or peri-infarcted area. Exogenous irisin treatment further increased the irisin staining in the I/R injured heart (Fig. 3a) The subcellular distribution of irisin in the injured cardiomyocytes from infarct area was observed, and it was noticed that there was overlapping pattern of endogenous irisin and Cox IV, a specific biomarker for mitochondria, where exogenous irisin treatment increased the accumulation of irisin in the mitochondria (Fig. 3b). Moreover, the targeting of irisin to the mitochondria of the cardiomyocytes in the infarct area was also confirmed by co-localization of MitoTracker Deep Red (red) and FITC-labelled irisin (green) (Fig. 3c). Denatured irisin (boiled for 10 minutes at 100°C, as control) did not show intracellular entry in cardiomyocytes after AR treatment, indicating the involvement of mitochondria in irisin-mediated cardioprotection.
Figure 3. Irisin targets mitochondria in infarct area of I/R-injured heart tissue.
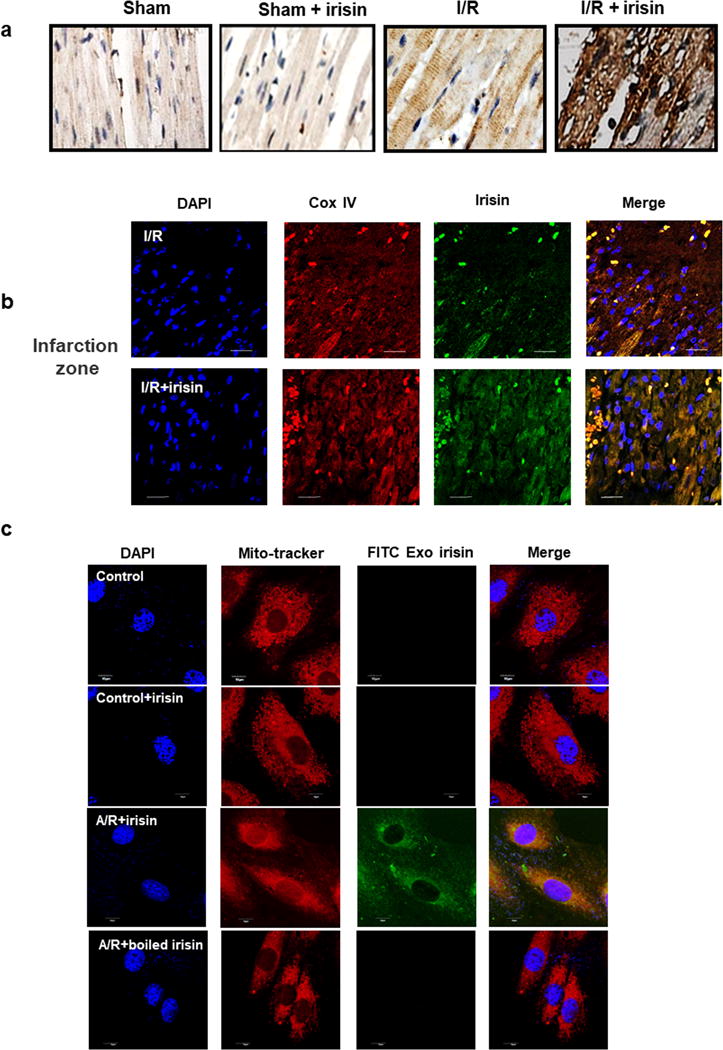
(a) IHC staining revealed concentration of irisin in the infarct area of rat heart. Hearts subjected to I/R alone only showed mild elevation of irisin in the infarct area. Rat hearts that were exposed to increasing endogenous irisin showed accumulating concentration of irisin in the infarct area. (b) Confocal microscopic imaging revealed the accumulation of irisin (green) in the I/R injured rat cardiomyocytes from infarct zone of heart tissue with or without exogenous irisin treatment, and co-localization with Cox IV, a specific biomarker for mitochondrial (red). The staining was repeated at least five times (Bar = 10 μm). (c) The mitochondria localization of exogenous FITC-labeled irisin in A/R treated H9c2 cells. H9c2 cells incubated with Mitotracker and FITC-conjugated irisin (FITC-Irisin, 0.1μg/ml). FITC-Irisin was taken up by cells following A/R treatment (middle). Boiled inactive irisin was not taken up by H9c2 cells under the same condition (bottom). Note FITC-irisin targeted to mitochondria in H9c2 cells after A/R treatment. (Scale bar=10μm).
To further identify the role of irisin targeting to mitochondria, the alteration of mitochondrial membrane potential (MMP) were evaluated by J-aggregate dye staining. Incubation of H9c2 cells with irisin (0.1 μg/ml) protected the loss of mitochondrial membrane potential since a significant increase in red/green fluorescence ratio was observed in A/R+irisin treated cells (Fig. 4a).
Figure 4. Irisin targets mitochondria-dependent oxidative stress pathway for cardioprotection.
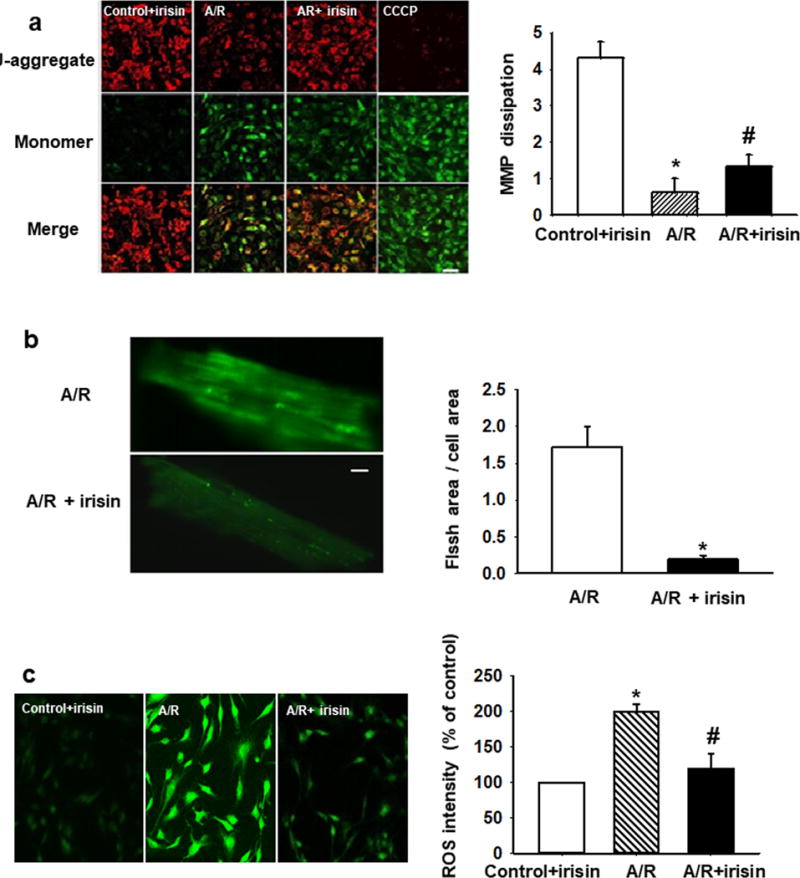
(a) Irisin treatment preserved MMP after A/R injury in H9c2 cells. Red fluorescence of JC-1 was reduced after A/R injury and could be partially rescued by irisin treatment (upper panels). Dissipation of MMP as determined by enhancement of green monomeric form of JC-1 could be partially rescued by irisin (middle panels). A mitochondrial uncoupling agent, CCCP (10mM), was used to completely dissipate the MMP. MMP was quantified as the ratio of red and green fluorescence of JC-1. Irisin treatment rescued A/R induced MMP dissipation. (n=4, *P<0.01 vs. control+irisin, #P<0.05 vs. A/R, Bar = 80 μm). (b) Mitochondria superoxide FLASH events from mouse cardiomyocytes subjected to A/R (upper panel), and with incubation of 100 ng/ml irisin in the extracellular solution (lower panel). Summation of 100 continuously recorded images of cpYFP was presented. Myocytes incubated with irisin show reduced FLASH signals. See supplementary movies for the dynamic changes in FLASH events. Quantitative analysis of mitochondrial FLASH events. n= 9-10 cells/group from 4 mice (*P<0.05 vs. A/R, Bar = 10 μm). (c) Total ROS production in H9c2 cells was quantified by DHE staining. Irisin treatment reduces A/R induced ROS (n=10, *P<0.01 vs. control+irisin, #P<0.05 vs. A/R, Bar = 80 μm).
In addition, a transgenic mouse model with targeted expression of cpYFP in mitochondria as a biosensor was used for direct measurement of superoxide signal produced by mitochondria31 cpYFP generated FLASH signals reflected the oxidative stress associated with changes in metabolic function of mitochondria21, 31. Cardiomyocytes derived from the cpYFP transgenic mice displayed little FLASH activity under basal conditions (data not shown), which is consistent with the observations of Wang et al31. As shown in Fig. 4b, A/R-treatment (16 hrs exposure to anoxia followed with 3 hrs reoxygenation) led to significant elevation of FLASH activity in the cardiomyocytes (see Supplemental Movies for the dynamic changes of FLASH signal in cardiomyocytes). Incubation with 100 ng/ml irisin prevented A/R-induced increase of FLASH activity in cardiomyocytes. In addition, elevated reactive oxygen species (ROS) were observed in cardiomyocytes subjected to A/R. The administration of irisin led to significant reduction in ROS activity (Fig. 4c). These results support the idea that irisin targets mitochondria-dependent oxidative stress pathway for cardio-protection.
Irisin interacts with SOD2 to preserve mitochondrial function during myocardial I/R
Superoxide dismutase (SOD) is an important endogenous antioxidant and provides protection against myocardial I/R32. Consistent with studies by other investigators, we found that I/R-treatment led to reduced enzymatic activity of superoxide dismutase (SOD) and administration of irisin could restore the compromised SOD activity in rat hearts that were subjected to I/R (Fig. 5a). Thus, the effect of irisin on protection of mitochondrial function appears to be linked to modulation of SOD function.
Figure 5. Protective effect of irisin on mitochondria-dependent oxidative stress is linked to modulation of SOD2.
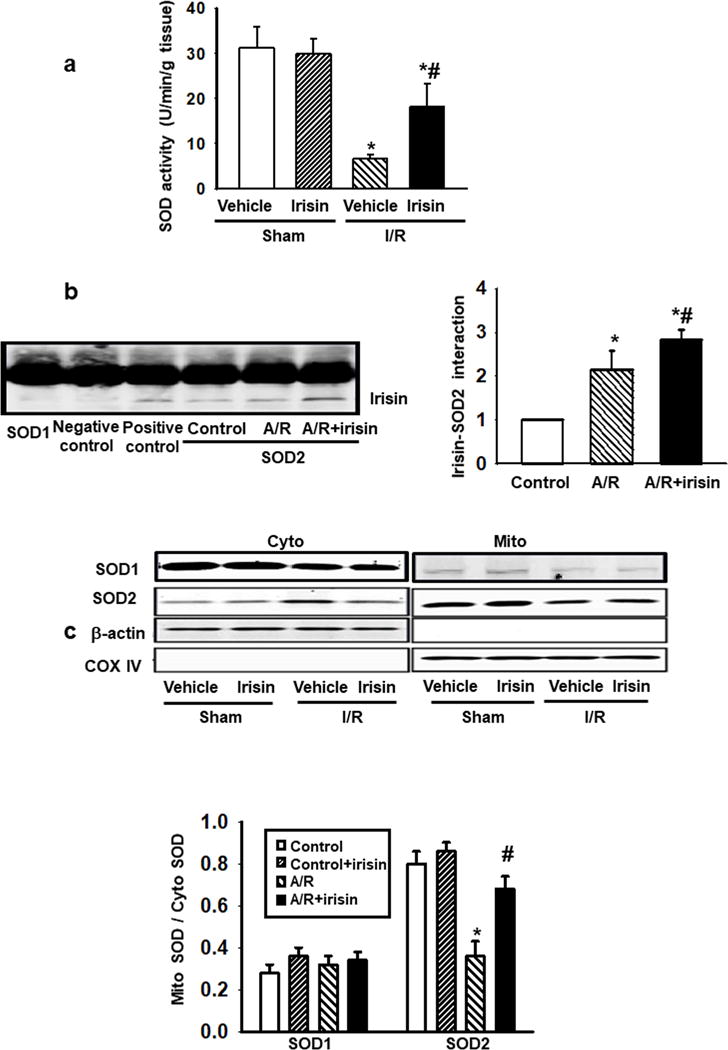
(a) Total SOD activity in heart tissue lysates was determined by nitroblue tetrazolium (NBT) method. Irisin treatment preserved SOD activity after I/R injury (n=10, *P<0.05 vs. sham control, #P<0.05 vs. I/R vehicle). (b) Co-immunoprecipitation assay was performed with H9c2 cell lysates with or without A/R injury. While irisin did not interact with SOD1, it could interact with SOD2. The interaction between irisin and SOD2 was enhanced in H9c2 cells subjected to A/R injury, and exogenous irisin treatment improved the interaction. Anti-SOD1 or anti-SOD2 antibody was used for immunoprecipitation, and anti-irisin antibody was used for immunoblotting. For negative control, rabbit IgG was used for immunoprecipitation. Anti-irisin antibody was used for both immunoprecipitation and immunoblot as positive control. (n=4, *P<0.05 vs. control, #P<0.05 vs. A/R). (c) SOD1 and SOD2 protein levels were determined by immunoblotting in mitochondrial (Mito) and cytosolic (Cyto) fractions of H9c2 cells. A/R treatment led to dissociation of SOD2 from mitochondria to cytosol, which could be partially prevented by irisin treatment. Cox IV and β-actin were used as house-keeping proteins for mitochondria and cytosol, respectively. The fraction of SOD1 (left panel) and SOD2 (right panel) in the mitochondria was summarized from multiple experiments in H9c2 cells following the various treatments (n=5, *P<0.01 vs. control, #P<0.01 vs. A/R group).
Cardiomyocytes contain three SOD subtypes: SOD1 - a cytosolic enzyme, SOD3 – an extracellular enzyme, and SOD2 - predominantly targeting the mitochondrial organelle.33 Co-immunoprecipitation assay demonstrated that irisin interacted with SOD2, but not with SOD1 (Fig. 5b) While the interaction of irisin and SOD2 was significantly increased in A/R cardiomyocytes, this increase of interaction was more noticeable in irisin-treated cardiomyocytes.
The different subcellular distribution of SOD2 was further examined using biochemical fractionation of mitochondria from the cytosol. As shown in Fig. 5c, increased dissociation of SOD2 from mitochondria to cytosol was observed in A/R-treated cardiomyocytes and the addition of irisin restored the mitochondria-localization of SOD2. The effects appeared to be specific to SOD2, as the subcellular distribution of SOD1 was not affected by A/R and irisin treatments. Summary data from multiple experiments demonstrated that irisin restored the mitochondria-targeting of SOD2 under conditions of A/R (Fig. 5c).
Based on these results, we conclude that functional interaction between irisin and SOD2 allows for maintenance of SOD2-targeting in mitochondria. These effects likely underlie the amelioration of I/R-induced oxidative stress in cardiomyocytes and improvement of mitochondrial function associated with MI.
DISCUSSION
Ischemia reperfusion injury contributes to deleterious outcomes in many vital organs such as heart, lungs, brain, and kidneys4, 34. Here we show that irisin protects heart injury through preservation of mitochondrial function by alleviating apoptotic cell death and reducing the mitochondrial oxidative stress. We further demonstrated that the cardioprotective effect of exogenous irisin is dependent on interaction with SOD2 and mitochondria.
Skeletal muscle has recently been recognized as an endocrine organ that not only contains metabolically important molecules, but also communicates with other tissues through the secretion of hormones, known as myokines, that are released into circulation during or immediately after physical activity35. Irisin is an exercise-induced hormone secreted by skeletal muscle that drives brown fat-like conversion of white adipose tissue, which improves systemic metabolism by increasing energy expenditure15. Due to these properties, irisin has emerged as an appealing therapeutic target for treatment of metabolic diseases36. In the present study, we found that irisin is an important myokine that protects heart from I/R injury.
There is compelling evidence that ROS plays an important role in myocardial I/R injury, and mitochondria are believed to be the major organelle that produces ROS in response to stress8. Elevated ROS production is known to cause an opening of mitochondrial permeability transition pore leading to dissipation of mitochondrial membrane potential. Subsequent activation of the apoptotic pathway eventually leads to cardiac cell death37, 38. Such dysfunction of mitochondria and ROS production constitute a vicious cycle that underlies the massive cardiac injury during I/R. Irisin-mediated alleviation of ROS production would interrupt this vicious cycle and reduce I/R injury to the heart. We present evidence that SOD2, the major mitochondrial antioxidant enzyme that regulates ROS production, is decreased during I/R treatment of the heart. The reduced SOD2 activity could be restored through intravenous administration of irisin. We found that exogenous irisin can enter cardiomyocytes to target SOD2 in mitochondria. Through interaction with irisin, the partition of SOD2 in the mitochondria is enhanced. Further examination of the functional irisin-SOD2 interaction will have significant translational value for therapeutic approaches for treatment of I/R-induced myocardial infarction.
Our study highlights the important endocrine function of skeletal muscle and opens a new treatment paradigm for prevention of cardiac diseases. Future studies will be required to elucidate the mechanisms that underlie the molecular recognition for irisin in the injured cardiomyocytes, and the down-stream signaling events that facilitate cardioprotection. Moreover, knowledge on the secretory and metabolic pathways for regulation of irisin levels in circulation will be important for therapeutic approaches to enhance systemic elevation of irisin as a preventative means for human diseases associated with chronic tissue injuries or mitochondrial dysfunction.
Sundarrajan et al.39 showed that exogenous irisin increased the diastolic volume, heart rate, and cardiac output in zebra fish via regulating the expressions of cardiovascular physiology-associated modulators such as myostatin a/b, troponin C, and troponin T2D. Furthermore, a recent study also reported the protective effect of irisin on myocardial I/R injury using the Langendorff perfused heart and cultured myocytes, which is ascribed to the increased SOD-1 expression and p38 phosphorylation, consequently resulting in the suppression of cardiomyocytes apoptosis40. However, the majority of SOD1 is localized in the cytosol, and only 1-2% of SOD1 is found in mitochondria33, 41. Since SOD2 is predominantly found in the mitochondria, it may play a more important role to regulate mitochondrial function. Our present in vivo and in vitro experiments showed irisin interacted with SOD2, but not with SOD1, and irisin restored the mitochondria-localization of SOD2. The effects appear to be specific for SOD2, as the subcellular distribution of SOD1 was not affected by A/R and irisin treatments.
In conclusion, our current data suggests that irisin plays a protective role against I/R injury to heart. Targeting the action of irisin in mitochondria presents a novel therapeutic intervention for myocardial infarction.
Supplementary Material
Acknowledgments
Funding
This project was supported by grants from the National Natural Science Foundation of China (31430043, 31730043), Innovation Team of the National Natural Science Foundation (81721001), China Postdoctoral Science Foundation funded project (2017M613431), Program for Changjiang Scholars and Innovative Research Team in University (IRT1216), and National Institutes of Health of the United States of America (R01-AR070752).
Footnotes
Conflict of interest
There are no competing interests to declare.
Supplemental Movies
Dynamic FLASH signals were showed in cardiomyocytes after anoxia during the period of reoxygenation. Images were acquired at a rate of 1 sec / frame.
References
- 1.Braunwald E, Kloner RA. Myocardial reperfusion: a double-edged sword? J Clin Invest. 1985;76:1713–1719. doi: 10.1172/JCI112160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 3.de Perrot M, Liu M, Waddell TK, Keshavjee S. Ischemia-reperfusion-induced lung injury. Am J Resp Crit Care. 2003;167:490–511. doi: 10.1164/rccm.200207-670SO. [DOI] [PubMed] [Google Scholar]
- 4.Carden DL, Granger DN. Pathophysiology of ischaemia-reperfusion injury. J Pathol. 2000;190:255–266. doi: 10.1002/(SICI)1096-9896(200002)190:3<255::AID-PATH526>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 5.Ng CS, Wan S, Arifi AA, Yim AP. Inflammatory response to pulmonary ischemia-reperfusion injury. Surg Today. 2006;36:205–214. doi: 10.1007/s00595-005-3124-2. [DOI] [PubMed] [Google Scholar]
- 6.Becker LB. New concepts in reactive oxygen species and cardiovascular reperfusion physiology. Cardiovasc Res. 2004;61:461–470. doi: 10.1016/j.cardiores.2003.10.025. [DOI] [PubMed] [Google Scholar]
- 7.Crimi E, Sica V, Williams-Ignarro S, Zhang H, Slutsky AS, Ignarro LJ, Napoli C. The role of oxidative stress in adult critical care. Free Radic Biol Med. 2006;40:398–406. doi: 10.1016/j.freeradbiomed.2005.10.054. [DOI] [PubMed] [Google Scholar]
- 8.Chen YR, Zweier JL. Cardiac mitochondria and reactive oxygen species generation. Circ Res. 2014;114:524–537. doi: 10.1161/CIRCRESAHA.114.300559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walters AM, Porter GA, Jr, Brookes PS. Mitochondria as a drug target in ischemic heart disease and cardiomyopathy. Circ Res. 2012;111:1222–1236. doi: 10.1161/CIRCRESAHA.112.265660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kramer JH, Misik V, Weglicki WB. Lipid peroxidation-derived free radical production and postischemic myocardial reperfusion injury. Ann NY Acad Sci. 1994;723:180–196. [PubMed] [Google Scholar]
- 11.Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361:1570–1583. doi: 10.1056/NEJMra0901217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eltzschig HK, Eckle T. Ischemia and reperfusion–from mechanism to translation. Nat Med. 2011;17:1391–1401. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferrer-Martinez A, Ruiz-Lozano P, Chien KR. Mouse PeP: a novel peroxisomal protein linked to myoblast differentiation and development. Dev Dyn. 2002;224:154–167. doi: 10.1002/dvdy.10099. [DOI] [PubMed] [Google Scholar]
- 14.Teufel A, Malik N, Mukhopadhyay M, Westphal H. Frcp1 and Frcp2, two novel fibronectin type III repeat containing genes. Gene. 2002;297:79–83. doi: 10.1016/s0378-1119(02)00828-4. [DOI] [PubMed] [Google Scholar]
- 15.Bostrom P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, Rasbach KA, Bostrom EA, Choi JH, Long JZ, Kajimura S, Zingaretti MC, Vind BF, Tu H, Cinti S, Hojlund K, Gygi SP, Spiegelman BM. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature. 2012;481:463–468. doi: 10.1038/nature10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cunha A. Basic research: Irisin–behind the benefits of exercise. Nat Rev Endocrino. 2012;8:195. doi: 10.1038/nrendo.2012.11. [DOI] [PubMed] [Google Scholar]
- 17.Sanchis-Gomar F, Lippi G, Mayero S, Perez-Quilis C, Garcia-Gimenez JL. Irisin: a new potential hormonal target for the treatment of obesity and type 2 diabetes. J Diabetes. 2012;4:196. doi: 10.1111/j.1753-0407.2012.00194.x. [DOI] [PubMed] [Google Scholar]
- 18.Villarroya F. Irisin, turning up the heat. Cell Metab. 2012;15:277–278. doi: 10.1016/j.cmet.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 19.Lee P, Linderman JD, Smith S, Brychta RJ, Wang J, Idelson C, Perron RM, Werner CD, Phan GQ, Kammula US, Kebebew E, Pacak K, Chen KY, Celi FS. Irisin and FGF21 are cold-induced endocrine activators of brown fat function in humans. Cell Metab. 2014;19:302–309. doi: 10.1016/j.cmet.2013.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin JK, Blackwood EA, Azizi K, Thuerauf DJ, Fahem AG, Hofmann C, Kaufman RJ, Doroudgar S, Glembotski CC. ATF6 Decreases Myocardial Ischemia/Reperfusion Damage and Links ER Stress and Oxidative Stress Signaling Pathways in the Heart. Circ Res. 2017;120:862–875. doi: 10.1161/CIRCRESAHA.116.310266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fang H, Chen M, Ding Y, Shang W, Xu J, Zhang X, Zhang W, Li K, Xiao Y, Gao F, Shang S, Li JC, Tian XL, Wang SQ, Zhou J, Weisleder N, Ma J, Ouyang K, Chen J, Wang X, Zheng M, Wang W, Cheng H. Imaging superoxide flash and metabolism-coupled mitochondrial permeability transition in living animals. Cell Res. 2011;21:1295–1304. doi: 10.1038/cr.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang WE, Yang D, Li L, Wang W, Peng Y, Chen C, Chen P, Xia X, Wang H, Jiang J, Liao Q, Li Y, Xie G, Huang H, Guo Y, Ye L, Duan DD, Chen X, Houser SR, Zeng C. Prolyl hydroxylase domain protein 2 silencing enhances the survival and paracrine function of transplanted adipose-derived stem cells in infarcted myocardium. Circ Res. 2013;113:288–300. doi: 10.1161/CIRCRESAHA.113.300929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Di Lisa F, Blank PS, Colonna R, Gambassi G, Silverman HS, Stern MD, Hansford RG. Mitochondrial membrane potential in single living adult rat cardiac myocytes exposed to anoxia or metabolic inhibition. J Physiol. 1995;486(Pt 1):1–13. doi: 10.1113/jphysiol.1995.sp020786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chouchani ET, Methner C, Nadtochiy SM, Logan A, Pell VR, Ding S, James AM, Cocheme HM, Reinhold J, Lilley KS, Partridge L, Fearnley IM, Robinson AJ, Hartley RC, Smith RA, Krieg T, Brookes PS, Murphy MP. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat Med. 2013;19:753–759. doi: 10.1038/nm.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 26.Flohe L, Otting F. Superoxide dismutase assays. Method Enzymol. 1984;105:93–104. doi: 10.1016/s0076-6879(84)05013-8. [DOI] [PubMed] [Google Scholar]
- 27.Linke A, Adams V, Schulze PC, Erbs S, Gielen S, Fiehn E, Mobius-Winkler S, Schubert A, Schuler G, Hambrecht R. Antioxidative effects of exercise training in patients with chronic heart failure: increase in radical scavenger enzyme activity in skeletal muscle. Circulation. 2005;111:1763–1770. doi: 10.1161/01.CIR.0000165503.08661.E5. [DOI] [PubMed] [Google Scholar]
- 28.Swift LM, Sarvazyan N. Localization of dichlorofluorescin in cardiac myocytes: implications for assessment of oxidative stress. Am J Resp Crit Care. 2000;278:H982–990. doi: 10.1152/ajpheart.2000.278.3.H982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, Wang X, Li K, Han P, Zheng M, Yin J, Wang W, Mattson MP, Kao JP, Lakatta EG, Sheu SS, Ouyang K, Chen J, Dirksen RT, Cheng H. Superoxide flashes in single mitochondria. Cell. 2008;134:279–290. doi: 10.1016/j.cell.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quinones M, Folgueira C, Sanchez-Rebordelo E, Al-Massadi O. Circulating Irisin Levels Are Not Regulated by Nutritional Status, Obesity, or Leptin Levels in Rodents. Mediat Inflamm. 2015;2015:620919. doi: 10.1155/2015/620919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Z, Tang X, Li Y, Leu C, Guo L, Zheng X, Zhu D. 20-Hydroxyeicosatetraenoic acid inhibits the apoptotic responses in pulmonary artery smooth muscle cells. Eur J Pharmacol. 2008;588:9–17. doi: 10.1016/j.ejphar.2008.03.045. [DOI] [PubMed] [Google Scholar]
- 32.Chen C, Lu W, Wu G, Lv L, Chen W, Huang L, Wu X, Xu N, Wu Y. Cardioprotective effects of combined therapy with diltiazem and superoxide dismutase on myocardial ischemia-reperfusion injury in rats. Life Sci. 2017;183:50–59. doi: 10.1016/j.lfs.2017.06.024. [DOI] [PubMed] [Google Scholar]
- 33.Zelko IN, Mariani TJ, Folz RJ. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med. 2002;33:337–349. doi: 10.1016/s0891-5849(02)00905-x. [DOI] [PubMed] [Google Scholar]
- 34.Collard CD, Gelman S. Pathophysiology, clinical manifestations, and prevention of ischemia-reperfusion injury. Anesthesiology. 2001;94:1133–1138. doi: 10.1097/00000542-200106000-00030. [DOI] [PubMed] [Google Scholar]
- 35.Iizuka K, Machida T, Hirafuji M. Skeletal muscle is an endocrine organ. JPharmacol Sci. 2014;125:125–131. doi: 10.1254/jphs.14r02cp. [DOI] [PubMed] [Google Scholar]
- 36.Roberts MD, Bayless DS, Company JM, Jenkins NT, Padilla J, Childs TE, Martin JS, Dalbo VJ, Booth FW, Rector RS, Laughlin MH. Elevated skeletal muscle irisin precursor FNDC5 mRNA in obese OLETF rats. Metabolism. 2013;62:1052–1056. doi: 10.1016/j.metabol.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim R, Emi M, Tanabe K, Murakami S, Uchida Y, Arihiro K. Regulation and interplay of apoptotic and non-apoptotic cell death. J Pathol. 2006;208:319–326. doi: 10.1002/path.1885. [DOI] [PubMed] [Google Scholar]
- 38.Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med. 2010;48:749–762. doi: 10.1016/j.freeradbiomed.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sundarrajan L, Yeung C, Hahn L, Weber LP, Unniappan S. Irisin regulates cardiac physiology in zebrafish. PLoS One. 2017;12:e0181461. doi: 10.1371/journal.pone.0181461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang H, Zhao YT, Zhang S, Dubielecka PM, Du J, Yano N, Chin YE, Zhuang S, Qin G, Zhao TC. Irisin plays a pivotal role to protect the heart against ischemia and reperfusion injury. J Cell Physiol. 2017;232:3775–3785. doi: 10.1002/jcp.25857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vande Velde C, Miller TM, Cashman NR. Cleveland DW Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria. Proc Natl Acad Sci U S A. 2008;105:4022–4027. doi: 10.1073/pnas.0712209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deng X, Huang W, Peng J, Zhu TT, Sun XL, Zhou XY, Yang H, Xiong JF, He HQ, Xu YH, He YZ. Irisin Alleviates Advanced Glycation End Products-Induced Inflammation and Endothelial Dysfunction via Inhibiting ROS-NLRP3 Inflammasome Signaling. Inflammation. 2018;41:260–275. doi: 10.1007/s10753-017-0685-3. [DOI] [PubMed] [Google Scholar]
- 43.Wu F, Song H, Zhang Y, Zhang Y, Mu Q, Jiang M, Wang F, Zhang W, Li L, Li H, Wang Y, Zhang M, Li S, Yang L, Meng Y, Tang D. Irisin Induces Angiogenesis in Human Umbilical Vein Endothelial Cells In Vitro and in Zebrafish Embryos In Vivo via Activation of the ERK Signaling Pathway. PLoS One. 2015;10:e0134662. doi: 10.1371/journal.pone.0134662. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.