

Abstract
Euglycemic diabetic ketoacidosis (DKA) has been recognized as a potentially fatal complication related to sodium-glucose cotransporter 2 (SGLT2) inhibitors. Herein, we report a patient of out-of-hospital cardiac arrest, with an initial cardiac rhythm of ventricular fibrillation, who was subsequently diagnosed with acute myocardial infarction, complicated with SGLT2 inhibitor-associated euglycemic DKA. The patient survived and achieved nearly full functional recovery. This report calls for increased attention to SGLT2 inhibitors’ fatal complications, as well as their proper use.
<Learning objective: Euglycemic diabetic ketoacidosis could develop in patients using sodium-glucose cotransporter 2 inhibitors. It is a potentially lethal complication and needs more attention. Physicians who prescribe this class of drugs should be aware of this complication. Meanwhile, research to elucidate patient characteristics that are more susceptible to this complication is urgently awaited.>
Keywords: Euglycemic diabetic ketoacidosis, Sodium-glucose cotransporter 2 inhibitor, Cardiac arrest, Myocardial infarction, Ventricular fibrillation
Introduction
Sodium-glucose cotransporter-2 (SGLT2) inhibitors are a new class of antidiabetic drug that have pleiotropic effects including improving cardiovascular outcomes [1]. Medicines of this class are known to have several adverse effects, including euglycemic diabetic ketoacidosis (DKA), which has been reported increasingly [2]. Here, we report a case of SGLT2 inhibitor-associated euglycemic DKA that was complicated with cardiac arrest from acute myocardial infarction.
Case report
A 49-year-old Asian man with a 1-year history of type 2 diabetes mellitus and vasospastic angina, whose body mass index was 22.1 kg/m2, suddenly lost consciousness while sightseeing, shortly after he complained of nausea. An automated external defibrillator was initiated 5 min later, without bystander cardiopulmonary resuscitation. On the basis of initial cardiac rhythm of ventricular fibrillation, the automated external defibrillator delivered 2 shocks. The emergency medical service arrived and started basic life support, and delivered 4 shocks. Return of spontaneous circulation was achieved after a total resuscitation time of 16 min. He was rushed to the emergency department (ED) of our hospital while unconscious.
Upon arrival at the ED, his Glasgow coma scale was E1V2M2. His blood pressure was 136/90 mmHg and heart rate was 85 beats/min. His respiration rate was 20 breaths/min, and peripheral oxygen saturation was 100% on 100% oxygen delivery. His initial 12-lead electrocardiograms showed ST-segment elevation in precordial leads, I, and aVL and reciprocal ST-segment depression in III and aVF (Fig. 1A). A transthoracic echocardiography demonstrated hypokinesis of basal to apical left ventricular (LV) anteroseptal wall. We, therefore, diagnosed him as having acute anteroseptal myocardial infarction. After intubation and a brain computed tomography ruling out an intracranial event, we initiated targeted temperature (34 °C) management (TTM). Emergency coronary angiography revealed a subtotal stenosis in proximal left anterior descending (LAD) artery under nitrate administration (Fig. 1B), which was most likely to be organic stenosis. We did not utilize intravascular imaging modalities, in order to achieve early revascularization to minimize the influence of hypoxic-ischemic encephalopathy. We subsequently performed percutaneous coronary intervention (PCI) successfully using an everolimus-eluting stent with a door-to-balloon time of 44 min, and gained good angiographic results with a Thrombolysis In Myocardial Infarction grade 3 flow (Fig. 1C). After the procedure, the patient was transferred to the intensive care unit (ICU), with 34 °C TTM being continued for 24 h.
Fig. 1.

Twelve-lead electrocardiograms on admission, and angiographic findings. The initial 12-lead electrocardiograms showed ST-segment elevation in V1–V6, I, and aVL, and reciprocal ST-segment depression in III and aVF (A) emergency coronary angiography revealed a subtotal occlusion of the proximal left anterior descending artery (B), which was then successfully stented (C).
In the first 24 h in the ICU, despite the fact that his blood pressure was maintained well without using catecholamine, and his lactate level was constantly below 2.0 mmol/L, the patient developed a high anion gap metabolic acidosis (Fig. 2A). The urinalysis showed that ketone was 3+, and other laboratory data 10 h after PCI are depicted in Table 1. We were informed that he had been taking aspirin 100 mg daily, isosorbide mononitrate 20 mg twice a day, nicorandil 5 mg 3 times a day, benidipine 4 mg twice a day, amlodipine 5 mg daily, pitavastatin 1 mg daily, metformin 500 mg 3 times a day, miglitol 50 mg 3 times a day, linagliptin 5 mg daily, and using isosorbide mononitrate 40 mg tape daily. With the evidence of ketonuria, ketonemia, the relatively low blood glucose level in spite of the severe ketoacidosis, and the fact that the patient was taking ipragliflozin 50 mg daily, an SGLT2 inhibitor, we concluded that he was experiencing SGLT2 inhibitor-associated euglycemic DKA, and started to correct it by 8.4% sodium bicarbonate. We did not initiate hypoglycemic therapy during the first 24 h because the blood glucose level was constantly less than 180 mg/dL (Fig. 2A). Shortly after the discontinuation of sodium bicarbonate on the 24th hour, the acidosis worsened temporarily, but improved favorably by extracellular fluid replacement solely (Fig. 2B). The creatine kinase (CK)-MB level declined with a peak level of 83 U/L 24 h after PCI, at which point we started rewarming. He was able to respond to simple verbal orders on post-PCI day 2, and was extubated 70 h after PCI, on post-PCI day 3. He had weakness in all extremities and hoarseness, but no disorientation or dysphagia. On post-PCI day 4, with his white blood cell count of 12,000/μL, and C-reactive protein level of 23.18 mg/dL, representing delayed inflammation in spite of aggressive antibiotic treatment with piperacillin–tazobactam, and vancomycin, he underwent a contrast computed tomography, which revealed a hepatic subcapsular hematoma in the caudate lobe caused by chest compression during cardiopulmonary resuscitation. This also explained the elevation of liver enzymes, as well as the discrepancy between CK and CK-MB (Table 1).
Fig. 2.

Clinical course during hospitalization after percutaneous coronary intervention (PCI). The patient developed high anion gap metabolic acidosis within 24 h after PCI, during the targeted temperature management (TTM), despite that his circulation was maintained in a stable condition (A). We corrected it with sodium bicarbonate replacement. After its discontinuation, acidosis worsened temporally (B). The creatine kinase (CK)-MB level started to decline 24 h after PCI, with a peak of 83 U/L. HCO3−, bicarbonate ion; HR, heart rate; mBP, mean blood pressure; pCO2, partial pressure of carbon dioxide.
Table 1.
Laboratory data on admission, 10 h after percutaneous coronary intervention, and on discharge.
| Variable | Reference range | On admission | 10 h after PCI | On discharge |
|---|---|---|---|---|
| Complete blood count | ||||
| Hemoglobin (g/dL) | 13.1–16.9 | 17.6 | 16.7 | 11.8 |
| Hematocrit (%) | 38.9–49.4 | 52.8 | 51.2 | 34.8 |
| White blood cell count (per mm3) | 3300–8700 | 17,300 | 27,100 | 8700 |
| Platelet count (per mm3) | 137,000–309,000 | 197,000 | 233,000 | 254,000 |
| Chemistry panel | ||||
| Albumin (g/dL) | 4.1–5.1 | 5.3 | – | 3.3 |
| Urea nitrogen (mg/dL) | 8.0–21.0 | 16.9 | – | 7.5 |
| Creatinine (mg/dL) | 0.61–1.04 | 0.81 | 0.55 | 0.51 |
| Total bilirubin (mg/dL) | 0.2–1.2 | 0.5 | – | 0.5 |
| Alkaline phosphatase (U/L) | 103–289 | 146 | – | 230 |
| Lactate dehydrogenase (U/L) | 118–223 | 1101 | 1061 | 282 |
| Aspartate aminotransferase (U/L) | 9–32 | 578 | 790 | 26 |
| Alanine aminotransferase (U/L) | 3–38 | 273 | 343 | 71 |
| γ-Glutamyltransferase (U/L) | 15–90 | 116 | – | – |
| Creatine kinase (U/L) | 57–218 | 89 | 461 | 100 |
| Creatine kinase-MB (U/L) | 0–30 | 28 | 66 | 9 |
| Sodium (mmol/L) | 137–149 | 140 | 140 | 142 |
| Potassium (mmol/L) | 3.7–5.0 | 3.9 | 4.1 | 3.7 |
| Chloride (mmol/L) | 102–110 | 104 | – | 105 |
| Glucose (mg/dL) | 80–110 | 134 | 171 | 166 |
| Hemoglobin A1c (%) | 4.6–6.2 | – | 7.5 | – |
| Troponin T (ng/mL) | 0.000–0.014 | 0.018 | – | – |
| Total ketone body (μmol/L) | 0–131 | – | 9185 | 376 |
| Acetoacetate (μmol/L) | 0–55 | – | 2400 | 55 |
| 3-Hydroxybutyrate (μmol/L) | 0–85 | – | 6785 | 247 |
| Arterial blood gas analysis | ||||
| Fractional inspired oxygen concentration (%) | – | 40.0 | 40.0 | – |
| pH | 7.350–7.450 | 7.258 | 7.111 | – |
| Partial pressure of arterial oxygen (Torr) | 80.0–100.0 | 159.0 | 76.9 | – |
| Partial pressure of arterial carbon dioxide (Torr) | 35.0–45.0 | 30.5 | 45.5 | – |
| Bicarbonate ion (μmol/L) | 22.0–28.0 | 13.4 | 14.6 | – |
| Lactate (mmol/L) | 0.5–1.6 | 1.2 | 1.7 | – |
PCI, percutaneous coronary intervention.
A transthoracic echocardiography on post-PCI day 4 demonstrated increased echogenicity and mild hypokinesis of mid to apical LV anteroseptal wall, an LV ejection fraction of 64.0% (modified Simpson's method), but neither significant valvular disease nor pulmonary hypertension.
On the same day, we received the angiographic findings 1 year prior to the event from a local hospital of his hometown (Fig. 3), which showed a moderate (50%) stenosis at rest, a total occlusion provoked by intra-coronary injection of 7 μg ergonovine, and a recovery to moderate stenosis after intra-coronary injection of isosorbide dinitrate, indicating vasospasm on organic lesion, at the site which was identical to the one where we stented, in proximal LAD.
Fig. 3.
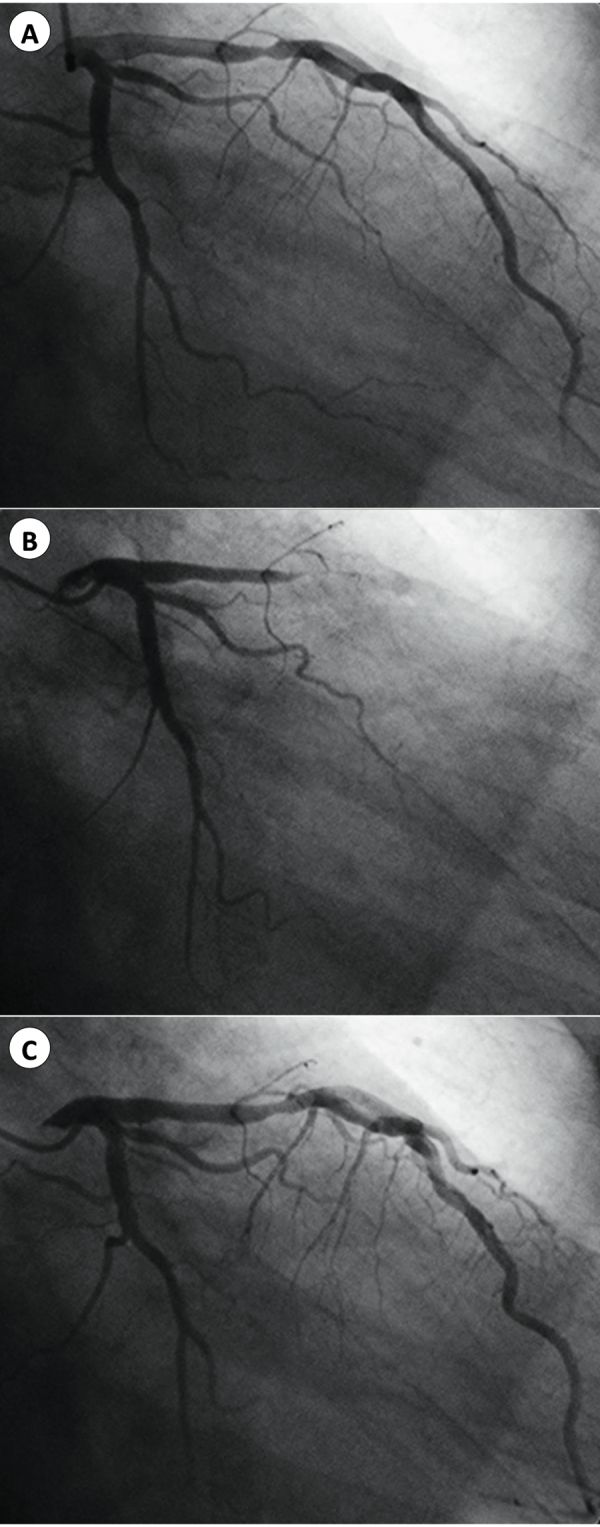
Coronary angiography obtained 1 year prior to the event. The patient's coronary angiographic findings 1 year previously demonstrated moderate stenosis in proximal left anterior descending artery at rest (A), a total occlusion provoked by intra-coronary ergonovine injection (B), and a recovery to moderate stenosis after intra-coronary isosorbide dinitrate injection (C).
The patient regained his muscular strength fully and could walk by himself on post-PCI day 5. He subsequently achieved full functional recovery except residual slight euphoria. He was then discharged on post-PCI day 10, with ipragliflozin discontinued. Upon discharge, a venous blood gas analysis showed a pH of 7.420 and a bicarbonate ion level of 26.3 mmol/L, urinalysis was negative for ketone. Other laboratory data on discharge are depicted in Table 1.
Discussion
SGLT2 inhibitors are well known as a new class of glucose-lowering medicines, which have pleiotropic effects such as a reduction in body weight and blood pressure [3], as well as a decrease in major adverse cardiovascular events, mainly through the reduction of heart failure [1]. These characteristics resulted in a large growth of prescriptions of SGLT2 inhibitors, which contributed to understanding of side effects of the new medicines, including an increase in urinary tract infection, genital infection, low-density lipoprotein cholesterol, and risk of bone fracture [3]. A serious one of them is euglycemic DKA, which has been reported by accumulated literature, and has been drawing much attention [2], [4]. This occurs in both type 1 and type 2 diabetes mellitus, and may be triggered by physical stress such as infection, surgery, alcohol intake, reduced calorie or fluid intake, and reduced insulin dose [4]. It is characterized by normal or mildly elevated blood glucose level. The U.S. Food and Drug Administration and European Medicines Agency have already issued warning documents concerning this potentially fatal complication. Patients with euglycemic DKA present with nausea, thirst, and lethargy, which makes these symptoms as warning signs [4]. The patient's complaint of nausea in this report, however, was immediately before loss of consciousness, which was probably due to acute hemodynamic collapse caused by acute myocardial infarction or ventricular fibrillation.
According to the limited literature, patients generally present to the hospital hours to days after the onset of the symptoms mentioned above. After the recognition of euglycemic DKA and the initiation of treatment including the cessation of SGLT2 inhibitors, fluid replacement, insulin, and bicarbonate ion replacement if needed, the acidosis improves. The time to pH normalization varies, depending on the severity and whether bicarbonate ion is replaced. Ketonemia usually improves in approximately 1 week [4], [5].
The mechanism of euglycemic DKA with SGLT2 inhibitors is proposed as follows: SGLT2 inhibitors lower glucose levels independently of insulin, decrease insulin level, and augment glucagon production, which accelerates lipolysis and beta-oxidation of fatty acids, resulting in excess ketogenesis [6]. This mechanism implies that patients with low insulin secretion are not good candidates to receive SGLT2 inhibitors. The present patient had a potentially low insulin secretion (homeostatic model assessment-β <38% on discharge), and took alcohol the night before presentation, which may have contributed to the development of euglycemic DKA.
There have been several reports of acute myocardial infarction complicated with DKA [7], [8]. Although the mechanism remains unclear, it is known that DKA precipitates a prothrombotic environment, secondary to hyperviscosity and increased coagulation activity [9]. Ketoacidosis also causes endothelial dysfunction, which may contribute to coronary vasospasm [10], [11]. In this patient, as shown in Table 1 and Fig. 2, the initial laboratory data, which were taken approximately 1 h after the onset, demonstrated a pH of 7.25, a bicarbonate level of 13.4 μmol/L, and a lactate level of 1.2 mmol/L. The low levels of pH and bicarbonate ion in the early phase indicate that acidosis had already developed before the onset of acute myocardial infarction. In addition, the maintained blood pressure and heart rate upon arrival at the ED and the constantly low lactate level during his clinical course, make it less likely that acute myocardial infarction and subsequent circulation collapse led to acidosis.
The PCI procedure in this patient was considered necessary by the medical team because of the existence of severe stenosis. Taking the potential contribution of vasospasm into consideration, the patient had already been undergoing optimal anti-spastic therapy, which made stenting as a choice of treatment. The patient and the medical team decided not to implant a cardioverter defibrillator, after a detailed discussion, given that although the only site where vasospasm was provoked has been stented, the risk remains. The patient would further discuss this issue with his primary cardiologist in the follow-up visits.
Euglycemic DKA has been recently reported as a potentially fatal complication of SGLT2 inhibitors. To the best of our knowledge, this is the first report that SGLT2 inhibitor-associated euglycemic DKA was complicated with cardiac arrest from acute myocardial infarction. This warrants more attention on the proper use of these medicines, as well as further investigation on this serious complication.
Conflict of interest
The authors declare that there is no conflict of interest.
Acknowledgments
The authors thank Drs Yoshikazu Kosugi and Yoshifumi Nakahara, from Aiseikai Yamashina Hospital for providing previous medical records including angiographic findings of the patient, and taking care of the patient after discharge.
References
- 1.Wu J.H., Foote C., Blomster J., Toyama T., Perkovic V., Sundstrom J., Neal B. Effects of sodium-glucose cotransporter-2 inhibitors on cardiovascular events, death, and major safety outcomes in adults with type 2 diabetes: a systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2016;4:411–419. doi: 10.1016/S2213-8587(16)00052-8. [DOI] [PubMed] [Google Scholar]
- 2.Rosenstock J., Ferrannini E. Euglycemic diabetic ketoacidosis: a predictable, detectable, and preventable safety concern with SGLT2 inhibitors. Diabetes Care. 2015;38:1638–1642. doi: 10.2337/dc15-1380. [DOI] [PubMed] [Google Scholar]
- 3.Vasilakou D., Karagiannis T., Athanasiadou E., Mainou M., Liakos A., Bekiari E., Sarigianni M., Matthews D.R., Tsapas A. Sodium-glucose cotransporter 2 inhibitors for type 2 diabetes: a systematic review and meta-analysis. Ann Intern Med. 2013;159:262–274. doi: 10.7326/0003-4819-159-4-201308200-00007. [DOI] [PubMed] [Google Scholar]
- 4.Peters A.L., Buschur E.O., Buse J.B., Cohan P., Diner J.C., Hirsch I.B. Euglycemic diabetic ketoacidosis: a potential complication of treatment with sodium-glucose cotransporter 2 inhibition. Diabetes Care. 2015;38:1687–1693. doi: 10.2337/dc15-0843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hayami T., Kato Y., Kamiya H., Kondo M., Naito E., Sugiura Y., Kojima C., Sato S., Yamada Y., Kasagi R., Ando T., Noda S., Nakai H., Takada E., Asano E. Case of ketoacidosis by a sodium-glucose cotransporter 2 inhibitor in a diabetic patient with a low-carbohydrate diet. J Diabetes Investig. 2015;6:587–590. doi: 10.1111/jdi.12330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singh A.K. Sodium-glucose co-transporter-2 inhibitors and euglycemic ketoacidosis: wisdom of hindsight. Indian J Endocrinol Metab. 2015;19:722–730. doi: 10.4103/2230-8210.167554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oo Y.H., Karam J.G., Resta C.A. Extreme insulin resistance in a patient with diabetes ketoacidosis and acute myocardial infarction. Case Rep Endocrinol. 2013;2013:520904. doi: 10.1155/2013/520904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Batra A.S., Acherman R.J., Wong P., Silka M.J. Acute myocardial infarction in a 12-year-old as a complication of hyperosmolar diabetic ketoacidosis. Pediatr Crit Care Med. 2002;3:194–196. doi: 10.1097/00130478-200204000-00021. [DOI] [PubMed] [Google Scholar]
- 9.Quigley R.L., Curran R.D., Stagl R.D., Alexander J.C., Jr. Management of massive pulmonary thromboembolism complicating diabetic ketoacidosis. Ann Thorac Surg. 1994;57:1322–1324. doi: 10.1016/0003-4975(94)91385-4. [DOI] [PubMed] [Google Scholar]
- 10.Close T.E., Cepinskas G., Omatsu T., Rose K.L., Summers K., Patterson E.K., Fraser D.D. Diabetic ketoacidosis elicits systemic inflammation associated with cerebrovascular endothelial cell dysfunction. Microcirculation. 2013;20:534–543. doi: 10.1111/micc.12053. [DOI] [PubMed] [Google Scholar]
- 11.Lanza G.A., Careri G., Crea F. Mechanisms of coronary artery spasm. Circulation. 2011;124:1774–1782. doi: 10.1161/CIRCULATIONAHA.111.037283. [DOI] [PubMed] [Google Scholar]