

Abstract
Non-alcoholic fatty liver disease (NAFLD) is now the most common cause of chronic liver diseases worldwide. It encompasses a spectrum of disorders ranging from isolated hepatic steatosis to nonalcoholic steatohepatitis (NASH), fibrosis, cirrhosis, and hepatocellular carcinoma. One of the key challenges in NAFLD is identifying which patients will progress. Epidemiological and genetic studies indicate a strong pattern of heritability that may explain some of the variability in NAFLD phenotype and risk of progression. To date, at least three common genetic variants in the PNPLA3, TM6SF2, and GCKR genes have been robustly linked to NAFLD in the population. The function of these genes revealed novel pathways implicated in both the development and progression of NAFLD. In addition, candidate genes previously implicated in NAFLD pathogenesis have also been identified as determinants or modulators of NAFLD phenotype including genes involved in hepatocellular lipid handling, insulin resistance, inflammation, and fibrogenesis. This article will review the current understanding of the genetics underpinning the development of hepatic steatosis and the progression of NASH. These newly acquired insights may transform our strategy to risk-stratify patients with NAFLD and to identify new potential therapeutic targets.
Keywords: NAFLD, NASH, genetics, PNPLA3, TM6SF2, GCKR, MBOAT7
Background
Non-alcoholic fatty liver disease (NAFLD) is the most common cause of chronic liver disease worldwide affecting up to 25% of the global population and a third of the US population[1–3]. Together with this growing epidemic, morbidity from NAFLD is on the rise with a 170% increase in cirrhosis caused by non-alcoholic steatohepatitis (NASH) on the liver transplant waitlist between 2004 and 2013 in the US. NASH cirrhosis is now the second leading indication for liver transplantation in the US[4–5].
The NAFLD epidemic has gone hand-in-hand with its major risk factor- insulin resistance- manifested by increasing rates of obesity and type 2 diabetes mellitus[6]. One might consider human fatty liver simply an acquired trait due to over-eating, similar to the process required to produce foie gras in ducks. However, NAFLD is a heterogeneous disease ranging from isolated hepatic steatosis to nonalcoholic steatohepatitis and cirrhosis[7] and progression seems to occur only in a subset of patients[8–9]. Growing evidence in the past decade points to a strong genetic contribution to both the development of NAFLD and its progression. Genetics show great promise in risk stratification and may lead to future therapeutic interventions[10]. This article seeks to summarize the current literature regarding NAFLD genetics and their potential utility in the management of NAFLD patients.
Heritability
A major observation indicating the heritability of NAFLD is the difference of its prevalence among ethnic groups. Two large multi-ethnic population studies in the US demonstrated that NAFLD was present at much higher rates in Hispanics compared to Caucasians, while African-Americans were relatively protected irrespective of insulin resistance and BMI[2,11]. Familial aggregation studies demonstrated that family members of overweight children with NAFLD were at higher risk of NAFLD compared to family members of overweight children without NAFLD[12]. Twin studies have also shown that up to 60% of the variability in serum ALT, a surrogate for liver fat content in the absence of alcohol or viral hepatitis, is genetically determined[13]. In another twin study using MRI proton-density fat fraction as a measurement of hepatic steatosis and MR elastography to determine hepatic fibrosis, both were highly correlated in monozygotic twins as compared to dizygotic twins[4,11]. After adjustment for age, ethnicity, and gender, the heritability of hepatic steatosis and hepatic fibrosis was 52% and 50%, respectively[14].
Genome-wide association studies (GWAS) have been used in the past decade to define the specific genetic mediators of this heritability. An rs738409 C>G variant in the patatin-like phospholipase domain-containing 3 (PNPLA3) gene, encoding an I148M mutation, was the first allele associated with intrahepatic fat content and appears to be a major genetic determinant of hepatic steatosis and the progression of fatty liver disease[15–16]. An rs58542926 C>T variant in the transmembrane 6 superfamily member 2 (TM6SF2) gene, encoding an E167K mutation, and an rs780094 C>T variant in the glucokinase regulator (GCKR) gene were later shown to be associated with both hepatic steatosis and the risk of progression to fibrosis[17–20]. More recently, the rs641738 C>T variant of the membrane bound O-acyltransferase domain-containing 7 (MBOAT7) gene was identified in alcoholic-related cirrhosis and subsequently confirmed to increase risk of hepatic steatosis and progressive liver disease in NAFLD[21–22]. Numerous other genetic variants involved in the pathogenesis of NAFLD, ranging from inflammatory response to insulin resistance and fibrogenesis, have also been implicated in NAFLD progression[10] (Table 1).
Tab.1.
Summary of genetic variants associated with NAFLD development and progression
| Gene | Variant | Function | Variant effect | MAF | Hepatic phenotype | Extrahepatic phenotype |
|---|---|---|---|---|---|---|
| PNPLA3 | rs738409 C>G | Lipid droplet remodeling | Impaired mobilization of FAs from lipid droplets through inhibition of other lipases, hepatic TG accumulation | 0.267 | ↑NAFLD ↑NASH ↑fibrosis ↑HCC |
↓CV risk No effect on IR |
| rs2294918 G>A | Decreased PNPLA3 production, attenuation of effect of I148M variant | 0.390 | ↓NAFLD ↓NASH |
|||
| TM6SF2 | rs58542926 C>T | VLDL secretion | Decreased VLDL secretion, hepatic TG accumulation | 0.067 | ↑NAFLD ↑NASH ↑fibrosis |
↓CV risk No effect on IR |
| GCKR | rs780094 A>G rs1260326 C>T |
Regulation of glucose influx to hepatocytes, de novo lipogenesis | Inability to regulate glucose influx into hepatocytes, increased de novo lipogenesis | 0.302 0.293 |
↑NAFLD ↑NASH ↑fibrosis |
No effect on CV risk ↓IR ↑CKD |
| MBOAT7 | rs641738 C>T | Catalyzes acyl chain remodeling of phosphatidyl- inositols, reduces free arachidonic acid levels |
Increased arachidonic acid levels, increased hepatic inflammation | 0.440 | ↑NAFLD ↑NASH ↑fibrosis ↑HCC |
No effect on CV or IR risk |
| HSD17B13 | rs72613567:TA | Unknown. Localizes to hepatocyte lipid droplets. | Decreased HSD17B13 and PNPLA3 production | 0.260 | ↓NASH ↓fibrosis |
|
| APOB | multiple | VLDL secretion | Abetalipoproteinemia | 0.01 | ↑NAFLD ↑NASH ↑fibrosis ↑HCC |
↓CV risk |
| MTTP | multiple | VLDL secretion | Hypobetalipoproteinemia | 0.01 | ↑NAFLD ↑NASH ↑fibrosis ↑HCC |
↓CV risk |
| LIPA | multiple | Hydrolysis of cholesteryl esters and LDL particles | Lysosomal acid lipase deficiency | 0.01 | ↑NAFLD ↑NASH ↑fibrosis |
↑CV risk |
| LPIN1 | rs13412852 C>T | Regulation of lipid metabolism | Reduced lipolysis, decreased flux of FAs to the liver | 0.205 | ↓NASH ↓fibrosis |
|
| FATP5 | rs56225452 G>A | Hepatocyte FA uptake | Increased hepatocyte FA uptake | 0.180 | ↑NASH | ↑IR |
| SOD2 | rs4880 C>T | Mitochondrial antioxidant | Increased oxidative stress | 0.411 | ↑fibrosis | |
| UCP2 | rs695366 G>A | Mitochondrial lipid metabolism | Increased UCP2 production, decreased oxidative stress | 0.264 | ↓NASH | |
| ENPP1 | rs1044498 A>C | Insulin signaling inhibitor | Increased inhibition of insulin signaling | 0.342 | ↑fibrosis | ↑IR |
| IRS1 | rs1801279 A>C | Insulin signaling | Decreased insulin signaling | 0.053 | ↑fibrosis | ↑IR |
| TRIB1 | rs2954021 G>A | Hepatic de novo lipogenesis | Increased hepatic TG | ↑NAFLD | ||
| IL28B | rs12979860 C>T | Innate immunity | Decreased IFN production | 0.356 | ↓NASH ↓fibrosis |
|
| MERTK | rs4374383 G>A | Innate immunity | Decreased hepatic stellate cell activation | 0.360 | ↓fibrosis | ↑IR |
| KLF6 | rs3750861 G>A | Activation of HSCs | Decreased HSC activation | 0.068 | ↓fibrosis | |
| TERT | multiple | Telomere maintenance | Accelerated hepatocyte senescence | 0.01 | ↑fibrosis |
MAF: minor allele frequency; IR: insulin resistance; CV: cardiovascular; CKD: chronic kidney disease; HSC: hepatic stellate cell.
PNPLA3
The PNPLA3 gene encodes a 481-amino acid membrane protein located in the endoplasmic reticulum and lipid droplets in hepatocytes and hepatic stellate cells (HSCs)[23]. Transcription of PNPLA3 is controlled by SREBP-1c and ChREBP, and is upregulated after feeding[23–24]. The PNPLA3 protein is post-transcriptionally modified by the presence of fatty acids to inhibit its degradation[23–24]. The rs738409 C>G polymorphism (I148M) in the PNPLA3 gene, first identified by GWAS in 2008, encodes a missense mutation that by far is the most important genetic determinant for hepatic fat content[15]. The carrier frequency of PNPLA3 I148M is as high as 49% in Hispanics, 23% in Caucasians, and 17% in African Americans in the Dallas Heart Study[15]. This genetic variant alone accounts for the ethnic difference in NAFLD prevalence.
In vitro, PNPLA3 acts as an acyltransferase that catalyzes the conversion of lysophosphatidic acid (LPA) to phosphatidic acid, while the I148M mutation renders a loss of enzymatic function[25–26]. However, the physiological function of PNPLA3 remains elusive. It is not clear whether the change in the enzyme activity contributes to the development of hepatic steatosis because PNPLA3 knock-out mice do no develop hepatic steatosis[26–28]. Recent studies suggested that PNPLA3 may be involved in lipid droplet remodeling in hepatocytes, where the I148M variant protein accumulates on the surface of lipid droplets by evading ubiquitination and impairs hydrolysis of triglyceride by lipases[26,29–31]. This hypothesis is also supported by the finding that PNPLA3 E434K, a variant that decreases PNPLA3 expression, can attenuate the effect of I148M on hepatic steatosis and steatohepatitis[32]. Interestingly, the PNPLA3 I148M variant is also present in HSCs, where the mutant allele is shown to activate HSCs independent of its role in hepatocytes[33–34].
Clinically, aside from causing intrahepatic triglyceride accumulation, the PNPLA3 I148M variant has also been shown to increase the risk of progressive liver disease. In the initial GWAS study by Romeo and coworkers, the I148M variant was associated with higher levels of alanine aminotransferase (ALT) indicating increased hepatic inflammation[15]. The PNPLA3 I148M variant was subsequently found to be associated with NASH, hepatic fibrosis, and hepatocellular carcinoma (HCC)[16,35–36]. The PNPLA3 I148M variant is also associated with increased risk of fibrosis progression and HCC in cirrhosis from hepatitis C and alcoholic liver disease independent of steatosis[35–37], suggesting a potential direct contribution of the variant to fibrogenesis and carcinogenesis that are unrelated to intrahepatic triglyceride accumulation[38].
TM6SF2
The TM6SF2 gene encodes a 351-amino acid protein with seven transmembrane domains expressed in the liver and intestine in humans[39]. The rs58542926 C>T (E167K) polymorphism in TM6SF2 was first found to be associated with NAFLD by GWAS in 2014 and encodes a missense protein resulting in loss-of-function[19]. The TM6SF2 E167K variant is present in 7.2% Europeans, 3.4% African Americans and 4.7% Hispanics. Although the function of TM6SF2 was not known at the time of this discovery, it was discovered shortly afterwards that the E167K variant impairs the lipidation and maturation of very low density lipoprotein (VLDL) in hepatocytes and chylomicrons in enterocytes, resulting in increased cellular triglyceride accumulation and decreased circulating triglyceride-rich lipoproteins[19,39–40]. Similar to the PNPLA3 I148M variant, the TM6SF2 E1267K variant not only increases the risk of steatosis, but has also been associated with increased risk of progressive liver disease and fibrosis[20]. This finding has also been observed in chronic hepatitis C patients with the TM6SF2 E1267K allele[41].
GCKR
GCKR plays an important role in hepatic glucose uptake through regulating the partitioning of GCK (glucokinase) between the cytosol and nucleus[42]. The P446L (rs1260326 C>T) variant of GCKR, first identified by GWAS in 2011, encodes a loss-of-function protein unable to inhibit glucokinase in response to fructose-6-phosphate[17,43]. Thus, the P446L variant of GCKR is associated with increased hepatic glucose uptake, which in turn may contribute to increased de novo lipogenesis and concomitantly decreased serum glucose and insulin levels[43]. The combination of PNPLA3 and GCKR minor alleles (referring to the less common variants, I148M and P446L, respectively) was shown to explain up to 30% of the liver fat content in obese children[44]. The GCKR P446L variant is also associated with an increased risk of fibrosis in NAFLD patients as well as elevated serum triglyceride levels[18].
MBOAT7
MBOAT7 (also known as lysophopshoplipid acyltransferase) catalyzes acyl chain remodeling of phosphatidylinositols, part of the Lands cycle, attaching arachidonic acid to lysophosphatidylinositol and reducing free arachidonic acid levels[45]. Arachidonic acid induces hepatocyte apoptosis, triggering hepatic inflammation and fibrosis[46–47]. Probably for this reason, the rs641738 C>T variant of MBOAT7, which results in decreased hepatic MBOAT7 expression, has not been observed to increase the risk of steatosis to the degree of PNPLA3 or TM6SF2. Rather, the rs641738 C>T variant of MBOAT7 is associated with an increase in hepatic inflammation and fibrosis in NAFLD[21,48–49]. The rs641738 C>T variant was first identified by GWAS in alcoholic liver disease in which it increases the risk of cirrhosis[22], and has been implicated in hepatitis B[50] and C[51]. Additionally, the rs641738 C>T variant has been tied to an increase in HCC risk in non-cirrhotics with NAFLD, as well as non-cirrhotic chronic hepatitis C and alcoholic liver disease[52].
HSD17B13
Most recently, exome-wide sequencing has identified the rs72613567:TA variant within hydroxysteroid 17-beta dehydrogenase 13 (HSD17B13), which was associated with a decreased risk of alcoholic liver disease, NASH, alcoholic cirrhosis, and NASH cirrhosis[53]. The HSD17B13 gene encodes a previously uncharacterized member of the hydroxysteroid 17-beta dehydrogenase family, and is primarily expressed in hepatocytes where the protein product is localizes to lipid droplets[54]. The precise function of HSD17B13 is currently unknown, but the splice variant results in in vitro loss of enzymatic function towards estradiol[53]. In a recent study of Abul-Husn et al., the rs72613567:TA variant was associated with a 30% decreased risk of NASH and a 49% decreased risk of NASH cirrhosis without an association with hepatic steatosis itself, suggesting a role in mitigating liver injury but not a role in intrahepatic triglyceride accumulation. Further study is required to confirm this association and the role of HSD17B13 in NAFLD pathogenesis.
Hepatic lipid metabolism
While PNPLA3 and TM6SF2 appear to be the most prominent population-wide determinants of hepatic steatosis, other relatively rare or less prominent genetic defects in intrahepatic lipid metabolism have been shown to cause fatty liver. Mutations in the genes governing hepatic processing and secretion of VLDL have been implicated in familial causes of NAFLD[55]. For instance, mutations within the apolipoprotein B (APOB) or proprotein convertase subtilisin kexin 9 (PCSK9) gene result in familial hypobetalipoproteinemia, characterized by low or absent plasma levels of apoB and LDL-C[56]. The apoB protein is responsible for the assembly and secretion of hepatic VLDL and intestinal chylomicrons. Loss-of-function or missense mutations within the APOB gene can result in a decrease in serum cholesterol and an increase in intrahepatic triglycerides that have been implicated in familial cases of steatohepatitis, cirrhosis, and hepatocellular carcinoma[57].
PCKS9 is a serine protease that enhances the degradation of LDL-receptors. Loss-of-function mutations within PCKS9 similarly result in decreased serum cholesterol, but do not seem to be associated with increased hepatic triglycerides[58]. Large phase Ⅲ clinical trials investigating monoclonal antibodies against PCSK9 to lower serum LDL similarly have not demonstrated any adverse hepatic side effects[59–60], though PCSK9 mutations may exacerbate liver disease in those with PNPLA3 and TM6SF2 mutations[61].
The microsomal triglyceride-transfer protein (MTTP) gene encodes a lipid transfer protein responsible for the recruitment of triglycerides during VLDL particle formation within the endoplasmic reticulum and may also serve as a chaperone assisting the folding of apoB in hepatocytes[62]. Mutations within the MTTP gene are responsible for abetalipoproteinemia, characterized by nearly undetectable plasma levels of LDL and apoB, as well as accumulation of triglycerides in the liver with subsequent cirrhosis[63–64]. Lomitapide is an inhibitor of MTTP marketed for the treatment of familial hypertriglyceridemia and acute pancreatitis. Long-term use of lomitapide has been associated with NASH cirrhosis[65].
Apolipoprotein C3 (apoC3) is a major constituent of VLDL particles and promotes triglyceride-rich VLDL assembly and secretion[66]. Several mutations within the APOC3 gene promoter have been associated with hypertriglyceridemia[67–68], and initial reports also indicated increased risk of fatty liver[69]. However, subsequent studies have failed to find an association between polymorphisms within the APOC3 promoter and progressive NAFLD[70–71]. Several variants within the coding sequence of apoC3 have been associated with hypotriglyceridemia[72–73] and hypertriglyceridemia[74], respectively. The relationship between these apoC3 coding sequence mutations and NAFLD is unclear. However, a recent study showed expression of the Gln38Lys variant in mice resulted in hepatic steatosis[75]. Since inhibition of apoC3 expression is currently being considered as a treatment of hypertriglyceridemia, one must keep in mind the potential risk of NAFLD and NASH[76].
Another rare familial cause of NASH cirrhosis is the deficiency of lysosomal acid lipase (LIPA), an autosomal recessive lysosomal storage disease caused by loss-of-function mutations within the LIPA gene. Lysosomal acid lipase is responsible for hydrolyzing cholesteryl esters, triglycerides, and LDL particles into free cholesterol and fatty acids. Its deficiency frequently results in death in infancy, though in adults it leads to hypercholesterolemia, cardiovascular disease, hepatic steatosis, and cirrhosis[77].
Fatty acid transport proteins (FATP) mediate hepatocyte uptake of free fatty acids. FATP5 increases hepatic free fatty acid uptake and silencing of FATP5 reverses steatosis in diet-induced NAFLD mice[78–79]. A variant (rs56225452) in the FATP5 promoter region regulating gene expression was associated with increased hepatic steatosis, higher ALT levels, and increased insulin resistance in the general population[80].
LPIN1 encodes a phosphatidate phosphatase expressed in adipose tissue and the liver where it acts as an inducible transcriptional coactivator to regulate fatty acid metabolism[81]. The rs12412852 polymorphism of LPIN1 was associated with a lower prevalence of NASH and hepatic fibrosis in a cohort of Italian children with NAFLD[82]. The mutation is thought to result in reduced lipolysis creating decreased flux of free fatty acids to the liver[82].
Oxidative stress and inflammatory response
Reactive oxidative species (ROS) produced with fatty acid beta oxidation in the mitochondria has been proposed as a major driver for NASH[83]. Several variations in genes involved in regulating mitochondrial redox status have been implicated in NASH progression. Uncoupling protein 2 (UCP2) regulates mitochondrial redox status by uncoupling oxidative phosphorylation[84]. A variation in the UCP2 promoter region (rs695366) increases UCP2 expression and is associated with decreased risk of NASH in patients with normal fasting glucose[85]. Similarly, another protein involved in mitochondrial protection from oxidative stress, the mitochondrial manganese-dependent superoxide dismutase (MnSOD), has been associated with protection from progressive NAFLD[86]. The rs4880 polymorphism in the SOD2 gene results in an increase in enzyme activity and was associated with a lower risk of NASH and fibrosis[86].
In addition to ROS activation, NASH is associated with activation of the innate immune response due to sterile inflammation and gut-derived bacterial products[87]. Interferon-λ3/λ4 (encoded by the IL28B gene) was first noted to be involved in hepatic innate immunity when the rs12979860 CC variant was associated with increased clearance of hepatitis C virus[88]. This variant leads to increased production of Interferon-λ3 and has been associated with increased steatohepatitis and fibrosis in NAFLD[89–90].
Polymorphisms in the promoter region of the TNFA gene encoding tumor necrosis factor-α have also been associated with the progression of NAFLD[91–92], though results have not been consistent[93–94] and may be due to linkage disequilibrium with other genes in the major human histocompatibility complex region[10].
MERTK encodes a tyrosine kinase that initiates the removal of dying cells by phagocytes and is involved in the activation of HSCs. The rs4374383 non-coding variant reduces MERTK expression and is associated with reduced fibrosis in both NAFLD and chronic hepatitis C infection[95–97].
Insulin resistance
Insulin resistance is closely associated with NAFLD pathophysiology and disease progression[98]. Outside of environmental factors that contribute to insulin resistance, genetic variations in the insulin signaling pathway have also been linked to NAFLD progression and fibrosis. In hepatocytes, insulin binds to the insulin receptor resulting in the activation of insulin receptor substrate-1 (IRS-1) and decreases glucose production. A loss-of-function mutation (rs1801278) in IRS1 results in hyperglycemia and has been associated with increased hepatic fibrosis[99]. A gain-of-function mutation in ectonucleotide pyrophosphatase/phosphodiesterase1 (ENPP1) inhibits insulin receptor activity, resulting in insulin resistance and accelerated liver fibrosis in a cohort of obese NAFLD patients[99].
Tribbles homolog1 (TRIB1) is a protein kinase involved in hepatic lipogenesis and glycogenesis[100]. Induction of TRIB1 expression increases plasma glucose and hepatic triglyceride production in a mouse model[101]. In humans, the rs2954021 polymorphism within TRIB1 has been associated with increased plasma triglycerides and the development of NAFLD[102].
Fibrogenesis
Several genetic polymorphisms associated with cell senescence have also been associated with fibrosis in NAFLD. For example, telomeres, which serve to prevent DNA damage and cell senescence with cell division, are maintained by the enzyme telomerase. Loss-of-function mutations in the telomerase reverse transcriptase (TERT) gene are associated with familial liver disease and accelerated development of cirrhosis and HCC in NAFLD and other etiologies of chronic liver disease[103–104]. In the same vein, p21 regulates the cell cycle by causing cell cycle arrest and cell senescence. The rs762623 SNP in the CDKN1A gene, which encodes p21, has been associated with progressive NAFLD and fibrosis in a cohort of NAFLD patients[105].
Hepatocyte senescence leads to hepatic fibrosis through the activation and proliferation of HSC[106]. Krueppel-like factor 6 (KLF6) is expressed by activated HSCs after liver injury resulting in collagen α1 transcription[107]. The rs3750861 variant of KLF6 results in alternative splicing and decreased activation of HSC after liver injury and reduced fibrosis[108]. In addition, the rs3750861 variant of KLF6 has been associated with decreased hepatic insulin resistance[109].
Hepatic iron deposition promotes fibrogenesis through multiple pathways including oxidative stress, subsequent mitochondrial dysfunction[110], and direct stimulation of HSC[111]. Genetic variations in the HFE, beta-globin, and TMPRSS6 genes predispose to hepatic iron accumulation and are associated with hepatic fibrosis in NAFLD patients[112–114].
Extrahepatic manifestations of NAFLD
In addition to modulating the hepatic phenotype of NAFLD, genetic variations play an important role in extrahepatic manifestations of the disease. NAFLD is an independent risk factor for the development of cardiovascular disease, the leading cause of death in NAFLD patients[115]. Given the intimate involvement of many NAFLD-related genetic variants in lipid and lipoprotein metabolism, it follows that these variations may also affect cardiovascular risk. Indeed, the TM6SF2 minor allele (ie E167K) which results in decreased hepatic VLDL secretion is associated with a lower cardiovascular risk, while the major allele increases total cholesterol and higher risk of myocardial infarction[116–117]. The PNPLA3 I148M variant, which has a somewhat lesser effect on plasma lipids, has also been associated with a degree of protection from cardiovascular disease[117–118]. The MBOAT7 minor allele, in which no effect on plasma lipids has been observed[21], also has a neutral effect on cardiovascular risk[117]. The GCKR rs1260326 variant has been shown to increase plasma triglycerides with no change in LDL or HDL levels and has no effect on cardiovascular risk[119]. Lysosomal acid lipase deficiency results in both increased serum cholesterol and hepatic triglycerides, greatly increasing cardiovascular risk[120]. While coronary artery disease is the leading cause of death for all-comer NAFLD patients, the genetics of NAFLD impact the cardiovascular risk in different ways, and a better understanding of an individual’s genetic underpinnings may also help to stratify their cardiovascular risk.
While acquired risk factors such as obesity lead to insulin resistance and both NAFLD and type 2 diabetes, any relationship beyond this is less clear. Similar to cardiovascular risk, when disentangling the link between NAFLD and type 2 diabetes, it is helpful to turn to genetics and the mechanism by which these genes exert their effects. For example, the minor alleles of both PNPLA3 and TM6SF2 result in hepatocyte lipid retention without any direct link to insulin signaling or glucose metabolism. Accordingly, neither the I148M nor E167K variant is associated with increased insulin resistance or incidence of type 2 diabetes[15,19,40,121]. Similarly, MBOAT7 that regulates circulating phosphatidylinositol composition, has no impact on insulin resistance or type 2 diabetes prevalence[21]. The GCKR P446L variant, on the other hand, results in the deregulation of hepatic glucose uptake, increased de novo lipogenesis, decreased serum glucose, and reduced insulin resistance[44,122]. Polymorphisms in the ENPP1 and IRS1 genes involved in insulin signaling result in insulin resistance and are associated with fibrosis in NAFLD[99] as well as risk of incident diabetes[123–124]. While NAFLD is often driven by insulin resistance, genetics predisposing to NAFLD largely do not impact insulin resistance directly.
NAFLD is also associated with chronic kidney disease[125]. The GCKR rs1260326 polymorphism has been associated with increased risk of chronic kidney disease in those with NAFLD, though the mechanism is not clear and results have not been replicated[126].
Genetic variations have proven useful in understanding not only the phenotypic variability of liver disease in NAFLD, but also in the pathophysiology behind its comorbidities. Such an understanding may also prove useful in the risk stratification of NAFLD patients with shared extrahepatic comorbidities.
Conclusions
Research in the past decade has shown that non-alcoholic fatty liver is a trait driven by both acquired and genetic causes, and has taught us important lessons. First of all, up to 50% of disease susceptibility and progression is heritable[14]. Major genetic determinants of hepatic steatosis in the population include PNPLA3 I148M, TM6SF2 E167K and GCKR P446L. These genes variances as well as less common genetic mutations linked to NAFLD highlights the importance of lipoprotein assembly, intrahepatic lipid handling, and glucose metabolism in the pathophysiology of NAFLD (Fig. 1). The genetics underlying the inflammation and fibrosis in NAFLD is less well-defined, mainly owing to a lack of large cohort studies with phenotyping and appropriate controls. Secondly, unfavorable genetics increases the disease susceptibility, but may not cause disease alone. This paradigm is most prominent in PNPLA3, where the I148M carriers are far more likely to develop NAFLD when they are obese, while lean carriers can still be disease-free. Last but not least, the presence of different genetic variances indicates that NAFLD is not one homogenous disease. Although they share the presence of fatty liver, their risk of progressive liver fibrosis and comorbidities may vary based on the genetic cause. This is exemplified by the impact on circulating lipoproteins by TM6SF2 E167K and to a lesser extent PNPLA3 I148M in NAFLD.
Fig.1.
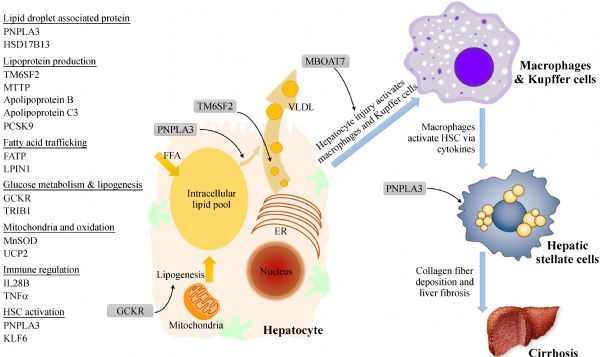
Genetic impact on hepatic steatosis, liver inflammation, and fibrosis in NAFLD.
Understanding the genetic underpinnings of NAFLD provides an exciting opportunity to refine our management of NAFLD patients. Although the most recent AASLD guidelines do not recommend testing for genetic variants in routine clinical care[127], the potential of such an approach has been demonstrated in several recent studies. Using genetics alone (PNPLA3, SOD2, KLF6, and LPIN1), one group was able to predict the presence of NASH in a cohort of children with NAFLD, though it was not useful in the prediction of fibrosis[128]. In one report, the minor allele of the IL28B gene along with clinical factors outperformed traditional non-invasive assessments in the prediction of fibrosis not just in NASH, but also in chronic viral hepatitis[129]. The combination of PNPLA3 genotype, AST level, and fasting insulin level has been shown to be useful in predicting the histologic presence of NASH in a cohort of NAFLD patients[130]. Similarly, we have shown that a combination of PNPLA3 and TM6SF2 genotype along with a lipoprotein-derived assessment of insulin resistance was able to predict both NAFLD activity and fibrosis (unpublished data). As our understanding of the role of genetics in NAFLD pathophysiology improves, these models show great promise not only in the risk stratification of the hepatic consequences of NAFLD, but may also prove useful in disentangling the extrahepatic consequences as well.
Perhaps the most exciting aspect of NAFLD genetics is the potential therapeutic implications. Downregulation of PNPLA3 I148M variant production by the E434K variant has been shown to attenuate the effect of the I148M variant on steatohepatitis[32]. Though largely in the theoretical stage of development, downregulation of the PNPLA3 I148M variant shows promise as a potential point of therapeutic intervention.
Genetic research over the past decade has provided valuable insight into the pathogenesis of NAFLD. Further refinement over the coming decade has the potential to transform the way we care for NAFLD patients with a personalized approach to monitoring tailored to an individual’s risk of fibrosis progression and treatment targeting the underlying mechanism of disease.
Acknowledgment
ZGJ is supported by an Alan Hofmann Clinical and Translational Research Award from AASLD.
References
- 1. Younossi ZM, Koenig AB, Abdelatif D, et al. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes[J]. Hepatology, 2016, 64(1): 73–84. [DOI] [PubMed] [Google Scholar]
- 2. Browning JD, Szczepaniak LS, Dobbins R, et al. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity[J]. Hepatology, 2004, 40(6): 1387–1395. [DOI] [PubMed] [Google Scholar]
- 3. Lazo M, Hernaez R, Eberhardt MS, et al. Prevalence of nonalcoholic fatty liver disease in the United States: The third national health and nutrition examination survey, 1988–1994[J]. Am J Epidemiol, 2013, 178(1): 38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wong RJ, Aguilar M, Cheung R, et al. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States[J]. Gastroenterology, 2015, 148(3): 547–555. [DOI] [PubMed] [Google Scholar]
- 5. Charlton MR, Burns JM, Pedersen RA, et al. Frequency and outcomes of liver transplantation for nonalcoholic steatohepatitis in the United States[J]. Gastroenterology, 2011, 141(4): 1249–1253. [DOI] [PubMed] [Google Scholar]
- 6. Marchesini G, Brizi M, Bianchi G, et al. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome[J]. Diabetes, 2001, 50(8): 1844–1850. [DOI] [PubMed] [Google Scholar]
- 7. Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease[J]. Hepatology, 2005, 41(6): 1313–1321. [DOI] [PubMed] [Google Scholar]
- 8. Teli MR, James OFW, Burt AD, et al. The natural history of nonalcoholic fatty liver: a follow-up study[J]. Hepatology, 1995, 22(6): 1714–1719. [PubMed] [Google Scholar]
- 9. Dam-Larsen S, Franzmann M, Andersen IB, et al. Long term prognosis of fatty liver: risk of chronic liver disease and death[J]. Gut, 2004, 53(5): 750–755 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dongiovanni P, Valenti L. Genetics of nonalcoholic fatty liver disease[J]. Metabolism, 2016, 65(8): 1026–1037. [DOI] [PubMed] [Google Scholar]
- 11. Guerrero R, Vega GL, Grundy SM, et al. Ethnic differences in hepatic steatosis: an insulin resistance paradox?[J]. Hepatology, 2009, 49(3): 791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schwimmer JB, Celedon MA, Lavine JE, et al. Heritability of nonalcoholic fatty liver disease[J]. Gastroenterology, 2009, 136(5): 1585–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Makkonen J, Pietiläinen KH, Rissanen A, et al. Genetic factors contribute to variation in serum alanine aminotransferase activity independent of obesity and alcohol: a study in monozygotic and dizygotic twins[J]. J Hepatol, 2009, 50(5): 1035–1042. [DOI] [PubMed] [Google Scholar]
- 14. Loomba R, Schork N, Chen CH, et al. Heritability of hepatic fibrosis and steatosis based on a prospective twin study[J]. Gastroenterology, 2015, 149(7): 1784–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Romeo S, Kozlitina J, Xing C, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease[J]. Nat Genet, 2008, 40(12): 1461–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Singal AG, Manjunath H, Yopp AC, et al. The effect of PNPLA3 on fibrosis progression and development of hepatocellular carcinoma: a meta-analysis[J]. Am J Gastroenterol, 2014, 109(3): 325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Speliotes EK. Yerges-Armstrong LM, Wu J, et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits[J]. PLoS Genet, 2011, 7(3): e1001324 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Petta S, Miele L, Bugianesi E, et al. Glucokinase regulatory protein gene polymorphism affects liver fibrosis in non-alcoholic fatty liver disease[J]. PLoS One, 2014, 9(2): e87523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kozlitina J, Smagris E, Stender S, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease[J]. Nat Genet, 2014, 46(4): 352–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu YL, Reeves HL, Burt AD, et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease[J]. Nat Commun, 2014, 5: 4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mancina RM, Dongiovanni P, Petta S, et al. The MBOAT7-TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of European descent[J]. Gastroenterology, 2016, 150(5): 1219–1230.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Buch S, Stickel F, Trépo E, et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis[J]. Nat Genet, 2015, 47(12): 1443–1448. [DOI] [PubMed] [Google Scholar]
- 23. Huang Y, He S, Li JZ, et al. A feed-forward loop amplifies nutritional regulation of PNPLA3[J]. Proc Natl Acad Sci U S A, 2010, 107(17): 7892–7897 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dubuquoy C, Robichon C, Lasnier F, et al. Distinct regulation of adiponutrin/PNPLA3 gene expression by the transcription factors ChREBP and SREBP1c in mouse and human hepatocytes[J]. J Hepatol, 2011, 55(1): 145–153. [DOI] [PubMed] [Google Scholar]
- 25. Kumari M, Schoiswohl G, Chitraju C, et al. Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase[J]. Cell Metab, 2012, 15(5): 691–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pingitore P, Pirazzi C, Mancina RM, et al. Recombinant PNPLA3 protein shows triglyceride hydrolase activity and its I148M mutation results in loss of function[J]. Biochim Biophys Acta, 2014, 1841(4): 574–580. [DOI] [PubMed] [Google Scholar]
- 27. Basantani MK, Sitnick MT, Cai L, et al. Pnpla3/Adiponutrin deficiency in mice does not contribute to fatty liver disease or metabolic syndrome[J]. J Lipid Res, 2011, 52(2): 318–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen W, Chang B, Li L, et al. Patatin-like phospholipase domain-containing 3/adiponutrin deficiency in mice is not associated with fatty liver disease[J]. Hepatology, 2010, 52(3): 1134–1142 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. He S, McPhaul C, LiJ Z, et al. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis[J]. J Biol Chem, 2010, 285(9): 6706–6715 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Smagris E, BasuRay S, Li J, et al. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis[J]. Hepatology, 2015, 61(1): 108–118 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. BasuRay S, Smagris E, Cohen JC, et al. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation[J]. Hepatology, 2017, 66(4): 1111–1124 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Donati B, Motta BM, Pingitore P, et al. The rs2294918 E434K variant modulates patatin-like phospholipase domain-containing 3 expression and liver damage[J]. Hepatology, 2016, 63(3): 787–798. [DOI] [PubMed] [Google Scholar]
- 33. Pingitore P, Dongiovanni P, Motta BM, et al. PNPLA3 overexpression results in reduction of proteins predisposing to fibrosis[J]. Hum Mol Genet, 2016, 25(23): 5212–5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bruschi FV, Claudel T, Tardelli M, et al. The PNPLA3 I148M variant modulates the fibrogenic phenotype of human hepatic stellate cells[J]. Hepatology, 2017, 65(6): 1875–1890. [DOI] [PubMed] [Google Scholar]
- 35. Trépo E, Nahon P, Bontempi G, et al. Association between the PNPLA3 (rs738409 C>G) variant and hepatocellular carcinoma: Evidence from a meta-analysis of individual participant data[J]. Hepatology, 2014, 59(6): 2170–2177. [DOI] [PubMed] [Google Scholar]
- 36. Valenti L, Alisi A, Galmozzi E, et al. I148M patatin-like phospholipase domain-containing 3 gene variant and severity of pediatric nonalcoholic fatty liver disease[J]. Hepatology, 2010, 52(4): 1274–1280. [DOI] [PubMed] [Google Scholar]
- 37. Valenti L, Rumi M, Galmozzi E, et al. Patatin-like phospholipase domain-containing 3 I148M polymorphism, steatosis, and liver damage in chronic hepatitis C[J]. Hepatology, 2011, 53(3): 791–799. [DOI] [PubMed] [Google Scholar]
- 38. Valenti L, Motta BM, Soardo G, et al. PNPLA3 I148M polymorphism, clinical presentation, and survival in patients with hepatocellular carcinoma[J]. PLoS One, 2013, 8(10): e75982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mahdessian H, Taxiarchis A, Popov S, et al. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content[J]. Proc Natl Acad Sci USA, 2014, 111(24): 8913–8918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Smagris E, Gilyard S, BasuRay S, et al. Inactivation of Tm6sf2, a gene defective in fatty liver disease, impairs lipidation but not secretion of very low density lipoproteins[J]. J Biol Chem, 2016, 291(20): 10659–10676 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Milano M, Aghemo A, Mancina RM, et al. Transmembrane 6 superfamily member 2 gene E167K variant impacts on steatosis and liver damage in chronic hepatitis C patients[J]. Hepatology, 2015, 62(1): 111–117. [DOI] [PubMed] [Google Scholar]
- 42. Raimondo A, Rees MG, Gloyn AL. Glucokinase regulatory protein: complexity at the crossroads of triglyceride and glucose metabolism[J]. Curr Opin Lipidol, 2015, 26(2): 88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Beer NL, Tribble ND, McCulloch LJ, et al. The P446L variant in GCKR associated with fasting plasma glucose and triglyceride levels exerts its effect through increased glucokinase activity in liver[J]. Hum Mol Genet, 2009, 18(21): 4081–4088 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Santoro N, Zhang CK, Zhao H, et al. Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents[J]. Hepatology, 2012, 55(3): 781–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gijón MA, Riekhof WR, Zarini S, et al. Lysophospholipid acyltransferases and arachidonate recycling in human neutrophils[J]. J Biol Chem, 2008, 283(44): 30235–30245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Serini S, Piccioni E, Merendino N, et al. Dietary polyunsaturated fatty acids as inducers of apoptosis: implications for cancer[J]. Apoptosis, 2009, 14(2): 135–152. [DOI] [PubMed] [Google Scholar]
- 47. Guicciardi ME, Gores GJ. Apoptosis: a mechanism of acute and chronic liver injury[J]. Gut, 2005, 54(7): 1024–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Luukkonen PK, Zhou Y, Hyötyläinen T, et al. The MBOAT7 variant rs641738 alters hepatic phosphatidylinositols and increases severity of non-alcoholic fatty liver disease in humans[J]. J Hepatol, 2016, 65(6): 1263–1265. [DOI] [PubMed] [Google Scholar]
- 49. Viitasalo A, Eloranta AM, Atalay M, et al. Association of MBOAT7 gene variant with plasma ALT levels in children: the PANIC study[J]. Pediatr Res, 2016, 80(5): 651–655. [DOI] [PubMed] [Google Scholar]
- 50. Thabet K, Chan HLY, Petta S, et al. The membrane-bound O-acyltransferase domain-containing 7 variant rs641738 increases inflammation and fibrosis in chronic hepatitis B[J]. Hepatology, 2017, 65(6): 1840–1850 . [DOI] [PubMed] [Google Scholar]
- 51. Thabet K, Asimakopoulos A, Shojaei M, et al. MBOAT7 rs641738 increases risk of liver inflammation and transition to fibrosis in chronic hepatitis C[J]. Nat Commun, 2016, 7: 12757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Donati B, Dongiovanni P, Romeo S, et al. MBOAT7 rs641738 variant and hepatocellular carcinoma in non-cirrhotic individuals[J]. Sci Rep, 2017, 7(1): 4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Abul-Husn NS, Cheng X, Li AH, et al. A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease[J]. N Engl J Med, 2018, 378(12): 1096–1106 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Su W, Wang Y, Jia X, et al. Comparative proteomic study reveals 17-HSD13 as a pathogenic protein in nonalcoholic fatty liver disease[J]. Proc Natl Acad Sci U S A, 2014, 111(31): 11437–11442 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kneeman JM, Misdraji J, Corey KE. Secondary causes of nonalcoholic fatty liver disease[J]. Therap Adv Gastroenterol, 2012, 5(3): 199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Welty FK. Hypobetalipoproteinemia and abetalipoproteinemia[J]. Curr Opin Lipidol, 2014, 25(3): 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cefalù AB, Pirruccello JP, Noto D, et al. A novel APOB mutation identified by exome sequencing cosegregates with steatosis, liver cancer, and hypocholesterolemia[J]. Arterioscler Thromb Vasc Biol, 2013, 33(8): 2021–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kotowski IK, Pertsemlidis A, Luke A, et al. A spectrum of PCSK9 alleles contributes to plasma levels of low-density lipoprotein cholesterol[J]. Am J Hum Genet, 2006, 78(3): 410–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Blom DJ, Hala T, Bolognese M, et al. A 52-week placebo-controlled trial of evolocumab in hyperlipidemia[J]. N Engl J Med, 2014, 370(19): 1809–1819. [DOI] [PubMed] [Google Scholar]
- 60. Robinson JG, Farnier M, Krempf M, et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events[J]. N Engl J Med, 2015, 372(16): 1489–1499. [DOI] [PubMed] [Google Scholar]
- 61. Di Filippo M, Vokaer B, Seidah NG. A case of hypocholesterolemia and steatosis in a carrier of a PCSK9 loss-of-function mutation and polymorphisms predisposing to nonalcoholic fatty liver disease[J]. J Clin Lipidol, 2017, 11(4): 1101–1105. [DOI] [PubMed] [Google Scholar]
- 62. Jiang ZG, Liu Y, Hussain MM, et al. Reconstituting initial events during the assembly of apolipoprotein B-containing lipoproteins in a cell-free system[J]. J Mol Biol, 2008, 383(5): 1181–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Collins JC, Scheinberg IH, Giblin DR, et al. Hepatic peroxisomal abnormalities in abetalipoproteinemia[J]. Gastroenterology, 1989, 97(3): 766–770. [DOI] [PubMed] [Google Scholar]
- 64. Braegger CP, Belli DC, Mentha G, et al. Persistence of the intestinal defect in abetalipoproteinaemia after liver transplantation[J]. Eur J Pediatr, 1998, 157(7): 576–578. [DOI] [PubMed] [Google Scholar]
- 65. Sacks FM, Stanesa M, Hegele RA. Severe hypertriglyceridemia with pancreatitis: thirteen years’ treatment with lomitapide[J]. JAMA Intern Med, 2014, 174(3): 443–447. [DOI] [PubMed] [Google Scholar]
- 66. Yao Z, Wang Y. Apolipoprotein C-Ⅲ and hepatic triglyceride-rich lipoprotein production[J]. Curr Opin Lipidol, 2012, 23(3): 206–212. [DOI] [PubMed] [Google Scholar]
- 67. Olivieri O, Stranieri C, Bassi A, et al. ApoC-Ⅲ gene polymorphisms and risk of coronary artery disease[J]. J Lipid Res, 2002, 43(9): 1450–1457. [DOI] [PubMed] [Google Scholar]
- 68. Guettier JM, Georgopoulos A, Tsai MY, et al. Polymorphisms in the fatty acid-binding protein 2 and apolipoprotein C-Ⅲ genes are associated with the metabolic syndrome and dyslipidemia in a South Indian population[J]. J Clin Endocrinol Metab, 2005, 90(3): 1705–1711. [DOI] [PubMed] [Google Scholar]
- 69. Petersen KF, Dufour S, Hariri A, et al. Apolipoprotein C3 gene variants in nonalcoholic fatty liver disease[J]. N Engl J Med, 2010, 362(12): 1082–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kozlitina J, Boerwinkle E, Cohen JC, et al. Dissociation between APOC3 variants, hepatic triglyceride content and insulin resistance[J]. Hepatology, 2011, 53(2): 467–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Valenti L, Nobili V, Al-Serri A, et al. The APOC3 T-455C and C-482T promoter region polymorphisms are not associated with the severity of liver damage independently of PNPLA3 I148M genotype in patients with nonalcoholic fatty liver[J]. J Hepatol, 2011, 55(6): 1409–1414 . [DOI] [PubMed] [Google Scholar]
- 72. Liu H, Labeur C, Xu CF, et al. Characterization of the lipid-binding properties and lipoprotein lipase inhibition of a novel apolipoprotein C-Ⅲ variant Ala23Thr[J]. J Lipid Res, 2000, 41(11): 1760–1771 . [PubMed] [Google Scholar]
- 73. von Eckardstein A, Holz H, Sandkamp M, et al. Apolipoprotein C-Ⅲ (Lys58----Glu). Identification of an apolipoprotein C-Ⅲ variant in a family with hyperalphalipoproteinemia[J]. J Clin Invest, 1991, 87(5): 1724–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Pullinger CR, Malloy MJ, Shahidi AK, et al. Hypertriglyceridemia in a large kindred of Mexican origin experimental subjects[J]. J Lipid Res, 1997, 38: 1833–1840. [PubMed] [Google Scholar]
- 75. Sundaram M, Curtis KR, Alipour MA, et al. T he apolipoprotein C-Ⅲ (Gln38Lys) variant associated with human hypertriglyceridemia is a gain-of-function mutation[J]. J Lipid Res, 2017, 58(11):2188–2196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bell TA, Graham MJ, Baker BF, et al. Therapeutic inhibition of apoC-Ⅲ for the treatment of hypertriglyceridemia[J]. Clin Lipidol, 2015, 10(2):191–203. [Google Scholar]
- 77. Reiner Ž, Guardamagna O, Nair D, et al. Lysosomal acid lipase deficiency--an under-recognized cause of dyslipidaemia and liver dysfunction[J]. Atherosclerosis, 2014, 235(1): 21–30. [DOI] [PubMed] [Google Scholar]
- 78. Hubbard B, Doege H, Punreddy S, et al. Mice deleted for fatty acid transport protein 5 have defective bile acid conjugation and are protected from obesity[J]. Gastroenterology, 2006, 130(4): 1259–1269. [DOI] [PubMed] [Google Scholar]
- 79. Doege H, Grimm D, Falcon A, et al. Silencing of hepatic fatty acid transporter protein 5 in vivo reverses diet-induced non-alcoholic fatty liver disease and improves hyperglycemia[J]. J Biol Chem, 2008, 283(32): 22186–22192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Auinger A, Valenti L, Pfeuffer M, et al. A promoter polymorphism in the liver-specific fatty acid transport protein 5 is associated with features of the metabolic syndrome and steatosis[J]. Horm Metab Res, 2010, 42(12): 854–859. [DOI] [PubMed] [Google Scholar]
- 81. Reue K, Brindley DN. Thematic review series: glycerolipids. Multiple roles for lipins/phosphatidate phosphatase enzymes in lipid metabolism[J]. J Lipid Res, 2008, 49(12): 2493–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Valenti L, Motta BM, Alisi A, et al. LPIN1 rs13412852 polymorphism in pediatric nonalcoholic fatty liver disease[J]. J Pediatr Gastroenterol Nutr, 2012, 54(5): 588–593. [DOI] [PubMed] [Google Scholar]
- 83. Caldwell SH, Swerdlow RH, Khan EM, et al. Mitochondrial abnormalities in non-alcoholic steatohepatitis[J]. J Hepatol, 1999, 31(3): 430–434. [DOI] [PubMed] [Google Scholar]
- 84. Berardi MJ, Chou JJ. Fatty acid flippase activity of UCP2 is essential for its proton transport in mitochondria[J]. Cell Metab, 2014, 20(3): 541–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Fares R, Petta S, Lombardi R, et al. The UCP2-866 >A promoter region polymorphism is associated with nonalcoholic steatohepatitis[J]. Liver Int, 2015, 35(5): 1574–1580. [DOI] [PubMed] [Google Scholar]
- 86. Al-Serri A, Anstee QM, Valenti L, et al. The SOD2 C47T polymorphism influences NAFLD fibrosis severity: evidence from case-control and intra-familial allele association studies[J]. J Hepatol, 2012, 56(2): 448–454 . [DOI] [PubMed] [Google Scholar]
- 87. Miele L, Valenza V, La Torre G, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease[J]. Hepatology, 2009, 49(6): 1877–1887. [DOI] [PubMed] [Google Scholar]
- 88. Thomas DL, Thio CL, Martin MP, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus[J]. Nature, 2009, 461(7265): 798–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Petta S, Grimaudo S, Cammà C, et al. IL28B and PNPLA3 polymorphisms affect histological liver damage in patients with non-alcoholic fatty liver disease[J]. J Hepatol, 2012, 56(6): 1356–1362. [DOI] [PubMed] [Google Scholar]
- 90. Eslam M, Hashem AM, Leung R, et al. Interferon-rs12979860 genotype and liver fibrosis in viral and non-viral chronic liver disease[J]. Nat Commun, 2015, 6: 6422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Tokushige K, Takakura M, Tsuchiya-Matsushita N, et al. Influence of TNF gene polymorphisms in Japanese patients with NASH and simple steatosis[J]. J Hepatol, 2007, 46(6): 1104–1110 . [DOI] [PubMed] [Google Scholar]
- 92. Valenti L, Fracanzani AL, Dongiovanni P, et al. Tumor necrosis factor α promoter polymorphisms and insulin resistance in nonalcoholic fatty liver disease[J]. Gastroenterology, 2002, 122(2): 274–280. [DOI] [PubMed] [Google Scholar]
- 93. Pastor IJ, Laso FJ, Romero A, et al. -238 G>A polymorphism of tumor necrosis factor alpha gene (TNFA) is associated with alcoholic liver cirrhosis in alcoholic Spanish men[J]. Alcohol Clin Exp Res, 2005, 29(11): 1928–1931. [DOI] [PubMed] [Google Scholar]
- 94. Gochee PA, Jonsson JR, Clouston AD, et al. Steatosis in chronic hepatitis C: association with increased messenger RNA expression of collagen I, tumor necrosis factor-alpha and cytochrome P450 2E1[J]. J Gastroenterol Hepatol, 2003, 18(4): 386–392. [DOI] [PubMed] [Google Scholar]
- 95. Petta S, Valenti L, Marra F, et al. MERTK rs4374383 polymorphism affects the severity of fibrosis in non-alcoholic fatty liver disease[J]. J Hepatol, 2016, 64(3): 682–690. [DOI] [PubMed] [Google Scholar]
- 96. RÜeger S, Bochud PY, Dufour JF, et al. Impact of common risk factors of fibrosis progression in chronic hepatitis C[J]. Gut, 2015, 64(10): 1605–1615. [DOI] [PubMed] [Google Scholar]
- 97. Musso G, Cassader M, De Michieli F, et al. MERTK rs4374383 variant predicts incident nonalcoholic fatty liver disease and diabetes: role of mononuclear cell activation and adipokine response to dietary fat[J]. Hum Mol Genet, 2017, 26(9): 1747–1758. [DOI] [PubMed] [Google Scholar]
- 98. Musso G, Cassader M, De Michieli F, et al. Nonalcoholic steatohepatitis versus steatosis: adipose tissue insulin resistance and dysfunctional response to fat ingestion predict liver injury and altered glucose and lipoprotein metabolism[J]. Hepatology, 2012, 56(3): 933–942. [DOI] [PubMed] [Google Scholar]
- 99. Dongiovanni P, Valenti L, Rametta R, et al. Genetic variants regulating insulin receptor signalling are associated with the severity of liver damage in patients with non-alcoholic fatty liver disease[J]. Gut, 2010, 59(2): 267–273. [DOI] [PubMed] [Google Scholar]
- 100. Bauer RC, Sasaki M, Cohen DM, et al. Tribbles-1 regulates hepatic lipogenesis through posttranscriptional regulation of C/EBPα[J]. J Clin Invest, 2015, 125(10): 3809–3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Ishizuka Y, Nakayama K, Ogawa A, et al. TRIB1 downregulates hepatic lipogenesis and glycogenesis via multiple molecular interactions[J]. J Mol Endocrinol, 2014, 52(2): 145–158. [DOI] [PubMed] [Google Scholar]
- 102. Kitamoto A, Kitamoto T, Nakamura T, et al. Association of polymorphisms in GCKR and TRIB1 with nonalcoholic fatty liver disease and metabolic syndrome traits[J]. Endocr J, 2014, 61(7): 683–689. [DOI] [PubMed] [Google Scholar]
- 103. Calado RT, Regal JA, Kleiner DE, et al. A spectrum of severe familial liver disorders associate with telomerase mutations[J]. PLoS One, 2009, 4(11): e7926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Hartmann D, Srivastava U, Thaler M, et al. Telomerase gene mutations are associated with cirrhosis formation[J]. Hepatology, 2011, 53(5): 1608–1617. [DOI] [PubMed] [Google Scholar]
- 105. Aravinthan A, Mells G, Allison M, et al. Gene polymorphisms of cellular senescence marker p21 and disease progression in non-alcohol-related fatty liver disease[J]. Cell Cycle, 2014, 13(9): 1489–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Aravinthan A, Scarpini C, Tachtatzis P, et al. Hepatocyte senescence predicts progression in non-alcohol-related fatty liver disease[J]. J Hepatol, 2013, 58(3): 549–556. [DOI] [PubMed] [Google Scholar]
- 107. Ratziu V, Lalazar A, Wong L, et al. Zf9, a Kruppel-like transcription factor up-regulated in vivo during early hepatic fibrosis[J]. Proc Natl Acad Sci USA, 1998, 95(16): 9500–9505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Miele L, Beale G, Patman G, et al. The Kruppel-like factor 6 genotype is associated with fibrosis in nonalcoholic fatty liver disease[J]. Gastroenterology, 2008, 135(1): 282–291.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Bechmann LP, Gastaldelli A, Vetter D, et al. Glucokinase links KrÜppel-like factor 6 to the regulation of hepatic insulin sensitivity in nonalcoholic fatty liver disease[J]. Hepatology, 2012, 55(4): 1083–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Lee HJ, Choi JS, Lee HJ, et al. Effect of excess iron on oxidative stress and gluconeogenesis through hepcidin during mitochondrial dysfunction[J]. J Nutr Biochem, 2015, 26(12): 1414–1423. [DOI] [PubMed] [Google Scholar]
- 111. Ruddell RG, Hoang-le D, Barwood JM, et al. Ferritin functions as a proinflammatory cytokine via iron-independent PKC-NF-regulated signalling in rat hepatic stellate cells[J]. Hepatology, 2010, 49(3): 887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Valenti L, Canavesi E, Galmozzi E, et al. Beta-globin mutations are associated with parenchymal siderosis and fibrosis in patients with non-alcoholic fatty liver disease[J]. J Hepatol, 2010, 53(5): 927–933. [DOI] [PubMed] [Google Scholar]
- 113. Valenti L, Fracanzani AL, Bugianesi E, et al. HFE genotype, parenchymal iron accumulation, and liver fibrosis in patients with nonalcoholic fatty liver disease[J]. Gastroenterology, 2010, 138(3): 905–912. [DOI] [PubMed] [Google Scholar]
- 114. Valenti L, Rametta R, Dongiovanni P, et al. The A736V TMPRSS6 polymorphism influences hepatic iron overload in nonalcoholic fatty liver disease[J]. PLoS One, 2012, 7(11): e48804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Targher G, Byrne CD, Lonardo A, et al. Non-alcoholic fatty liver disease and risk of incident cardiovascular disease: A meta-analysis[J]. J Hepatol, 2016, 65(3): 589–600. [DOI] [PubMed] [Google Scholar]
- 116. Holmen OL, Zhang H, Fan Y, et al. Systematic evaluation of coding variation identifies a candidate causal variant in TM6SF2 influencing total cholesterol and myocardial infarction risk[J]. Nat Genet, 2014, 46(4): 345–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Simons N, Isaacs A, Koek GH, et al. PNPLA3, TM6SF2, and MBOAT7 genotypes and coronary artery disease[J]. Gastroenterology, 2017, 152(4): 912–913. [DOI] [PubMed] [Google Scholar]
- 118. Tang CS, Zhang H, Cheung CYY, et al. Exome-wide association analysis reveals novel coding sequence variants associated with lipid traits in Chinese[J]. Nat Commun, 2015, 6: 10206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Kozian DH, Barthel A, Cousin E, et al. Glucokinase-activating GCKR polymorphisms increase plasma levels of triglycerides and free fatty acids, but do not elevate cardiovascular risk in the Ludwigshafen Risk and Cardiovascular Health Study[J]. Horm Metab Res, 2010, 42(7): 502–506. [DOI] [PubMed] [Google Scholar]
- 120. Fouchier SW, Defesche JC. Lysosomal acid lipase A and the hypercholesterolaemic phenotype[J]. Curr Opin Lipidol, 2013, 24(4): 332–338. [DOI] [PubMed] [Google Scholar]
- 121. Sookoian S, Castaño GO, Scian R, et al. Genetic variation in transmembrane 6 superfamily member 2 and the risk of nonalcoholic fatty liver disease and histological disease severity[J]. Hepatology, 2015, 61(2): 515–525. [DOI] [PubMed] [Google Scholar]
- 122. Valenti L, Alisi A, Nobili V. Unraveling the genetics of fatty liver in obese children: additive effect of P446L GCKR and I148M PNPLA3 polymorphisms[J]. Hepatology, 2012, 55(3): 661–663. [DOI] [PubMed] [Google Scholar]
- 123. Grarup N, Urhammer SA, Ek J, et al. Studies of the relationship between the ENPP1 K121Q polymorphism and type 2 diabetes, insulin resistance and obesity in 7,333 Danish white subjects[J]. Diabetologia, 2006, 49(9): 2097–2104 . [DOI] [PubMed] [Google Scholar]
- 124. Jellema A, Zeegers MPA, Feskens EJM, et al. Gly972Arg variant in the insulin receptor substrate-1 gene and association with Type 2 diabetes: a meta-analysis of 27 studies[J]. Diabetologia, 2003, 46(7): 990–995. [DOI] [PubMed] [Google Scholar]
- 125. Musso G, Gambino R, Tabibian JH, et al. Association of non-alcoholic fatty liver disease with chronic kidney disease: a systematic review and meta-analysis[J]. PLoS Med, 2014, 11(7): e1001680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Hishida A, Takashima N, Turin TC, et al. GCK, GCKR polymorphisms and risk of chronic kidney disease in Japanese individuals: data from the J-MICC Study[J]. J Nephrol, 2014, 27(2): 143–149. [DOI] [PubMed] [Google Scholar]
- 127. Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases[J]. Hepatology, 2018, 67(1): 328–357. [DOI] [PubMed] [Google Scholar]
- 128. Nobili V, Donati B, Panera N, et al. A 4-polymorphism risk score predicts steatohepatitis in children with nonalcoholic fatty liver disease[J]. J Pediatr Gastroenterol Nutr, 2014, 58(5): 632–636. [DOI] [PubMed] [Google Scholar]
- 129. Eslam M, Hashem AM, Romero-Gomez M, et al. FibroGENE: A gene-based model for staging liver fibrosis[J]. J Hepatol, 2016, 64(2): 390–398 . [DOI] [PubMed] [Google Scholar]
- 130. Hyysalo J, Männistö VT, Zhou Y, et al. A population-based study on the prevalence of NASH using scores validated against liver histology[J]. J Hepatol, 2014, 60(4): 839–846. [DOI] [PubMed] [Google Scholar]