

Publisher's Note: There is a Blood Commentary on this article in this issue.
Key Points
Polyreactive IgM mediates complement activation by PF4/heparin complexes via the classical pathway.
Complement activation by PF4/heparin complexes varies widely among healthy donors.
Abstract
The mechanisms by which exposure to heparin initiates antibody responses in many, if not most, recipients are poorly understood. We recently demonstrated that antigenic platelet factor 4 (PF4)/heparin complexes activate complement in plasma and bind to B cells. Here, we describe how this process is initiated. We observed wide stable variation in complement activation when PF4/heparin was added to plasma of healthy donors, indicating a responder “phenotype” (high, intermediate, or low). Proteomic analysis of plasma from these healthy donors showed a strong correlation between complement activation and plasma immunoglobulin M (IgM) levels (r = 0.898; P < .005), but not other Ig isotypes. Complement activation response to PF4/heparin in plasma displaying the low donor phenotype was enhanced by adding pooled IgM from healthy donors, but not monoclonal IgM. Depletion of IgM from plasma abrogated C3c generation by PF4/heparin. The complement-activating features of IgM are likely mediated by nonimmune, or natural, IgM, as cord blood and a monoclonal polyreactive IgM generate C3c in the presence of PF4/heparin. IgM facilitates complement and antigen deposition on B cells in vitro and in patients receiving heparin. Anti-C1q antibody prevents IgM-mediated complement activation by PF4/heparin complexes, indicating classical pathway involvement. These studies demonstrate that variability in plasma IgM levels correlates with functional complement responses to PF4/heparin. Polyreactive IgM binds PF4/heparin, triggers activation of the classical complement pathway, and promotes antigen and complement deposition on B cells. These studies provide new insights into the evolution of the heparin-induced thrombocytopenia immune response and may provide a biomarker of risk.
Visual Abstract
Introduction
Autoantibodies to platelet factor 4 (PF4)/heparin develop in 25% to 50% of patients exposed to heparin during cardiopulmonary bypass1-5 and cause heparin-induced thrombocytopenia (HIT) in a subset of patients. This striking propensity for antibody formation in clinic settings such as cardiac surgery remains unexplained.
The high incidence of heparin sensitization is difficult to reconcile with known mechanisms of major histocompatibility complex-restricted antigen presentation. One potential explanation derives from our recent findings showing robust complement activating activities of ultralarge complexes of PF4/heparin (ULCs).6 PF4/heparin ULCs activate complement in plasma in a heparin-dependent manner, both in vitro and in patients receiving heparin therapy. Complement activation by PF4/heparin ULCs elicits binding of C3 fragments to antigen and facilitates antigen deposition on circulating B cells via the complement receptor 2/CD21.6 As binding of complement-coated antigen to CD21 potentiates its immunogenicity 1000- to 10 000-fold,7 complement activation and subsequent binding of PF4/heparin to B cells may represent early, sensitizing events in recipients of heparin.
How PF4/heparin complexes activate complement is unknown. Complement can be activated by the classical, alternative, or lectin pathways individually or in combination. Several lines of evidence implicate involvement of the classical pathway in HIT. Studies performed in the 1960s and 1970s showed that mixtures of polycations and polyanions, such as protamine (PRT)/heparin and lysozyme/DNA, activate the classical pathway of complement, possibly through a nonimmune mechanism.8-14 More recently, anti-PF4/heparin reactive immunoglobulin M (IgM) has been identified in healthy donor blood. Zheng and colleagues were first to identify IgM-secreting B cells in the peripheral blood of all donors tested.15 In these studies, anti-PF4/heparin IgM from healthy donors reacted preferentially with PF4/heparin complexes compared with PF4, and reactive B cells were found at a fairly high frequency (0.1-1.0 per 1000 B cells) in the peripheral blood mononuclear population. Krauel and colleagues also found high levels of anti-PF4/heparin IgM autoreactive B cells in the peripheral blood of healthy donors, as well as circulating anti-PF4/heparin IgM in infants 1 to 3 months of age and anti-PF4/heparin antibody-secreting cells in cord blood samples.16 These investigators speculated that anti-PF4/heparin reactivity was likely encoded by the “natural IgM” repertoire of antibodies.16
In mice and humans, IgM occurs either as a membrane-bound monomer on B cells (as a part of B-cell receptor) or as a secreted, pentameric protein in plasma, sIgM.17 The repertoire of sIgM can be further divided into natural or immune on the basis of antigen-binding specificities. Natural IgM is named as such because it can be detected in normal quantities in mice grown under antigen/germ-free conditions, likely arises from endogenous antigens, displays reactivity to a wide range of seemingly unrelated self and foreign antigens, and exhibits germline-encoded variable heavy- and light-chain genes.18,19 Immune IgM, in contrast, represents an antigen-specific immune response to pathogens or external antigens with limited cross-reactivity and presence of highly mutated variable gene regions.18,20
The polyreactivity of natural IgM endows it with diverse sentinel functions in health and disease.17 In infection, natural IgM facilitates antigen-specific immunity by binding pathogens, triggering complement activation and transporting antigen via noncognate B cells to splenic subcompartments.21 Mice lacking natural IgM exhibit defective antigen trapping of particulate antigen, impaired germinal center formation, increased susceptibility to bacterial and viral pathogens, and defective T-cell-dependent immunity.22,23 Intriguingly, these defects mirror the phenotypes seen with deficiencies of classical pathway components and/or the complement receptor CD21,24-26 suggesting an interconnectedness of pathways involving natural IgM, complement, and CD21.
In the studies that follow, we show that nonimmune, naturally occurring IgM mediates complement activation by PF4/heparin complexes and promotes antigen deposition on B cells. In addition, our studies suggest that preexposure levels of plasma IgM may constitute a stable biomarker for the risk for sensitization and possible development of HIT.
Materials and methods
Materials
Recombinant human PF4 was purified as described.27 UFH was from Elkins-Sinn Inc. (Cherry Hill, NJ). Unless specified, other chemicals, buffers, and tissue culture reagents were purchased from Millipore Sigma (St. Louis, MO). IgM and IgG from healthy donor plasma and IgM from plasma from patients with myeloma was purchased from Athens Research and Technology (Athens, GA). Intravenous immunoglobulin was purchased from Grifols (Los Angeles, CA). The following antibodies were used: anti-C1q (Cell Sciences, Inc., Newburyport, MA), anti-C3c (Quidel, San Diego, CA), anti-MBL (R&D Systems, Minneapolis, MN), and murine IgG1 isotype control (Invitrogen, Carlsbad, CA), fluorescently conjugated anti-human CD19 and conjugated streptavidin (eBioscience, San Diego, CA), and fluorescently conjugated goat anti-human IgM (Jackson Labs, West Grove, PA). Monoclonal antibody KKO (IgG2bκ recognizing PF4/heparin),28 ADA (IgG3 recognizing PRT sulfate/heparin),29,30 and 2E4 (monoclonal IgM with polyreactive specificities to single-stranded DNA, β-galactosidase, and other antigens)31 were developed, purified, and isolated in the laboratory according to published methods. PF4/heparin-specific IgM was isolated using beads coated with PF4 bound to heparin immobilized on diamino-dipropylamine agarose (Thermo Fisher Scientific, Waltham, MA), as previously described.32
Blood samples
Blood from healthy donors or patients receiving heparin therapy was collected into citrate with written consent using an IRB-approved protocol (Duke IRB#: Pro00010740). Human subjects were enrolled in accordance with the Declaration of Helsinki. Human umbilical cord blood was obtained as discarded clinical samples under an IRB exempt provision (Duke IRB#: Pro00047355). Where indicated, studies were performed in whole blood or 100% plasma from healthy donors.
Immunoglobulin levels
Total immunoglobulins (IgG, IgA, and IgM) in serum were quantified by the Duke University Hospital Clinical Immunology Laboratory by rate nephelometry.
Antigen-C3c capture ELISA assay and specificity ELISA
Antigen-C3c capture enzyme-linked immunosorbent assay (ELISA) was performed as described in supplemental Data, available on the Blood Web site.30 For studies involving Igs (IgM, IgG, myeloma IgM, monoclonal polyreactive IgM), ethylenediaminetetraacetic acid (EDTA), ethylene glycol-bis (β-aminoethyl ether)-N,N,N',N'-tetraacetic acid (EGTA), magnesium chloride (MgCl2), or classical/lectin pathway, reagents were added to plasma before incubating with antigen.
Antigen specificity was determined against various antigens (bovine serum albumin, PF4, PF4/heparin, PRT, PRT/heparin, lysozyme [Lys], Lys/heparin, or heparin alone), using protein concentrations of 10 µg/mL and heparin (0.2-1.3 U/mL, based on optimal antigenic protein:heparin ratios) by ELISA. Bound IgM was detected with goat anti-human IgM (μ-chain specific) peroxidase-conjugated antibody by colorimetric detection.6
Flow cytometry studies
Flow-based studies of antigen, complement, or IgM binding to B cells were performed as described.6 Patient samples were processed without the addition of exogenous antigen. Cells were analyzed using a BD FACS Canto Flow cytometer (BD Biosciences, Franklin Lakes, NJ). Signals from a minimum of 10 000 cells were acquired from each sample. Analyses were performed using FCS express software (Version 5 Flow Research Edition; De Novo Software).
IgM studies
Goat anti-human IgM (µ-chain specific) agarose beads were used to deplete IgM from plasma. Anti-IgM or control agarose beads were incubated with plasma, and depleted plasma supernatant was analyzed in the antigen-C3c capture assay described here.
Proteomic analysis
Plasma from donors identified as having a high, intermediate, or low complement activity in the antigen-C3c capture ELISA assay were submitted to the Duke Proteomics and Metabolomics Shared Resource (https://genome.duke.edu/cores-and-services/proteomics-and-metabolomics). Proteomic data were analyzed using published protocols.33 Principal component analysis, statistical tests, and agglomerative clustering were performed using the Rosetta Elucidator.
Statistics
Data are expressed as mean ± standard deviation (SD). Significance was calculated using Student t test or 1-way ANOVA. Descriptive statistics (means/standard deviations/range are used to describe continuous variables. Normality was assessed using the Shapiro-Wilk/Anderson-Darling tests. Correlations between normally distributed continuous variables are conducted using Pearson correlations, and correlations between non-normally distributed variables are conducted using Spearman correlations. Statistical significance is assessed at α = 0.05. No adjustment is made for multiple testing. Statistical analyses were performed using the SAS 9.4 statistical software (SAS Institute Inc., Cary, NC) or GraphPad Prism (Graph Pad Software Version 7.0).
Results
Complement responses to PF4/heparin complexes among healthy donors defines a donor phenotype
In the course of developing a capture immunoassay to detect complement activation by PF4/heparin complexes,30 we noted marked heterogeneity in C3c generation when we studied plasmas from different healthy donors. As shown in Figure 1A, complement activation in this assay occurs when plasma from healthy donors with no prior history of heparin exposure (n = 10) was incubated with PF4/heparin, but not with buffer or PF4 alone or heparin alone. Complement activation in response to PF4/heparin was highly variable, with some donors expressing high reactivity (eg, donor 2) while others expressed intermediate (eg, donors 1 and 3) and/or low (eg, donor 4) levels of complement activation. Intriguingly, for a given donor, responses to PF4/heparin constituted a stable phenotype that remained high, intermediate, or low over time (up to ∼1.7 years; Figure 1B). Differences in complement activation among high, intermediate, or low responders correlated with the amount of PF4/heparin antigen and C3c deposited on B cells by flow cytometry (supplemental Figure 1A-B). The fact that the immunoassay is performed using isolated plasma led us to ask whether these differences in donor phenotype are governed by circulating plasma components.
Figure 1.

Complement activation in response to PF4/heparin complexes among healthy donors defines a donor phenotype. (A) Plasma from healthy donors (n = 10) was incubated with buffer, PF4 ± heparin, or heparin alone, and binding of C3c to PF4/heparin complexes was measured by antigen-C3c capture ELISA assay. The graph shows the anti-C3c absorbance of donor plasma incubated with different antigens. Each symbol represents an individual donor. Results are shown from a representative experiment performed a minimum of 3 times, with multiple donors in each experiment. (B) Complement activation by PF4/heparin of donors (1, 2, 3, and 4) was determined during a period of 626 days (∼1.7 years) and normalized to donor 1, who was studied at all points. The graph shows percentage PF4/heparin complement activation relative to donor 1 on different days. **P < .0001.
Donor phenotype correlates with plasma IgM levels
To determine the serologic basis for the donor phenotype, we first examined the plasma proteome of 8 donors displaying high (n = 3), intermediate (n = 2), or low (n = 3) reactivity in the C3c-antigen capture assay. As shown in supplemental Table 1, mass spectrometry identified 5 proteins that showed significant correlation with high/intermediate vs low donors (supplemental Table 1 shows highest to lowest significance in P value for high/intermediate vs low): IgM µ chain C region (40 peptides quantified; 4-fold increase; P = .001), complement C1q subcomponent subunit B (10 peptides quantified; 1-fold increase; P = .01), complement factor H-related protein 4 (1 peptide quantified; 2-fold increase; P = .031), complement C1q subcomponent subunit A (4 peptides quantified; 1-fold increase; P = .033), and complement factor D (4 peptides quantified; 1-fold decrease; P = .034). On the basis of data showing the greatest peptide coverage for IgM (40 peptides) and associated significant P values, we examined the correlation between complement activation with donor IgM in more detail. As shown in Figure 2A, by mass spectrometry, an individual’s IgM levels showed a strong correlation with complement activation (r = 0.898; P < .005; Pearson’s correlation).
Figure 2.

Complement activation by PF4/heparin correlates with plasma/serum IgM levels. (A) Proteomic analysis of plasmas with a high, intermediate, or low complement response phenotype shows strong correlation with plasma IgM. Graph shows IgM protein intensity determined by proteomic analysis (x-axis) and complement activation response to PF4/heparin, as measured by the antigen-C3c capture ELISA assay (y-axis). (B-D) Serum immunoglobulin levels (IgM, IgG, and IgA) from 29 healthy donors were measured in the clinical laboratory and correlated with an individual’s complement activation response to PF4/heparin, as measured by the antigen-C3c capture ELISA assay. Graphs show correlation of complement activation (y-axis) as a function of immunoglobulin levels (x-axis). Each symbol in the graph represents an individual donor. Complement activation values were normalized to an intermediate donor studied in parallel.
To affirm these findings and examine the influence of other immunoglobulins, we tested a larger cohort (n = 29) of healthy individuals for complement activation response to PF4/heparin and measured corresponding IgM, IgG, and IgA levels in our clinical laboratory. As shown in Figure 2B-D, we again noted a strong correlation between complement activation by PF4/heparin and total IgM (0.82; P < .0001; by Spearman correlation as IgM levels were not normally distributed), but not IgG or IgA levels, in the same samples (IgG: r = −0.36; P = .05; IgA: r = −0.22; P = not significant; by Pearson’s correlation for normally distributed data).
Plasma IgM mediates complement activation by PF4/heparin complexes
The studies shown in Figure 2 demonstrate a strong correlation between an individual’s plasma IgM levels and the extent of complement activation response to PF4/heparin, but they do not show that IgM is required. To investigate the involvement of IgM, we augmented or depleted IgM from the plasma of donors with low or intermediate reactivity, respectively. Addition of commercial IgM (0-1000 µg/mL; isolated and pooled from ∼20 healthy donors) to the plasma of 2 low phenotype donors caused a dose-dependent increase in complement activation (Figure 3A; ∼10-fold increase in C3c generation seen with 1000 µg/mL IgM vs 0 IgM; P < .0001). Neither polyclonal IgG (0-5000 µg/mL; Figure 3A) nor monoclonal IgM (0-1000 µg/mL) restored complement activation at any concentration tested (Figure 3A), even when higher IgG concentrations were tested to mimic the 5- to 10-fold higher levels of IgG in plasma. Conversely, removal of IgM from plasma diminished C3c generation. Specifically, when plasma from an intermediate phenotype donor was depleted of IgM, there was marked loss of complement activation in response to PF4/heparin (Figure 3B) compared with the same plasma treated with control beads (Figure 3B; P < .0001). Furthermore, repleting IgM (400 µg/mL) in plasma devoid of IgM rescued complement activation by PF4/heparin (Figure 3B; P < .0001 compared with no added IgM). As expected, addition of similar amounts of IgM to plasma incubated with control beads enhanced complement activation (Figure 3B).
Figure 3.

Plasma IgM mediates complement activation by PF4/heparin complexes. (A) Commercial IgM (0-1000 μg/mL; filled symbols) or IgG (0-5000 μg/mL; open symbols) or monoclonal myeloma IgM (0-1000 μg/mL; hatched symbols) was added to the plasmas of 2 donors with a low complement response type (circle/square), and complement activation by PF4/heparin was measured by the antigen-C3c capture ELISA assay. Graph shows complement activation (y-axis) as a function of added immunoglobulin concentration. (B) Plasma with an intermediate donor phenotype was incubated with anti-IgM or control beads, followed by the addition of buffer, PF4/heparin, or PF4/heparin +400 µg/mL IgM, and complement activation was measured by the antigen-C3c capture ELISA assay. Graph shows complement activation (y-axis) in control or IgM depleted plasma (x-axis). (C) Plasma with a low donor phenotype was incubated with varying antigen concentrations (PF4; 0-25 μg/mL + heparin; 0-0.25 U/mL) and IgM (0-800 μg/mL), and complement activation was measured by the antigen-C3c capture ELISA assay. Graph shows complement activation response at varying IgM concentrations (y-axis) as a function of PF4/heparin concentrations (x-axis). *P < .005; **P < .0001. Results are shown from a representative experiment involving 3 donors tested on 3 different occasions.
We next varied the concentrations of IgM and PF4/heparin to mirror changes likely to occur in the clinical setting. To do so, we used plasma from a donor with low IgM levels and tested the effects of increasing IgM (0-800 µg/mL) and PF4/heparin concentrations (PF4, 2.5-25 µg/mL and heparin, 0.025-0.25 U/mL) at a fixed molar protein:heparin ratio of 6.6. As shown in Figure 3C, complement activation was more dependent on changes in IgM levels than in PF4/heparin. At levels of IgM less than 200 µg/mL, complement activation was fairly insensitive to levels of PF4/heparin. In contrast, when the IgM was higher than 400 µg/mL, lower PF4/heparin ratios (7.5:0.075 and 10:0.1, respectively) sufficed to activate complement. Together, these findings demonstrate that complement activation by PF4/heparin complexes is dependent on IgM.
Polyreactive, naturally occurring IgM mediates complement activation by PF4/heparin
The findings that plasma containing IgM from individual healthy donors without prior heparin exposure (Figures 1 and 2) and that pooled IgM from healthy donors (Figure 3) activate complement in response to PF4/heparin suggests possible involvement of natural IgM. To investigate the role of natural IgM, we first examined the antigen-binding specificities of commercial donor IgM, which can be assumed to reflect little to no contribution from individuals who have been exposed to heparin. Binding of pooled IgM (or plasma dilutions of individual donors) to a panel of heparin-binding proteins was measured in the presence or absence of added heparin. As seen in Figure 4A, commercial IgM showed broad reactivity to a variety of antigens. In general, antigen reactivity was higher in the presence of heparin. Plasma from specific donors with high (supplemental Figure 2A) and intermediate (supplemental Figure 2B) IgM levels also showed broad reactivity with multiple antigens, but reactivity to individual antigens varied slightly from commercial IgM. As expected, the low IgM donor showed minimal reactivity to all antigens tested (supplemental Figure 2C). To exclude a subpopulation of PF4/heparin-specific IgM within a polyclonal IgM pool, commercial IgM was subjected to affinity purification using a PF4/heparin column. As shown in supplemental Figure 3, affinity-purified IgM did not differ from unfractionated IgM with respect to binding various antigens, thus excluding the presence or role for antigen-specific IgM in complement activation. Moreover, polyreactive IgM also activated complement in the presence of PRT/heparin complexes. As shown in supplemental Figure 4A, PRT/heparin complexes activated complement in plasma containing IgM, but not in plasma depleted of IgM. As with PF4/heparin, addition of polyclonal IgM to IgM-depleted plasma restored complement activation (supplemental Figure 4A).
Figure 4.

Polyreactive IgM mediates complement activation by PF4/heparin. (A) Antigen-specificity of commercial IgM was determined using microtiter plates coated with various antigens. The graph shows the binding (y-axis) of various concentrations of commercial IgM (1.25-80.0 µg/mL) to different antigens. (B) Complement activation by polyreactive monoclonal IgM, 2E4, in the plasma of 2 donors (low complement activation phenotype; circle/square) in response to buffer (open symbols), PF4 alone (hatched symbols), heparin alone (half-filled symbols) and PF4/heparin (filled symbols) as measured by the antigen-C3c capture ELISA assay. Graph shows complement activation (y-axis) as a function of added polyreactive IgM concentrations. Results are shown from a representative experiment involving 3 donors tested on 3 different occasions. **P < .0001, relative to no polyreactive IgM added. (C) Whole blood from the cord blood of a baby was incubated with buffer or PF4 ± heparin and binding of PF4/heparin (KKO) or C3c to the B cells was determined by flow cytometry. Histograms show the representative results from 2 different experiments with 2 different cord blood samples.
We then sought to confirm or exclude existence of natural IgM using 2 independent approaches. First we examined the complement activating activity of a monoclonal IgM antibody, 2E4, with broad specificities analogous to natural IgM. 2E4 recognizes diverse endogenous antigens (eg, single-stranded DNA β-galactosidase, insulin) and several strains of streptococci.31 The addition of 2E4 to the plasma of 2 donors with low complement reactivity initiated an antibody-dependent increase in complement activation in the presence of PF4/heparin (P < .0001 at 10 and 50 μg/mL 2E4), but not after addition of PF4 alone, heparin alone, or buffer (Figure 4B). As with PF4/heparin, 2E4 activated complement in response to PRT/heparin complexes, but not when plasma was incubated with PRT alone (supplemental Figure 4B). Next, we examined complement activation in response to PF4/heparin in cord blood, which is enriched in natural IgM.34 Because IgM levels in cord blood are less than 10% of adult levels, which is insufficient to activate complement in the plasma-based C3c immunoassay, we used the more sensitive flow-based assay. Cord blood incubated with PF4/heparin increased antigen (Figure 4C, top panel) and C3c (Figure 4C, bottom panel) on B cells (light blue line) compared with incubation with PF4 (yellow line) or buffer alone (purple line). These data further support the concept that naturally occurring IgM mediates complement activation by PF4/heparin complexes.
Complement activation by IgM is mediated through the classical pathway
The complement system can be activated by the alternative, classical, and/or lectin pathways. To first investigate the alternative pathway in PF4/heparin-mediated complement activation, we performed differential chelation studies using EDTA and EGTA, wherein the alternative pathway, sensitive to Mg2+, is inhibited by EDTA, but not EGTA.35 As shown in Figure 5A, addition of EDTA or EGTA to plasma before addition of PF4/heparin eliminated complement activation. Further, Mg2+ supplementation of EGTA-treated plasma did not rescue complement activation by PF4/heparin. Similar results were obtained in whole blood assay using flow cytometry (supplemental Figure 5). To examine involvement of the lectin and classical pathways, plasma or whole blood was preincubated with monoclonal antibodies to C1q or MBL or isotype controls, and complement activation responses to PF4/heparin were assessed by immunoassay (Figure 5B) or flow cytometry (supplemental Figure 6). Anti-C1q inhibited complement activation by IgM, whereas anti-MBL antibodies or mouse isotype control did not. In addition, in data not shown, we excluded involvement of individual lectin proteins, ficolin-2 and ficolin-3 in complement activation by PF4/heparin complexes. Mass spectrometry data accompanying Figure 2A did not show correlation of lectin proteins with complement activation phenotype, nor was functional inhibition of ficolin-2 associated with loss of complement activation in our immunoassay (data not shown). These studies establish that complement is activated by PF4/heparin through the classical complement pathway.
Figure 5.

PF4/heparin activate complement by classical pathway. (A) Plasma from a healthy donor was incubated with EDTA (10 mM) or EGTA (10 mM) ± MgCl2 (10 mM) or with buffer before incubating with PF4/heparin and complement activation was measured by the antigen-C3c capture ELISA assay. The y-axis shows the complement activation in different incubation conditions. ***P < .0001. (B) Plasma from a healthy donor was incubated with various concentration of anti-C1q antibody, anti-MBL antibody, or control antibody (0-100 μg/mL) before adding PF4/heparin and complement activation by PF4/heparin was determined by the antigen-C3c capture ELISA assay. The y-axis shows the complement activation in presence of various antibodies. *P < .05; **P < .001; ***P < .0001 compared with no antibody added condition. Results are shown from a representative experiment involving 3 donors tested on 3 different occasions.
Plasma IgM colocalizes with complement and antigen on B cells in vitro and in patients receiving heparin
Circulating IgM facilitates antigen transport of particulate antigen through colocalizing with antigen and complement on the surface of noncognate B cells.21,36 To examine whether IgM colocalizes with PF4/heparin antigen and complement on B cells, we examined surface IgM on B cells before and after addition of antigen. Consistent with previous findings,6 incubation of whole blood with buffer or PF4 alone is not associated with antigen or complement deposition on B cells. Under these same conditions, basal levels of IgM were detected on B cells, likely as a result of binding of anti-IgM antibody to surface B-cell receptor. In contrast, when whole blood was incubated with PF4/heparin ULCs, there was a marked shift in fluorescent signals for complement and PF4/heparin, as well as IgM (Figure 6A-B). This increase in IgM binding is likely a result of plasma-derived antibody (as opposed to surface IgM), as addition of excess heparin reduced PF4/heparin and IgM fluorescence to baseline (Figure 6A-B).
Figure 6.

Plasma IgM colocalizes with PF4/heparin and C3 fragments on B cells in healthy donors and patients on heparin therapy. (A-B) Whole blood from a representative healthy donor was incubated with buffer or PF4 (25 μg/mL) ± heparin (0.25 U/mL) and binding of C3c, PF4/heparin, and IgM on B cells was determined by flow cytometry. Binding of C3c/anti-PF4/heparin (KKO)/IgM to B cells is shown with and without PF4 ± heparin by histogram overlays in normal and excess heparin wash conditions (A) and as mean fluorescent intensity (B). Results are shown from a representative experiment involving 3 donors tested on 3 different occasions. (C-D) Binding of C3c, PF4/heparin, and IgM on B cells in the circulation of heparinized patients was determined by flow cytometry. Binding of C3c/anti-PF4/heparin (KKO)/IgM to B cells is shown pre- and postheparin exposure in the patient by overlay histograms (C) and as mean fluorescence intensity (D). Results are shown from 1 representative patient out of 3 patients studied. *P < .005; **P < .0001.
To explore the clinical relevance of these observations, we examined B cells from patients for colocalization of IgM with PF4/heparin before and after exposure to heparin. As shown in Figure 6C, there was no PF4/heparin, minimal C3c, and basal expression of IgM on B cells before receiving heparin (Figure 6C [magenta histogram] and D [magenta columns]). By 9 hours after initiating heparin, increased binding of PF4/heparin, complement, and IgM was seen on circulating B cells (Figure 6C [steel blue histogram] and D [steel blue columns]). Taken together, these studies show IgM colocalizes with PF4/heparin and complement fragments on circulating B cells, and increased bound IgM is likely plasma-derived.
Discussion
The HIT antigen consists of a complex between cationic PF4 and anionic heparin that generate an array of charged motifs that have similarities to key features of microbial pattern recognition molecules.37 As shown in our recent work, these properties endow the PF4/heparin ULC with robust complement activating properties.6
Here, we delineate the mechanism by which PF4/heparin complexes activate complement. We observed wide, but stable, variation in IgM levels in healthy donors that closely correlate with their complement activating responses to PF4/heparin. Our data show that polyreactive IgM binds PF4/heparin, triggers activation of the classical complement pathway, and promotes antigen and complement deposition on B cells. Natural IgM mediates this process, and complement activation by IgM can be attenuated by classical pathway inhibitors.
A major finding from our study is that natural IgM, rather than immune IgM, is likely responsible for PF4/heparin-mediated complement activation. First, plasmas from healthy donors with no prior heparin exposure activate complement in an IgM-dependent manner (Figures 1-3). In individuals with low IgM, polyclonal commercial IgM (derived from the plasma of ∼20 healthy donors), but not monoclonal IgM derived from a myeloma patient’s plasma or IgG (Figure 3A), enhances the complement activation response to PF4/heparin. Commercial IgM and individual donor IgM display broad reactivity with multiple unrelated antigens (Figure 4A; supplemental Figure 2). Moreover, a monoclonal antibody with polyreactivity (pAb2E4) to bacterial antigens (Figure 4B)31 as well as cord blood plasma (Figure 4C), which contains mostly natural IgM,34,38 activates complement in response to PF4/heparin as well. Thus, our data suggest that polyreactive IgM that develops in response to early encounters with endogenous39,40 or exogenous antigens41,42 with features of PF4/heparin-like molecules likely contribute to host immunity, allowing for rapid development of antigen-specific antibodies that mediate HIT.
Our studies may also help to reconcile seemingly disparate observations on the contributions of innate and adaptive immunity to the development of HIT antibodies. Whereas several studies, using athymic mice and mice depleted of CD4+ T cells, indicate an absolute requirement for T cells,43,44 other studies show T-cell independence. In these latter studies, mice lacking marginal zone B cells,45 B lymphocytes belonging to the innate immune system,46 were unable to develop antibodies after PF4/heparin immunization. These findings led the authors to speculate that HIT was likely T-cell independent. However, as marginal zone B cells are an important source of polyreactive IgM necessary for complement activation, antigen transport, and subsequent adaptive immunity, the requirements for marginal zone B cells are in keeping with their role in bridging innate and adaptive immunity.
The pentameric structure of circulating IgM facilitates high avidity interactions with antigen and allows IgM to have a 1000-fold greater affinity for the classical pathway component C1q compared with IgG.17 Complement activation is most robust when IgMs (either immune or nonimmune) bind to particulate antigens and undergo conformational change to initiate binding of C1q.47 Our data are consistent with these reports, as PF4/heparin complexes by virtue of its charge and size48 behave as particulate antigen, promote IgM binding, and enable complement activation and antigen deposition on B cells (Figure 6A-B). The observations that circulating B cells from heparinized patients show similar colocalization of IgM, antigen, and complement (Figure 6C-D) provide not only valuable clinical confirmation of in vitro data but also validate this IgM-mediated pathway as an important mechanism of immune activation in HIT and suggest plasma IgM levels may provide a biomarker for the risk for seroconversion.
Our studies also identify IgM and the classical pathway as potential diagnostic and/or therapeutic targets in HIT. In healthy donors, donor phenotypes of complement activation, which correlate with IgM levels (Figure 2A), remain stable over time (Figure 1B). In addition, studies shown in Figure 3C indicate a threshold effect for IgM not seen with PF4/heparin. Although additional studies are needed to establish the stability of IgM levels over time both in healthy donors and in patients experiencing infection or inflammation, measurement of IgM at time of heparin exposure may identify patients at low or high risk for sensitization. Last, we show that the classical pathway can be a therapeutic target in HIT. Disruption of IgM-C1q interactions prevent PF4/heparin-mediated complement activation, whereas inhibition of the alternative pathway or MBL activity has no effect (Figure 5). Targeted inhibitors of C1 q/r/s complex such as anti-C1s therapy or broader complement targets such as Cp40,49 a peptide inhibitor of C3, could potentially be used to prevent HIT seroconversions.
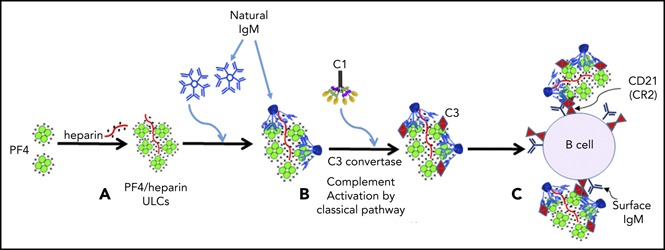
In conclusion, our studies support the following model of complement activation by PF4/heparin complexes. Under physiological conditions, circulating PF4, IgM, and C1 do not associate. Once heparin is administered at concentrations that generate PF4/heparin ULCs in the form of particulate antigen, preexisting IgM binds to PF4/heparin ULCs and undergoes a conformational change (Figure 7A) that initiates binding of C1q followed by activation of the C1 complex. Activation of the classical pathway culminates in activation of the C3 convertase, incorporation of the C3 fragments onto PF4/heparin ULCs (Figure 7B), and subsequent binding of IgM/C3-coated antigen to B cells via complement receptor 2 (Figure 7C). Prospective studies in patients receiving heparin therapy will be necessary to define the threshold amounts of IgM and/or PF4/heparin necessary to initiate complement activation and validate the relevance of this mechanism for subsequent HIT antibody formation.
Figure 7.

Mechanism of complement activation by PF4/heparin complexes. (A) Heparin displaces PF4 to form ULCs. (B) Polyreactive natural IgM from plasma binds to ULCs, changes conformation and binds C1q to activate the classical pathway of complement activation. (C) IgM and complement-coated antigen binds to B cells via complement receptor 2.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank the Duke Proteomics and Metabolomics Shared Resource for performing proteomic studies, Duke Cancer Center Flow facility for use of shared flow cytometer instruments, and the Duke University Hospital Clinical Immunology Laboratory for measuring serum immunoglobulins. The authors thank Garnett Kelsoe for scientific input.
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grants P01HL110860 (L.R., M.P., D.B.C., and G.M.A.), R01HL136512 (G.M.A.), R01HL139448 (M.P. and D.B.C.), R01HL142122 (D.B.C.), R01HL128895 (D.B.C.), P01HL040387 (D.B.C.), and K08HL127183 (G.M.L.); American Heart Association grant 17GRNT33460445 (L.R.); and Foundation for Society for Maternal-Fetal Medicine/American Association of Obstetricians and Gynecologists Foundation Research Training Scholarship (J.B.G.)
Footnotes
Data from this manuscript were presented in abstract form at the 59th annual meeting of the American Society of Hematology, Atlanta, GA, 11 December 2017, the 58th annual meeting of the American Society of Hematology, San Diego, CA, 3 December 2016, as well as the Thrombosis and Hemostasis Societies of North America 2018 Annual Meeting, San Diego, CA, 10 March 2018.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: S.K. and G.M.A. provided conception and design; S.K., J.R., L.R., A.J., G.M.L., J.B.G., S.G., A.L.N., M.P., D.B.C., and G.M.A. provided study materials or patients; S.K., J.R., and A.J. provided collection and assembly of data; S.K., J.R., L.R., A.J., G.M.L., M.K., M.F., M.P., D.B.C., and G.M.A. provided data analysis and interpretation; S.K., M.F., M.P., D.B.C., and G.M.A. provided manuscript writing; and S.K., J.R., L.R., A.J., G.M.L., J.B.G., S.G., A.L.N., M.K., M.F., M.P., D.B.C., and G.M.A. provided final approval of manuscript.
Conflict-of-interest disclosure: G.M.A. has an awarded patent for KKO (US Application NO 60/143,536); G.M.A., M.P., and D.B.C. have pending intellectual property applications. The remaining authors declare no competing financial interests.
Correspondence: Gowthami M. Arepally, Division of Hematology, DUMC Box 3486, Room 356 A, Alex H. Sands Building, Research Dr, Durham, NC 27710; e-mail: arepa001@mc.duke.edu.
REFERENCES
- 1.Trossaërt M, Gaillard A, Commin PL, Amiral J, Vissac AM, Fressinaud E. High incidence of anti-heparin/platelet factor 4 antibodies after cardiopulmonary bypass surgery. Br J Haematol. 1998;101(4):653-655. [DOI] [PubMed] [Google Scholar]
- 2.Pouplard C, May MA, Iochmann S, et al. . Antibodies to platelet factor 4-heparin after cardiopulmonary bypass in patients anticoagulated with unfractionated heparin or a low-molecular-weight heparin: clinical implications for heparin-induced thrombocytopenia. Circulation. 1999;99(19):2530-2536. [DOI] [PubMed] [Google Scholar]
- 3.Lee GM, Welsby IJ, Phillips-Bute B, Ortel TL, Arepally GM. High incidence of antibodies to protamine and protamine/heparin complexes in patients undergoing cardiopulmonary bypass. Blood. 2013;121(15):2828-2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bakchoul T, Zöllner H, Amiral J, et al. . Anti-protamine-heparin antibodies: incidence, clinical relevance, and pathogenesis. Blood. 2013;121(15):2821-2827. [DOI] [PubMed] [Google Scholar]
- 5.Pouplard C, Leroux D, Rollin J, Amiral J, May MA, Gruel Y. Incidence of antibodies to protamine sulfate/heparin complexes incardiac surgery patients and impact on platelet activation and clinical outcome. Thromb Haemost. 2013;109(6):1141-1147. [DOI] [PubMed] [Google Scholar]
- 6.Khandelwal S, Lee GM, Hester CG, et al. . The antigenic complex in HIT binds to B cells via complement and complement receptor 2 (CD21). Blood. 2016;128(14):1789-1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dempsey PW, Allison ME, Akkaraju S, Goodnow CC, Fearon DT. C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science. 1996;271(5247):348-350. [DOI] [PubMed] [Google Scholar]
- 8.Leonard CG, Thorne CB. Studies on the non-specific precipitation of basic serum proteins with gamma-glutamyl polypeptides. J Immunol. 1961;87:175-188. [PubMed] [Google Scholar]
- 9.Willoughby WF, Ford RT, Shin HS. Complement activation by lysozyme-DNA complexes. J Immunol. 1973;111:296-297. [Google Scholar]
- 10.Rent R, Ertel N, Eisenstein R, Gewurz H. Complement activation by interaction of polyanions and polycations. I. Heparin-protamine induced consumption of complement. J Immunol. 1975;114(1 Pt 1):120-124. [PubMed] [Google Scholar]
- 11.Loos M, Bitter-Suermann D. Mode of interaction of different polyanions with the first (C1,C1) the second (C2) and the fourth (C4) component of complement. IV. Activation of C1 in serum by polyanions. Immunology. 1976;31(6):931-934. [PMC free article] [PubMed] [Google Scholar]
- 12.Gewurz H. Nonimmune activation of C: two new phenomena. J Immunol. 1973;111(1):305. [Google Scholar]
- 13.Claus DR, Siegel J, Petras K, Skor D, Osmand AP, Gewurz H. Complement activation by interaction of polyanions and polycations. III. Complement activation by interaction of multiple polyanious and polycations is the presence of C-reactive protein. J Immunol. 1977;118(1):83-87. [PubMed] [Google Scholar]
- 14.Bruins P, te Velthuis H, Eerenberg-Belmer AJ, et al. . Heparin-protamine complexes and C-reactive protein induce activation of the classical complement pathway: studies in patients undergoing cardiac surgery and in vitro. Thromb Haemost. 2000;84(2):237-243. [PubMed] [Google Scholar]
- 15.Zheng Y, Wang AW, Yu M, et al. . B-cell tolerance regulates production of antibodies causing heparin-induced thrombocytopenia. Blood. 2014;123(6):931-934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krauel K, Schulze A, Jouni R, et al. . Further insights into the anti-PF4/heparin IgM immune response. Thromb Haemost. 2016;115(4):752-761. [DOI] [PubMed] [Google Scholar]
- 17.Ehrenstein MR, Notley CA. The importance of natural IgM: scavenger, protector and regulator. Nat Rev Immunol. 2010;10(11):778-786. [DOI] [PubMed] [Google Scholar]
- 18.Baumgarth N, Tung JW, Herzenberg LA. Inherent specificities in natural antibodies: a key to immune defense against pathogen invasion. Springer Semin Immunopathol. 2005;26(4):347-362. [DOI] [PubMed] [Google Scholar]
- 19.Haury M, Sundblad A, Grandien A, Barreau C, Coutinho A, Nobrega A. The repertoire of serum IgM in normal mice is largely independent of external antigenic contact. Eur J Immunol. 1997;27(6):1557-1563. [DOI] [PubMed] [Google Scholar]
- 20.Boes M. Role of natural and immune IgM antibodies in immune responses. Mol Immunol. 2000;37(18):1141-1149. [DOI] [PubMed] [Google Scholar]
- 21.Link A, Zabel F, Schnetzler Y, Titz A, Brombacher F, Bachmann MF. Innate immunity mediates follicular transport of particulate but not soluble protein antigen. J Immunol. 2012;188(8):3724-3733. [DOI] [PubMed] [Google Scholar]
- 22.Boes M, Esau C, Fischer MB, Schmidt T, Carroll M, Chen J. Enhanced B-1 cell development, but impaired IgG antibody responses in mice deficient in secreted IgM. J Immunol. 1998;160(10):4776-4787. [PubMed] [Google Scholar]
- 23.Ochsenbein AF, Fehr T, Lutz C, et al. . Control of early viral and bacterial distribution and disease by natural antibodies. Science. 1999;286(5447):2156-2159. [DOI] [PubMed] [Google Scholar]
- 24.Cutler AJ, Botto M, van Essen D, et al. . T cell-dependent immune response in C1q-deficient mice: defective interferon gamma production by antigen-specific T cells. J Exp Med. 1998;187(11):1789-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fischer MB, Ma M, Goerg S, et al. . Regulation of the B cell response to T-dependent antigens by classical pathway complement. J Immunol. 1996;157(2):549-556. [PubMed] [Google Scholar]
- 26.Ahearn JM, Fischer MB, Croix D, et al. . Disruption of the Cr2 locus results in a reduction in B-1a cells and in an impaired B cell response to T-dependent antigen. Immunity. 1996;4(3):251-262. [DOI] [PubMed] [Google Scholar]
- 27.Rauova L, Hirsch JD, Greene TK, et al. . Monocyte-bound PF4 in the pathogenesis of heparin-induced thrombocytopenia. Blood. 2010;116(23):5021-5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arepally GM, Kamei S, Park KS, et al. . Characterization of a murine monoclonal antibody that mimics heparin-induced thrombocytopenia antibodies. Blood. 2000;95(5):1533-1540. [PubMed] [Google Scholar]
- 29.Lee GM, Joglekar M, Kuchibhatla M, et al. . Serologic characterization of anti-protamine/heparin and anti-PF4/heparin antibodies. Blood Adv. 2017;1(11):644-651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khandelwal S, Johnson AM, Liu J, et al. . Novel immunoassay for complement activation by PF4/heparin complexes. Thromb Haemost. 2018;118(8):1484-1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou Z-H, Zhang Y, Hu Y-F, Wahl LM, Cisar JO, Notkins AL. The broad antibacterial activity of the natural antibody repertoire is due to polyreactive antibodies. Cell Host Microbe. 2007;1(1):51-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suh JS, Aster RH, Visentin GP. Antibodies from patients with heparin-induced thrombocytopenia/thrombosis recognize different epitopes on heparin: platelet factor 4. Blood. 1998;91(3):916-922. [PubMed] [Google Scholar]
- 33.Reidel B, Thompson JW, Farsiu S, Moseley MA, Skiba NP, Arshavsky VY. Proteomic profiling of a layered tissue reveals unique glycolytic specializations of photoreceptor cells. Mol. Cell. Proteomics. 2011;10(3):M110.002469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grönwall C, Silverman GJ. Natural IgM: beneficial autoantibodies for the control of inflammatory and autoimmune disease. J Clin Immunol. 2014;34(S1 Suppl 1):S12-S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fine DP, Marney SR Jr, Colley DG, Sergent JS, Des Prez RM. C3 shunt activation in human serum chelated with EGTA. J Immunol. 1972;109(4):807-809. [PubMed] [Google Scholar]
- 36.Youd ME, Ferguson AR, Corley RB. Synergistic roles of IgM and complement in antigen trapping and follicular localization. Eur J Immunol. 2002;32(8):2328-2337. [DOI] [PubMed] [Google Scholar]
- 37.Krarup A, Mitchell DA, Sim RB. Recognition of acetylated oligosaccharides by human L-ficolin. Immunol Lett. 2008;118(2):152-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mouthon L, Lacroix-Desmazes S, Nobrega A, Barreau C, Coutinho A, Kazatchkine MD. The self-reactive antibody repertoire of normal human serum IgM is acquired in early childhood and remains conserved throughout life. Scand J Immunol. 1996;44(3):243-251. [DOI] [PubMed] [Google Scholar]
- 39.Guilbert B, Dighiero G, Avrameas S. Naturally occurring antibodies against nine common antigens in human sera. I. Detection, isolation and characterization. J Immunol. 1982;128(6):2779-2787. [PubMed] [Google Scholar]
- 40.Thornton BP, Vĕtvicka V, Ross GD. Natural antibody and complement-mediated antigen processing and presentation by B lymphocytes. J Immunol. 1994;152(4):1727-1737. [PubMed] [Google Scholar]
- 41.Krauel K, Pötschke C, Weber C, et al. . Platelet factor 4 binds to bacteria, [corrected] inducing antibodies cross-reacting with the major antigen in heparin-induced thrombocytopenia. Blood. 2011;117(4):1370-1378. [DOI] [PubMed] [Google Scholar]
- 42.Krauel K, Weber C, Brandt S, et al. . Platelet factor 4 binding to lipid A of gram-negative bacteria exposes PF4/heparin-like epitopes. Blood. 2012;120(16):3345-3352. [DOI] [PubMed] [Google Scholar]
- 43.Suvarna S, Rauova L, McCracken EK, et al. . PF4/heparin complexes are T cell-dependent antigens. Blood. 2005;106(3):929-931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zheng Y, Yu M, Padmanabhan A, et al. . Critical role of CD4 T cells in PF4/heparin antibody production in mice. Blood. 2015;125(11):1826-1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zheng Y, Yu M, Podd A, et al. . Critical role for mouse marginal zone B cells in PF4/heparin antibody production. Blood. 2013;121(17):3484-3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cerutti A, Cols M, Puga I. Marginal zone B cells: virtues of innate-like antibody-producing lymphocytes. Nat Rev Immunol. 2013;13(2):118-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Czajkowsky DM, Shao Z. The human IgM pentamer is a mushroom-shaped molecule with a flexural bias. Proc Natl Acad Sci USA. 2009;106(35):14960-14965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Suvarna S, Espinasse B, Qi R, et al. . Determinants of PF4/heparin immunogenicity. Blood. 2007;110(13):4253-4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Y, Shao D, Ricklin D, et al. . Compstatin analog Cp40 inhibits complement dysregulation in vitro in C3 glomerulopathy. Immunobiology. 2015;220(8):993-998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.