

Abstract
Background: The tumor microenvironment has recently emerged as a new target of anticancer chemotherapy. Selective activation of anticancer chemotherapy in the tumor microenvironment would further reduce the toxicity of anticancer drugs toward normal tissues. Fibroblast activation protein (FAP) is known to be selectively overexpressed on cancer-associated fibroblasts (CAFs) in the tumor microenvironment. Here, we designed an anticancer chemotherapeutic system based on promelittin, a peptide toxin that is selectively converted from an inactive form to the pore-forming melittin upon cleavage by FAP in the tumor microenvironment.
Methods: We conjugated promelittin-containing FAP-cleavable sequences to pegylated phospholipids and anchored them to reduced graphene oxide (rGO) nanosheets. The resulting nanosheets, PL-rGO, were tested for hemolysis and used for doxorubicin delivery. In vitro cocultures and in vivo tumor growth (n = 5 mice per group) with tissue immunostaining were used to test the selective activation of anticancer chemotherapy by FAP expressed on CAFs.
Results: FAP-specific hemolytic activity of PL-rGO was observed in cocultures of CAFs and HT29 cells but not in HT29 cells alone. Doxorubicin-loaded PL-rGO (Dox/PL-rGO) showed 3.4-fold greater cell-killing efficacy (compared with free Dox in the CAF/HT29 coculture system, effects that were not observed in HT29 cells alone). Intravenously administered Dox/PL-rGO reduced the growth of HT29 tumors more effectively than other treatments (Dox/PL-rGO: mean = 200.6 mm3, 95% confidence interval [CI] = 148.7 to 252.5 mm3; free Dox: mean = 697.0 mm3, 95% CI = 646.9 to 747.1 mm3, PL: mean = 565.0 mm3, 95% CI = 550.5 to 579.6 mm3; Dox/rGO: mean = 637.6 mm3, 95% CI = 619.5 to 655.7 mm3; PL-rGO: mean = 464.4 mm3, 95% CI = 433.0 to 495.8 mm3). Immunostaining of tumor tissues revealed that survival of CAFs and HT29 cells was lowest in the group treated with Dox/PL-rGO.
Conclusions: The demonstration of selective activation of PL-rGO by FAP on CAFs suggests that PL-rGO may serve as a tumor microenvironment–responsive anticancer chemotherapy system.
The tumor microenvironment has recently emerged as a new target of anticancer chemotherapy. This microenvironment—a complex system composed of many cell types, including fibroblasts, endothelial cells, and immune cells (1)—has been reported to contribute to cancer progression and dissemination (2). Accordingly, altering the tumor microenvironment could be an efficient strategy for isolating and destroying cancer cells and disrupting the crosstalk between cancer cells and their microenvironment.
Among various cells in the tumor microenvironment, fibroblasts have received particular attention for microenvironment-targeted anticancer therapy. It has been reported that fibroblasts in the tumor microenvironment, termed cancer-associated fibroblasts (CAFs), are different from those in normal tissues. CAFs, which are known to promote tumor growth and invasion (3,4), are frequently observed in various human cancers, including breast (5), lung (6), pancreatic (7), and colon cancers (8).
Fibroblast activation protein (FAP) has been reported to be selectively overexpressed on the membranes of CAFs. FAP, a type II integral membrane glycoprotein, is a serine protease of the dipeptidyl peptidase IV family that is uniquely capable of cleaving the Pro-Xxx amino acid bond (9). Although FAP is not detectable on most normal fibroblasts, it is observed on CAFs in more than 90% of breast (5), lung (6), and colon carcinomas (8). This CAF-selective overexpression of FAP can be exploited in the design of a tumor microenvironment–responsive nanotherapeutic agent that is only active after proteolytic degradation by FAP.
Melittin, a water-soluble, 26-amino-acid amphipathic peptide derived from the venom of the honeybee Apis mellifera, is a nonspecific cytolytic peptide that attacks all lipid membranes. Because it is nonspecific, systemic administration of melittin results in toxicity, limiting its therapeutic applicability (10). This limitation could be circumvented by activating melittin selectively in tumor microenvironments, for example, by FAP, an approach that would require a suitable peptide engineering and delivery system.
Reduced graphene oxide (rGO) nanosheets have received considerable attention for their potential to deliver various anticancer drugs (11–13) and imaging agents (14) to tumor tissues. Thanks to their planar regions, the surfaces of rGO nanosheets are readily loaded with drugs and various functional surface–coating materials, such as phospholipid (15) and cholesterol (13) derivatives.
In this study, we sought to design a tumor microenvironment–activated nanomedicine with enhanced anticancer therapeutic efficacy. To take advantage of the selective overexpression of FAP on CAFs, we designed a FAP-activated promelittin lipid derivative (PL) and loaded it onto rGO nanosheets containing doxorubicin (Dox). Here, we tested whether the FAP-specific cleavage of PL by CAFs could activate the anticancer effect of Dox in the tumor microenvironment upon systemic administration of Dox-containing PL-rGO nanosheets.
METHODS
Preparation of PL-rGO Nanosheets
The surface of rGO nanosheets was coated with PL by mixing rGO nanosheets (1 mg/mL) in triple-distilled water (TDW) with an equivalent volume of PL solution (2 mg/mL) at a PL:rGO weight ratio of 2:1. In some experiments, Dox was loaded onto plain rGO or PL-rGO nanosheets. Dox-loaded rGO (Dox/rGO) and Dox-loaded PL-rGO (Dox/PL-rGO) were prepared by adding 1 mL of Dox solution (0.1 mg/mL; Sigma-Aldrich) to 1 mL of rGO or PL-rGO nanosheets, respectively, in TDW and incubating for 10 minutes at room temperature. Unloaded free Dox was then removed using a PD-10 desalting column (GE Healthcare, Piscataway, NJ).
Hemolysis Assay
HT29 cells (1 × 106 cells), isolated CAFs (1 × 106 cells), or HT29 cells cocultured with CAFs (2 × 106 cells total) were incubated with 10 μg of PL, 1 μg of purified FAP (R&D Systems Inc., Minneapolis, MN), and purified matrix metalloproteinase 9 (MMP9; R&D Systems Inc.) in the presence of mouse red blood cells (mRBCs; 2×104 cells/mL) for one hour at 37 °C. mRBCs suspended in PBS alone were used as a zero hemolysis control, and mRBCs in the presence of 1% Triton X-100 (USB Corp., Cleveland, OH) served as the 100% hemolysis control. After centrifugation at 1000 × g for 10 minutes, supernatants containing nonlysed mRBCs were transferred, in triplicate, to a clear, flat-bottomed, 96-well polystyrene plate (SPL Life Sciences). The degree of hemolysis was assessed by measuring the absorbance of samples at 540 nm using a Molecular Devices Spectra Max Plus automatic plate reader.
In Vitro Antitumor Efficacy Study Of Dox/PL-Rgo
The in vitro antitumor efficacy of Dox delivered on rGO nanosheets was tested using cell viability assays. HT29 cells (3 × 104 cells/well) or HT29 cells plus CAFs, treated with or without siFAP (9 × 104 cells/well), were seeded onto 24-well plates and then treated the next day with PL-rGO or Dox/PL-rGO at a concentration of 12.5 μg/mL rGO and 1 μM Dox. After removing the Dox-containing culture medium, rGO or PL-rGO (rGO concentration, 12.5 μg/mL) was added to the cells. After incubating for three hours, culture media were replaced with fresh media and cells were maintained for 24 hours. Cell viability was measured using a Cell Counting Kit-8 (CCK-8; Dojindo, Tokyo, Japan) or a live cell–staining assay (LIVE/DEAD Viability Assay; Molecular Probes, Eugene, OR).
Assessment of In Vivo Antitumor Activity
The in vivo antitumor efficacy of Dox/PL-rGO was tested using HT29 tumor–bearing mice. Five-week-old female athymic nude mice were subcutaneously injected in the dorsal right side with 5× 106 HT29 cells. When tumor volumes reached 120 mm3, mice were intravenously administered Dox (1 mg/kg), PL (10 mg/kg), Dox/rGO (1 mg/kg Dox, rGO 5 mg/kg), PL-rGO (10 mg/kg PL, 5 mg/kg rGO), or Dox/PL-rGO (1 mg/kg Dox, 10 mg/kg PL, 5 mg/kg rGO) every other day for a total of three injections. Twenty-four days after tumor inoculation, tumor tissues were fixed in 10% formalin and embedded in paraffin blocks for immunofluorescence staining. Antitumor effects were determined by immunostaining tumor tissue sections (4 μm thick) with an anti-FAP or anti-CD133 antibody.
Statistical Analysis
Two-sided analysis of variance (ANOVA), with Student-Newman-Keuls post hoc tests, was used for the statistical evaluation of experimental data. A P value of less than .05 was considered statistically significant. All error bars in the figures represent 95% confidence interval (CIs). All statistical analyses were done using SPSS software (version 23; Chicago, IL).
RESULTS
Design and Characterization of Dox/PL-Rgo Nanosheets
Surfaces of rGO nanosheets were coated with PL and loaded with Dox, as depicted in Figure 1A. A possible working mechanism of Dox/PL-rGO is illustrated in Figure 1B. The promelittin moiety of Dox/PL-rGO nanosheets has a FAP-cleavable peptide sequence that is activated by FAP protease, which is overexpressed on CAFs in the tumor microenvironment, liberating the pore-forming peptide melittin. The formation of pores in tumor cells and cells of the tumor microenvironment by diffused melittin is predicted to increase the uptake of Dox/PL-rGO and enhance antitumor efficacy.
Figure 1.

Nanostructure of doxorubicin-loaded promelittin lipid derivative–reduced graphene oxide (Dox/PL-rGO) nanosheets and a schematic depiction of the hypothesized mechanism underlying their action. A) The promelittin lipid derivative, PL, containing a fibroblast activation protein (FAP)–cleavable sequence, was anchored onto rGO nanosheets. The resulting PL-rGO was further loaded with Dox, yielding Dox/PL-rGO. B) Schematic depiction of the presumed mechanism of Dox/PL-rGO. Activation of the promelittin moiety of PL-rGO by FAP overexpressed on cancer-associated fibroblasts releases melittin. Diffusion of melittin to surrounding tumor cells and cells in the tumor microenvironment promotes formation of pores in the membrane, increasing the cellular uptake of Dox-loaded rGO nanosheets and enhancing anticancer efficacy. CAFs = cancer-associated fibroblasts; Dox = doxorubicin; FAP = fibroblast activation protein; PL = promelittin lipid derivative; rGO = reduced graphene oxide.
Anchoring of PL and subsequent loading of Dox onto rGO nanosheets did not statistically significantly affect the mean size of rGO nanosheets (PL-rGO: mean = 94.6 nm, 95% CI = 93.3 to 95.9 nm, P = .80; Dox/PL-rGO: mean = 91.2 nm, 95% CI = 88.7 to 93.8 nm, P = .25) (Figure 2A). Zeta potential values, on the other hand, decreased upon surface coating of PL (mean = 9.4 mV, 95% CI = 5.6 to 13.2 mV, P = .001), but increased after loading of cationic Dox onto PL-rGO nanosheets (mean = 18.5 mV, 95% CI = 16.3 to 20.7 mV, P = .02) (Figure 2B). The loading efficiencies of PL onto rGO were measured by phosphate assay (16). The loading efficiencies of PL onto rGO statistically significantly (P < .001) increased as the weight ratios of PL to rGO decreased from 20:1 to 2:1 (Supplementary Figure 1, available online). There were no statistically significant differences (P = .65) in loading efficiencies of PL onto rGO between the ratios of 2:1 (mean = 95.3%, 95% CI = 92.5% to 98.1%) and 1:1 (mean = 94.5%, 95% CI = 92.8% to 96.3%). Thus, a PL-rGO weight ratio of 2:1 was chosen for further experiments in this study.
Figure 2.

Characterization of doxorubicin-loaded promelittin lipid derivative–reduced graphene oxide (Dox/PL-rGO) nanosheets. A) The sizes of rGO, PL-rGO, and Dox/PL-rGO nanosheets were measured by dynamic light scattering. Error bars represent 95% confidence intervals (CIs). B) Zeta potential values for rGO, PL-rGO, and Dox/PL-rGO nanosheets were determined by laser Doppler microelectrophoresis at an angle of 22° using an ELS-8000 instrument (*P < .05; †P < .01). Error bars represent 95% CIs. Two-sided analysis of variance, along with Student-Newman-Keuls post hoc tests, was used for statistical analysis. C) Adsorption of Dox onto rGO or PL-rGO nanosheets was analyzed at various time points by fluorometry. D) The loading efficiencies of Dox on rGO or PL-rGO were determined by measuring the quenching of Dox upon adsorption to rGO nanosheets. Error bars represent 95% CIs. Dox = doxorubicin; PL = promelittin lipid derivative; rGO = reduced graphene oxide.
The adsorption of Dox onto rGO or PL-rGO nanosheets (Dox:rGO weight ratio of 5:1) was complete within one minute (Figure 2C). The fluorescence intensity of Dox incorporated into rGO nanosheets decreased to 13% of its original intensity at 60 seconds after physical mixing. Similarly, the fluorescence intensity of Dox incorporated into PL-rGO nanosheets rapidly decreased to 2% of its original intensity at 60 seconds after physical mixing. When Dox was loaded onto rGO or PL-rGO at a Dox:rGO weight ratio of 5:1 for 10 minutes, mean values of Dox loading efficiencies were 96.3% (95% CI = 95.6% to 97.0%) and 97.5% (95% CI = 96.2% to 98.4%), respectively (Figure 2D).
FAP Expression-Dependent Hemolytic Activity of PL-Rgo Nanosheets
The expression levels of FAP differed between HT29 cells (Figure 3A) and CAFs (17) (Figure 3B). Flow cytometry revealed that 90.3% of CAFs were positive for FAP expression (Figure 3B), whereas only 5.1% of HT29 cells were FAP-positive (Figure 3A). To test whether the promelittin moiety of PL or PL-rGO could be selectively cleaved to pore-forming melittin by exogenous FAP or by FAP expressed on CAFs, we used a mouse red blood cell (mRBC) hemolysis assay. mRBCs were lysed by PL (Figure 3C) and PL-rGO (Figure 3E) after treatment with FAP, but not after treatment with matrix metalloproteinase (MMP)-9. Compared with untreated cells or MMP9-treated cells, FAP treatment induced statistically significantly greater hemolysis of mRBCs by PL (P < .001) (Figure 3C) and PL-rGO (P < .001) (Figure 3E). Notably, mRBC lysis by PL (P < .001) (Figure 3D) and PL-rGO (P < .001) (Figure 3F) was observed in a coculture system of CAFs and HT29 cells, but not with HT29 cells alone.
Figure 3.
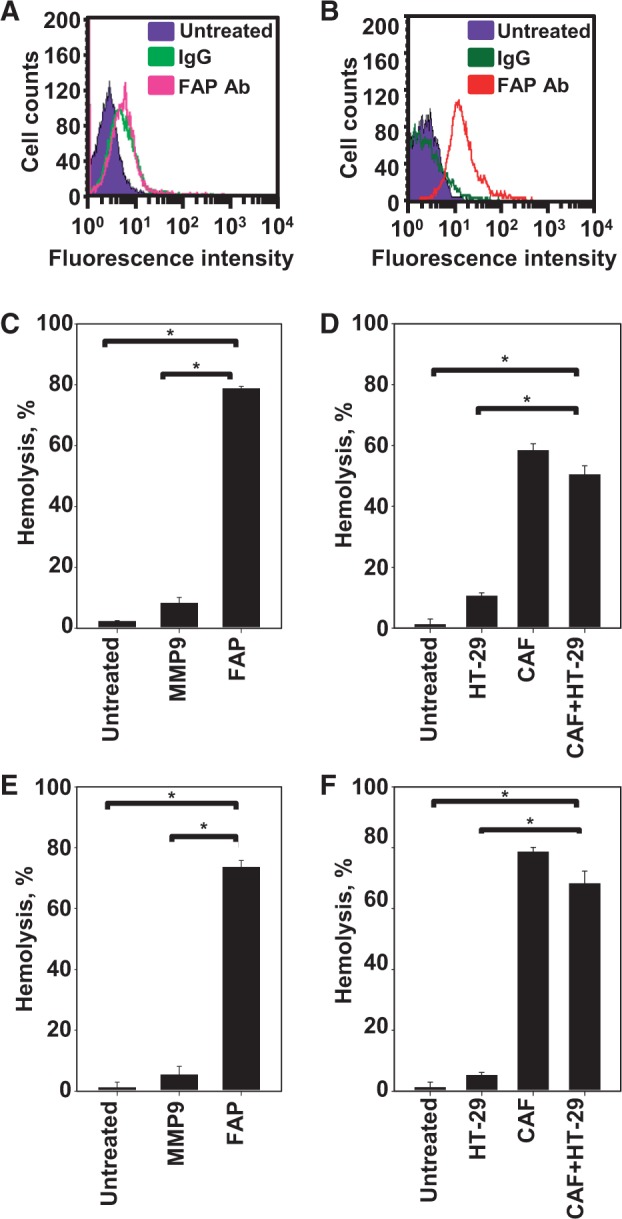
Fibroblast activation protein (FAP) expression and hemolysis of mRBCs. HT29 cells (A) and cancer-associated fibroblasts (CAFs) (B) were stained with an anti-FAP antibody. Fluorescence-positive cells were measured by flow cytometry. Promelittin lipid derivative (PL) (C) and PL–reduced graphene oxide (rGO) (E) were treated with MMP9 or FAP. HT29 cells alone or cocultured with CAFs were treated with PL (D) or PL-rGO (F). The hemolytic activity of PL (C and D) and PL-rGO (E and F) against mouse red blood cells was quantified by hemolysis assay (n = 3; *P < .001). Error bars represent 95% confidence intervals. Two-sided analysis of variance, along with Student-Newman-Keuls post hoc tests, was used for statistical analysis. CAFs = cancer-associated fibroblasts; FAP = fibroblast activation protein; IgG = immunoglobulin G; MMP9 = matrix metallopeptidase 9.
Cellular Uptake of PL-Rgo Nanosheets in the Presence of FAP-Positive CAFs
To demonstrate enhanced cellular uptake of PL-rGO, we first established FAP knockdown CAFs by transfection with siFAP (18). Treatment of CAFs with siFAP decreased the mRNA levels of FAP (mean = 30.9%, 95% CI = 27.1% to 34.8%, P < .001) (Supplementary Figure 2A, available online) compared with control levels. Immunoblot showed a reduction in FAP protein expression (Supplementary Figure 2B, available online). By contrast, treatment of CAFs with siGFP (control) did not decrease FAP mRNA or protein. In monocultures of CAFs or HT29 cells, cellular uptake of PL-rGO nanosheets was selectively enhanced in FAP-positive CAFs but not in FAP-negative HT29 cells (Figure 4A) or FAP knockdown CAFs (Figure 4B), in which uptake of fluorescent dye-labeled PL-rGO was not different from that of fluorescent dye-labeled rGO nanosheets. In contrast, cellular uptake of fluorescent dye-labeled PL-rGO was greater than that of fluorescent dye-labeled rGO in HT29 cells cocultured with FAP-positive CAFs (Figure 4C).
Figure 4.
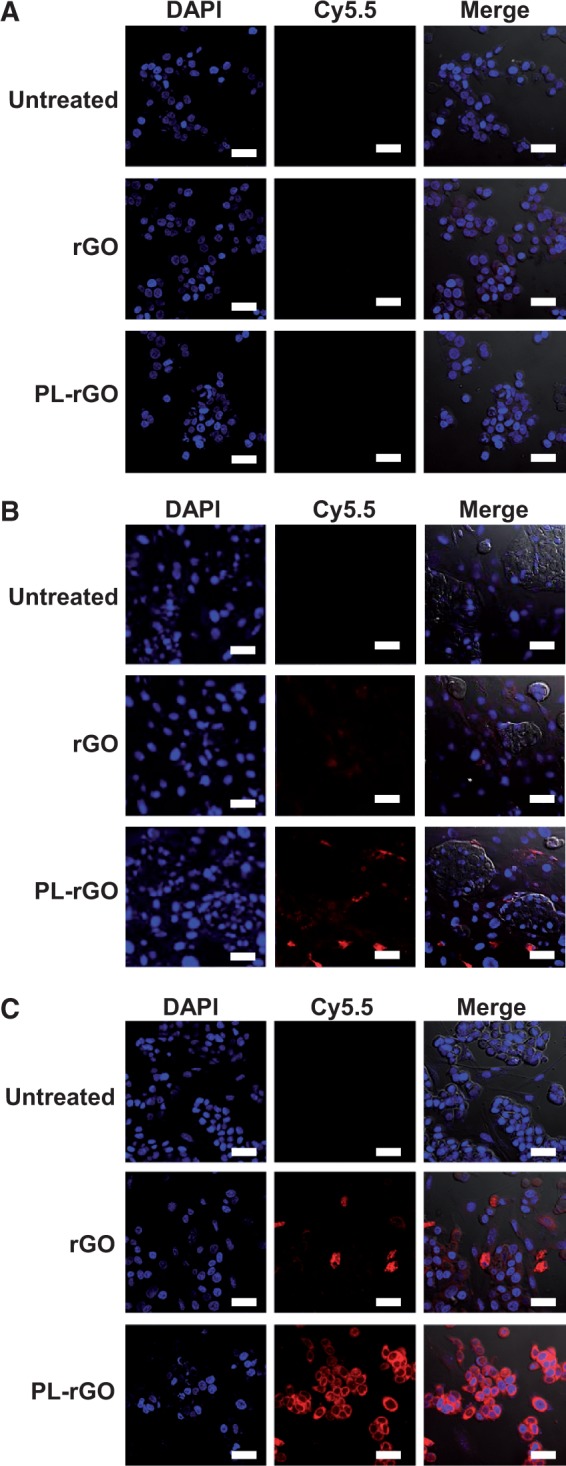
Uptake of promelittin lipid derivative (PL)–reduced graphene oxide (rGO) nanosheets in fibroblast activation protein (FAP)–positive, FAP-negative, and FAP knockdown cells. A) HT29 cells were left untreated or were treated with fluorescent lipid dye-labeled rGO or PL-rGO. B) Cocultured FAP knockdown cancer-associated fibroblasts (CAFs) and HT29 cells were left untreated or were treated with fluorescent lipid dye-labeled rGO or PL-rGO. C) Cocultured CAFs and HT29 cells were left untreated or were treated with fluorescent lipid dye-labeled rGO or PL-rGO. After incubation for one hour, cellular fluorescence was observed by confocal microscopy. Scale bar = 20 μm. PL = promelittin lipid derivative; rGO = reduced graphene oxide.
In Vitro Anticancer Efficacy of Dox/PL-Rgo Nanosheets
We tested whether the increased uptake of PL-rGO translates to enhanced efficacy of Dox-loaded PL-rGO in vitro. The anticancer activity of Dox/PL-rGO was enhanced in the CAF/HT29 cell coculture system but not in FAP-negative HT29 cells (Figure 5A), where anticancer activity did not statistically significantly differ among all groups tested (Dox: mean = 90.5%, 95% CI = 88.2% to 92.8%, P = .91; PL: mean = 93.8%, 95% CI = 93.1% to 94.5%, P = .81; Dox + rGO: mean = 91.5%, 95% CI = 90.3% to 92.6%, P = .98; Dox/rGO: mean = 93.9%, 95% CI = 91.3% to 96.6%, P = .85; PL-rGO: mean = 91.3%, 95% CI = 90.5% to 92.1%, P = .92; Dox + PL-rGO: mean = 89.7%, 95% CI = 84.8% to 94.6%, P = .86). The anticancer activity of Dox/PL-rGO was 2.7- and 1.5-fold greater in CAF/HT29 cell cocultures than in HT29 cells and FAP knockdown CAF/HT29 cells transfected with siFAP, respectively. Dox/PL-rGO (mean = 33.3%, 95% CI = 32.4% to 34.3%) exerted 3.4- and 2.1-fold greater cell-killing effect compared with free Dox (mean = 80.1%, 95% CI = 78.9% to 81.3%) and Dox/rGO (mean = 68.9%, 95% CI = 66.0% to 71.8%) in CAF/HT29 cell cocultures. Moreover, Dox/PL-rGO showed statistically significantly greater anticancer effect (P < .001) compared with sequential treatment with Dox plus PL-rGO (mean = 52.0%, 95% CI = 50.8% to 53.2%) in CAF/HT29 cell cocultures (Figure 5A). A Live/Dead fluorescence assay system revealed similar results, showing that Dox/PL-rGO exerted notable cell-killing effects selectively in cocultured CAFs and HT29 cells (Figure 5D), but not in HT29 cells alone (Figure 5B) or in FAP knockdown CAF/HT29 cells (Figure 5C).
Figure 5.

In vitro anticancer efficacy of doxorubicin (Dox) delivered using promelittin lipid derivative (PL)–reduced graphene oxide (rGO) nanosheets. A) HT29 cells alone or cocultured with fibroblast activation protein (FAP) knockdown cancer-associated fibroblasts (CAFs) or FAP-positive CAFs were treated for three hours with Dox, PL, Dox + PL/rGO, Dox/rGO, PL-rGO, Dox + PL-rGO, or Dox/PL-rGO. Untreated cells were used as a control. After further incubating for 24 hours, cell viability was measured by cell counting kit-8 assay. Error bars represent 95% confidence intervals. (*P < .01; †P < .001). Two-sided analysis of variance, along with Student-Newman-Keuls post hoc tests, was used for statistical analysis. Live HT29 cells (B), FAP-knockdown CAFs cocultured with HT29 cells (C), or FAP-positive CAFs cocultured with HT29 cells (D) were stained with calcein-AM and analyzed by fluorescence microscopy. Scale bar = 250 μm. CAF = cancer-associated fibroblast; Dox = doxorubicin; FAP = fibroblast activation protein; PL = promelittin lipid derivative; rGO = reduced graphene oxide.
Cytotoxicity of Free Dox and Dox/PL-Rgo Nanosheets in Normal Fibroblasts
The differential cytotoxicity of free Dox and Dox/PL-rGO was investigated in normal fibroblasts (19). Free Dox showed a concentration-dependent cytotoxic effect in normal fibroblasts (Supplementary Figure 3, available online). In contrast, Dox/PL-rGO nanosheets did not statistically significantly induce cytotoxicity in normal fibroblasts compared with control cells (1 μM Dox on PL-rGO: mean = 91.5%, 95% CI = 88.8% to 94.2%, P = .08; 5 μM Dox on PL-rGO: mean = 91.8%, 95% CI = 89.8% to 93.8%, P = .06; 10 μM Dox on PL-rGO: mean = 90.2%, 95% CI = 85.6% to 94.8%, P = .05). Treatment of normal fibroblasts with 10 μM free Dox decreased cell viability (mean = 67.0%, 95% CI = 60.7% to 73.3%, P < .001) compared with control cells.
In Vivo Tumor Tissue Accumulation and Penetration of PL-Rgo Nanosheets
We next evaluated the tumor distribution of DSPE-PEG5000-Cy5.5 lipid-labeled rGO and PL-rGO following in vivo administration. At 24 hours postdose (Figure 6A), the accumulation of fluorescence at tumor sites was greater in mice treated with PL-rGO than in those treated with rGO. This greater fluorescence intensity of fluorescent PL-rGO in tumor tissues was maintained at 72 hours (Figure 6B) postinjection. At this latter time point, photon count measurements revealed that the tumor retention of DSPE-PEG5000-Cy5.5-labeled PL-rGO nanosheets (mean = 12.3 x 103 photon counts, 95% CI = 9.3 to 15.3 x 103 photon counts) was three-fold higher than that of DSPE-PEG5000-Cy5.5-labeled rGO nanosheets (mean = 4.0 x 103 photon counts, 95% CI = 3.2 to 4.8 x 103 photon counts) (Figure 6B). The in vivo tumor penetration of DSPE-PEG5000-Cy5.5 lipid-labeled rGO and PL-rGO was further evaluated using eXplore Optix imaging with 3D analysis. At one day post dose, a total of eight Z-stack images were captured at a 1 mm interval from the bottom of tumor tissues treated with DSPE-PEG5000-Cy5.5 lipid-labeled rGO (Figure 6C) or DSPE-PEG5000-Cy5.5 lipid-labeled PL-rGO (Figure 6D). The ratio of fluorescent Cy5.5-positive z-stack images over total images was greater for DSPE-PEG5000-Cy5.5 lipid-labeled PL-rGO (mean = 0.9, 95% CI = 0.7 to 1.0), compared with DSPE-PEG5000-Cy5.5 lipid-labeled rGO (mean = 0.5, 95% CI = 0.3 to 0.6).
Figure 6.

Biodistribution and tumor tissue penetration of promelittin lipid derivative (PL)-reduced graphene oxide (rGO) nanosheets. HT29 tumor–bearing mice (5 per group) were intravenously injected with DSPE-PEG5000-Cy5.5 lipid-labeled rGO or PL-rGO. After 24 hours (A), the in vivo distribution of various rGO nanosheets was visualized using a molecular imaging system. B) Total photon counts of tumor sites relative to those in the untreated group were quantified 72 hours postdose using an in vivo imaging system (*P < .01; †P < .001). Error bars represent 95% confidence intervals. Two-sided analysis of variance, along with Student-Newman-Keuls post hoc tests, was used for statistical analysis. One day postdose, the tumor penetration depth of DSPE-PEG5000-Cy5.5 lipid-labeled rGO (C) or PL-rGO (D) was analyzed using eXplore Optix system software. Y-axis represents the z-axis depth (mm) from the bottom (zero value) of tumor tissues. PL = promelittin lipid derivative; rGO = reduced graphene oxide.
In Vivo Antitumor Efficacy of Dox/PL-Rgo Nanosheets
The in vivo antitumor efficacy of Dox/PL-rGO was evaluated in an HT29 tumor xenograft model. On day 24 after tumor inoculation, tumor volumes were statistically significantly smaller (P < .001) in mice treated with Dox/PL-rGO (mean = 200.6 mm3, 95% CI = 148.7 to 252.5 mm3) than in those treated with free Dox (mean = 697.0 mm3, 95% CI = 646.9 to 747.1 mm3), free PL (mean = 565.0 mm3, 95% CI = 550.5 to 579.6 mm3), Dox/rGO (mean = 637.6 mm3, 95% CI = 619.5 to 655.7 mm3), or PL-rGO (mean = 464.4 mm3, 95% CI = 433.0 to 495.8 mm3) (Figure 7A). Consistent with this, tumor weights on day 24 were the lowest in mice treated with Dox/PL-rGO (Figure 7B). The inhibition of tumor weight growth by Dox/PL-rGO (mean = 155.0 mg, 95% CI = 99.0 to 211.0 mg) was 4.4- and 3.6-fold higher than that by free Dox (mean = 685.0 mg, 95% CI = 661.0 to 709.0 mg) and Dox/rGO (mean = 555.2 mg, 95% CI = 483.2 to 627.2 mg) , respectively. Immunofluorescence staining showed that the anti-CD133-positive HT29 (Figure 7C) and anti-FAP-positive CAF (Figure 7D) cell population was lowest in tumor tissues of mice treated with Dox/PL-rGO.
Figure 7.

In vivo antitumor efficacy of doxorubicin-loaded promelittin lipid derivative–reduced graphene oxide (Dox/PL-rGO) nanosheets and immunofluorescence staining of tumor tissues. HT29 tumor–bearing mice (5 per group) were intravenously administered Dox (1 mg/kg), PL (10 mg/kg), Dox/rGO (1 mg/kg Dox, 5 mg/kg rGO), PL-rGO (10 mg/kg PL, 5 mg/kg rGO), or Dox/PL-rGO (1 mg/kg Dox, 10 mg/kg PL, 5 mg/kg rGO) every other day for a total of three injections. A) Tumor volumes were measured in each group (***P < .001). Error bars represent 95% confidence intervals (CIs). Two-sided analysis of variance (ANOVA), along with Student-Newman-Keuls post hoc tests, was used for statistical analysis. B) On day 24, tumor tissues were extracted and weighed (*P < .001). Error bars represent 95% CIs. Two-sided ANOVA, along with Student-Newman-Keuls post hoc tests, was used for statistical analysis. Immunofluorescence staining of tumor tissues extracted on day 24 after tumor inoculation was performed using anti-CD133 (C) and anti–fibroblast activation protein antibodies (D). E) Merged image. Scale bar = 250 μm. Dox = doxorubicin; FAP = fibroblast activation protein; PL = promelittin lipid derivative; rGO = reduced graphene oxide.
In Vivo Toxicity of Dox/PL-Rgo Nanosheets
The in vivo toxicity of Dox/PL-rGO nanosheets was evaluated after a single injection or three repeated injections. Compared with untreated group, there were no statistically significant alterations in blood urea nitrogen (single administration: P = .65; repeated administrations: P = .72) (Supplementary Figure 4, A and C, available online) or creatinine (single administrations: P = .10; repeated administrations: P = .90) (Supplementary Figure 4, B and D, available online) after injections of Dox/PL-rGO (1 mg/kg Dox, 10 mg/kg PL, 5 mg/kg rGO), and all hematological parameters were within the normal range (Supplementary Tables 1 and 2, available online).
Discussion
This study demonstrated that PL-rGO is activated by FAP expressed on CAFs in the tumor microenvironment and is capable of enhancing the cellular delivery and antitumor efficacy of Dox. PL, containing an FAP-cleavable peptide sequence, exerted an RBC lysis action following treatment with FAP, but not MMP. Moreover, HT29 cells cocultured with CAFs, but not HT29 cells alone, enhanced the antitumor effects of Dox/PL-rGO compared with Dox/rGO. Dox/PL-rGO was capable of reducing the growth of tumors to a greater degree than other treatments.
Dox/PL-rGO was prepared by physical adsorption of Dox and PL onto rGO nanosheets. The high planar surface area of rGO allows the adsorption of fatty acid chains and aromatic drugs, resulting in a high loading capacity (20,21). Because promelittin was conjugated to pegylated phospholipid in PL, the phospholipid portion of PL may confer binding affinity to rGO nanosheets through hydrophobic interactions (22). Dox has been reported to bind rGO nanosheets via π-π stacking interactions (23). Similar to our observation, a previous study reported that the maximum adsorption of Dox onto GO nanosheets occurred within 1.5 minutes after the addition of Dox (24).
Promelittin is selectively cleaved by FAP to melittin, which in turn lyses RBCs (25). Unlike FAP, MMP9, which is secreted by CAFs (26), did not support the hemolytic function of PL-rGO. The specific susceptibility of PL and PL-rGO to FAP suggests that activation of PL in the coculture system is attributable to cleavage of promelittin by FAP expressed on CAFs.
In this study, we could not test the in vivo efficacy of free Dox together with PL-rGO in tumor-xenografted mouse model. Because of the rapid adsorption of free Dox onto rGO, physical mixing essentially resulted in the formation of Dox/PL-rGO. In the cell culture system, we could avoid the formation of Dox/PL-rGO by sequential treatment with free Dox and PL-rGO. However, the blood circulation of systemically administered Dox would make it difficult to prevent the possible formation of Dox/PL-rGO even after sequential treatment. Nevertheless, our PL-rGO system may have merit in that it enables the selective activation of cytotoxic drugs in the tumor microenvironment, thereby minimizing the side effects of the cytotoxic drug on normal tissues. The tumor microenvironment has recently been reported to express drug-metabolizing enzymes and to contribute to the chemoprotection of tumor cells. In acute myeloid leukemia, the expression of cytochrome p450 (CYP)-26, a retinoid-metabolizing enzyme, in the tumor microenvironment was shown to reduce the activity of all-trans retinoic acid and protect acute myeloid leukemia cells (27). Expression of CYP3A4 in human bone marrow–derived stromal cells has been shown to reduce the sensitivity of multiple myeloma and leukemia cells to bortezomib and etoposide (28). An inhibitor of CYP was also shown to overcome the drug resistance of multiple myeloma cells (28). Compared with free Dox, Dox/PL-rGO is expected to be less susceptible to CYP, owing to lower enzyme accessibility to Dox adsorbed onto rGO nanosheets. The reduced metabolism of Dox on PL-rGO in the tumor microenvironment may result in an increase in the drug sensitivity of tumor cells. Moreover, it is possible that dual loading of Dox and CYP inhibitors on PL-rGO could further enhance the sensitivity of tumor cells to Dox.
One limitation of the Dox/PL-rGO system might be the tumor-type dependence of its selective activation. In this study, we used a colon cancer xenograft model for selective activation of PL-rGO by FAP expressed on CAFs. CAFs are known to be one of the major cell types in the microenvironment of solid tumors (29). FAP-expressing CAFs have also recently been reported in various hematopoietic tumors, such as multiple myeloma (30,31). Because the population of CAFs would differ depending on the type of tumor, the activity of FAP in the tumor microenvironment would be expected to vary, possibly limiting the type of tumors in which Dox/PL-rGO can be selectively activated. Although we tested the in vivo activity of Dox/PL-rGO in a xenografted mouse model, further studies will be needed to confirm the activity of Dox/PL-rGO in the human tumor microenvironment for clinical trials.
Funding
This work was supported by a research grant from the Ministry of Science, Information and Communication Technology and Future Planning, Republic of Korea (NRF-2015R1A2A1A01005674).
Notes
The study sponsors had no role in the design of the study; the collection, analysis, or interpretation of the data; the writing of the manuscript; or the decision to submit the manuscript for publication.
Supplementary Material
References
- 1. De Veirman K, Rao L, De Bruyne E, et al. Cancer associated fibroblasts and tumor growth: focus on multiple myeloma. Cancers (Basel). 2014;6(3):1363–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sounni NE, Noel A. Targeting the tumor microenvironment for cancer therapy. Clin Chem. 2013;59(1):85–93. [DOI] [PubMed] [Google Scholar]
- 3. Orimo A, Gupta PB, Sgroi DC, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121(3): 335–348. [DOI] [PubMed] [Google Scholar]
- 4. Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell. 2010;17(2):135–147. [DOI] [PubMed] [Google Scholar]
- 5. Tchou J, Kossenkov AV, Chang L, et al. Human breast cancer associated fibroblasts exhibit subtype specific gene expression profiles. BMC Med Genomics. 2012;5:39–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liao Y, Ni Y, He R, Liu W, Du J. Clinical implications of fibroblast activation protein-α in non-small cell lung cancer after curative resection: a new predictor for prognosis. J Cancer Res Clin Oncol. 2013;139(9):1523–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shi M, Yu DH, Chen Y, et al. Expression of fibroblast activation protein in human pancreatic adenocarcinoma and its clinicopathological significance. World J Gastroenterol. 2012;18(8):840–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Henry LR, Lee HO, Lee JS, et al. Clinical implications of fibroblast activation protein in patients with colon cancer. Clin Cancer Res. 2007;13(6):1736–1741. [DOI] [PubMed] [Google Scholar]
- 9. Brennen WN, Isaacs JT, Denmeade SR. Rationale behind targeting fibroblast activation protein-expressing carcinoma-associated fibroblasts as a novel chemotherapeutic strategy. Mol Cancer Ther. 2012;11(2):257–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gajski G, Garaj-Vrhovac V. Melittin: a lytic peptide with anticancer properties. Environ Toxicol Pharmacol. 2013;36(2):697–705. [DOI] [PubMed] [Google Scholar]
- 11. Liu J, Cui L, Losic D. Graphene and graphene oxide as new nanocarriers for drug delivery applications. Acta Biomater. 2013;9(12):9243–9257. [DOI] [PubMed] [Google Scholar]
- 12. Feng L, Wu L, Qu X. New horizons for diagnostics and therapeutic applications of graphene and graphene oxide. Adv Mater. 2013;25(2):168–186. [DOI] [PubMed] [Google Scholar]
- 13. Miao W, Shim G, Kang CM, et al. Cholesteryl hyaluronic acid-coated, reduced graphene oxide nanosheets for anti-cancer drug delivery. Biomaterials. 2013;34(37):9638–9647. [DOI] [PubMed] [Google Scholar]
- 14. Miao W, Shim G, Kim G, et al. Image-guided synergistic photothermal therapy using photoresponsive imaging agent-loaded graphene-based nanosheets. J Control Release. 2015;211:28–36. [DOI] [PubMed] [Google Scholar]
- 15. Hou C, Quan H, Duan Y, Zhang Q, Wang H, Li Y. Facile synthesis of water-dispersible Cu2O nanocrystal-reduced graphene oxide hybrid as a promising cancer therapeutic agent. Nanoscale. 2013;5(3):1227–1232. [DOI] [PubMed] [Google Scholar]
- 16. Niu G, Cogburn B, Hughes J. Preparation and characterization of doxorubicin liposomes. Methods Mol Biol. 2010;624:211–219. [DOI] [PubMed] [Google Scholar]
- 17. Castelló-Cros R, Cukierman E. Stromagenesis during tumorigenesis: characterization of tumor-associated fibroblasts and stroma-derived 3D matrices. Methods Mol Biol. 2009;522:275–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Verma SC, Agarwal P, Krishnan MY. Primary mouse lung fibroblasts help macrophages to tackle Mycobacterium tuberculosis more efficiently and differentiate into myofibroblasts up on bacterial stimulation. Tuberculosis. 2016;97:172–180. [DOI] [PubMed] [Google Scholar]
- 19. Wang H, Wu Q, Liu Z, et al. Downregulation of FAP suppresses cell proliferation and metastasis through PTEN/PI3K/AKT and Ras-ERK signaling in oral squamous cell carcinoma. Cell Death Dis. 2014;5:e1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chowdhury SM, Surhland C, Sanchez Z, et al. Graphene nanoribbons as a drug delivery agent for lucanthone mediated therapy of glioblastoma multiforme. Nanomedicine. 2014;11(1):109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Miao W, Shim G, Lee S, Lee S, Choe YS, Oh YK. Safety and tumor tissue accumulation of pegylated graphene oxide nanosheets for co-delivery of anticancer drug and photosensitizer. Biomaterials. 2013;34(13):3402–3410. [DOI] [PubMed] [Google Scholar]
- 22. Robinson JT, Tabakman SM, Liang Y, et al. Ultrasmall reduced graphene oxide with high near-infrared absorbance for photothermal therapy. J Am Chem Soc. 2011;133(17):6825–6831. [DOI] [PubMed] [Google Scholar]
- 23. Yang X, Zhang X, Liu Z, Ma Y, Huang Y, Chen Y. High-efficiency loading and controlled release of doxorubicin hydrochloride on graphene oxide. J Phys Chem C. 2008;112(45):17554–17558. [Google Scholar]
- 24. Wu S, Zhao X, Li Y, et al. Adsorption properties of doxorubicin hydrochloride onto graphene oxide: equilibrium, kinetic and thermodynamic studies. Materials. 2013;6(5):2026–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. LeBeau AM, Brennen WN, Aggarwal S, Denmeade SR. Targeting the cancer stroma with a fibroblast activation protein-activated promelittin protoxin. Mol Cancer Ther. 2009;8(5):1378–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cirri P, Chiarugi P. Cancer associated fibroblasts: the dark side of the coin. Am J Cancer Res. 2011;1(4):482–497. [PMC free article] [PubMed] [Google Scholar]
- 27. Su M, Alonso S, Jones JW, et al. All-trans retinoic acid activity in acute myeloid leukemia: role of cytochrome P450 enzyme expression by the microenvironment. PLoS One. 2015;10(6):e0127790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Alonso S, Su M, Jones JW, et al. Human bone marrow niche chemoprotection mediated by cytochrome P450 enzymes. Oncotarget. 2015;6(17):14905–14912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kakarla S, Song XT, Gottschalk S. Cancer-associated fibroblasts as targets for immunotherapy. Immunotherapy. 2012;4(11):1129–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Frassanito MA, Rao L, Moschetta M, et al. Bone marrow fibroblasts parallel multiple myeloma progression in patients and mice: in vitro and in vivo studies. Leukemia. 2014;28(4):904–916. [DOI] [PubMed] [Google Scholar]
- 31. Raffaghello L, Vacca A, Pistoia V, Ribatti D. Cancer associated fibroblasts in hematological malignancies. Oncotarget. 2015;6(5):2589–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.