

Abstract
The extracellular matrix (ECM) is a salient feature of all solid tissues within the body. This complex, acellular entity is composed of hundreds of individual molecules whose assembly, architecture and biomechanical properties are critical to controlling the behaviour and phenotype of the different cell types residing within tissues. Cells are the basic unit of life and the core building block of tissues and organs. At their simplest, they follow a set of rules, governed by their genetic code and effected through the complex protein signalling networks that these genes encode. These signalling networks assimilate and process the information received by the cell to control cellular decisions that govern cell fate. The ECM is the biggest provider of external stimuli to cells and as such is responsible for influencing intracellular signalling dynamics. In this review, we discuss the inclusion of ECM as a central regulatory signalling sub‐network in computational models of cellular decision making, with a focus on its role in diseases such as cancer.
Linked Articles
This article is part of a themed section on Translating the Matrix. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.1/issuetoc
Abbreviations
- DDR2
discoidin domain receptor tyrosine kinase 2
- ECM
extracellular matrix
- EGFR
EGF receptor
- FAK
focal adhesion kinase
- HSC
hepatic stellate cells
- MKK
MAPK kinase
- MLC
myosin light chain
- ODE
ordinary differential equation
- RhoA
Ras homologue gene family, member A
- RTK
receptor tyrosine kinase
- Src
SRC proto‐oncogene, non‐receptor tyrosine kinase
Dynamics of extracellular matrix homeostasis
The extracellular matrix (ECM) makes up the complex network of macromolecules that surrounds cells and tissues and not only provides structural and mechanical support but is also fundamentally important in modulating cellular activity via the activation or suppression of intracellular signalling pathways (Lu et al., 2012; Skhinas and Cox, 2017). Regulatory cues from the external environment can significantly influence intracellular signalling and cell behaviour in response to a cell's surroundings. Conversely, alterations in signalling programmes can result in changes in the ECM and the downstream ECM‐mediated signalling networks. Importantly, the timescales over which ECM remodelling and rewiring of intracellular signalling networks occur, the former being long term with the latter being almost immediate, allow both short‐ and long‐term regulation of cellular behaviour in tissues and organs. The caveat to this is that dysregulation of ECM homeostasis either resulting in or due to altered signalling programmes creates a positive feed‐forward loop which can drive disease progression in a range of disorders such as chronic degenerative and autoimmune diseases, fibrosis and malignancies (Bonnans et al., 2014).
Throughout life, the ECM in most tissues is continually remodelled, undergoing degradation, balanced by synthesis and deposition. However, this process is dysregulated in a range of diseases. Alterations of the ECM can arise from aberrant expression or turnover of matrix components, such as collagen, fibronectin and hyaluronan, or altered post‐translational modifications, such as collagen crosslinking by lysyl oxidase (Cox et al., 2015; Chang et al., 2017), lysyl hydroxylase and transglutaminase activity (Wells, 2008), or proteolytic matrix degradation. These ECM changes occur in both normal and diseased tissue, and their effect on intracellular signalling pathways is critical in the regulation of function and phenotype of cells residing within these tissues. Moreover, due to the relative longevity of ECM molecules, extracellular cues from the ECM may have a chronic impact on the activation or suppression of intracellular signalling programmes. As such, we propose that the ECM should be considered as a key regulatory sub‐network in signalling networks that modulate both short‐ and long‐term cell response, behaviour, phenotype and drug response.
The ECM as a modulator of intracellular signalling
ECM components serve as ligands for cell surface receptors that activate a range of signalling pathways in healthy and diseased tissues. In fact, the complete removal of all extracellular ligands leads to anoikis, defined as apoptosis induced by inadequate or inappropriate cell–matrix interactions. Therefore, the ECM can be seen a key regulator of almost all intracellular signalling within living cells. Of the ECM engaging receptors, mechano‐sensing integrins are one of the most extensively studied families due to their involvement in cell–matrix interactions and wound repair, as well as their role in degenerative cartilage based diseases, fibrosis and cancer (Humphries et al., 2006; Desgrosellier and Cheresh, 2010). During wound healing, integrin heterodimers on the surface of cells are activated by ECM components such as laminin, collagen, fibronectin and tenascin (fully reviewed by Humphries et al., 2006) to induce multiple signalling pathways. It should be noted that in addition to the absolute amount of ECM components affecting how signalling pathways are activated, the ratios of different ECM components to one another will also modulate global cellular response as a result of the complex reciprocal feedback and feed‐forward loops present within most signalling networks. Thus, cells need to integrate multiple non‐binary signals in a decision‐making process in order to effect a response. This is important, not only because of the differential activation of receptors as ECM component ratios change but also because, as the ECM components change, the global 3D ECM organization is also affected and the spatial topology surrounding cells and tissues thus changes (Mayorca‐Guiliani et al., 2017). The ensuing alterations in ECM assembly will therefore alter the distribution of solid‐state ECM ligands and can result in the localization and compartmentalization of intracellular signalling.
The process of ECM‐mediated extracellular–intracellular signalling regulation typically occurs through transmembrane gatekeeper proteins such as the integrins. For example, integrin engagement with specific ECM molecules leads to integrin activation and the subsequent recruitment, assembly and phosphorylation of intracellular signalling complexes, including protein tyrosine kinase (TK) 2 [focal adhesion kinase (FAK)] and its associated TK, SRC proto‐oncogene, non‐receptor tyrosine kinase (Src). Both FAK and Src are heavily involved in adhesion dynamics and mediating cell motility in response to the ECM (Figure 1A) (for full review, see Mitra et al., 2005). FAK/Src activation can then go on to modulate activity of the MAPK (Figure 1B) and PI3K/Akt (PKB) pathways (Figure 1C), both of which mediate cell proliferation, differentiation and migration through transcriptional regulation. The downstream cellular response is typically the expansion and remodelling of the micro‐environment. For example, during wound repair, MAPK activation and signalling through downstream effector proteins – MAPK3 (ERK1) and MAPK1 (ERK2) – induces stromal cell proliferation and cell motility which are required for the wounding response (Matsubayashi et al., 2004). Similarly, PI3K/Akt activation has also been linked with platelet signalling, an important initiator of the repair process (Guidetti et al., 2015) along with cell adhesion (Luo et al., 2014). Collectively, ECM‐mediated integrin activation and its downstream signalling pathways increase cell proliferation, survival and migration of cells (Bonnans et al., 2014) and thus work to establish and maintain a micro‐environment conducive to tissue repair.
Figure 1.
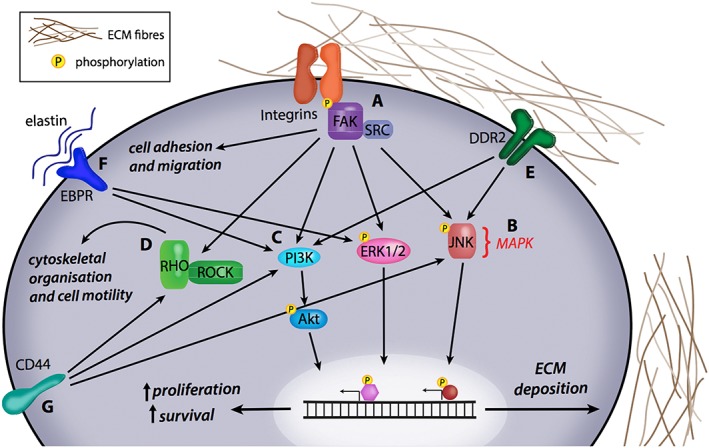
The effect of changes in ECM‐mediated signalling pathways. (A) Integrin activation by ECM components such as collagen, fibronectin, tenascin and laminin can phosphorylate the FAK/Src pathway and lead to downstream changes in cell adhesion and migration. Integrin‐mediated FAK/Src activation is also central to the activation of (B) MAPK signalling via ERK1/2 and JNK phosphorylation as well as (C) PI3K/Akt activation, leading to increased proliferation and cell survival through changes in transcriptional gene regulation. (D) FAK/Src activation can also mediate Rho/Rho‐associated kinase (ROCK) signalling, which affects cytoskeletal organization and cell motility in normal and diseased tissue states. (E) Fibrillar collagen in the ECM activates DDR2, which can mediate the MAPK and PI3K/Akt signalling pathways to influence gene expression and subsequently cellular behaviour. (F) Elastin binding to its receptor elastin binding protein receptor can influence PI3K/Akt signalling and MAPK signalling via ERK1/2. (G) Hyaluronan, another component of ECM remodelling, activates CD44 receptor, which can also affect Rho‐ROCK signalling.
Dysregulation of ECM‐mediated signalling networks in disease
The signalling networks activated during developmental processes are typically tightly regulated, switching on and off as required. However, they are also dysregulated in various disease states, either through genetic mutation or aberrant activation via extracellular cues. For example, excessive deposition and remodelling of ECM components in response to injury or damage can result in fibrosis (Cox et al., 2013; Cox and Erler, 2016). Chronic fibrosis in the liver leads to cirrhosis, characterized by a significant loss of liver function due to the presence of excessive scar tissue (Schuppan and Afdhal, 2008). It is typically driven by the deregulation of NF‐κB and TGFβ signalling in hepatic stellate cells (HSCs). These HSC‐derived myofibroblasts are responsible for the increased secretion and deposition of matrix components such as collagen and basement membrane proteins (Friedman et al., 1985), which then act to modulate signalling networks in all of the other cell types resident within the fibrosing liver. Similarly, idiopathic pulmonary fibrosis is characterized by dysregulation of, among others, the TGF‐β, MAPK and PI3K/Akt signalling pathways (Makarev et al., 2016), leading to aberrant tissue repair and extensive ECM remodelling which leads to the clinical loss of lung function. The activation of these pathways, which occurs in both forms of fibrosis, will enhance cell survival and proliferation (Wells, 2008) and acts to hyper‐activate a pro‐fibrotic cellular phenotype thus perpetuating further ECM remodelling and changes in signalling networks.
Of note, fibrosis is a characteristic of almost all solid tumours (Cox and Erler, 2016), with the establishment of a fibrotic micro‐environment considered to be essential in tumour cell survival and cancer progression at the primary tumour and secondary sites of many cancer types. In particular, activation of the integrin‐mediated FAK/Src pathway by a stiffer tumour micro‐environment promotes signalling network rewiring to drive cancer cell proliferation in primary colorectal and breast tumours (Baker et al., 2013; Cox et al., 2013) and is also involved in cell migration via changes in cell‐to‐cell contact and impaired vasculature (Potter et al., 2005). In parallel, related signalling networks including the small GTPase Rho and its associated kinase, Rho‐associated coiled‐coil containing protein kinase 1 (Rho kinase), are both involved in cytoskeletal organization and cell migration. Rho kinase activation increases myosin light chain (MLC) phosphorylation by supressing MLC phosphatase, which is necessary for the establishment and maintenance of stress fibres and focal adhesions. Additionally, Rho kinase also phosphorylates another kinase, LIMK, which inactivates the actin depolymerizing factor cofilin, and, together with Rac1 and downstream effector protein mDia1, is involved in actin polymerization, stress fibre formation and cell migration (Amano et al., 2010). This signalling network is typically under tight regulation but can also be hyper‐activated by ECM remodelling. Aberrant activation of this signalling network leads to increased tumour invasiveness in pancreatic ductal adenocarcinoma (Rath et al., 2017) and squamous cell carcinoma (Figure 1D) (Samuel et al., 2011).
In contrast to excessive ECM deposition, excessive ECM turnover is caused by aberrant expression or activity of matrix‐degrading enzymes. MMPs, ADAMs, hyaluronidases, plasminogen and cathepsins have been observed in cancer and forms of chronic tissue degradation. A range of MMPs including MMP1, MMP2 and MMP9 have been shown to be involved in both the promotion and inhibition of cancer progression in breast and lung cancer, through their effect on ECM remodelling and downstream effects on intracellular signalling networks (Egeblad and Werb, 2002). For example, in osteoarthritis, aberrant production of fibronectin, versican and laminin induces altered integrin‐mediated FAK/Src signalling and subsequent increase in MMP2 and MMP9 expression, therefore resulting in loss of matrix integrity and increased ECM degradation (Prasadam et al., 2013). In a similar manner, up‐regulated cytokine production associated with rheumatoid arthritis causes increased expression and clustering of integrin receptors, followed by activation of their signalling pathways, including ERK, JNK subfamily, FAK/Src and PI3K pathways. This leads to the increased production of matrix‐degrading enzymes such as MMP1 and MMP3 (Lowin and Straub, 2011). In particular, the phosphorylation of JNK subfamily in synovial fibroblasts has been linked with increased expression of collagenases consistent with the chronic ECM degradation seen in rheumatoid arthritis (Han et al., 1999). In addition to integrin‐mediated signalling programmes, the activation of a range of other ECM receptors can facilitate the transduction of extracellular cues in healthy and diseased tissue states. An in‐depth discussion of all of these ECM components and receptors is beyond the scope of this review; however, we have included some of these in Figure 1 such as the collagen binding receptor, DDR2, (Figure 1E), the elastin binding protein receptors (Figure 1F) and the hyaluronan receptor CD44 (Figure 1G).
Mapping signalling networks using computational models
As mentioned above, these signalling transduction networks are the means by which a cell conveys external stimuli from the cell membrane to the nucleus or processes internal signals pertaining to cell stress and metabolic status (Kolch et al., 2015). These networks are analogous to complex logic circuits built from simple modular components that allow the accurate and reliable transduction of signalling through a cell (Kholodenko, 2006; Murray and Miller, 2015). Gaining an understanding of the structure of signalling transduction networks represents the coming era of precision medicine strategies, as improvements in computing resources allow the efficient analysis of large datasets involving complex dynamic processes within a timeframe that allows for clinical benefit (Kolch et al., 2015; Barbolosi et al., 2016). Mathematical modelling is invaluable in this regard, as it is impossible to intuitively grasp the dynamics of signalling transduction networks because of their complex structures and nonlinear dynamics, which include extensive built‐in redundancies, feedback loops and crosstalk. To aid the reader in the following section, a number of the specific terms used to describe these mathematical modelling concepts are further expanded upon in Table 1.
Table 1.
Definitions of relevant mathematical modelling concepts
| Term | Definition |
|---|---|
| Bi‐stability | The property of a system having two stable equilibrium states that it is capable of rapidly switching between |
| Feedback loop | A signalling component wherein system outputs are routed back to previous elements of the system as inputs, thereby altering behaviour of the entire network |
| Mechanistic model | A mathematical model that uses comprehensive information about network components, wiring structure, and kinetics to simulate the behaviour of a signalling network as a whole |
| Network node | A point of signal redistribution or termination |
| Ordinary differential equation | A differential equation that contains one or more functions of a single independent variables. In the context of a protein signalling network, this variable might be the expression level or activity of a specific protein |
| Redundancy | Replication of critical functions of a system, increasing its robustness |
| Statistical model | A mathematical model that uses available experimental data to visualize network topologies |
| Ultrasensitivity | An output response that is more sensitive to stimulus than the Michaelis‐Menten response |
Current strategies in network biology encompass many different modelling approaches ranging from simple statistics to complex in‐depth descriptions of dynamic behaviour. These modelling approaches differ in their level of abstraction, data requirements and predictive power but can be broadly separated into two categories: descriptive statistical models and predictive mechanistic models (Halasz et al., 2016). Statistical models are derived to create relationships between signalling nodes that best describe the available experimental data (Terfve et al., 2015; Chitforoushzadeh et al., 2016; Hill et al., 2016). Conceptually, these models are essentially maps of signalling transduction networks that allow direct comparison between healthy cells and those with aberrant signalling (Krogan et al., 2015). It has been proposed that these models could be employed to interpret high‐volume genomic datasets, identifying the network structures responsible for driving a diseased phenotype, rather than individual genes (Krogan et al., 2015). This approach would encourage the development of therapeutic agents to consider their effects on broader network structures, rather than individual targets. This concept has demonstrated its effectiveness, particularly within the field of cancer medicine, but it is not without its drawbacks. Although statistical models are useful tools for visualizing network topologies, they are limited in their ability to predict functional properties of these networks (Klinger et al., 2013; Halasz et al., 2016).
Understanding the consequences of changes in these networks is fundamental to effective treatment, and as such systems biologists have developed mechanistic models that use comprehensive information about the network's components, wiring structure and kinetics in order to predict functional properties and perturbation outcomes (Kolch et al., 2015; Halasz et al., 2016). Such dynamic modelling has revealed that emerging systems‐level properties, such as activity pulses, ultrasensitivity and bistability are critical mechanisms by which cells can robustly control their behaviour in uncertain environments (Fey et al., 2016). These predictive dynamic models are the next stage in tracking the progression of complex diseases that are generally nonlinear, time‐dependent processes that cannot be adequately assessed using traditional static biomarkers (Kolch and Fey, 2017).
By considering the spatiotemporal aspects of subcellular signalling and the effects of noise on signal transduction that traditional biomarkers typically fail to account for, mechanistic models provide a deeper understanding of the cellular processes involved in disease progression or drug response (Fey et al., 2015; Halasz et al., 2016). Numerous existing studies have demonstrated the efficacy of these models as prognostic tools and highlighted several key advantages that they provide. By capitalizing on a computational model's ability to rapidly iterate different conditions, it is possible to consider the effects of various combinations of therapeutic agents on disease progression and determine which combination of drugs will have the most beneficial effect on an individual patient's treatment (Zhao et al., 2014). Mechanistic models are also an effective method of removing bias from the development of treatment strategies, as they can be used to contextualize biological data that initially appears to be counter‐intuitive, and are capable of analysing systems whose complexity precludes the use of smaller‐scale studies (Lindner et al., 2017). Critically, these models can also make use of information that is not prognostically significant until placed within the context of its cognate network and use this additional information to more effectively assess the status of the disease. This approach has already been used to understand the time and dose dependencies of drug‐induced cancer cell death and to predict clinical outcomes of cancer patients (Fey et al., 2015; Lindner et al., 2017).
As these models evolve to become more sophisticated, their clinical viability becomes increasingly apparent. The field of cancer medicine in particular has proven to be fertile ground for this approach, where using network models as biomarkers could potentially broaden the range of effective prognostic markers available to clinicians. A significant example of this is kinase signalling in cancer medicine. The protein kinase domain is the most frequently mutated in cancer cells, making it an attractive therapeutic target (Fleuren et al., 2016). However, owing to the complex, plastic and adaptable nature of phosphorylation networks, targeting an individual node typically results in the rapid development of acquired resistance in the tumour, as built‐in redundancies and crosstalk compensate for the signal perturbation. Considering these networks in a more holistic manner and developing mathematical models to describe the signalling events that cannot be intuitively understood can facilitate the rational design of kinase‐targeting therapies. A particularly exciting application for these models is to anticipate and pre‐empt resistance.
MAPK signalling is an important focal point in cancer medicine. While there have been many attempts to target these pathways, their inherent plasticity affords cancer cells an avenue to acquire resistance to targeted therapies. Computational modelling offers the ability to mitigate the effects of this plasticity by prescribing more nuanced therapeutic options. An existing model of the ERK subfamily pathway demonstrates the ability of a computational model to iteratively simulate different network conditions, such as RAS, B‐Raf and EGF receptor (EGFR) mutations, which are commonly observed to drive a cancer phenotype (Orton et al., 2009). This particular model identified the Rap1 pathway as a potential therapeutic target. A similar approach involved testing B‐Raf and KRAS mutations in a model of the RAS–ERK and PI3K/mechanistic target of rapamycin pathways informed by FRET image analysis identified effective combinations of MEK and PI3K inhibitors (Fujita et al., 2014). More recently, we have devised an ordinary differential equation (ODE)‐based mathematical model of the reaction kinetics of JNK subfamily activity to predict clinical outcomes in individual neuroblastoma patients that can be adapted to compare and predict the efficacy of different therapeutic agents (Fey et al., 2015).
Computational models of the EGFR family have also demonstrated the ability of mathematical analyses to identify novel therapeutic strategies. The inceptive models of these pathways were developed by constructing the topological structure of the signalling cascade and quantifying the relationships between all the molecular species therein (Kholodenko et al., 1999). Developing a model in this manner allows for the assessment of how cells process information, such as cues from the ECM, and the prediction of response outputs. Cancer is a dynamic disease, however, and ignoring the cellular context such as the ECM in these models ultimately limits their effectiveness. Existing mathematical models can be adapted to improve their usefulness. For example, by incorporating data on phosphatase dynamics into an EGFR pathway computational model, novel therapeutic targets and combination therapies have been designed, in an effort to overcome resistance to receptor TK (RTK) inhibitors (Nguyen et al., 2013). In this same vein, a model can be adapted to incorporate crosstalk with, for instance, EGFR and the insulin receptor (InsR) signalling. The crosstalk between EGFR and InsR signalling is exceedingly complex, with multiple points of interaction. By adapting an existing EGFR model to include InsR signalling, we are able to simulate the dynamics of these two pathways in concert and identify critical nodes to prescribe combinations of therapeutic targets and predict their effects (Borisov et al., 2009). As research in this field continues, network modelling strategies and concepts evolve. At present, there are not only models that incorporate crosstalk with other signalling cascades (Borisov et al., 2009; Frohlich et al., 2015) but also physiological conditions, such as temperature (Moehren et al., 2002). In the future, these models could be used clinically to assess an individual patient's condition and predict combinations of therapeutic targets that would provide the best treatment options, a concept that is already being verified in vitro (Borisov et al., 2009; Nguyen et al., 2013). It is apparent that the predictive power of these networks improves as they incorporate different sets of data. As they become increasingly comprehensive, their clinical potential increases accordingly. We argue that the inclusion of ECM interactions within these models will further significantly improve their accuracy and therefore greatly improve their clinical potential.
Potential applications for modelling of matrix signalling
As these computational models of signalling pathways are increasingly applied to precision medicine and rationalized drug re‐purposing (Kolch et al., 2015), the gulf in our understanding of how signalling pathways behave in vitro and in vivo is becoming more apparent. A major difference between the in vitro analysis of signalling pathways and the in vivo reality is the manner in which cultured cells attach to their synthetic micro‐environment and the fundamental influence this has on pathway activation, cell behaviour and drug response.
Physiologically, the ECM not only provides structural support and a cellular attachment point, but these interactions with specific ECM components also direct discrete cellular responses through activation of integrin‐mediated signalling pathways. Within the cancer micro‐environment, these interactions and cellular responses are perturbed not only by compositional alterations in the ECM but also by physical alterations that influence the density, and stiffness, of the ECM. Increased ECM stiffness is known to be a fundamental property in malignant transformation (Levental et al., 2009). Accordingly, a number of experimental models have demonstrated that cancer cells are more proliferative, invasive and drug‐resistant when grown on, or within, increasingly rigid matrices (Baker et al., 2013; Cox et al., 2013). The signalling pathways involved in sensing and responding to this increased ECM stiffness are now beginning to be characterized, and here, we argue that the inclusion of this ECM‐induced signalling within pharmacologically relevant models of pathway activation will be a key advance necessary for their successful use as a tool for precision medicine.
As described above, the emergent properties of network simulations are determined by the structure of the network in question. That is, the experimentally determined wiring between components that takes into account known mechanisms of activation, inactivation and regulatory crosstalk. Below, we will describe three separate, although in no way exhaustive, examples of how ECM interactions are partly responsible for re‐programming and re‐wiring the drug‐induced signalling response, and how the inclusion of these aspects of ECM signalling could improve current modelling approaches.
Receptor tyrosine kinase (RTK) clustering induced by matrix interactions
Aberrant RTK activity is known to drive tumour progression in a number of tumour types (Lemmon and Schlessinger, 2010). Accordingly, a number of therapeutic approaches have been devised to target RTK activity, either through neutralizing antibodies or small molecule kinase inhibitors. As a general therapeutic mechanism, targeting RTK activity will usually elicit a strong initial response, but relapse invariably occurs due to a number of avenues of therapeutic resistance (Kennedy et al., 2016).
Several computational models have been developed that can predict the behaviour of RTKs and their downstream signalling pathways (Ryu et al., 2015; Adlung et al., 2017), including those that have been adapted to predict optimal combination therapies in an attempt to overcome resistance to RTK inhibitors. Invariably, these models are developed and calibrated under standard conditions using data derived from cancer cell lines grown on tissue culture plastic. However, it is becoming clear that both the activity and substrate specificity of RTKs can be strongly influenced by ECM interactions, controlled by ECM biochemistry and stiffness, suggesting that these considerations should be included in future modelling approaches.
Under tissue culture conditions with specifically tuneable ECM stiffness, many growth factor receptors, including EGFR, cluster within focal adhesions on stiff substrates but remain diffusely distributed on soft substrates (Wang et al., 1998; Sieg et al., 2000; Umesh et al., 2014). ECM stiffness is known to promote tumour progression by causing integrins to cluster within focal adhesions (Paszek et al., 2005; Levental et al., 2009), and direct interactions between these RTKs and integrins are thought to facilitate this localized enrichment (Wang et al., 1998; Sieg et al., 2000; Cabodi et al., 2004). This spatial clustering of RTK activity on stiff substrates alters the balance between ligand concentration and receptor auto‐phosphorylation, which ultimately amplifies ligand‐induced RTK signalling (Ichinose et al., 2004; Levental et al., 2009; Stabley et al., 2013). Additionally, the localization of RTKs within focal adhesions on stiff substrates promotes signalling through FAK, which increases cell migration (Sieg et al., 2000), while FAK is also required for RTK‐mediated oncogenic transformation (Benlimame et al., 2005). Taken together, this suggests that extracellular cues from the surrounding ECM may not only amplify the signalling response from these receptors but also qualitatively change the functional outcome of RTK activation by altering substrate phosphorylation.
A recent study demonstrated a similar concept by modelling the effect of ECM concentration and composition upon the kinetics of integrin binding and clustering (Hudson et al., 2017). In the same way, the concentration‐dependent dynamics and substrate specificity of RTK signalling in response to changing ECM conditions are key computational parameters required for future model development. These model characteristics will strongly influence network dynamics by modulating the strength and direction of feedback and thereby alter both the functional outcome of RTK signalling and the sensitivity of individual network nodes to therapeutic inhibition. Therefore, modelling studies that include ECM stiffness and composition as model parameters are likely to yield more accurate predictions about how these potent oncogenes can be efficiently targeted. Future studies are likely to benefit from an understanding of the differing activation kinetics and substrate specificity of RTKs on soft versus stiff matrices, including a detailed model of the altered feedback structures and fragile nodes under these contrasting conditions.
Spatial aspects of small GTPase signalling
While ECM stiffness can specifically promote the clustering of RTKs within focal adhesions, the clustering of signalling proteins at the plasma membrane is a general mechanism necessary for fine‐tuning the response of a number of signalling pathways (Garcia‐Parajo et al., 2014). The spatial co‐localization of many individual molecules is thought to reduce the inherent noise within signalling pathways by overcoming the thresholds required for analogue to digital conversions (Harding and Hancock, 2008). This phenomenon has been empirically demonstrated for the small GTPase Ras, which can form nanoclusters of approximately seven molecules (Hancock and Parton, 2005). In this spatial arrangement, each nanocluster functions as an individual switch and allows the generation of a graded output of ERK signalling by activating nanoclusters proportionally to the concentration of EGF (Tian et al., 2007).
A similar mechanism has also been proposed for the formation of Rac1 nanoclusters, where the spatial regulation of Rac1 at the edges of lamellipodia is controlled by gradients of nanoclusters containing 50–100 molecules (Remorino et al., 2017). No such observation has yet been made for the related GTPase, RhoA, although both FRET‐biosensor imaging and computational studies have been employed to describe the complex spatial regulation of Rac1 and RhoA (Timpson et al., 2011; Johnsson et al., 2014). The complex spatial interplay between these two mutually antagonistic small GTPases is crucial for a number of processes associated with the migration of epithelial cells (Ridley et al., 2003), and they therefore play a key role in tumour invasion. Despite their prominent role in a number of tumour types, a strategy for the effective pharmacological inhibition of these small GTPases remains elusive. Therefore extensive efforts have been undertaken to identify potential therapeutic strategies that exploit the complex networks that regulate the oncogenic function of GTPases (Hetmanski et al., 2016a).
The tightly localized switching between Rac1 and RhoA signalling, which is necessary for the coordinate cycles of actin polymerization and depolymerization that promote efficient cell migration, is regulated by a complex network of guanine nucleotide exchange factors, GTPase activating proteins and kinases. Therefore a number of modelling approaches have been adopted in order to investigate the interplay between Rac1 and RhoA signalling (Hetmanski et al., 2016a). This has included Boolean models that have defined the network logic associated with mutually antagonistic Rac1 and RhoA signalling (Hetmanski et al., 2016b) and dynamic ODE‐based models that describe the spatiotemporal dynamics and bistability of Rac1 and RhoA signalling (Tsyganov et al., 2012; Nikonova et al., 2013; Byrne et al., 2016).
Interestingly, another consequence of the integrin clustering associated with increasing ECM stiffness is the promotion of RhoA signalling to its downstream effector Rho‐associated kinase, and a subsequent increase in cytoskeletal contractility, a loss of cell polarity, increased cell growth and migration (Paszek et al., 2005; Keely, 2011). The malignant phenotype associated with increased ECM stiffness and altered ECM composition has also been associated with Rac1‐driven PI3K signalling (Chaudhuri et al., 2014). Clearly, the spatiotemporal regulation of RhoA and Rac1 will be heavily influenced by the specific interactions occurring between epithelial cells and the ECM. An increased understanding of how ECM interactions alter both the network logic and switch‐like nature of RhoA and Rac1 activation is likely to yield more accurate predictions of cell behaviour across a variety of substrates and potentially highlight specific avenues of therapeutic intervention for these elusive drug targets.
Altered MAPK equilibrium states
A number of elegant models have been developed to describe the dynamics of MAPK signalling, in which a key regulatory mechanism is the negative feedback and crosstalk that occurs through both rapid, phosphorylation‐based inhibition of upstream signalling components, and slower transcriptional induction of negative regulators (Kholodenko et al., 2010; Bluthgen, 2015; Ryu et al., 2015). This dynamic network rewiring strongly regulates the temporal kinetics of each discrete MAPK but also generates feedback‐based network structures that can influence the sensitivity and dynamic behaviour of all MAPK pathways (Fey et al., 2012).
Experimentally, MAPK pathways are usually modelled under serum‐starved conditions, followed by rapid pathway activation through the addition of growth factors or cell attachment/spreading. In these cases, a spike of activity is observed which initiates a number of negative feedback processes, and the signal decays over time. However, in contrast to these experimental conditions, cells normally exist within a native micro‐environment of constant stimulation by growth factors, cytokines, ECM interactions and mechano‐sensory input. Therefore, while many models can accurately predict the acute dynamics of MAPK activation, in reality, most cells will maintain an equilibrium state of MAPK activity that represents a delicate balance between ERK, JNK and p38 activation.
Where these models of acute activation can be of particular use is in the prediction of drug‐induced signalling in cancer cells (Fey et al., 2015). However, to fully understand the response of cells within a complex physiological setting, we need to progress to a point where these models can accurately represent the baseline network state of cells in a native 3D environment before a reliable prediction can be made about drug‐induced signalling. As ECM‐mediated integrin signalling is known to activate each MAPK pathway within different contexts (see above), the exposure of cells to a specific set of ECM components is likely to generate a distinct state of MAPK equilibrium that would heavily influence any acute, drug‐induced signalling. For example, integrin engagement with the ECM is known to activate both ERK and PI3K/Akt, both of which can inhibit drug‐induced activation of apoptotic JNK signalling (Fey et al., 2015). For ERK signalling, this can occur through ERK‐dependent up‐regulation of the JNK‐specific phosphatases (Katagiri et al., 2005; Cagnol and Rivard, 2013), whereas Akt can inhibit the kinases directly upstream of JNK, namely, MAPK kinase (MKK) 4 and MKK7 (Fey et al., 2015) (Figure 2A). Therefore, an increase in the baseline ERK and Akt activation (driven through altered ECM inputs) would repress the drug‐induced, switch‐like activation of JNK (Figure 2B). In this way, a fibrotic tumour micro‐environment rich in stiff ECM components could reduce drug sensitivity by repressing apoptotic signalling through this stress‐activated MAPK pathway (Figure 2C).
Figure 2.
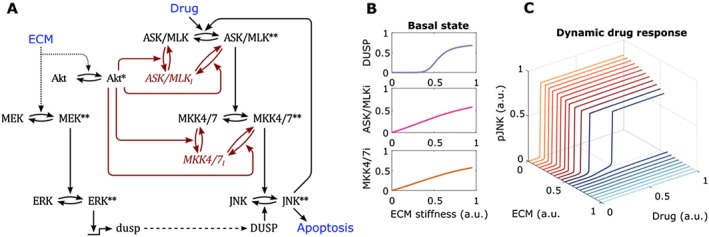
Computational model of the interplay between the ECM and drug activated MAPK‐JNK signalling network (A) Scheme outlining how the ECM induced activation of ERK and PI3K/Akt influences the JNK signalling network. Black arrows represent activating biochemical reactions, red arrows represent inhibitory reactions. * and ** represents single and double phosphorylation events respectively on kinases. Red text represents model species in the inhibited state (also denoted by i ). (B) Simulated input/output curves showing the relationship between ECM stiffness and the basal state of both DUSP (dual‐specificity phosphatase) expression and the inhibited forms of ASK/MLK (apoptosis signal regulating kinase / mixed lineage kinase) and MKK4/7. (C) Simulated input/output curves showing the changes in ultrasensitivity of drug induced JNK activation as ECM stiffness is increased.
As we have previously modelled the theoretical consequences of this MAPK pathway crosstalk (Fey et al., 2012), it is possible that, in the future, tumour‐specific models could be adapted to take into account the ECM composition and stiffness of each tumour. This would generate an established, baseline, equilibrium state from which personalized predictions could be reliably made.
Conclusions
At their simplest, cells follow a set of rules governed by their genetic code. These rules, which are executed by the protein‐based signalling networks that the genes encode, control the assimilation of information and decision‐making processes that shape a cell's response to their surroundings. Under normal situations, each independent cellular decision results in the emergent phenomena of correct organ or tissue function. Homeostasis of the ECM is a fundamental regulator of cell and tissue behaviour, providing the majority of extracellular signalling cues that feed into these cellular decision‐making processes. In many tissue diseases, aberrant ECM provides signalling cues that significantly alter cellular decision‐making processes, typically to the detriment of the organ or individual. As yet, we still do not fully understand the importance and role of the ECM as a key signalling sub‐network in these processes, and only through a bottom‐up modelling approach to dissecting intracellular signalling, which includes the ever‐present ECM, will we begin to deepen our understanding of how to tackle these complex tissue diseases from a therapeutic perspective.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b).
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
T.R.C. is supported by a Susan G. Komen for the Cure catalyst award and the National Health and Medical Research Council (NHMRC). J.N.S. is supported by the Cancer Institute NSW (CINSW). D.R.C. is a Cancer Institute NSW fellow (13/FRL/1‐02) and is supported by the NHMRC. J.F.H. is a recipient of an Australian Postgraduate Award.
Hastings, J. F. , Skhinas, J. N. , Fey, D. , Croucher, D. R. , and Cox, T. R. (2019) The extracellular matrix as a key regulator of intracellular signalling networks. British Journal of Pharmacology, 176: 82–92. 10.1111/bph.14195.
Contributor Information
David R Croucher, Email: d.croucher@garvan.org.au.
Thomas R Cox, Email: t.cox@garvan.org.au.
References
- Adlung L, Kar S, Wagner MC, She B, Chakraborty S, Bao J et al (2017). Protein abundance of AKT and ERK pathway components governs cell type‐specific regulation of proliferation. Mol Syst Biol 13: 904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano M, Nakayama M, Kaibuchi K (2010). Rho‐kinase/ROCK: a key regulator of the cytoskeleton and cell polarity. Cytoskeleton (Hoboken) 67: 545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker AM, Bird D, Lang G, Cox TR, Erler JT (2013). Lysyl oxidase enzymatic function increases stiffness to drive colorectal cancer progression through FAK. Oncogene 32: 1863–1868. [DOI] [PubMed] [Google Scholar]
- Barbolosi D, Ciccolini J, Lacarelle B, Barlesi F, Andre N (2016). Computational oncology – mathematical modelling of drug regimens for precision medicine. Nat Rev Clin Oncol 13: 242–254. [DOI] [PubMed] [Google Scholar]
- Benlimame N, He Q, Jie S, Xiao D, Xu YJ, Loignon M et al (2005). FAK signaling is critical for ErbB‐2/ErbB‐3 receptor cooperation for oncogenic transformation and invasion. J Cell Biol 171: 505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluthgen N (2015). Signaling output: it's all about timing and feedbacks. Mol Syst Biol 11: 843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnans C, Chou J, Werb Z (2014). Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol 15: 786–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borisov N, Aksamitiene E, Kiyatkin A, Legewie S, Berkhout J, Maiwald T et al (2009). Systems‐level interactions between insulin‐EGF networks amplify mitogenic signaling. Mol Syst Biol 5: 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne KM, Monsefi N, Dawson JC, Degasperi A, Bukowski‐Wills JC, Volinsky N et al (2016). Bistability in the Rac1, PAK, and RhoA signaling network drives actin cytoskeleton dynamics and cell motility switches. Cell Syst 2: 38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabodi S, Moro L, Bergatto E, Boeri Erba E, Di Stefano P, Turco E et al (2004). Integrin regulation of epidermal growth factor (EGF) receptor and of EGF‐dependent responses. Biochem Soc Trans 32: 438–442. [DOI] [PubMed] [Google Scholar]
- Cagnol S, Rivard N (2013). Oncogenic KRAS and BRAF activation of the MEK/ERK signaling pathway promotes expression of dual‐specificity phosphatase 4 (DUSP4/MKP2) resulting in nuclear ERK1/2 inhibition. Oncogene 32: 564–576. [DOI] [PubMed] [Google Scholar]
- Chang J, Lucas MC, Leonte LE, Garcia‐Montolio M, Singh LB, Findlay AD et al (2017). Pre‐clinical evaluation of small molecule LOXL2 inhibitors in breast cancer. Oncotarget 8: 26066–26078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri O, Koshy ST, Branco da Cunha C, Shin JW, Verbeke CS, Allison KH et al (2014). Extracellular matrix stiffness and composition jointly regulate the induction of malignant phenotypes in mammary epithelium. Nat Mater 13: 970–978. [DOI] [PubMed] [Google Scholar]
- Chitforoushzadeh Z, Ye Z, Sheng Z, LaRue S, Fry RC, Lauffenburger DA et al (2016). TNF‐insulin crosstalk at the transcription factor GATA6 is revealed by a model that links signaling and transcriptomic data tensors. Sci Signal 9: ra59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox TR, Bird D, Baker AM, Barker HE, Ho MW, Lang G et al (2013). LOX‐mediated collagen crosslinking is responsible for fibrosis‐enhanced metastasis. Cancer Res 73: 1721–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox TR, Erler JT (2016). Fibrosis and cancer: partners in crime or opposing forces? Trends Cancer 2: 279–282. [DOI] [PubMed] [Google Scholar]
- Cox TR, Rumney RM, Schoof EM, Perryman L, Hoye AM, Agrawal A et al (2015). The hypoxic cancer secretome induces pre‐metastatic bone lesions through lysyl oxidase. Nature 522: 106–110. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Desgrosellier JS, Cheresh DA (2010). Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer 10: 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeblad M, Werb Z (2002). New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer 2: 161–174. [DOI] [PubMed] [Google Scholar]
- Fey D, Croucher DR, Kolch W, Kholodenko BN (2012). Crosstalk and signaling switches in mitogen‐activated protein kinase cascades. Front Physiol 3: 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fey D, Halasz M, Dreidax D, Kennedy SP, Hastings JF, Rauch N et al (2015). Signaling pathway models as biomarkers: patient‐specific simulations of JNK activity predict the survival of neuroblastoma patients. Sci Signal 8: ra130. [DOI] [PubMed] [Google Scholar]
- Fey D, Kuehn A, Kholodenko BN (2016). On the personalised modelling of cancer signalling. IFAC‐PapersOnLine 49: 312–317. [Google Scholar]
- Fleuren ED, Zhang L, Wu J, Daly RJ (2016). The kinome 'at large' in cancer. Nat Rev Cancer 16: 83–98. [DOI] [PubMed] [Google Scholar]
- Friedman SL, Roll FJ, Boyles J, Bissell DM (1985). Hepatic lipocytes: the principal collagen‐producing cells of normal rat liver. Proc Natl Acad Sci U S A 82: 8681–8685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohlich H, Bahamondez G, Gotschel F, Korf U (2015). Dynamic Bayesian network modeling of the interplay between EGFR and Hedgehog signaling. PLoS One 10: e0142646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y, Komatsu N, Matsuda M, Aoki K (2014). Fluorescence resonance energy transfer based quantitative analysis of feedforward and feedback loops in epidermal growth factor receptor signaling and the sensitivity to molecular targeting drugs. FEBS J 281: 3177–3192. [DOI] [PubMed] [Google Scholar]
- Garcia‐Parajo MF, Cambi A, Torreno‐Pina JA, Thompson N, Jacobson K (2014). Nanoclustering as a dominant feature of plasma membrane organization. J Cell Sci 127: 4995–5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidetti GF, Canobbio I, Torti M (2015). PI3K/Akt in platelet integrin signaling and implications in thrombosis. Adv Biol Regul 59: 36–52. [DOI] [PubMed] [Google Scholar]
- Halasz M, Kholodenko BN, Kolch W, Santra T (2016). Integrating network reconstruction with mechanistic modeling to predict cancer therapies. Sci Signal 9: ra114. [DOI] [PubMed] [Google Scholar]
- Han Z, Boyle DL, Aupperle KR, Bennett B, Manning AM, Firestein GS (1999). Jun N‐terminal kinase in rheumatoid arthritis. J Pharmacol Exp Ther 291: 124–130. [PubMed] [Google Scholar]
- Hancock JF, Parton RG (2005). Ras plasma membrane signalling platforms. Biochem J 389: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding AS, Hancock JF (2008). Using plasma membrane nanoclusters to build better signaling circuits. Trends Cell Biol 18: 364–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetmanski JHR, Schwartz JM, Caswell PT (2016a). Rationalizing Rac1 and RhoA GTPase signaling: a mathematical approach. Small GTPases: 1–6. 10.1080/21541248.2016.1218406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetmanski JHR, Zindy E, Schwartz JM, Caswell PT (2016b). A MAPK‐driven feedback loop suppresses Rac activity to promote RhoA‐driven cancer cell invasion. PLoS Comput Biol 12: e1004909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill SM, Heiser LM, Cokelaer T, Unger M, Nesser NK, Carlin DE et al (2016). Inferring causal molecular networks: empirical assessment through a community‐based effort. Nat Methods 13: 310–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson SV, Dolin CE, Poole LG, Massey VL, Wilkey D, Beier JI et al (2017). Modeling the kinetics of integrin receptor binding to hepatic extracellular matrix proteins. Sci Rep 7: 12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphries JD, Byron A, Humphries MJ (2006). Integrin ligands at a glance. J Cell Sci 119: 3901–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinose J, Murata M, Yanagida T, Sako Y (2004). EGF signalling amplification induced by dynamic clustering of EGFR. Biochem Biophys Res Commun 324: 1143–1149. [DOI] [PubMed] [Google Scholar]
- Johnsson AK, Dai Y, Nobis M, Baker MJ, McGhee EJ, Walker S et al (2014). The Rac‐FRET mouse reveals tight spatiotemporal control of Rac activity in primary cells and tissues. Cell Rep 6: 1153–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katagiri C, Masuda K, Urano T, Yamashita K, Araki Y, Kikuchi K et al (2005). Phosphorylation of Ser‐446 determines stability of MKP‐7. J Biol Chem 280: 14716–14722. [DOI] [PubMed] [Google Scholar]
- Keely PJ (2011). Mechanisms by which the extracellular matrix and integrin signaling act to regulate the switch between tumor suppression and tumor promotion. J Mammary Gland Biol Neoplasia 16: 205–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy SP, Hastings JF, Han JZ, Croucher DR (2016). The under‐appreciated promiscuity of the epidermal growth factor receptor family. Front Cell Dev Biol 4: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kholodenko BN (2006). Cell‐signalling dynamics in time and space. Nat Rev Mol Cell Biol 7: 165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kholodenko BN, Demin OV, Moehren G, Hoek JB (1999). Quantification of short term signaling by the epidermal growth factor receptor. J Biol Chem 274: 30169–30181. [DOI] [PubMed] [Google Scholar]
- Kholodenko BN, Hancock JF, Kolch W (2010). Signalling ballet in space and time. Nat Rev Mol Cell Biol 11: 414–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinger B, Sieber A, Fritsche‐Guenther R, Witzel F, Berry L, Schumacher D et al (2013). Network quantification of EGFR signaling unveils potential for targeted combination therapy. Mol Syst Biol 9: 673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolch W, Fey D (2017). Personalized computational models as biomarkers. J Pers Med 7: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolch W, Halasz M, Granovskaya M, Kholodenko BN (2015). The dynamic control of signal transduction networks in cancer cells. Nat Rev Cancer 15: 515–527. [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Lippman S, Agard DA, Ashworth A, Ideker T (2015). The cancer cell map initiative: defining the hallmark networks of cancer. Mol Cell 58: 690–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon MA, Schlessinger J (2010). Cell signaling by receptor tyrosine kinases. Cell 141: 1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT et al (2009). Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 139: 891–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner AU, Salvucci M, Morgan C, Monsefi N, Resler AJ, Cremona M et al (2017). BCL‐2 system analysis identifies high‐risk colorectal cancer patients. Gut 66: 2141–2148. [DOI] [PubMed] [Google Scholar]
- Lowin T, Straub RH (2011). Integrins and their ligands in rheumatoid arthritis. Arthritis Res Ther 13: 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P, Weaver VM, Werb Z (2012). The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol 196: 395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Li C, Xu T, Liu W, Ba X, Wang X et al (2014). PI3K is involved in beta1 integrin clustering by PSGL‐1 and promotes beta1 integrin‐mediated Jurkat cell adhesion to fibronectin. Mol Cell Biochem 385: 287–295. [DOI] [PubMed] [Google Scholar]
- Makarev E, Izumchenko E, Aihara F, Wysocki PT, Zhu Q, Buzdin A et al (2016). Common pathway signature in lung and liver fibrosis. Cell Cycle 15: 1667–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubayashi Y, Ebisuya M, Honjoh S, Nishida E (2004). ERK activation propagates in epithelial cell sheets and regulates their migration during wound healing. Curr Biol 14: 731–735. [DOI] [PubMed] [Google Scholar]
- Mayorca‐Guiliani AE, Madsen CD, Cox TR, Horton ER, Venning FA, Erler JT (2017). ISDoT: in situ decellularization of tissues for high‐resolution imaging and proteomic analysis of native extracellular matrix. Nat Med 23: 890–898. [DOI] [PubMed] [Google Scholar]
- Mitra SK, Hanson DA, Schlaepfer DD (2005). Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol 6: 56–68. [DOI] [PubMed] [Google Scholar]
- Moehren G, Markevich N, Demin O, Kiyatkin A, Goryanin I, Hoek JB et al (2002). Temperature dependence of the epidermal growth factor receptor signaling network can be accounted for by a kinetic model. Biochemistry 41: 306–320. [DOI] [PubMed] [Google Scholar]
- Murray BW, Miller N (2015). Durability of kinase‐directed therapies – a network perspective on response and resistance. Mol Cancer Ther 14: 1975–1984. [DOI] [PubMed] [Google Scholar]
- Nguyen LK, Matallanas D, Croucher DR, von Kriegsheim A, Kholodenko BN (2013). Signalling by protein phosphatases and drug development: a systems‐centred view. FEBS J 280: 751–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikonova E, Tsyganov MA, Kolch W, Fey D, Kholodenko BN (2013). Control of the G‐protein cascade dynamics by GDP dissociation inhibitors. Mol Biosyst 9: 2454–2462. [DOI] [PubMed] [Google Scholar]
- Orton RJ, Adriaens ME, Gormand A, Sturm OE, Kolch W, Gilbert DR (2009). Computational modelling of cancerous mutations in the EGFR/ERK signalling pathway. BMC Syst Biol 3: 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A et al (2005). Tensional homeostasis and the malignant phenotype. Cancer Cell 8: 241–254. [DOI] [PubMed] [Google Scholar]
- Potter MD, Barbero S, Cheresh DA (2005). Tyrosine phosphorylation of VE‐cadherin prevents binding of p120‐ and beta‐catenin and maintains the cellular mesenchymal state. J Biol Chem 280: 31906–31912. [DOI] [PubMed] [Google Scholar]
- Prasadam I, Farnaghi S, Feng JQ, Gu W, Perry S, Crawford R et al (2013). Impact of extracellular matrix derived from osteoarthritis subchondral bone osteoblasts on osteocytes: role of integrinbeta1 and focal adhesion kinase signaling cues. Arthritis Res Ther 15: R150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath N, Morton JP, Julian L, Helbig L, Kadir S, McGhee EJ et al (2017). ROCK signaling promotes collagen remodeling to facilitate invasive pancreatic ductal adenocarcinoma tumor cell growth. EMBO Mol Med 9: 198–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remorino A, De Beco S, Cayrac F, Di Federico F, Cornilleau G, Gautreau A et al (2017). Gradients of Rac1 nanoclusters support spatial patterns of Rac1 signaling. Cell Rep 21: 1922–1935. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G et al (2003). Cell migration: integrating signals from front to back. Science 302: 1704–1709. [DOI] [PubMed] [Google Scholar]
- Ryu H, Chung M, Dobrzynski M, Fey D, Blum Y, Lee SS et al (2015). Frequency modulation of ERK activation dynamics rewires cell fate. Mol Syst Biol 11: 838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel MS, Lopez JI, McGhee EJ, Croft DR, Strachan D, Timpson P et al (2011). Actomyosin‐mediated cellular tension drives increased tissue stiffness and beta‐catenin activation to induce epidermal hyperplasia and tumor growth. Cancer Cell 19: 776–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuppan D, Afdhal NH (2008). Liver cirrhosis. Lancet 371: 838–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, Damsky CH et al (2000). FAK integrates growth‐factor and integrin signals to promote cell migration. Nat Cell Biol 2: 249–256. [DOI] [PubMed] [Google Scholar]
- Skhinas JN, Cox TR (2017). The interplay between extracellular matrix remodelling and kinase signalling in cancer progression and metastasis. Cell Adh Migr. 10.1080/19336918.2017.1405208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stabley D, Retterer S, Marshall S, Salaita K (2013). Manipulating the lateral diffusion of surface‐anchored EGF demonstrates that receptor clustering modulates phosphorylation levels. Integr Biol (Camb) 5: 659–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terfve CD, Wilkes EH, Casado P, Cutillas PR, Saez‐Rodriguez J (2015). Large‐scale models of signal propagation in human cells derived from discovery phosphoproteomic data. Nat Commun 6: 8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian T, Harding A, Inder K, Plowman S, Parton RG, Hancock JF (2007). Plasma membrane nanoswitches generate high‐fidelity Ras signal transduction. Nat Cell Biol 9: 905–914. [DOI] [PubMed] [Google Scholar]
- Timpson P, McGhee EJ, Morton JP, von Kriegsheim A, Schwarz JP, Karim SA et al (2011). Spatial regulation of RhoA activity during pancreatic cancer cell invasion driven by mutant p53. Cancer Res 71: 747–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsyganov MA, Kolch W, Kholodenko BN (2012). The topology design principles that determine the spatiotemporal dynamics of G‐protein cascades. Mol Biosyst 8: 730–743. [DOI] [PubMed] [Google Scholar]
- Umesh V, Rape AD, Ulrich TA, Kumar S (2014). Microenvironmental stiffness enhances glioma cell proliferation by stimulating epidermal growth factor receptor signaling. PLoS One 9: e101771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Weaver VM, Petersen OW, Larabell CA, Dedhar S, Briand P et al (1998). Reciprocal interactions between beta1‐integrin and epidermal growth factor receptor in three‐dimensional basement membrane breast cultures: a different perspective in epithelial biology. Proc Natl Acad Sci U S A 95: 14821–14826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells RG (2008). The role of matrix stiffness in regulating cell behavior. Hepatology 47: 1394–1400. [DOI] [PubMed] [Google Scholar]
- Zhao B, Pritchard JR, Lauffenburger DA, Hemann MT (2014). Addressing genetic tumor heterogeneity through computationally predictive combination therapy. Cancer Discov 4: 166–174. [DOI] [PMC free article] [PubMed] [Google Scholar]