

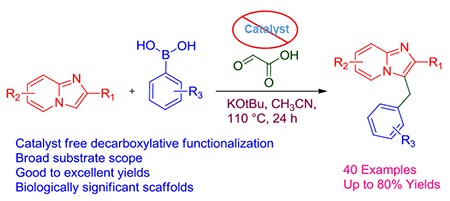
Abstract
Multicomoponenet reactions (MCRs) are robust tools for the rapid synthesis of complex, small molecule libraries for use in drug discovery and development. By utilizing MCR chemistry, we developed a protocol to functionalize the C-3 position of imidazo[1,2-a]pyridine through a three component, decarboxylation reaction involving imidazo[1,2-a]pyridine, glyoxalic acid, and boronic acid.
Graphical Abstract
Introduction
Multicomponent reactions (MCRs) facilitate the rapid generation of diverse, chemical libraries with complex form and function, which have vast use in drug discovery and development.1 MCRs represent a powerful tool to expeditiously expand chemical diversity and to reach novel, chemical space. Because of their utility in medicinal chemistry, MCRs have become an essential tool to increase hit-to-lead efficiency while decreasing time exhausted in the medicinal chemistry iterative cycle.2 The advantages of MCRs include one-pot synthesis, mild reaction conditions, and post-MCR functionalization to help constrain rotatable bonds and resolve stereochemistry.3 In an effort to develop MCR chemistries to help expand drug chemotypes, we designed a novel decarboxylative, Petasis-like three component reaction to functionalize the C-3 position of imidazo[1,2-a]pyridine.
The imidazo[1,2-a]pyridine core is found in pharmaceuticals and natural products that possess a broad range of biological and pharmacological activities such as anticancer,4 antibacterial,5 anti-viral,6 antifungal,7 antiprotozoal,8 anti-inflammatory and antiulcer.9 Imidazo[1,2-a]pyridines such as alpidem, necopidem, and saripidem are marketed as anxiolytic drugs10 and zolpidem is used to treat insomnia.11 Another derivative, minodronic acid, is used to treat osteoporosis12 and olprinone for heart failure.13 In particular, imidazo[1,2-a]pyridine derivatives are important for exploratory drug discovery research, which has led to the identification of novel kinase inhibitors with activities against PI3K, p38, Nek2, and NFkB inducing kinase.14 Owing to the biological importance of functionalized imidazo[1,2-a]pyridines, a variety of synthetic strategies have been developed to functionalize the heterocycle.15 The strategies require metal catalysis, multiple steps, and molar equivalence of oxidants. Therefore, rapid, operationally- simplistic, and eco-friendly methods are still needed to functionalize imidazo[1,2-a]pyridines for use in drug discovery.
Because of the biological importance of C-3 substituted imidazo[1,2-a]pyridines and the nucleophilic nature of the C-3 carbon, numerous C-H functionalization reactions, such as sulfonylation, arylation, amination, carbonylation, annulation and oxidative homocoupling, have been developed in recent years to enhance C-3 diversity.16 One-pot methods are available to construct 2,3-disubstituted imidazo[1,2-a]pyridines but all require metal catalysis.15a,17 Direct arylomethylations have been disclosed but are relatively rare, and neither methodology is catalyst free (Figure 1).18 Accordingly, functionalizing imidazo[1,2-a]pyridines through an operationally-simplistic, catalyst free reaction from commercially available starting materials is highly warranted.
Figure 1:
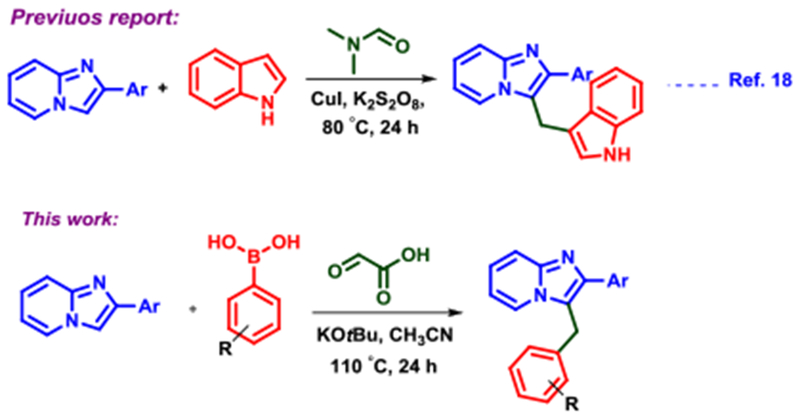
Literature precedence for C-3 functionalization of imidazo[1,2-a]pyridines
In the current protocol, we disclose an economical, catalyst free, eco-friendly MCR for the construction of aryl methane derivatives of imidazo[1,2-a]pyridine using commercially available boronic acids. The MCR was strategically designed to rapidly expand accessible chemotypes with an imidazo[1,2-a]pyridine core.
Results and discussion
We began our investigation for arylomethylation with 2-(4-methoxyphenyl)imidazo[1,2-a]pyridine 1a, glyoxylic acid 2a, and 4-methoxy phenylbronic acid 3a in dimethyl formamide (DMF) as solvent at 100 °C for 12 h. With these conditions, only 10% of the desired product 3aa was obtained, while the major products were a mixture of the imidazopyridine/glyoxylic acid adduct 3aab and the non-decarboxylated form of the desired product 3aac (Table 1, Entry 1).
Table 1:
Optimization of reaction conditionsa
 | |||||||
|---|---|---|---|---|---|---|---|
| S.No | Solvent | Promoter | Tempe. (°C) | Time (h) | % of Yieldb (3aa) | % of Yieldb (3aab) | % of Yieldb (3aac) |
| 1 | DMF | - | 100 | 12 | 10 | 50 | 15 |
| 2 | DMF | - | 120 | 24 | 30 | 60 | 20 |
| 3 | DMF | - | 110 | 24 | 40 | 50 | 30 |
| 4 | Dioxane | - | 110 | 24 | 30 | 25 | 40 |
| 5 | EtOH | - | 110 | 24 | - | 55 | 30 |
| 6 | EtOH/ H2O |
- | 110 | 24 | - | 50 | 25 |
| 7 | CH3CN | - | 110 | 24 | 40 | 50 | - |
| 8 | CH3CN | pTSA | 110 | 24 | 20 | 50 | - |
| 9* | CH3CN | KOtBu | 110 | 24 | 75 | 10 | - |
| 10 | DMF | KOtBu | 110 | 24 | 60 | 20 | - |
| 11 | CH3CN | NaOtBu | 110 | 24 | 65 | 30 | - |
| 12 | CH3CN | Cs2CO3 | 110 | 24 | 45 | 40 | - |
| 13 | CH3CN | KOtBu | 110 | 12 | 50 | 40 | |
| 14# | CH3CN | KOtBu | 110 | 24 | 60 | 40 | |
| 15 | DMF | PTSA | 110 | 24 | - | 50 | - |
| 16 | Toulene | KOtBu | 110 | 24 | - | 40 | - |
| 17 | DCE | KOtBu | 110 | 24 | - | 40 | - |
Reaction conditions: 1a (1 mmol), 2a (1.5 mmol), 3a (1.5 mmol)
Base, 1 mmol;
Base, 1.5 mmol; solvent (4.0 mL).
Isolated yield.
The yield of the desired product 3aa increased to 30% when the reaction was extended to 24 hours and heated to 120 °C (Table 1, Entry 2). This suggests that the desired decarboxylation of intermediate 3aac could be achieved at higher temperatures with longer reaction durations. Even with more aggressive conditions, however, satisfactory yields of 3aa were not obtained. Various solvents were investigated to help increase transformation but all failed to improve yield other than acetonitrile, which furnished 3aa in 40% yield (Table 1, Entry 7). Employment of p-toluenesulfonic acid to activate intermediate 3ab did not have a positive impact on overall yield (Table 1, Entry 8). Further investigation with various bases produced a dramatic improvement in yield (Table 1, Entry 9–14). With 1 eq. of KOtBu the isolated yield of the desired product 3aa increased to 75% (Table 1, Entry 9). Reaction optimization clearly indicated that a strong base, non-nucleophilic base is necessary to promote reaction progression and, to efficiently decarboxylate intermediate 3aac, an aprotic solvent is necessary.
With the optimized conditions in hand, we sequentially examined the substrate scope using commercially available boronic acids (Figure 2).
Figure 2:

Arylomethylation substrate scope with various boronic acids and imidazo[1,2-a]pyridinesa,b
aReactions were conducted with 1.0 mmol of 1a-1h, 1.5 mmol of 2a, 1.5 mmol of 3a-3s and 1.0 mmol of KOtBu in CH3CN at 110 °C for 24 h. bIsolated yields.
It was identified that electron donating boronic acids furnished good to excellent yields, while electron withdrawing boronic acids gave lower yields. However, strongly electron withdrawing boronic acids, such as -NO2, and -CN, were unable to produce the desired product. This suggests that electron density is important for product conversion, and the reaction is robust enough to handle mild electron withdrawing functionality. We further examined aryl boronic acids for transformation, such as bezothiophene and benzofuran (Figure 2, 3ap and 3aq). Both were successful in producing the desired product in good yields, but monocyclic hetero aryl boronic acids, such as pyridine, furan, and thiophene, did not furnish the desired product likely from delocalized electron density. The hindered substituted boronic acid 3an exhibited good transformation suggesting that steric effects do not have a large impact on product conversion.
Further, we diversified the imidazo[1,2-a]pyridine core using both electron rich and deficient functionalities and, among them, electron donating groups furnished excellent to good yields compared to their electron deficient counterparts (Figure 3). We expanded the imidazo[1,2-a]pyridine core to test transformation with more complex derivatives and observed excellent product conversion as seen with examples 4a-4n (Figure 3).
Figure 3:

More diverse imidazopyridines for arylomethylationa,b
aReactions were conducted with 1.0 mmol of 1j-1n & 1t-1x, 1.5 mmol of 2a, 1.5 mmol of 3a,3i & 3l and 1.0 mmol of KOtBu in CH3CN at 110 °C for 24 h. bIsolated yields.
Proposed reaction mechanism
Based on previous reports19 and control experiments, we proposed a mechanism for the arylomethylation reaction, which consists of a Petasis-like mechanism followed by decarboxylation to generate the desired product (Figure 3). To support the mechanism, we completed extensive control experiments and identified molecular ion peaks that correspond to intermediates 3aab and 3aac (See Supplemental Information). With this evidence, we proposed that the reaction initiates from the nucleophilic attack of glyoxylic acid by imidazo[1,2-a]pyridine to afford the stable intermediate 3aab through A. Under high temperature and basic conditions, the boronic acid can complex with intermediate 3aab to generate B. The resulting adduct is converted to intermediate C through phenyl migration to the benzylic position of imidazo[1,2-a]pyridine. The final step is decarboxylation of C to generate the desired arylomethylated product. It is important to note that intermediate 3aab can be isolated and converted to the desired product with addition of base and boronic acid, supporting the proposed mechanism (See Supplemental Information).
Biological Evaluation in Cancer Cell Lines
Products from the arylomethylation were screened against cancer cell lines to help identify novel chemotypes that impair cancer cell growth. The cell lines employed for the studies were HCC827 (EGFR-driven), LC-2/ad (RET-driven), and H460 (non-oncogene). We identified that 3aa, which was the first compound synthesized, did not exhibit strong activity against any cell line tested (GI50 > 1 μM). From screening the entire library of the arylomethylation series, it was identified that compounds 4g and 4d were able to potently inhibit cancer cell growth and exhibited sub-micromolar growth inhibition (GI50) values. The most active compound, 4g, exhibited a GI50 on LC-2/ad cells of 0.063 ± 0.029 μM. The compound exhibited some selectivity between H460 and LC-2/ad, suggesting 4g may be more active against RET-driven cell lines. Further studies are underway to determine the exact mechanism by which 4g and 4d elicit antiproliferative effects.
Conclusions
In conclusion, we have developed an innovative, catalyst free route to access distinctly substituted, C-3 arylomethylation derivatives of imidazo[1,2-a]pyridines. For ease of use, the method was developed to utilize commercially available boronic acids and glyoxylic acid, through a three component Petasis-like reaction followed by decarboxylation in one pot. The current protocol improves on prior methods, which all require the use of a metal catalyst and oxidizing agents. By using this methodology, we have achieved broad substrate scope with 39 variously substituted analogues, and we also evaluated antiproliferative activity in cancer cell lines. From the study, 4g and 4d were identified as promising hit, anticancer candidates, which support the use of this new methodology to identify novel, bioactive molecules.
Supplementary Material
Figure 4:

Proposed reaction mechanism
Table 3:
Antiproliferative activity and selectivity of 3aa, 4g, and 4da
| Cell GI50 (μM) | |||
|---|---|---|---|
| Comp. | H460 | HCC827 | LC-2/ad |
| 3aa | >1 | >1 | >1 |
| 4g | 0.15 ± 0.01 | 0.60 ± 0.18 | 0.063 ± 0.029 |
| 4d | 0.39 ± 0.07 | 2.03 ± 0.56 | 0.28 ± 0.040 |
GI50 values are expressed in μM units and are the results of three independent experiments.
Acknowledgements
This work was supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number “P20 GM109005”. H.L. was supported by the grants NIH 1R01CA194094-010 and 1R01CA197178-01A1 and UAMS start-up funding. B.F. was supported by the UAMS Seeds of Science cancer research grant, the UAMS College of Pharmacy Seed Grant, and an ATA/Thyca research grant.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.(a) Weber L, Curr. Med. Chem, 2002, 9, 2085–93 [DOI] [PubMed] [Google Scholar]; (b) Akritopoulou-Zanze I, Curr. Opin. Chem. Biol, 2008, 12, 324–331 [DOI] [PubMed] [Google Scholar]; (b) Naresh G, Kant R and Narender T, Org. Lett, 2014, 16, 4528–4531 [DOI] [PubMed] [Google Scholar]; (c) Ashitha KT, Praveen Kumar V, Fathimath Salfeena CT and Sasidhar BS, J. Org. Chem, 2018, 83, 113–124 [DOI] [PubMed] [Google Scholar]; (d) Naresh G, Kant R and Narender T, J. Org. Chem, 2014, 79, 3821–3829 [DOI] [PubMed] [Google Scholar]; (e) Naresh G and Narender T, RSC Adv, 2014, 4, 11862–11866 [Google Scholar]
- 2.(a) Hulme C, in Multicomponent Reactions, Wiley-VCH Verlag GmbH & Co; KGaA, Weinheim, FRG, 2005, 311–341 [Google Scholar]; (b) V Magedov I and Kornienko A, Chem. Heterocycl. Compd, 2012, 48, 33–38 [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Slobbe P, Ruijter E and Orru RVA, Medchemcomm, 2012, 3, 1189 [Google Scholar]; (d) Nagesh N, Raju G, Srinivas R, Ramesh P, Damoder reddy M and Rajireddy Ch., Biochimica et Biophysica Acta 2015, 1850, 129–140. [DOI] [PubMed] [Google Scholar]; (e) Lei J, Meng J-P, Tang D-Y, Frett B, Chen Z-Z and Xu Z-G, Mol. Divers, 2018, 22, 503–516. [DOI] [PubMed] [Google Scholar]
- 3.(a) Touré BB and Hall DG, Chem. Rev, 2009, 109, 4439–4486 [DOI] [PubMed] [Google Scholar]; (b) Schlegel M and Schneider C, Org. Lett, 2018, 20, 3119–3123 [DOI] [PubMed] [Google Scholar]; (c) Lin W, Zheng Y-X, Xun Z, Huang Z-B and Shi D-Q, ACS Comb. Sci, 2017, 19, 708–713 [DOI] [PubMed] [Google Scholar]; (d) Naresh G, Kant R and Narender T, Org. Lett, 2015, 17, 3446–3449 [DOI] [PubMed] [Google Scholar]; (e) Erb W, Neuville L and Zhu J, J. Org. Chem, 2009, 74, 3109–3115. [DOI] [PubMed] [Google Scholar]; (f) Sharma N, Li Z, Sharma UK and Van der Eycken EV, Org. Lett, 2014, 16, 3884–3887. [DOI] [PubMed] [Google Scholar]
- 4.(a) El-Sayed WM, Hussin WA, Al-Faiyz YS and Ismail MA, Eur. J. Pharmacol, 2013, 715, 212–218 [DOI] [PubMed] [Google Scholar]; (b) Kamal A, Reddy JS, Ramaiah MJ, Dastagiri D, Bharathi EV, Prem Sagar MV, Pushpavalli SNCVL, Ray P and Pal-Bhadra M, Medchemcomm, 2010, 1, 355 [Google Scholar]; (c) Kim O, Jeong Y, Lee H, Hong S-S and Hong S, J. Med. Chem, 2011, 54, 2455–2466 [DOI] [PubMed] [Google Scholar]
- 5.(a) Shukla NM, Salunke DB, Yoo E, Mutz CA, Balakrishna R and David SA, Bioorg. Med. Chem, 2012, 20, 5850–5863 [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Al-Tel TH, Al-Qawasmeh RA and Zaarour R, Eur. J. Med. Chem, 2011, 46, 1874–1881. [DOI] [PubMed] [Google Scholar]
- 6.(a) Feng S, Hong D, Wang B, Zheng X, Miao K, Wang L, Yun H, Gao L, Zhao S and Shen HC, ACS Med. Chem. Lett, 2015, 6, 359–62 [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Véron J-B, Allouchi H, Enguehard-Gueiffier C, Snoeck R, Andrei G, De Clercq E and Gueiffier A, Bioorg. Med. Chem, 2008, 16, 9536–9545. [DOI] [PubMed] [Google Scholar]
- 7.Rival Y, Grassy G, Taudou A and Ecalle R, Eur. J. Med. Chem, 1991, 26, 13–18. [Google Scholar]
- 8.Ismail MA, Arafa RK, Wenzler T, Brun R, Tanious FA, Wilson WD and Boykin DW, Bioorg. Med. Chem, 2008, 16, 683–691. [DOI] [PubMed] [Google Scholar]
- 9.(a) Lacerda RB, de Lima CKF, da Silva LL, Romeiro NC, Miranda ALP, Barreiro EJ and Fraga CAM, Bioorg. Med. Chem, 2009, 17, 74–84 [DOI] [PubMed] [Google Scholar]; (b) Kaminski JJ, Doweyko J AM. Med. Chem,1997, 40, 427–436. [DOI] [PubMed] [Google Scholar]
- 10.(a) George PG, Rossey G, Sevrin M, Arbilla S, Depoortere H and Wick AELERS,. Monograph Ser. 1993, 8, 49. [Google Scholar]; (b) Depoortere H and George P, US 5064836, 1991 [Google Scholar]; (c) Sanger DJ, Behav. Pharmacol. 1995, 6, 116. [PubMed] [Google Scholar]
- 11.Du B, Shan A, Zhong X, Zhang Y, Chen D and Cai K, Am. J. Med. Sci, 2014, 347, 178–182. [DOI] [PubMed] [Google Scholar]
- 12.Sorbera LA, Castañer J and Leeson PA, Drugs Future, 2002, 27, 935. [Google Scholar]
- 13.Uemura Y, Tanaka S, Ida S and Yuzuriha T, J. Pharm. Pharmacol, 1993, 45, 1077–81. [DOI] [PubMed] [Google Scholar]
- 14.(a) Rupert KC, Henry JR, Dodd JH, Wadsworth SA, Cavender DE, Olini GC, Fahmy B and Siekierka JJ, Bioorg. Med. Chem. Lett, 2003, 13, 347–50 [DOI] [PubMed] [Google Scholar]; (b) Xi J-B, Fang Y-F, Frett B, Zhu M-L, Zhu T, Kong Y-N, Guan F-J, Zhao Y, Zhang X-W, Li H, Ma M-L and Hu W, Eur. J. Med. Chem, 2017, 126, 1083–1106 [DOI] [PubMed] [Google Scholar]; (c) Demchenko YN, Brents LA, Li Z, Bergsagel LP, McGee LR and Kuehl MW, Oncotarget, 2014, 5, 4554–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Chernyak N and Gevorgyan V, Angew. Chemie Int. Ed., 2010, 49, 2743–2746 [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chioua M, Soriano E, Infantes L, Jimeno ML, Marco-Contelles J and Samadi A, European J. Org. Chem, 2013, 2013, 35–39 [Google Scholar]; (c) Huo C, Tang J, Xie H, Wang Y and Dong J, Org. Lett, 2016, 18, 1016–1019 [DOI] [PubMed] [Google Scholar]; (d) Shakoor SMA, Agarwal DS, Kumar A and Sakhuja R, Tetrahedron, 2016, 72, 645–652 [Google Scholar]; (e) Xie Y, Wu J, Che X, Chen Y, Huang H and Deng G-J, Green Chem, 2016, 18, 667–671. [Google Scholar]
- 16.(a) Guo Y-J, Lu S, Tian L-L, Huang E-L, Hao X-Q, Zhu X, Shao T and Song M-P, J. Org. Chem, 2018, 83, 338–349 [DOI] [PubMed] [Google Scholar]; (b) Choy PY, Luk KC, Wu Y, So CM, Wang L and Kwong FY, J. Org. Chem, 2015, 80, 1457–1463 [DOI] [PubMed] [Google Scholar]; (c) Kielesiński Ł, Tasior M and Gryko DT, Org. Chem. Front, 2015, 2, 21–28 [Google Scholar]; (d) Mondal S, Samanta S, Jana S and Hajra A, J. Org. Chem, 2017, 82, 4504–4510 [DOI] [PubMed] [Google Scholar]; (d) Shakoor SMA, Agarwal DS, Kumar A and Sakhuja R, Tetrahedron, 2016, 72, 645–652 [Google Scholar]; (e) Samanta S, Mondal S, Santra S, Kibriya G and Hajra A, J. Org. Chem, 2016, 81, 10088–10093. [DOI] [PubMed] [Google Scholar]
- 17.DiMauro EF and Kennedy JM, J.Org. Chem, 2007, 72, 1013–1016. [DOI] [PubMed] [Google Scholar]
- 18.Mondal S, Samanta S, Santra S, Bagdi AK and Hajra A, Adv. Synth. Catal, 2016, 358, 3633–3641. [Google Scholar]
- 19.Naskar D, Neogi S, Roy A and Mandal AB, Tetrahedron Lett, 2008, 49, 6762–6764. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.