

ABSTRACT
Immunogenicity is a key factor capable of influencing the efficacy and safety of therapeutic antibodies. A recently developed method called MHC-associated peptide proteomics (MAPPs) uses liquid chromatography/mass spectrometry to identify the peptide sequences derived from a therapeutic protein that are presented by major histocompatibility complex class II (MHC II) on antigen-presenting cells, and therefore may induce immunogenicity. In this study, we developed a MAPPs technique (called Ab-MAPPs) that has high throughput and can efficiently identify the MHC II-presented peptides derived from therapeutic antibodies using magnetic nanoparticle beads coated with a hydrophilic polymer in the immunoprecipitation process. The magnetic beads could identify more peptides and sequence regions originating from infliximab and adalimumab in a shorter measurement time than Sepharose beads, which are commonly used for MAPPs. Several sequence regions identified by Ab-MAPPs from infliximab corresponded to immunogenic sequences reported by other methods, which suggests the method’s high potential for identifying significant sequences involved in immunogenicity. Furthermore, our study suggests that the Ab-MAPPs method can recognize the difference of a single amino acid residue between similar antibody sequences with different levels of T-cell proliferation activity and can identify potentially immunogenic peptides with high binding affinity to MHC II. In conclusion, Ab-MAPPs is useful for identifying the immunogenic sequences of therapeutic antibodies and will contribute to the design of therapeutic antibodies with low immunogenicity during the drug discovery stage.
KEYWORDS: immunogenicity, MAPPs, MHC-associated peptide proteomics, magnetic beads, therapeutic antibodies, identification, MS, mass spectrometry, MHC II, major histocompatibility complex class II, T cell epitopes
Introduction
The recent surge in the development and launch of therapeutic antibodies across a broad range of indications can be attributed to their specific binding to target molecules and the lower toxicity that the specificity gives them compared to small-molecule drugs. However, one recognized shortcoming of therapeutic antibodies and other therapeutic protein products is their immunogenicity.1–3 Immunogenicity typically refers to the ability of a therapeutic product to provoke an immune response in a patient, such as the production of anti-drug antibodies (ADAs). Several health authorities have issued guidelines or guidance on the assessment of immunogenicity4,5 because of the disadvantages posed to patients by diminished drug efficacy and safety.6,7 The efficacy of a therapeutic antibody is diminished when ADAs that develop in response to the antibody affect its binding to the target molecule and its pharmacokinetic profile.7 For instance, previous studies have shown that the percentage of non-responders to infliximab or adalimumab therapy is higher among patients who have developed ADAs.8–10 The impact of immunogenicity on safety has also been documented, with reports of hypersensitivity and anaphylactic shock resulting from a type I or a type III allergy in patients treated with therapeutic antibodies and other therapeutic proteins.11,12 For these reasons, if a new therapeutic protein causes serious immunogenicity, it can lead to development being discontinued, so careful consideration of this issue is warranted.
The causes of immunogenic response to therapeutic proteins include patient-related factors, such as genetic background and immunity acquired from diseases and concomitant therapy, and product-related factors, such as the amino acid sequence of the drug.6,7 Although the amino acid sequences of therapeutic antibodies are derived from antibodies naturally occurring in the body, ADAs may develop in response to antigen-specific sequences in the complementarity-determining regions (CDRs) or, in the case of chimeric antibodies, to sequences derived from other animal species.13,14 Recent advances in the development of therapeutic antibodies have allowed amino acid sequences to be optimized to bestow desirable pharmacokinetic and physicochemical properties, and antibodies can also be highly engineered to achieve various functions. Because these engineered antibodies have amino acid sequences distinct from those of naturally occurring antibodies, it is reasonable to assume that they pose an increased risk of immunogenicity. The emergence of severe immunogenicity during clinical development is not only detrimental to patients, but can also be time-consuming and costly for companies because it reduces the feasibility of the clinical trial and can potentially derail the development process. It is therefore essential to assess the immunogenic potential of antibody candidates and minimize the possibility of immunogenicity conferred by amino acid sequences in the drug discovery stage.
ADAs are produced in response to treatment with therapeutic proteins by two known mechanisms: T cell–dependent and T cell–independent activation of B cells.15 T cell–dependent activation is considered to be the more important mechanism of the two because it produces IgG-class ADAs and develops memory B cells that produce higher-affinity, more robust, and more sustained antibody responses.7 In the T cell–dependent pathway, a therapeutic protein is first internalized by antigen-presenting cells (APCs), such as dendritic cells (DCs), and then degraded into short peptides. Some of these peptides are presented on major histocompatibility complex (MHC) class II (MHC II) molecules of the APCs, and the resulting peptide–MHC II complexes are then recognized by specific helper T cells. The activation of these cells triggers the activation of B cells, eventually leading to the production of ADAs.6,16 The structure of the therapeutic protein, particularly the amino acid sequence, has a large impact on the initial process of this immunogenic pathway, up to presentation on MHC II molecules.16 The presence of T cell epitopes (peptides presented on MHC II that are capable of activating T cells) in the amino acid sequence of a therapeutic antibody has a significant implication in determining the immunogenic potential of the antibody. Establishing a system to predict and assess T cell epitopes in amino acid sequences is therefore crucial in the development of therapeutic antibodies with low immunogenicity.
Potential T cell epitopes are generally predicted using in silico tools. The affinity and stability of T cell epitopes that bind to MHC II pockets are mainly defined by core residues limited in length to 9 to 10 amino acids,7 so in silico predictive tools are used to make computational predictions based on the available sequence data of MHC II-bound peptides. Several commercial in silico tools that predict T cell epitopes using their own unique algorithms have been described in the literature.7,17–19 However, it has been reported that in silico tools may be over-predictive and may not be able to predict immunodominant T cell epitopes properly.20,21 The literature on predicting T cell epitopes has also described two in vitro assays using synthetic peptides of 9 to 20 amino acids that are found in the amino acid sequences of therapeutic proteins to be evaluated for immunogenicity: 1) a binding assay to evaluate the binding affinity to MHC II,22 and 2) a T cell assay to evaluate the potential for inducing T cell activation.23 These in vitro assays are capable of comprehensively evaluating the whole sequence of a therapeutic protein through multiple analysis with libraries of overlapping peptides (known as epitope mapping). However, the assay process is highly labor-intensive and fails to consider actual intracellular processing events.20
Another technique used to detect MHC-presented peptides is liquid chromatography–mass spectrometry (LC/MS), a very powerful analytical tool in the fields of proteomics and peptidomics. Its use in the detection of MHC-presented peptides began in the field of vaccine development, where it was used to search for antigens presented on MHC class I.24,25 The analysis of peptides presented on MHC II that are associated with immunogenicity has recently come to be known as MHC-associated peptide proteomics (MAPPs).20 MAPPs has been practically applied to identifying T cell epitopes in birch pollen allergen and recombinant coagulation factor VIII product;26,27 however, there is still very little in the literature about the application of MAPPs to therapeutic antibodies.28–30 In the MAPPs technique, a therapeutic protein is co-incubated with DCs, the main type of APC, and the peptide fragments presented on MHC II molecules are then extracted and purified before identifying the amino acid sequences with LC/MS.20 As these peptides are naturally processed and presented on MHC II molecules, the MAPPs technique can better detect genuine T cell epitopes than in silico methods or binding assays using synthetic peptides. We therefore surmised that MAPPs would be useful for identifying the sequences of therapeutic antibodies responsible for inducing immunogenic responses. However, since immunogenic responses of therapeutic antibodies would be weaker than those of pathogens, allergens, and vaccines,30 a highly sensitive evaluation system is required. Additionally, as mentioned above, to produce therapeutic antibodies with low immunogenicity, it is important to evaluate the immunogenicity from the drug discovery stage, so the test system should ideally be capable of evaluating many drug candidates in a short period while using as few cells as possible.
With this in mind, we developed a highly sensitive, high-throughput MAPPs system for identifying the T cell epitopes of therapeutic antibodies (hereafter referred to as Ab-MAPPs). In the MAPPs procedures for immunoprecipitation (IP) and purification of peptide–MHC II complexes, we compared the commonly-used Sepharose beads27,31 with magnetic nanoparticle beads coated with a hydrophilic polymer, polyglycidyl methacrylate (pGMA) (FG magnetic beads). FG magnetic beads have high dispersibility and low non-specific binding of biomolecules; consequently, they have been reported to achieve good recovery of targets at a high level of purity in the affinity purification process.32 As model therapeutic antibodies, we used infliximab and adalimumab because they show high frequency of ADA incidence in clinical practice.33,34 We then compared the peptides and sequence regions derived from the antibodies when using each type of bead, and examined the effects of the LC gradient profile on the level of identification. By comparing the sequence regions identified in this study with those previously described in the literature, we evaluated the validity of the peptides identified by our proposed method.
Furthermore, to determine from a more practical perspective whether our proposed method can identify actual sequences with immunogenic potential, we compared the results of identifying the MHC II-presented peptides from two of our proprietary antibodies that, apart from two mutations, have very similar sequences. We then performed a binding assay to major MHC II allele types for the peptide sequences containing one of the mutations and a helper T cell assay for the antibodies, and examined their relevance to the results obtained by Ab-MAPPs. Based on our findings, we evaluated the usefulness of Ab-MAPPs in identifying T cell epitopes for the development of therapeutic antibodies with low immunogenicity.
Results
Comparison of the beads used for IP when identifying peptides presented on MHC II
Infliximab-derived peptide sequences identified by MAPPs using FG magnetic beads and the conventional Sepharose beads are shown in Figure 1.A and C. As reported in previous MAPPs studies,26,28 multiple length variants sharing the same MHC II-binding core sequence were observed (a set of variants is called a cluster), so the number of identified peptides per amino acid position is also summarized in heatmaps (Figure 1.B) and D)). In the samples from two donors, more peptides and clusters from the heavy and light chains of infliximab were identified when using FG magnetic beads than when using Sepharose beads (Table 1).
Figure 1.
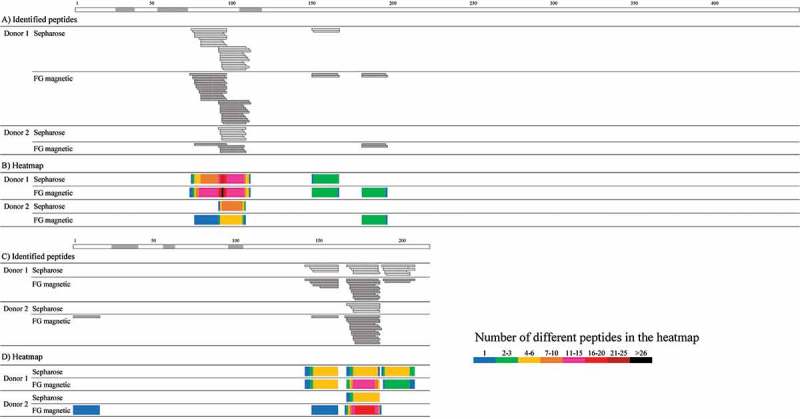
Peptide sequences in the A) heavy and C) light chains of infliximab identified by MAPPs using Sepharose or FG magnetic beads. The number of the identified peptides per amino acid position for the B) heavy and D) light chains are shown in heatmaps in which the cell colors represent the number of identified peptides (see key). The regions shown in gray above the information for the heavy and light chains represent CDR regions.35 Peptide-MHC II complexes were obtained from infliximab-pulsed DCs differentiated from 5.4 × 106 monocytes from two donors using Sepharose or FG magnetic beads coupled with almost the same amount of anti-HLA-DR antibody in the IP process overnight or for 2 hours of mixing time, respectively.
Table 1.
Summary of the number of infliximab- and adalimumab-derived peptides and clusters identified by MAPPs using Sepharose or FG magnetic beads.
| VH region |
CH region |
VL region |
CL region |
Total |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Infliximab and Adalimumab | LC gradient time (h) | Cluster | Peptide | Cluster | Peptide | Cluster | Peptide | Cluster | Peptide | Cluster | Peptide | |
| Infliximab | ||||||||||||
| Donor 1 | Sepharose | 1 | 2 | 23 | 1 | 2 | 0 | 0 | 3 | 15 | 6 | 40 |
| |
FG magnetic |
1 |
2 |
28 |
2 |
4 |
0 |
0 |
3 |
19 |
7 |
51 |
| Donor 2 | Sepharose | 1 | 1 | 7 | 0 | 0 | 0 | 0 | 1 | 5 | 2 | 12 |
| |
FG magnetic |
1 |
2 |
5 |
1 |
2 |
1 |
1 |
2 |
17 |
6 |
25 |
| Adalimumab | ||||||||||||
| Donor 3 | Sepharose | 1 | 2 | 24 | 0 | 0 | 2 | 3 | 1 | 7 | 5 | 34 |
| 2 | 3 | 30 | 0 | 0 | 3 | 6 | 1 | 6 | 7 | 42 | ||
| FG magnetic | 1 | 3 | 33 | 0 | 0 | 3 | 11 | 1 | 10 | 7 | 54 | |
| |
|
2 |
3 |
34 |
0 |
0 |
3 |
10 |
1 |
2 |
7 |
46 |
| Donor 4 | Sepharose | 1 | 1 | 17 | 1 | 2 | 0 | 0 | 1 | 1 | 3 | 20 |
| 2 | 1 | 21 | 2 | 2 | 0 | 0 | 0 | 0 | 3 | 23 | ||
| FG magnetic | 1 | 1 | 21 | 1 | 1 | 0 | 0 | 1 | 1 | 3 | 23 | |
| 2 | 1 | 23 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 23 | ||
VH, CH, VL, and CL represent variable and constant regions of the heavy chain and variable and constant regions of the light chain, respectively.
Furthermore, DC samples pulsed with adalimumab were measured by LC/MS and compared at different LC gradient times of about 1 and 2 hours. Adalimumab-derived peptide sequences identified by MAPPs using FG magnetic beads and Sepharose beads are shown in heatmaps (Figure 2). When the results from the two types of beads were compared at the same LC gradient time (1 hour), more peptides and clusters of adalimumab tended to be identified with FG magnetic beads than with Sepharose beads, especially in Donor 3 (Table 1). By extending the gradient time up to 2 hours, the number of peptides and clusters identified using the Sepharose beads grew larger and was close to the results obtained using FG magnetic beads, but the number identified using the FG magnetic beads did not increase at the longer time.
Figure 2.
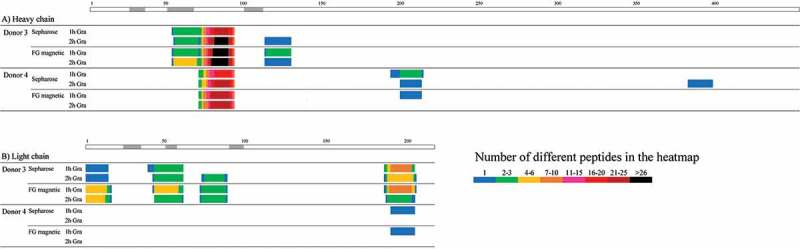
Heatmaps of the number of the peptides per amino acid position identified for the A) heavy and B) light chains of adalimumab by MAPPs using Sepharose or FG magnetic beads at different LC gradient times. The regions shown in gray above the information for the heavy and light chains represent CDR regions.36 Peptide-MHC II complexes were obtained from adalimumab-pulsed DCs differentiated from 5.4 × 106 monocytes from two donors using the two types of beads coupled with almost the same amount of anti-HLA-DR antibody in the IP process, with mixing overnight. The obtained peptide mixture was separated on a C18 reversed-phase nano-capillary column with a 2%-37% acetonitrile gradient containing 0.5% acetic acid for approximately 1 hour (69 min) or 2 hours (138 min).
A portion of each LC/MS sample from DCs pulsed with infliximab was loaded on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). As a result of silver-staining to evaluate the degree of purification by IP, the samples pretreated with Sepharose beads showed more and thicker bands than those with FG magnetic beads (Figure 3). Furthermore, the amount of contamination interfering with the identification of MHC II-presented peptides was verified by comparing the total ion chromatograms (TICs) taken from the MAPPs samples obtained using the two types of beads. The background level in the sample pretreated with Sepharose beads was considerably higher than with FG magnetic beads, especially in the range of the retention time at which hydrophobic peptides were eluted (Figure 4a and 4b). Accordingly, the mass chromatograms of the peptides identified only by FG magnetic beads showed that these peptides were buried in the background noise in the sample pretreated with Sepharose beads (Figure 4c-4f).
Figure 3.
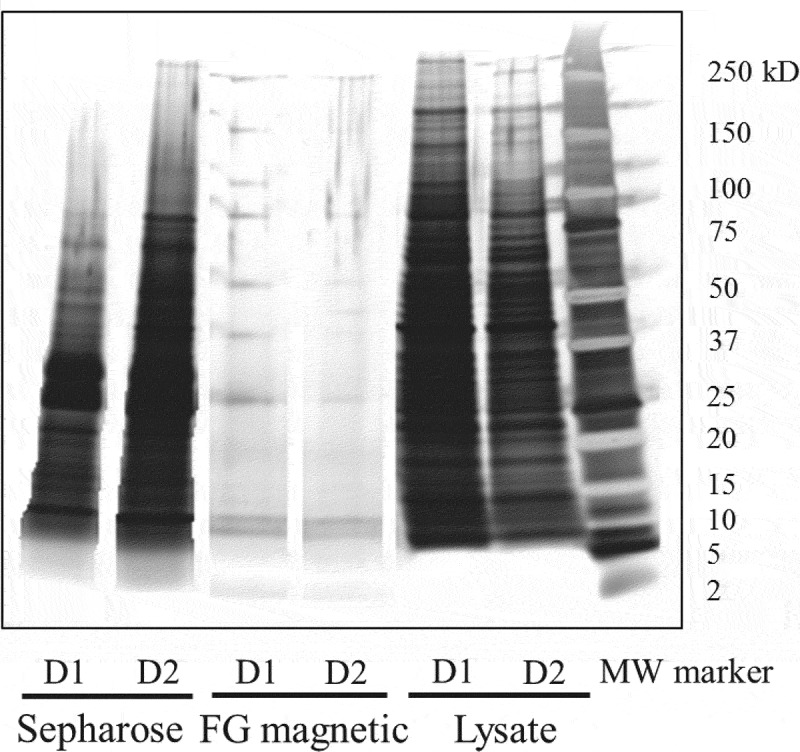
SDS-PAGE/silver staining of MAPPs samples pretreated with Sepharose or FG magnetic beads. MAPPs samples were the same as the samples used to obtain data in Figure 1. Approximately one third of the volume of the MAPPs samples obtained in the IP process was applied. Lysate samples before IP were applied at 1 μg/lane (approximately 0.2%-0.3% of the total volume of the lysates). D1 and D2 mean Donors 1 and 2, respectively.
Figure 4.
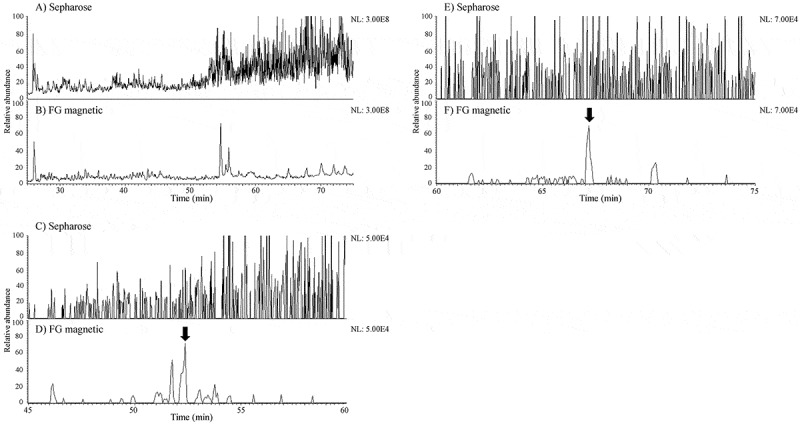
Typical TICs of MAPPs samples obtained by A) Sepharose or B) FG magnetic beads and mass chromatograms of the peptides identified only by FG magnetic beads. Mass chromatograms of the heavy chain FR3 peptide of infliximab in the samples obtained by C) Sepharose and D) FG magnetic beads and of the light chain FR1 peptide of infliximab in the samples obtained by E) Sepharose and F) FG magnetic beads from Donor 2. The arrows represent the peaks of the peptides concerned.
Characterization of Ab-MAPPs using FG magnetic beads
The number of MHC II-presented peptides identified by Ab-MAPPs in samples obtained from different amounts of lysate were compared. Lysates of DCs differentiated from 1.4, 2.7, and 5.4 × 106 monocytes were pretreated and then measured in the same batch of LC/MS. The sequence regions that were identified for infliximab were almost the same in each sample from the same donor, although some failures in the identification of the sequence regions were observed in the samples from 1.4 × 106 monocytes. As shown in Figure 5, the number of peptides identified in each cluster, as well as their peak intensity (data not shown), tended to increase according to the amount of lysate used.
Figure 5.
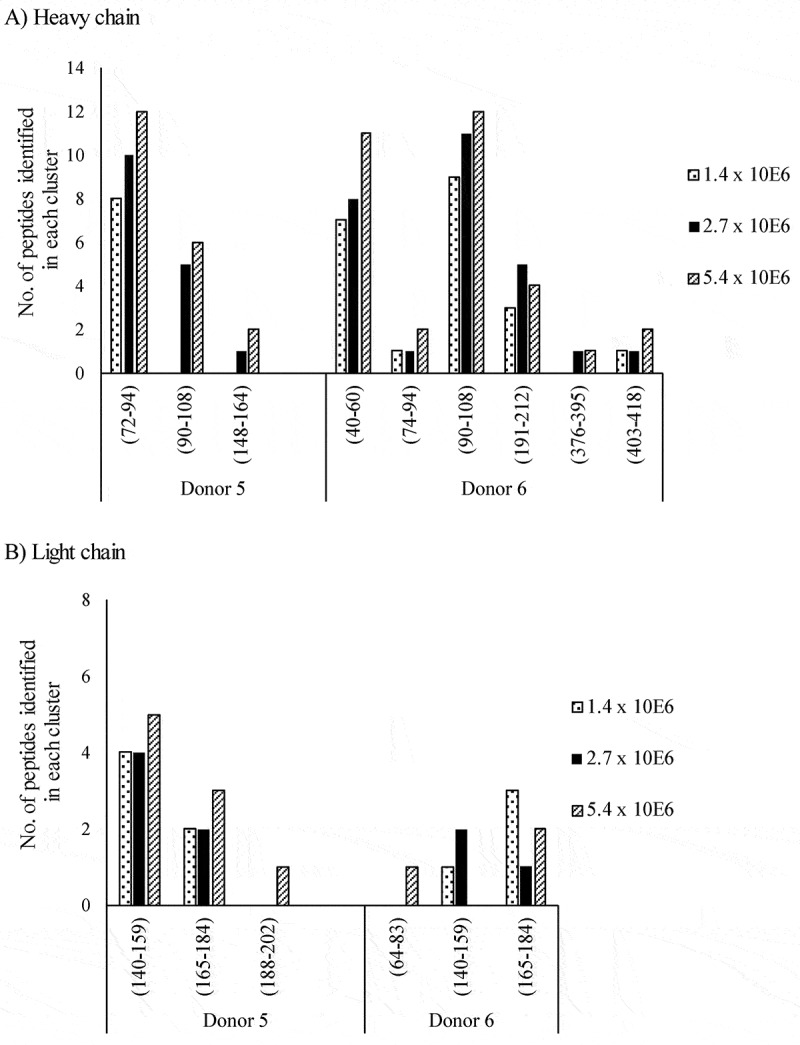
The number of peptides identified in each cluster for the A) heavy and B) light chains of infliximab obtained from different amounts of DCs using FG magnetic beads on Ab-MAPPs. Peptide-MHC II complexes were obtained from infliximab-pulsed DCs differentiated from 1.4, 2.7, or 5.4 × 106 monocytes using FG magnetic beads coupled with the anti-HLA-DR antibody in the IP process for 2 hours of mixing time.
To verify the reproducibility of the Ab-MAPPs assay, three infliximab-pulsed samples taken from each of three donors were immunoprecipitated and measured on different days. As a result, the sequence regions identified for infliximab were almost the same between the three samples derived from one donor, in all three donors tested (Figure 6). In addition, there was little difference in the number of peptides identified in each cluster between samples from the same donor. No infliximab-derived peptides were identified in the control samples that had not been pulsed with infliximab.
Figure 6.
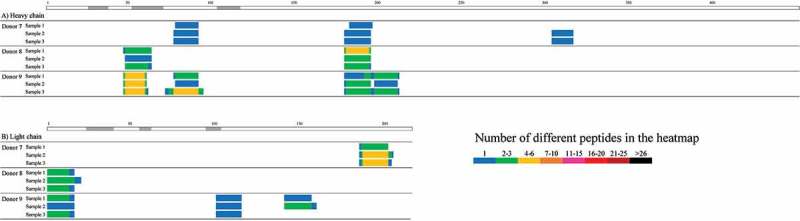
Ab-MAPPs heatmaps of the number of peptides identified for the A) heavy and B) light chains of infliximab in three individual samples pretreated and measured on different days to evaluate the reproducibility. Peptide-MHC II complexes were obtained from infliximab-pulsed DCs differentiated from 2.7 × 106 monocytes using FG magnetic beads coupled with the anti-HLA-DR antibody that were immunoprecipitated overnight.
Comparison of Ab-MAPPs with in silico prediction for a model therapeutic antibody
Sequence regions of infliximab identified by Ab-MAPPs were compared with sequences in the antibody that were predicted by the in silico tool EpiMatrix to have the potential to bind to human leukocyte antigen (HLA)-DR molecules (Figure 7). MHC II-binding core sequences for almost all of the peptides identified by Ab-MAPPs appeared to overlap with the sequences predicted in silico.
Figure 7.

Comparison of the sequences in A) heavy and B) light chains of infliximab that were identified by Ab-MAPPs with those predicted in silico or those reported to induce T cell responses. Data from Ab-MAPPs represent the sequences identified in each donor evaluated. Data from the in silico tool represent the amino acid positions of sequences predicted as hits in two or more (dark blue) and in one (pale blue) out of four allele types possessed by the donors of the infliximab samples used for Ab-MAPPs and available for prediction by EpiMatrix (HLA-DRB1*0401, 0701, 1101, and 1301). Sequences in the variable regions of infliximab that have a potential to induce T cell responses were reported by Hellendoorn et al.37 (1) and Hamze et al.38 (2), based on data using 30 and 15 donors, respectively. The regions shown in gray above the information for the heavy and light chains represent CDR regions.35.
Peptides in the variable regions of infliximab reported in literature to induce T cell proliferation37,38 are also shown in Figure 7. Many of the sequence regions identified by Ab-MAPPs corresponded to the peptides that are known to activate T cells.
Monoclonal antibodies with similar sequences evaluated by Ab-MAPPs, a binding assay, and a helper T cell assay
Heatmaps of Ab-MAPPs for two of our proprietary monoclonal antibodies (mAb1 and mAb2) that have very similar sequences are shown in Figure 8. We evaluated samples from 10 donors that include a variety of HLA-DRB1 allele types and cover most of the major types (Table S1),39 because we presumed that more donors were needed to compare two antibodies with such similar sequences. As the figure shows, their sequences are only different by two amino acid positions. In the presumed framework region 3 (FR3) of the heavy chain containing one of the mutations, many peptides were identified in eight of the 10 donors tested for mAb1, whereas no peptides were identified in any of 10 donors for mAb2. In sequence regions other than the above heavy chain FR3, the peptides identified in the two mAbs were similar. No peptides derived from the two antibodies were identified in control samples from any of the four donors tested, which were not pulsed with either antibody.
Figure 8.
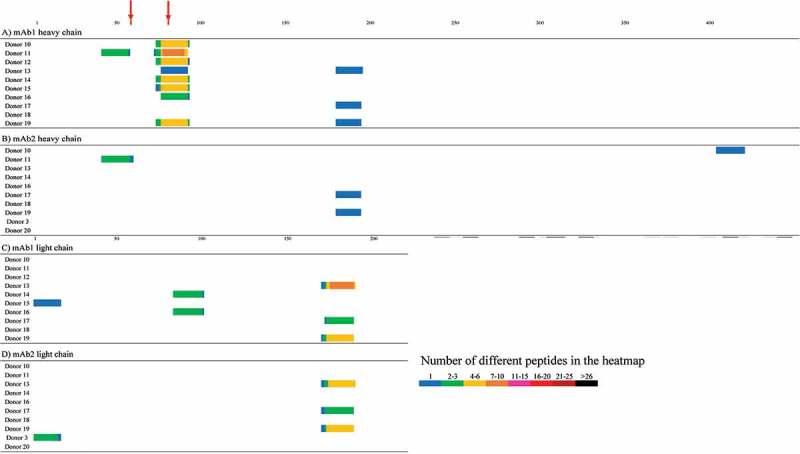
Ab-MAPPs heatmaps of the number of the identified peptides in the heavy chains of A) mAb1 and B) mAb2 and light chains of C) mAb1 and D) mAb2. Peptide-MHC II complexes were obtained from mAb1- or mAb2-pulsed DCs differentiated from 2.7 × 106 monocytes using FG magnetic beads coupled with the anti-HLA-DR antibody in the IP process for 2 hours of mixing time. The red cells indicate the mutation points between the two mAbs.
The peptides of the heavy chain FR3 region for mAb1 and mAb2 that contain one mutation, SKNTVYLQIDSLRAEDTA and SKNTVDLQIDSLRAEDTA, were synthesized, and their binding affinity to eight major HLA-DR allele types (DRB1*0101, 0301, 0401, 0701, 0801, 1101, 1301, 1501) was evaluated. As a result of a competition-based binding assay with purified HLA-DR molecules, the IC50 values of the mAb1 peptide were equal to or less than 10 μM for the seven allele types other than *0801, while the mAb2 peptide showed larger IC50 than 10 μM for the seven allele types other than *0101, suggesting that the mAb1 peptide binds considerably more potently to major HLA-DR allele types than the mAb2 peptide (Table 2).
Table 2.
Binding affinity of mAb1 and mAb2 FR3 peptides to major HLA-DRB1 allele types. Binding of the evaluated peptides is expressed as the inhibition of binding of the labeled standard peptide.
| IC50 (nM) | ||||||||
|---|---|---|---|---|---|---|---|---|
| HLA-DRB1 | *0101 | *0301 | *0401 | *0701 | *0801 | *1101 | *1301 | *1501 |
| mAb1 peptide | 57 | 10,396 | 202 | 1,379 | >100,000 | 1,914 | 1,967 | 3,336 |
| mAb2 peptide | 5,583 | 46,529 | >100,000 | >100,000 | >100,000 | >100,000 | >100,000 | >100,000 |
The potential of mAb1 and mAb2 to induce T cell proliferation was evaluated in a helper T cell assay. As an index of T cell proliferation activity, the stimulation index (SI) was estimated in 10 to 20 donors for the number of bromodeoxyuridine-positive (BrdU+) helper T cells when cluster of differentiation (CD) 8-negative (CD8−) and CD25low peripheral blood mononuclear cells (PBMCs) were cultured with the test antibody (Figure 9). The degree of T cell proliferation by the negative control etanercept, which was reported to show a low frequency of ADA incidence in clinic,33,40 was totally dulled, but the positive control humanized A33 (hA33), which was reported to show a high frequency of ADA incidence in clinical practice,16 induced more intense T cell proliferation. These results confirm that the helper T cell assay appropriately evaluated the immunogenic potential. In the assay, SI values of mAb1 were estimated at a similar level to the positive control; on the other hand, mAb2 showed reduced SI values compared to mAb1, but slightly higher than the negative control.
Figure 9.
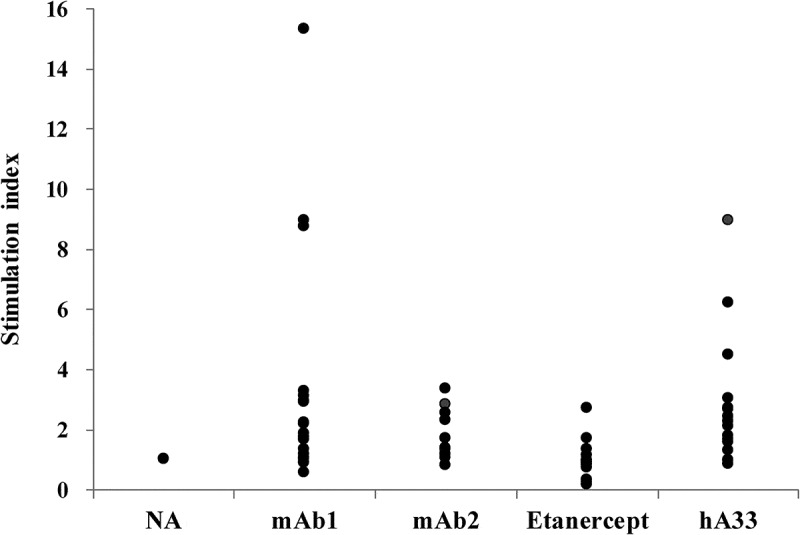
T cell proliferation activity of mAb1 and mAb2 in the helper T cell assay. The antibodies were added at 100 μg/mL to CD8− CD25low PBMCs of 10 to 20 donors and cultivated for 5, 6, or 7 days. Stimulation index represents the maximum value of ratios of the number of BrdU+ helper T cells in the antibody-treated sample to that in the no-antigen (NA) sample through the cultivation period for each donor.
Discussion
Immunogenicity is a key factor capable of influencing the efficacy and safety of therapeutic antibodies.6 We developed the Ab-MAPPs system with higher sensitivity and throughput so that it can be applied to immunogenic research on therapeutic antibodies in the drug discovery stage.
IP and purification of peptide–MHC complexes using beads bound to an anti-MHC antibody has been used in the past as an effective technique for detecting MHC-presented peptides.24,27,28,41 Because these assays can be made more sensitive by pretreatment that efficiently recovers targets at a high purity, the purity and efficiency achieved by FG magnetic beads was compared with that achieved by Sepharose beads. As a result of a LC/MS analysis of the MHC II-presented peptides extracted and purified from DCs, more peptides and sequence regions originating from the model antibodies, infliximab and adalimumab, were identified by the FG magnetic beads bound with anti-HLA-DR antibodies than by the Sepharose beads (Figure 1, 2; Table 1). In particular, the number of sequence regions identified is critical because candidate sequences for T cell epitopes could be overlooked. Additionally, the number of peptides and regions identified by Sepharose beads tended to increase according to the length of gradient time, and a longer LC gradient time appeared to be needed for Sepharose beads to obtain results comparable to FG magnetic beads. One of the benefits of FG magnetic beads is that their surface is coated with pGMA polymer. This hydrophilic polymer provides FG magnetic beads with their characteristic features of high dispersibility and low non-specific binding of biomolecules,32 which means that a higher signal-to-noise ratio can be achieved by efficiently capturing target molecules in the IP process and reducing contamination. Actually, as shown by the results of SDS-PAGE/silver staining (Figure 3) and TICs (Figure 4a and 4b), the surface-treated FG magnetic beads had significantly less contamination by non-specific binding than the Sepharose beads, which have a porous surface. The influence of contamination appears to be well illustrated by the fact that almost all of the peptides identified only by FG magnetic beads were observed in the range of the retention time at which the two types of beads produced very different background levels. When using Sepharose beads, contaminant proteins and peptides could interfere with the ionization and spectral matching in tandem MS for target peptides, which may reduce the number of identified peptides and obstruct attempts to shorten the measurement time. Additionally, in our preliminary study that compared FG magnetic beads with another type of magnetic bead from a different manufacturer, the number of identified peptides derived from infliximab was reduced (data not shown), suggesting that not all magnetic beads can detect MHC II-presented peptides well. The above findings suggest that a higher-throughput assay technique that is better at identifying MHC II-presented peptides can be developed using FG magnetic beads than using Sepharose beads. Practically speaking, FG magnetic beads not only reduce the measurement time, but are also easy to use in experiments, which makes them suitable for treating many samples in the drug discovery stage, whereas Sepharose beads need more complex handling than the magnetic beads.
To further characterize our proposed technique of using FG magnetic beads, samples pretreated from different amounts of lysate obtained from DCs pulsed with infliximab were analyzed in the same LC/MS batch to evaluate the MHC II-presented peptides. The results showed that almost all of the same regions were identified for the same donors regardless of lysate amount, and that the number of identified peptides (Figure 5) and their peak intensities (data not shown) tended to increase with the amount of lysate. We also performed IP and LC/MS assays on three separate infliximab samples on separate days, and were able to identify nearly all of the same sequences in the same donors on each occasion (Figure 6), thereby demonstrating good reproducibility between batches. Proteomics techniques generally do not possess good reproducibility, but the above findings suggest that our proposed technique is robust and is also capable of indicating relative amounts of MHC II-presented peptides in DC samples when comparing their amounts in the same cluster. In terms of the number of cells used in our assay, the samples pretreated with DCs obtained from 1.4 × 106 monocytes did include sporadic identification failures, such as a failure to identify the peptides straddling the FR3 and CDR3 of the heavy chain in Donor 5. However, there were virtually no differences in the identified sequence regions between the samples derived from 2.7 × 106 and 5.4 × 106 monocytes (Figure 5), which suggests that 2.7 × 106 monocytes (equivalent to approximately 1 × 106 DCs based on 30%–50% of differentiation rate from monocytes) would enable Ab-MAPPs to identify MHC II-presented sequences appropriately. The number of cells needed for Ab-MAPPs is less than that previously reported for MAPPs assays using Sepharose beads (5 × 106 or more of DCs27,28,31). Moreover, whereas previous studies describe the use of fresh PBMCs,28,30,31 we demonstrated the feasibility of using frozen PBMCs as described in Materials and Methods. Our Ab-MAPPs system identified most of the sequence regions reported recently by Hamze et al. for infliximab by a MAPPs technique using fresh PBMCs,38 although the number of donors used in our experiments was limited. Because the Ab-MAPPs system can evaluate immunogenicity using only a few frozen cells, it would certainly be practical for analyzing multiple antibodies in the drug discovery phase.
Another crucial question that needs to be answered is whether the MAPPs system including our proposed method is actually capable of identifying immunogenic sequences that are clinically significant. Although the problems caused by therapeutic antibodies with high immunogenicity are known, there are very few reports in the literature that describe attempts to identify T cell epitopes in the amino acid sequence of immunogenic therapeutic antibodies. As indicated in Figure 7, the sequence regions causing immunogenicity in the amino acid sequence of infliximab are supported by data from T cell assays using numerous overlapping synthetic peptides from the variable regions of the heavy and light chains, and also by reported data for several modified antibodies confirmed by T cell assay to have reduced immunogenicity.37,38 Since T cell assays have been reported as a tool for predicting immunogenic potency in clinical settings to some extent,30 it is highly likely that the above-mentioned sequences are in fact clinically relevant T cell epitopes. Our Ab-MAPPs data on infliximab was obtained from a limited number of donors, but it identified multiple sequence regions corresponding to the potential T cell epitopes mentioned above. This would suggest that our test system is indeed capable of identifying significant immunogenic sequences, although further verification may be needed using other therapeutic antibodies after their immunogenic sequences have been revealed in the future. Additionally, the disparity in identifying these immunogenic sequences of infliximab between FG magnetic and Sepharose beads (Figure 1) would point to the usefulness of our proposed method using FG magnetic beads.
In silico tools have often been used to predict T cell epitopes. In this study, the comparison of the MHC II-presented sequence regions identified shows that many of the infliximab-derived sequences that were isolated using Ab-MAPPs were also predicted by the in silico tool. EpiMatrix is a tool that predicts MHC-presented peptides by calculating the MHC binding affinity of peptides with nine residues in their protein sequence, based on the pocket profiles of MHC molecules.19,42 So, this finding would suggest that Ab-MAPPs is very good at identifying peptides with high binding affinity to MHC II. Furthermore, the MAPPs technique can directly identify peptides naturally presented on MHC II and comprehensively evaluate whole sequences (not only the CDR regions) of engineered therapeutic antibodies more easily than other in vitro methods. On the other hand, in vitro assays using a synthetic peptide library, such as a T cell assay23 and an ELISpot assay,43 can observe T cell responses to the peptide sequences. In silico tools have advantages in time- and cost-efficiency when obtaining information on potential T cell epitopes presented by many different HLA allele types because they can rapidly predict computationally, without doing experiments that use cell materials, whereas in vitro assays including Ab-MAPPs have a practical limitation of the comprehensiveness of HLA allele types to get a broad coverage of the population. In addition, some in silico tools can predict whether the responding T cells will be effector or regulatory.44 Therefore, we consider that Ab-MAPPs would be effective for designing therapeutic antibodies with low immunogenicity, and using it in conjunction with in silico tools and other in vitro assays will increase its efficiency.
To further demonstrate the reliability of Ab-MAPPs in identifying T cell epitopes, we used the technique to identify the MHC II-presented peptides in two of our proprietary antibodies (mAb1 and mAb2) with very similar sequences. Although the antibody sequences differ by only two amino acid residues, a drastic difference was observed in the Ab-MAPPs results. In other words, due to the presence of a single mutation in the heavy chain FR3 region, the peptides containing this mutation position were identified as the main presented region in almost all of the evaluated donor samples for mAb1, but were not identified in any of the donor samples for mAb2 (Figure 8). By synthesizing the two peptides from the FR3 region and estimating their binding affinities to the major HLA-DR allele types, we found that the FR3 peptide in mAb2 had lower binding affinity than that in mAb1 to almost all of the eight evaluated allele types (Table 2). Given the fact that the FR3 region of mAb1 retains its animal-derived sequence while the FR3 region of mAb2 has a humanized sequence, these binding assay results demonstrate that Ab-MAPPs possesses the following two important features: 1) The system could recognize the difference in a single residue and identify a sequence of non-human origin likely to induce an immunogenic response; and 2) The identification results would be consistent with the HLA-DR binding affinity.
Furthermore, by taking into account data from the helper T cell assay for the two very similar mAbs, we evaluated how useful Ab-MAPPs would be for attenuating the immunogenicity of therapeutic antibodies. The helper T cell assay evaluated the T cell-activating potential and revealed that mAb1 induced the same or greater activation as the positive control, whereas the activation by mAb2 was considerably diminished (Figure 9). While these two antibodies have not been used in clinical practice, the helper T cell assays yielded results (data not shown) that are largely consistent with reported clinical findings on immunogenicity,6,30 so mAb1 could be predicted to induce a high level of immunogenicity if it were used in clinical treatment. We suspect that the difference in the T cell-activating potential of the two antibodies despite their remarkably similar sequences can most likely be attributed to the sequence region in which the mutation is contained. As mentioned above, our proposed Ab-MAPPs system detected a major difference in the peptide presentation of this altered region, which suggests that Ab-MAPPs was able to properly identify the immunogenic sequence of mAb1. The finding of our study illustrates that the comparison of sequences identified by Ab-MAPPs and T cell-activating potentials found in a helper T cell assay between antibodies with sequential similarity may be a useful approach for identifying the sequences responsible for immunogenicity. In addition, apart from the FR3 region, the sequence regions identified from mAb1 and mAb2 by Ab-MAPPs were considerably coincident, as would be expected from their close sequence similarity. However, the presumed FR3-CDR3 region in the light chain of mAb2 was not identified, even though the two antibodies have identical light chain sequences. Although the precise reason why it could not be identified is not clear, it might be caused by changes in the proteolytic cleavage pattern and presenting priority, which are affected by heavy chain modification in the steric structure of the antibody. In this evaluation, we used samples from 10 donors that included a variety of HLA-DRB1 allele types and covered most of the major types (Table S1).39 We think that it could be a better way to meticulously cover the major allele types, especially in the drug discovery stage, because designing efficiently therapeutic antibodies with low immunogenicity is required in that stage. By accumulating more data on therapeutic antibodies in the future, we anticipate that our system will contribute to more precise evaluation and more efficient development of therapeutic antibodies with low immunogenicity.
In conclusion, we succeeded in developing an Ab-MAPPs technique to identify MHC II-presented peptides derived from therapeutic antibodies. The technique was suggested to have high identification performance, high throughput, and good reproducibility, resulting from the use of magnetic nanoparticle beads, which produce a very low level of contamination due to non-specific binding, in the IP and purification processes. Several sequence regions from infliximab (which causes high frequency of ADA incidence in clinical practice) that were identified using our Ab-MAPPs technique corresponded to the T cell epitopes obtained by other methods, which suggests that our proposed method would be capable of identifying significant T cell epitopes involved in immunogenicity. A comparison of antibodies with similar sequences suggested that our Ab-MAPPs technique could recognize the difference in a single residue and generate identification results that are consistent with HLA-DR binding affinity. These findings suggest that the Ab-MAPPs technique is a useful tool for identifying the immunogenic sequences of therapeutic antibodies, and we expect that the use of this technique during drug discovery will contribute to the development of therapeutic antibodies with low immunogenicity.
Materials and methods
Ethics statement
The use of human-derived materials in this research was approved by the Research Ethics Committee of Chugai Pharmaceutical Co., Ltd. In our preliminary studies, we found the use of cryopreserved PBMCs for the MAPPs assay to be feasible, whereas it was difficult to use cryopreserved PBMCs for the helper T cell assay because it caused the attenuation of T cell activation responses (data not shown). Since there is a limit to the number of volunteers in our company, we used cryopreserved PBMCs only for the MAPPs assay. The peripheral blood samples for the helper T cell assay were donated by 20 healthy Japanese volunteers after written informed consent was obtained, and the samples were anonymized.
Antibodies and peptides evaluated
Model therapeutic antibodies and fusion protein, infliximab, adalimumab, and etanercept, were purchased from Centocor, Inc. (Horsham, PA; Remicade®), Abbott Laboratories (Chicago, IL; Humira®), and Pfizer Inc. (New York, NY; Enbrel®), respectively. An immunogenic model antibody, hA33, was prepared by Life Technologies Corp. according to its previously disclosed amino acid sequence.45 Two of our proprietary antibody variants, mAb1 and mAb2, were expressed transiently in HEK293 cells transfected with plasmids encoding the heavy and light chains and purified from culture supernatants using protein A affinity chromatography and gel permeation chromatography as in a common method.
Synthetic peptides evaluated in the binding assay were prepared by GenScript Corp.
MAPPs assay
Preparation of mature DCs pulsed with therapeutic antibodies
Cryopreserved PBMCs used in the MAPPs assay were purchased from Lonza Ltd. (Basel, Switzerland). The HLA-DRB1 allele types of the donors for the PBMCs are summarized in Table S1. CD14+ mononuclear cells (1.4 to 5.4 × 106 cells) were obtained from cryopreserved PBMCs using anti-CD14 microbeads (Miltenyi Biotec GmbH, 130–050-201) according to the manufacturer’s protocol and differentiated into immature DCs in reference to a previous report.28 Immature DCs were matured by adding lipopolysaccharide (Sigma-Aldrich Co., LLC., 5886) at 1 μg/mL and loaded with the therapeutic antibody to be evaluated.
Immobilization of anti-HLA-DR antibodies on the beads for IP
To isolate peptide-MHC II complexes by IP, an anti-human HLA-DR antibody, clone G46-6 (Becton, Dickinson and Company, 555809) was immobilized on two types of beads. CNBr-activated Sepharose 4B beads (GE Healthcare UK Ltd., 17–0430-01) or FG NHS magnetic beads (Tamagawa Seiki Co, Ltd., TAS8848N1141) were used to covalently immobilize the anti-HLA-DR antibody at a ratio of 1 mg/100 mg beads or 0.5 mg/10 mg beads, respectively, according to the manufacturers’ protocols. The binding ratio of the added antibody was estimated with Coomassie Plus reagent (Thermo Fisher Scientific Inc., 1856210) by comparing the protein concentration of the supernatant after immobilization with that of the antibody solution before immobilization. Binding ratios of Sepharose and FG magnetic beads were more than 90% and 70%-80%, respectively.
Isolation of HLA-DR-associated peptides
To immunoprecipitate the peptide-MHC II complexes, the two types of beads coupled with the anti-HLA-DR antibody were used. Mature DCs derived from 1.4 to 5.4 × 106 of monocytes were lysed in lysis buffer (20 mM Tris buffer (pH 7.8) containing 5 mM MgCl2, 1% Triton X-100 (Roche Diagnostics, 11332481001), and protease inhibitors) for 1 hour at 4°C. After centrifugation, the lysate was incubated with anti-HLA-DR antibody coupled to either Sepharose or FG magnetic beads, using almost the same amount of anti-HLA-DR antibody with each type of bead. For samples treated with Sepharose beads, after incubation overnight at 4°C, the beads were washed several times with lysis buffer and phosphate-buffered saline containing 0.1% Zwittergent 3–12 (Merck KGaA, 693015) and ultrapure water in turn, by reference to a previous method.28 For samples treated with FG magnetic beads, after incubation for 2 hours to overnight at 4°C, the beads were washed several times with binding/wash buffer described in the manufacturer’s protocol and ultrapure water in turn. After washing, peptides were eluted from the beads by adding 0.1% trifluoroacetic acid (Thermo Fisher Scientific, Inc., 28904) at 37°C and dried with a centrifugal concentrator, Plus 5305C (Eppendorf AG, Hamburg, Germany).
Identification of HLA-DR-associated peptides with LC/MS
The peptide mixtures obtained were reconstituted in acidic water containing 2% acetonitrile, and approximately one-third of the sample volume was injected onto a nano-LC/MS system. Chromatographic separation was achieved using Ultimate 3000 RSLCnano system (Thermo Fisher Scientific, Inc., Waltham, MA) with a C18 reversed phase nano-capillary column (3 μm, 75 μm x 120 mm) (Nikkyo Technos Co., Ltd., Tokyo, Japan), and the eluate was ionized under nano-electrospray, positive-ion mode and measured using an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific, Inc.). Peptides were separated on the column with a 2%-37% acetonitrile gradient containing 0.5% acetic acid for 69 or 138 min (only when evaluating the effects of gradient time for adalimumab samples) at 250 nL/min of flow rate. MS data acquisition was performed by data-dependent scan mode. Collision-induced dissociation in the ion-trap was performed for doubly and triply charged precursor ions selected from each full scan in the Orbitrap.
Peptides were identified with Sequest search algorithm against Swiss-prot human database containing the sequence of the evaluated therapeutic antibody using Proteome Discoverer software ver. 1.4 (Thermo Fisher Scientific, Inc.). Peptides showing more than 2.3 or 2.8 of cross-correlation value (Xcorr) for doubly or triply charged ions, respectively, and less than 0.1 of the delta cross-correlation (dCn) were considered as true hits, by reference to a previous report.28
SDS-PAGE/silver staining
The MAPPs samples prepared from DCs of Donors 1 and 2 using Sepharose or FG magnetic beads were subjected to SDS-PAGE analysis. Aliquots of the LC/MS samples equivalent to approximately one-third of the sample volume finally obtained through the pretreatment process were applied. For the comparison with the LC/MS samples, lysate samples before IP were also applied at 1 μg/lane (approximately 0.2%-0.3% of the total volume of the lysates). Each sample added Tris-glycine SDS buffer containing 12% mercaptoethanol was applied on a 4%-20% polyacrylamide gel (Bio-Rad Laboratories, Inc., 456–1096) for the electrophoresis. The gel was stained by a conventional silver staining method using silver stain II kit Wako (Wako Pure Chemical Industries, Ltd., 291–50301). The image was captured with an image analyzer LAS-4000 (Fujifilm Corp., Tokyo, Japan), and protein contaminants were compared between the samples prepared with Sepharose and FG magnetic beads.
Helper T cell assay
A helper T cell assay was conducted using fresh PBMCs from healthy volunteers as described below. In order to guarantee the assay results, hA33 was selected as a positive control and etanercept as a negative control, which were reported to show high and low frequency of ADA incidence in clinical practice, respectively.16,33,40
Cell culture
Purified CD8− CD25low PBMCs were used for cell culture and measurement of the proliferation activity of CD4+ T cells. PBMCs were isolated from the peripheral blood of 10 to 20 healthy volunteers by density-gradient centrifugation with Ficoll-Paque PLUS (GE Healthcare UK Ltd., 17–1440-03), and CD8+ PBMCs were removed using a magnetic cell-sorting system with beads conjugated with antibodies against CD8 (Life Technologies Corp., 11147D) according to the manufacturer’s instructions. The sorted CD8− PBMCs were incubated with beads conjugated with antibodies against CD25 (Life Technologies Corp., 11157D) to remove CD25high cells. Because it was reported that the presence of CD4+ CD25high cells inhibited antigen-induced activation responses by CD4+ CD25− responder T cells,46 we removed CD25high cells when we evaluated the effector potential, so that their regulatory effects would be attenuated. The sorted CD8− CD25low PBMCs were seeded at 2 × 106 cells/mL/well in AIM-V Medium (Life Technologies Corp., 12055–91) containing 3% AB-serum (Sigma-Aldrich Co., LLC., H3667), and incubated in a humidified atmosphere of 5% CO2 at 37°C for two hours. After addition of test antibodies at 100 μg/mL, the cells were cultured for a total of 7 days under 5% CO2 at 37°C (20, 20, 10, 14 or 16 donors for no antigen, mAb1, mAb2, etanercept, or hA33 group, respectively).
Measurement of T cell proliferation
The proliferation activity of helper T cells was measured with BrdU+ cell proliferation using a BrdU Flow kit (Becton, Dickinson and Company, 559619) and flow cytometry instrument, BD FACSCanto II (Becton, Dickinson and Company). After 5, 6, and 7 days of the cultivation explained above, aliquots of cells in each well were transferred, and BrdU was added. After 1-day cultivation, the cells were stained with anti-human CD3 antibody (Becton, Dickinson and Company, 557597), anti-human CD4 antibody (Becton, Dickinson and Company, 550630), and anti-human CD14 antibody (Becton, Dickinson and Company, 558121), and BrdU+ helper T cells in the subset of CD4+ CD3+ CD14− cells were measured in the flow cytometry analysis. FlowJo ver. 7.6.2 software (Tomy Digital Biology Co., Ltd., Tokyo, Japan) was used for data analysis. The SI value was determined as the ratio of the number of BrdU+ helper T cells of the test antibody-treated sample to that of the untreated sample. The maximum SI in all of the cultivation time points was used as the index of the helper T cell proliferation activity for each donor sample.
Binding assay
The binding affinity of peptides to MHC II was evaluated by EpiVax, Inc. in a competition-based HLA binding assay, as reported previously.39 Briefly, unlabeled peptides to be evaluated were resolved in dimethyl sulfoxide and then added at 0.1 to 100 μM of the final concentrations to 0.1 μM of biotinylated standard peptide and 50 nM of purified HLA-DR molecules. After incubation for 24 hours at 37°C to reach steady equilibrium, peptide-HLA-DR complexes were captured on enzyme-linked immunosorbent assay plates coated with an anti-HLA-DR antibody, clone L243 (Bio X Cell, Inc., BE0160). After wash and incubation with Europium-labeled Streptavidin (Perkin-Elmer Inc., 1244–360) for 1 hour at ambient temperatures, time-resolved fluorescence to measure the bound labeled standard peptides was assessed by a SpectraMax M5 plate reader (Molecular Devices, LLC., Sunnyvale, CA). All assays were performed in triplicate. Binding affinity was evaluated for eight common allele types representing more than 90% of human populations: DRB1*0101, 0301, 0401, 0701, 0801, 1101, 1301, and 1501.39 Binding of the evaluated peptides was expressed as the percent inhibition of the labeled standard peptide, and the concentration of the evaluated peptides at which it inhibited 50% of the binding of the labeled standard peptide (IC50) was calculated.
In silico prediction
Peptides in the sequences of the therapeutic antibodies to have a potential to bind to HLA-DR were predicted by EpiMatrix (EpiVax, Inc.), an in silico tool. The Z-scores, which indicate the potential to bind to each major HLA-DR allele type (HLA-DRB1*0101, 0301, 0401, 0701, 0801, 1101, 1301, and 1501), of overlapping 9-mer peptides in the test sequence were estimated, and the peptide sequences showing a Z score of more than 1.64 were considered hits.
Abbreviations
- ADA
anti-drug antibody
- APC
antigen-presenting cell
- BrdU
bromodeoxyuridine
- CD
cluster of differentiation
- CDR
complementarity-determining region
- DC
dendritic cell
- FR
framework region
- hA33
humanized A33
- HLA
human leukocyte antigen
- IP
immunoprecipitation
- LC
liquid chromatography
- mAb
monoclonal antibody
- MAPPs
MHC-associated peptide proteomics
- MHC
major histocompatibility complex
- MHC II
major histocompatibility complex class II
- MS
mass spectrometry
- PBMC
peripheral blood mononuclear cell
- pGMA
polyglycidyl methacrylate
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- SI
stimulation index
- TIC
total ion chromatogram
Acknowledgments
The authors would like to sincerely thank Dr. Tatiana Pimenova, Dr. Heather Hinton, Dr. Anja Langenkamp, Dr. Thomas Weiser, Dr. Thomas Singer, and their coworkers of Immunosafety group of Roche Pharmaceutical Research and Early Development for their enormous supports and helpful advices on establishing the Ab-MAPPs technique. We would like to express our gratitude to Frances Terry, MPH and Anne S. De Groot, MD of EpiVax for obtaining very significant data in the binding assay. We are deeply grateful to Mr. Yasuhiko Nakata, Ms. Ayumi Maeno, Ms. Sachie Hiranabe, Ms. Motoko Okamoto, Mr. Atsushi Ohba, and their coworkers of Chugai Research Institute for Medical Science, Inc. for their technical supports of generating and preparing the mAb1 and mAb2. We would like to express our appreciation to Dr. Tomoyuki Igawa, Dr. Masayuki Mishima, Dr. Zenjiro Sampei, Mr. Taichi Kuramochi, and Dr. Kunihiro Hattori of Chugai Pharmaceutical for their insightful suggestions on planning the studies and interpreting the results. We also thank Mr. Daniel Anley and Ms. Sally Matsuura of Chugai Pharmaceutical for their scientific writing assistance of this paper.
Disclosure statement
The binding assay was conducted in EpiVax, Inc. under the finance from Chugai Pharmaceutical Co., Ltd.
Supplemental material
Supplemental data for this article can be accessed here.
References
- 1.Bhogal N. Immunotoxicity and immunogenicity of biopharmaceuticals: design concepts and safety assessment. Curr Drug Saf. 2010;5:293–307. PMID:20615176 https://www.ncbi.nlm.nih.gov/pubmed/20615176. [DOI] [PubMed] [Google Scholar]
- 2.Buttel IC, Voller K, Schneider CK. Immunogenicity and its impact on benefit/risk considerations in the authorization of biopharmaceuticals. Curr Drug Saf. 2010;5:287–292. PMID:20615175 https://www.ncbi.nlm.nih.gov/pubmed/20615175. [DOI] [PubMed] [Google Scholar]
- 3.Kloks C, Berger C, Cortez P, Dean Y, Heinrich J, Jensen LB, Koppenburg V, Kostense S, Kramer D, Spindeldreher S, et al. A fit-for-purpose strategy for the risk-based immunogenicity testing of biotherapeutics: a European industry perspective. J Immunol Methods. 2015;417:1–9. PMID:25602137 https://www.ncbi.nlm.nih.gov/pubmed/25602137. [DOI] [PubMed] [Google Scholar]
- 4.European Medicines Agency, Committee for Medicinal Products for Human Use (CHMP) Guideline on immunogenicity assessment of monoclonal antibodies intended for in vivo clinical use. [Online] http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128688.pdf.
- 5.US Food and Drug Administration, Center for Drug Evaluation and Research (CDER) and Center for Biologics Evaluation and Research (CBER) Guidance for industry; immunogenicity assessment for therapeutics protein products. [Online] https://www.fda.gov/downloads/drugs/guidances/ucm338856.pdf.
- 6.Kuriakose A, Chirmule N, Nair P. Immunogenicity of biotherapeutics: causes and association with posttranslational modifications. J Immunol Res. 2016;Epub; PMID:27437405 https://www.ncbi.nlm.nih.gov/pubmed/27437405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jawa V, Cousens LP, Awwad M, Wakshull E, Kropshofer H, De Groot AS. T-cell dependent immunogenicity of protein therapeutics: preclinical assessment and mitigation. Clin Immunol. 2013;149:534–555. PMID:24263283 https://www.ncbi.nlm.nih.gov/pubmed/24263283. [DOI] [PubMed] [Google Scholar]
- 8.West RL, Zelinkova Z, Wolbink GJ, Kuipers EJ, Stokkers PCF, Van Der Woude CJ. Immunogenicity negatively influences the outcome of adalimumab treatment in Crohn’s disease. Aliment Pharmacol Ther. 2008;28:1122–1126. PMID:18691349 https://www.ncbi.nlm.nih.gov/pubmed/18691349. [DOI] [PubMed] [Google Scholar]
- 9.Bartelds GM, Krieckaert CLM, Nurmohamed MT, Van Schouwenburg PA, Lems WF, Twisk JWR, Dijkmans BAC, Aarden L, Wolbink GJ. Development of antidrug antibodies against adalimumab and association with disease activity and treatment failure during long-term follow-up. JAMA. 2011;305:1460–1468. PMID:21486979 https://www.ncbi.nlm.nih.gov/pubmed/21486979. [DOI] [PubMed] [Google Scholar]
- 10.Pascual-Salcedo D, Plasencia C, Ramiro S, Nuno L, Bonilla G, Nagore D, Ruiz Del Agua A, Martinez A, Aarden L, Martin-Mola E, et al. Influence of immunogenicity on the efficacy of long-term treatment with infliximab in rheumatoid arthritis. Rheumatology (Oxford). 2011;50:1445–1452. PMID:21427177 https://www.ncbi.nlm.nih.gov/pubmed/21427177. [DOI] [PubMed] [Google Scholar]
- 11.Niimi S. Effects of immunogenicity of biopharmaceuticals to pharmacokinetics, efficacy, and safety, and its mitigation strategy (Japanese). Pharm Med Device Regul Sci Soc Japan. 2013;44:114–122. http://ci.nii.ac.jp/naid/40019586829. [Google Scholar]
- 12.Koren E, Zuckerman LA, Mire-Sluis AR. Immune response to therapeutic proteins in human – clinical significance, assessment and prediction. Curr Pharm Biotechnol. 2002;3:349–360. PMID:12463417 https://www.ncbi.nlm.nih.gov/pubmed/12463417. [DOI] [PubMed] [Google Scholar]
- 13.Hwang WYK, Foote J. Immunogenicity of engineered antibodies. Methods. 2005;36:3–10. PMID:15848070 https://www.ncbi.nlm.nih.gov/pubmed/15848070. [DOI] [PubMed] [Google Scholar]
- 14.Harding FA, Stickler MM, Razo J, DuBridge R. The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions. MAbs. 2010;2:256–265. PMID:20400861 https://www.ncbi.nlm.nih.gov/pubmed/20400861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zubler RH. Naïve and memory B cells in T-cell-dependent and T-independent responses. Springer Semin Immunopathol. 2001;23:405–419. PMID:11826617 https://www.ncbi.nlm.nih.gov/pubmed/11826617. [DOI] [PubMed] [Google Scholar]
- 16.Van Walle I, Gansemans Y, Parren PW, Stas P, Lasters I. Immunogenicity screening in protein drug development. Expert Opin Biol Ther. 2007;7:405–418. PMID:17309332 https://www.ncbi.nlm.nih.gov/pubmed/17309332. [DOI] [PubMed] [Google Scholar]
- 17.Bian H, Hammer J. Discovery of promiscuous HLA-II-restricted T cell epitopes with TEPITOPE. Methods. 2004;34:468–475. PMID:15542373 https://www.ncbi.nlm.nih.gov/pubmed/15542373. [DOI] [PubMed] [Google Scholar]
- 18.Nielsen M, Lundegaard C, Lund O. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinformatics. 2007;8:238 PMID:17608956 https://www.ncbi.nlm.nih.gov/pubmed/17608956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Groot AS, McMurry J, Moise L. Prediction of immunogenicity: in silico paradigms, ex vivo and in vivo correlates. Curr Opin Pharamacol. 2008;8:620–626. PMID:18775515 https://www.ncbi.nlm.nih.gov/pubmed/18775515. [DOI] [PubMed] [Google Scholar]
- 20.Kropshofer H, Singer T. Overview of cell-based tools for pre-clinical assessment of immunogenicity of biotherapeutics. J Immunotoxicol. 2006;3:131–136. PMID:18958693 https://www.ncbi.nlm.nih.gov/pubmed/18958693. [DOI] [PubMed] [Google Scholar]
- 21.Niimi S. Immunogenicity prediction methods of biopharmaceuticals (Japanese). Pharm Med Device Regul Sci Soc Japan. 2013;44:26–35. http://ci.nii.ac.jp/naid/40019556786. [Google Scholar]
- 22.Tangri S, Mothe BR, Eisenbraun J, Sidney J, Southwood S, Briggs K, Zinckgraf J, Bilsel P, Newman M, Chesnut R, et al. Rationally engineered therapeutic proteins with reduced immunogenicity. J Immunol. 2005;174:3187–3196. PMID:15749848 https://www.ncbi.nlm.nih.gov/pubmed/15749848. [DOI] [PubMed] [Google Scholar]
- 23.Jahn-Schmid B, Radakovics A, Luttkopf D, Scheurer S, Vieths S, Ebner C, Bohle B. Bet v 1 (142-156) is the dominant T-cell epitope of the major birch pollen allergen and important for cross-reactivity with Bet v 1-related food allergens. J Allergy Clin Immunol. 2005;116:213–219. PMID: 15990797 https://www.ncbi.nlm.nih.gov/pubmed/15990797. [DOI] [PubMed] [Google Scholar]
- 24.Van Der Heeft E, Ten Hove GJ, Herberts CA, Meiring HD, Van Els CAC, De Jong APJM. A microcapillary column switching HPLC-electrospray ionization MS system for the direct identification of peptides presented by major histocompatibility complex class I molecules. Anal Chem. 1998;70:3742–3751. PMID: 9751018 https://www.ncbi.nlm.nih.gov/pubmed/9751018. [DOI] [PubMed] [Google Scholar]
- 25.Poland GA, Ovsyannikova IG, Johnson KL, Naylor S. The role of mass spectrometry in vaccine development. Vaccine. 2001;19:2692–2700. PMID:11257411 https://www.ncbi.nlm.nih.gov/pubmed/11257411. [DOI] [PubMed] [Google Scholar]
- 26.Mutschlechner S, Egger M, Briza P, Wallner M, Lackner P, Karle A, Vogt AB, Fischer GF, Bohle B, Ferreira F. Naturally processed T cell-activating peptides of the major birch pollen allergen. J Allergy Clin Immunol. 2010;125:711–718. PMID:20132976 https://www.ncbi.nlm.nih.gov/pubmed/20132976. [DOI] [PubMed] [Google Scholar]
- 27.Van Haren SD, Wroblewska A, Herczenik E, Kaijen PH, Ruminska A, Ten Brinke A, Meijer AB, Voorberg J. Limited promiscuity of HLA-DRB1 presented peptides derived of blood coagulation factor VIII. PLoS One. 2013;8:e80239 PMID:24244658 https://www.ncbi.nlm.nih.gov/pubmed/24244658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rombach-Riegraf V, Karle AC, Wolf B, Sorde L, Koepke S, Gottlieb S, Krieg J, Djidja MC, Baban A, Spindeldreher S, et al. Aggregation of human recombinant monoclonal antibodies influences the capacity of dendritic cells to stimulate adaptive T-cell responses in vitro. PLoS One. 2014;9:e86322 PMID:24466023 https://www.ncbi.nlm.nih.gov/pubmed/24466023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xue L, Hickling T, Song R, Nowak J, Rup B. Contribution of enhanced engagement of antigen presentation machinery to the clinical immunogenicity of a human interleukin (IL)-21 receptor-blocking therapeutic antibody. Clin Exp Immunol. 2016;183:102–113. PMID:26400440 https://www.ncbi.nlm.nih.gov/pubmed/26400440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karle A, Spindeldreher S, Kolbinger F. Secukinumab, a novel anti-IL-17A antibody, shows low immunogenicity potential in human in vitro assays comparable to other marketed biotherapeutics with low clinical immunogenicity. MAbs. 2016;8:536–550. PMID:26817498 https://www.ncbi.nlm.nih.gov/pubmed/26817498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karle AC, Oostingh GJ, Mutschlechner S, Ferreira F, Lackner P, Bohle B, Fischer GF, Vogt AB, Duschl A. Nitration of the pollen allergen bet v 1.0101 enhances the presentation of bet v 1-derived peptides by HLA-DR on human dendritic cells. PLos One. 2012;7:e31483 PMID:22348091 https://www.ncbi.nlm.nih.gov/pubmed/22348091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nishio K, Masaike Y, Ikeda M, Narimatsu H, Gokon N, Tsubouchi S, Hatakeyama M, Sakamoto S, Hanyu N, Sandhu A, et al. Development of novel magnetic nano-carriers for high-performance affinity purification. Colloids Surf B Biointerfaces. 2008;64:162–169. PMID:18313904 https://www.ncbi.nlm.nih.gov/pubmed/18313904. [DOI] [PubMed] [Google Scholar]
- 33.Hsu L, Armstrong AW. Anti-drug antibodies in psoriasis: a critical evaluation of clinical significance and impact on treatment response. Expert Rev Clin Immunol. 2013;9:949–958. PMID:24128157 https://www.ncbi.nlm.nih.gov/pubmed/24128157. [DOI] [PubMed] [Google Scholar]
- 34.Krieckaert CLM, Bartelds GM, Lems WF, Wolbink GJ. The effect of immunomodulators on the immunogenicity of TNF-blocking therapeutic monoclonal antibodies: a review. Arthritis Res Ther. 2010;12:217 PMID:21029481 https://www.ncbi.nlm.nih.gov/pubmed/21029481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fang J, Doneanu C, Alley WR Jr., Yu YQ, Beck A, Chen W. Advanced assessment of the physicochemical characteristics of Remicade and Inflectra by sensitive LC/MS techniques. MAbs. 2016;8:1021–1034. PMID:27260215 https://www.ncbi.nlm.nih.gov/pubmed/27260215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang X, Das TK, Singh SK, Kumar S. Potential aggregation prone regions in biotherapeutics: a survey of commercial monoclonal antibodies. MAbs. 2009;1:254–267. PMID:20065649 https://www.ncbi.nlm.nih.gov/pubmed/20065649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hellendoorn K, Baker M, Carr FJ. TNF alpha-binding polypeptide compositions and methods. United States Patent, Patent No.: US7754853B2; 2004filed; [Online] http://patft.uspto.gov/netacgi/nph-Parser?Sect2=PTO1&Sect2=HITOFF&p=1&u=/netahtml/PTO/search-bool.html&r=1&f=G&l=50&d=PALL&RefSrch=yes&Query=PN/7754853.
- 38.Hamze M, Meunier S, Karle A, Gdoura A, Goudet A, Szely N, Pallardy M, Carbonnel F, Spindeldreher S, Mariette X, et al. Characterization of CD4 T cell epitopes of infliximab and rituximab identified from healthy donors. Front Immunol. 2017;8:500 PMID:28529511 https://www.ncbi.nlm.nih.gov/pubmed/28529511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moise L, Song C, Martin WD, Tassone R, De Groot AS, Scott DW. Effect of HLA DR epitope de-immunization of Factor VIII in vitro and in vivo. Clin Immunol. 2012;142:320–331. PMID:22222093 https://www.ncbi.nlm.nih.gov/pubmed/22222093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anderson P, Louie J, Lau A, Broder M. Mechanisms of differential immunogenicity of tumor necrosis factor inhibitors. Curr Rheumatol Rep. 2005;7:3–9. PMID:15760575 https://www.ncbi.nlm.nih.gov/pubmed/15760575. [DOI] [PubMed] [Google Scholar]
- 41.Costantino CM, Spooner E, Ploegh HL, Hafler DA. Class II MHC self-antigen presentation in human B and T lymphocytes. PLoS One. 2012;7:e29805 PMID:22299025 https://www.ncbi.nlm.nih.gov/pubmed/22299025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weber CA, Mehta PJ, Ardito M, Moise L, Martin B, De Groot AS. T cell epitope: friend or foe? Immunogenicity of biologics in context. Adv Drug Deliv Rev. 2009;61:965–976. PMID:19619593 https://www.ncbi.nlm.nih.gov/pubmed/19619593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mazor R, Vassall AN, Eberle JA, Beers R, Weldon JE, Venzon DJ, Tsang KY, Benhar I, Pastan I. Identification and elimination of an immunodominant T-cell epitope in recombinant immunotoxins based on Pseudomonas exotoxin A. Proc Natl Acad Sci USA. 2012;109:E3597–603. PMID:23213206 https://www.ncbi.nlm.nih.gov/pubmed/23213206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Groot AS, Moise L, McMurry JA, Wambre E, Van Overtvelt L, Moingeon P, Scott DW, Martin W. Activation of natural regulatory T cells by IgG Fc-derived peptide “Tregitopes. Blood. 2008;112:3303–3311. PMID:18660382 https://www.ncbi.nlm.nih.gov/pubmed/18660382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.King DJ, Adair JR, Owens RJ. Humanized antibodies directed against A33 antigen. International Patent, Application No.: PCT/GB93/02529; 1993filed; [Online] http://www.google.co.in/patents/WO1994013805A1?cl=en&hl=ja.
- 46.Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+ CD25high regulatory cells in human peripheral blood. J Immunol. 2001;167:1245–1253. PMID:11466340 https://www.ncbi.nlm.nih.gov/pubmed/11466340. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.