

ABSTRACT
Protein-based biotherapeutics are produced in engineered cells through complex processes and may contain a wide variety of variants and post-translational modifications that must be monitored or controlled to ensure product quality. Recently, a low level (~1–5%) impurity was observed in a number of proteins derived from stably transfected Chinese hamster ovary (CHO) cells using mass spectrometry. These molecules include antibodies and Fc fusion proteins where Fc is on the C-terminus of the construct. By liquid chromatography-mass spectrometry (LC-MS), the impurity was found to be ~1177 Da larger than the expected mass. After tryptic digestion and analysis by LC-MS/MS, the impurity was localized to the C-terminus of Fc in the form of an Fc sequence extension. Targeted higher-energy collision dissociation was performed using various normalized collision energies (NCE) on two charge states of the extended peptide, resulting in nearly complete fragment ion coverage. The amino acid sequence, SLSLSPEAEAASASELFQ, obtained by the de novo sequencing effort matches a portion of the vector sequence used in the transfection of the CHO cells, specifically in the promoter region of the selection cassette downstream of the protein coding sequence. The modification was the result of an unexpected splicing event, caused by the resemblance of the commonly used GGU codon of the C-terminal glycine to a consensus splicing donor. Three alternative codons for glycine were tested to alleviate the modification, and all were found to completely eliminate the undesirable C-terminal extension, thus improving product quality.
Keywords: monoclonal antibody, Fc-fusion, Fc-extension, mass spectrometry, de novo sequencing, alternative splicing
Introduction
The production of recombinant proteins in mammalian cells has become well-established using robust cloning, expression, purification, and analytical methodologies. Still, unexpected post-translational modifications (PTMs) or protein product variants can occur. These may include unusual or unexpected glycosylation,1,2 oxidation,3,4 deamidation,5,6 glycation,7 phosphorylation,8 sulfation,9 S-thiolation,10 sequence extensions,11-13 and more.
Liquid chromatography coupled with mass spectrometry or tandem mass spectrometry (LC-MS and LC-MS/MS, respectively) have become the first choice in the analytical “tool kit” for identifying discrepancies that result in differences in mass due to covalent modifications (such as PTMs) or truncation. Confident identification of these changes is also aided by MS instrumentation with high resolution and mass accuracy,14 such as electrospray ionization – time of flight (ESI-TOF) instruments.15 Even higher mass accuracy may be obtained with certain instrumentation such as the Orbitrap16 or Fourier transform – ion cyclotron resonance (FT-ICR).17 High resolution instrumentation, together with MS/MS capabilities, has made MS an indispensable tool in protein therapeutics characterization,18 as evident by its ubiquitous usage in recent biologics license applications.19
The sensitivity of LC-MS, and more importantly the dynamic range by which a minor impurity can be detected in the presence of the main component at much higher concentration, may be limited by the complexity of the starting molecule. In many cases, heterogeneity from glycosylation or the large size of the molecule may further affect the ability to detect and quantify low level modifications. Fortunately, there are a number of methods that can simplify and clarify these mass complexities. For example, enzymatic deglycosylation (with PNGase F, sialidase, or O-glycanase) has been useful for identifying modifications initially missed due to complex glycosylation.2 Another way to reduce complexity is to reduce multi-chain proteins (e.g., antibody, Fc-fusion) into individual chains prior to analysis. The immunoglobulin-degrading enzyme from Streptococcus pyogenes (IdeS) clips below the hinge in many antibody species to yield F(ab’)2, Fc, and even F(ab) if mild reduction is included.20,21 Limited Lys-C digestion has proven useful in IgG1 monoclonal antibodies (mAbs) to generate Fc and Fab fragments.22-24 Any of these simplification strategies may have utility in increasing the analytical sensitivity and dynamic range with the goal of identifying variants and PTMs.
Recent examples of protein modification due to sequence extension that were determined through MS include those published by Kotia,11 Zhang,12 and Scott.13 Kotia observed N-terminal sequence extensions and clips in a mAb expressed in Chinese hamster ovary (CHO) cells as the result of incomplete cleavage of the signal peptide.11 This ragged clipping may be eliminated through in silico screening of signal peptide sequences using programs such as SignalP.25 In a different mAb, Zhang discovered that a single base-pair mutation (TAA-> GAA) changed a stop codon into a Glu residue,12 and more importantly, further extended the molecule as the stop codon is no longer present at this point. This read-through yielded a 1237 Da mass increase and incorporated light chain vector sequence into the molecule. Theoretically, errors in translation could also yield the same result. However, both the high level of the extended version (~14%) and DNA sequencing results pointed to mutation as the main cause. Similarly, Scott found mass extensions of 1047 Da or 3815 Da after the C-terminal Gly of the heavy chain in two mAb clones expressed in CHO-K1.13 These extensions were attributed to rearrangement of the DNA construct such that light chain vector sequence was incorporated into the C-terminus of the heavy chain (HC). This third example utilized de novo sequencing (identification of primary sequence without pre-existing reference sequence) to obtain the amino acid sequence of the extended peptide.
De novo sequencing has also been used to identify an unexpected 46 amino acid sequence extension in recombinant protein G using both “top down” and “bottom up” MS-based approaches.26 The unexpected sequence included a His tag, α-N-gluconoylation and α-N-phosphogluconoylation PTMs. “Top down” analyses were performed using matrix-assisted laser desorption/ionization (MALDI)-in source decay (ISD) MS, while “bottom up” experiments involved tryptic digests of the protein using a MALDI quadrupole ion trap (QIT) time of flight (ToF) mass spectrometer (MALDI-QIT ToF).
Here, we describe the discovery of a low-level sequence extension near the C-terminus of Fc in multiple protein products expressed in a stable CHO system. Using peptide mapping with high-resolution MS and de novo sequencing, the primary sequence of the extension was elucidated. This was determined to be the result of incorporating vector sequence into the translated protein through alternative splicing, due to the resemblance of the glycine codon GGU to consensus splicing donors. The extension is completely eliminated by choosing other Gly codons.
Results
Discovery of +1177 Da species in LC-MS
The presence of the + 1177 Da species was first observed in a reduced Fc-fusion protein produced from a stable CHO pool (Figure 1A). The discovery of this low-level species was aided by the lower mass of the protein, as well as the reduced heterogeneity compared to an intact monoclonal antibody. Later, this species was also observed on a number of antibody molecules produced by the same CHO expression system (Figure 1B). The levels were estimated at ~1–5% by comparing the MS signal of the unmodified and extended products from deconvoluted LC-MS spectra.
Figure 1.

Panel A: Deconvoluted mass of the Fc-fusion protein by LC-MS. Panel B: Deconvoluted mass of a non-reduced antibody (no N-linked glycan). Panel C: Deconvoluted mass of a reduced heavy chain (No N-linked glycan). All samples demonstrate the +1177 Da species.
With a mass difference this large, our first hypothesis was that this species may be caused by uncommon glycosylation. Further analyses were performed with the antibody to localize the modification to protein chains or domains. Reduced mass measurement was conducted, which localized the modification to the heavy chain (Figure 1C). Limited Lys-C digestion22-24 was also performed, which further localized the modification to the Fc (data not shown). These findings were consistent with our discovery on the Fc-fusion protein, and all point to modification somewhere on the Fc.
Peptide mapping
In order to further localize the modification, tryptic digestion was performed, followed by LC-MS/MS on a Q-Exactive Plus, a hybrid quadrupole-Orbitrap mass spectrometer. The LC-MS/MS results described below indicated that the +1177 Da was localized to the C-terminus of the fusion protein.
The C-terminal lysine residue of the protein is commonly removed (des-K) from recombinant proteins produced in mammalian cells.27,28 The des-K C-terminal Fc peptide of SLSLSPG has a signal of m/z 660.36 (monoisotopic [M + H]+) and elutes at T42.70 minutes by LC-MS/MS (Figure 2A). To test the hypothesis of a C-terminal peptide extension, we looked for low m/z b-series ions from SLSLSPG that should also exist in a C-terminally extended product. A peptide with doubly and triply charged ions at m/z 918.95 and 612.97 elutes at T61.83 min in the same analysis and yields similar low m/z b-ions (Figure 2B). Comparing the higher collision energy dissociation29 (HCD) MS/MS spectra of the two peptides, we observed common b-series fragment ions of m/z 201.12 (b2), 288.15 (b3), 401.24 (b4), and 488.27 (b5), suggesting a relationship between the two peptides (Figure 2C and 2D). The deconvoluted masses (monoisotopic [M]) for these two species are 659.35 Da and 1835.88 Da, a difference of 1176.53 Da, thus confirming that the peptide at T61.83 minutes is an extended or modified version of the C-terminal peptide.
Figure 2.
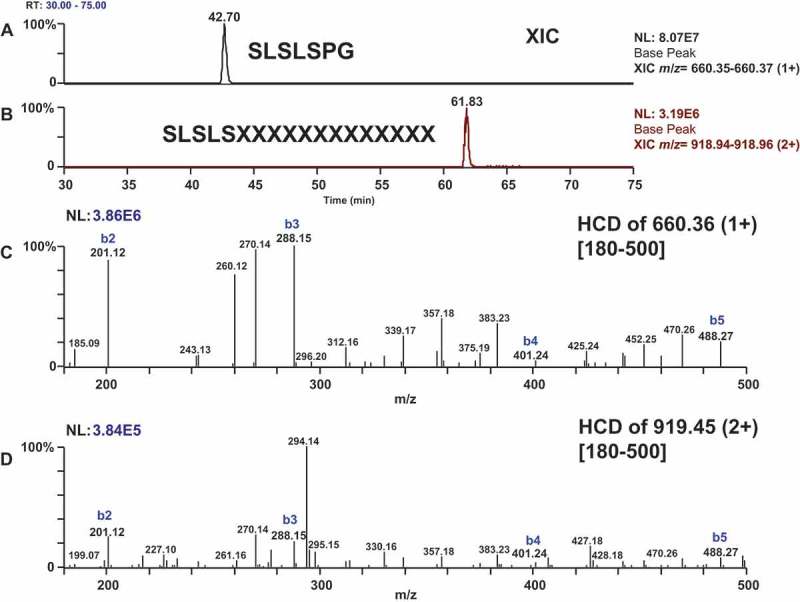
XIC of m/z 660.35–660.37 [Panel A] (des K C-terminal peptide SLSLSPG) and 919.44–919.46 [Panel B] (a related, but extended peptide). low m/z b-ions 660.36 [Panel C] and m/z 919.45 [Panel D] are common between both peptides, suggesting a relationship between the two spectra. Deconvolution of both peptides reveals a mass difference of 1176.53 Da, consistent with the mass increase observed at the protein chain level.
The initial HCD spectra for the extended C-terminal peptide were insufficient for de novo sequencing for two reasons: 1) lower abundance of the precursor ion, and hence lower signal-to-noise ratio in the MS/MS spectra; and 2) non-optimal collision energy, and hence lack of high m/z fragments. We used a targeted MS/MS analysis to test alternative collision energies at NCE 20 and 32 (in addition to 27), and determined that MS/MS of m/z 919.43 using NCE of 20 gave the best MS/MS spectra that can be used for de novo sequencing. All HCD data with this condition were averaged from T61.6–62.1 minutes, then deconvoluted using Xtract and displayed as monoisotopic [M + H]+ (Figure 3).
Figure 3.
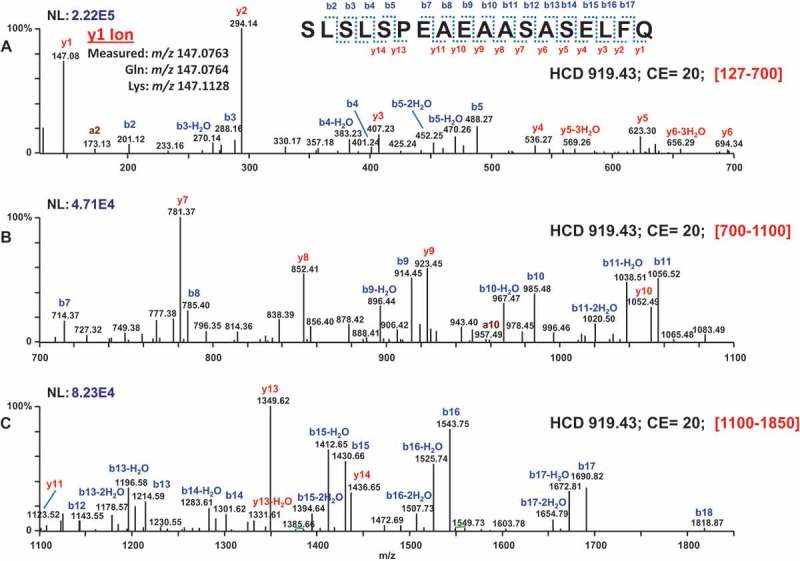
Targeted HCD of 919.43 using a normalized collision energy of 20. Spectra from T61.6–62.1 min were averaged, deconvoluted using Xtract, and displayed as monoisotopic [M + H]+ . Panel A: [127–700], Panel B: [700–1100], Panel C: [1100–1850]. De novo sequencing yielded the peptide SLSLSPEAEAASASELFQ, with the observed fragment ions labeled in the below sequence.
De novo sequencing
De novo sequencing is the process of constructing the sequences of unknown peptides and proteins from MS/MS data. This is frequently performed by software,30,31 but often times manually, through matching b-, y-, and other fragment ion data into linear stretches of amino acids.32 If the sequences are already in a database, automated search algorithms (e.g., Mascot)33 based on matching spectra to known sequences are the better choice. However, unknown sequences or those with unusual or unexpected PTMs may benefit from manual de novo analysis or de novo software programs such as PEAKS.31 Based on manual inspection, the deconvolved fragment ion spectra was sufficient to propose that the modified peptide sequence was SLSLSPEAEAASASELFQ, with the expected uncertainty of Leu/Ile at each position with a mass difference of 113.08 Da (identical for Leu and Ile), as most MS techniques cannot distinguish between these two isobaric amino acids. Because the first two isobaric Leu/Ile in the peptide sequence were from Fc, they could confidently be assigned as Leu. The last Leu/Ile could only be assigned after matching the sequence to that from the vector (see “Unanticipated splicing as a possible cause” section below). Experimental assignment, which could involve collecting the peptide for Edman sequencing or accessing an Orbitrap Fusion Tribrid instrument for an MS3 ETD/HCD experiment,34-36 was therefore no longer necessary.
Unanticipated splicing as a possible cause
The de novo sequencing results of SLSLSPEAEAASASELFQ appear to result from an in-frame fusion of the C-terminus of the human Fc protein (SLSLSPGK) with the sequence EAEAASASELFQ. The presence of this sequence is not contained within the coding sequence, nor could it be explained by a simple read-through of the stop codon, as the 3ʹ nucleotide sequence does not encode for amino acids matching the sequence identified by de novo sequencing. We then looked for any open reading frames (ORF) present in the vector to attempt to match sequences encoding for this peptide. The sequence of EAEAASASELFQ matches an ORF of EAEAASASELFQK* (* as the stop codon) within our expression vectors in a downstream promoter that drives expression of a puromycin selection cassette, about 500bp from the Fc stop codon (Figure 4). Similar to the C-terminal Lys residue on human Fc, the Lys residue on this extended terminal product was also post-translationally removed before the protein was secreted into the medium. The possibility of a low-level deletion or rearrangement was ruled out, as we also observed this extension on other molecules produced from this vector and expression system. Upon closer examination of the sequence at the DNA level, it was discovered that the normal C-terminal human Fc amino acid sequence, SLSLSPGK* (* as the stop codon), is encoded by the RNA sequence CUGUCUCCGGGUAAAUAG, which matches a near consensus mRNA splice donor (AG/GURAGU, where R = A or G) (see Alberts et al,37 p. 349). Within this putative splice donor, the donor invariant sequence GU falls within the glycine codon GGU. It was also discovered that the promoter derived peptide sequence, EAEAASASELFQK, was contained within the following mRNA sequence:
Figure 4.

Aberrant splicing is the cause of the +1177 Da Fc-extension.
CUGACUAAUUUUUUUUAUUUAUGCAG/AGGCCGAGGCCGCCUCUGCCUCUGAGCUAUUCCAGAAGUAG.
The underlined sequence mimics a near consensus mRNA splice acceptor (YYYYYYYYNCAG/G, where Y = C or U, N = any nucleotide). The sequence CUGAC is found within close proximity of the splice acceptor site and could serve as a putative branch point (with the consensus sequence of YURAC).
Remediation of unexpected splicing and complete elimination of the +1177 Da species
Based on the mechanism proposed above, a number of options were considered to eliminate the splice variant contaminant. With the discovery of the splice donor, the splice acceptor, and branch point sequences within our expression vectors containing human Fc sequences, it was postulated that elimination of one or all of these near consensus sequences should remove the unexpected splice forms, and therefore the spliced protein variant contaminant. Additionally, since the antibody expression cassette ends with a polyadenylation (pA) signal, the unexpected splice event is only possible when the expression cassette message fails to terminate at the pA site. Therefore, an alternative strategy exists wherein the use of an alternative polyadenylation (pA) signal that is significantly more efficient at terminating the expression cassette message would also have the desired effect by removing the discovered promoter branch point and acceptor site sequences from the expression cassette message.
Even though there are many possible solutions, their feasibility and effectiveness differ. As the branch point and acceptor site sequences are embedded within the promoter, mutations within this sequence to eliminate the aberrant splice events may have required undesirable vector engineering. Optimizing the pA signal would have required similar effort with no guarantee of success. Therefore, our efforts were directed to the splice donor found within the human Fc sequence, specifically the donor invariant sequence GU, which falls within the glycine codon GGU.
The use of alternative codons for glycine would not disrupt the normal peptide sequence, but would eliminate the splicing donor, and therefore eliminate the Fc-extension species. Glycine is coded by four possible codons: GGA, GGU, GGC and GGG. We tested this theory by expressing both the Fc-fusion protein, as well as the antibody, with all four possible glycine codons and subjecting the recombinant products to LC-MS and LC-MS/MS analyses.
As expected, the GGU codon products retained the low-level Fc-extension species, while the other three codons completely eliminated this undesired by-product (Figure 5A-D). In order to confirm this finding with better dynamic range, we performed peptide mapping (Figure 5E-H). With the knowledge of the cause of the modification, we opted for a simpler digestion condition, with less sample manipulation steps. Specifically, we used Lys-C digestion without any reduction and alkylation steps because the C-terminal peptide is not associated with any disulfide bonds, and has a lysine as the clipping site for both the Lys-C used here and the trypsin used in earlier studies. The LC-MS conditions were also slightly changed: column temperature was raised from 30°C to 50°C, and mobile phase modifier was changed from formic acid to trifluoroacetic acid (TFA). These changes were responsible for the retention time shift from Figure 2 to Figure 5 (panels 5E-H).
Figure 5.

Left column: Deconvoluted mass spectra of a reduced Fc-fusion protein using different glycine codons for the final glycine of Fc (Y axis zoomed 10X). Panel A: Glycine codon GGU. Panel B: Glycine codon GGA. Panel C: Glycine codon GGC. Panel D: Glycine codon GGG. The results clearly show that all three alternative codons eliminate the Fc extension/aberrant splicing. Right column: XIC of the C-terminal peptides from a Lys-C digest of the same protein constructs. SLSLSPEAEAASASELFQ elutes at ~70.2 min. The degraded form of the extension peptide SLSLSPEAEAASASEL is also observed at ~63.9 min.
In panels 5E-H, extracted ion chromatograms (XIC) of peptides SLSLSPG m/z [660.35–660.37] (~T54 min.), SLSLSPEAEAASASELFQ m/z [918.94–918.96] (T70.16 min.), and SLSLSPEAEAASASEL m/z [781.37–781.39] (T63.92 min) were produced. The peptide signals were expanded 25x to better display Fc extended products, and to illustrate that they only exist with the GGU codon (panel E). It should be noted that the proteolytic digests were performed on samples after 2–3 months of storage at 4°C after the initial LC-MS was performed. It was clear that some C-terminal degradation of SLSLSPEAEAASASELFQ to SLSLSPEAEAASASEL (loss of 1–2 C-terminal amino acids) has occurred, confirmed by re-analyzing the samples by LC-MS (data not shown). Overall, the results from LC-MS/MS confirm the findings at the protein level that the Fc extension products are exclusive to constructs that use the GGU codon at this glycine site.
In order to select one codon from the three “clean” codons, we also compared the titer of the production, with the caveat that titer at such small scale can be prone to fluctuations. The titer was consistent between the various codon choices, with values of 101–108 mg/L. Ultimately, GGC was selected as the final glycine codon because it is listed as having the highest occurrence in the mammalian codon usage table under CHO.38,39
Discussion
Targeted high resolution HCD enables de novo sequencing
Prior to high mass accuracy HCD fragmentation becoming available, low m/z fragment ions would often be lost in ion trap instruments due to the 1/3 rule (i.e., fragment ions less than 1/3 of the precursor ion m/z cannot be trapped).40 Having the full complement of low m/z b- and y- series fragment ions made it much easier to establish a relationship between the unmodified and extended forms of the peptide. Additionally, the accurate mass of the y1 ion (m/z 147.0763) clearly indicated that the modified peptide contained a Gln (m/z 147.0764, 0.7ppm error), rather than a Lys (m/z 147.1128, 250ppm error) at the C-terminus of the tryptic-derived peptide. The combination of targeting the extended peptide and using lower NCE values resulted in higher m/z fragment ions (> 1300) that were integral in obtaining the unknown peptide sequence. Many of these fragments were not observed in the original MS/MS data. In the past, deciphering an unexpected low-level modification such as this might have required the offline harvesting of large amounts of peptide in order to get sufficient material for Edman degradation. This would be time consuming, but nevertheless feasible, provided there was no chromatographic co-elution with other unrelated components. De novo sequencing is still possible using older low-resolution MS instrumentation (LTQ or 3D ion traps); however, the low mass accuracy of the resulting precursor and fragment ions often leads to ambiguous or even mistaken amino acid assignments. With high resolution/high mass accuracy MS instrumentation, errors in de novo sequencing are more evident, especially if 5 ppm or greater differences exist between the theoretical and observed precursor masses.
Germline GGU codon for the c-terminal glycine acts as a near consensus splice donor site
The GGU codon for the glycine near the C-terminus of the Fc is germline and is present in the majority of the entries in the international ImMunoGeneTics information system (IMGT).41 It is therefore expected that this codon arrangement is widely used. The GGU codon combines with the C-terminal Fc lysine codon and the stop codon to form the splice donor site (Figure 4).
The presence of the splicing donor site alone is not sufficient to cause the splicing event that leads to Fc-extension. Other conditions, such as the presence of a splicing branch site and accepter site in the sequence after the stop codon for Fc, are also necessary. However, since it is a standard practice to include introns into expression vectors to augment expression titers,42 this condition can be met in many cases.
As splicing events occur at the RNA level, a long transcript that contains not only the Fc coding sequence, as well as the vector backbone, downstream promoter, and puromycin cassette, is also necessary. Such a transcript should typically be prevented by transcriptional termination and poly-adenylation of the transcript driven by the poly A signal present at the 3ʹ end of the Fc stop codon. The poly A signal acts to terminate transcription through the pause site and release the transcriptional machinery, and may lead to occlusion of downstream genes.43,44 Inefficient transcript termination is known to happen at a low frequency and is dependent on the strength of the poly A signal and pause sites in the 3ʹ untranslated regions of the gene.43,45 In the case study presented, we observe the low level (~1–5%) extension of the Fc product, which could be explained by rare and inefficient transcriptional termination, followed by efficient splicing of the near consensus splice sites present in the resulting transcription. If the splice consensus sites are not present in such a read-through transcript, the Fc translation would terminate at the intended stop codon and no additional amino acids would be appended to the protein of interest, regardless of the presence of the longer transcripts. Indeed, we demonstrate here that removal of the donor site through a codon change of the glycine codon eliminates the identified extension. We therefore highly recommend that the codon for the glycine near the C-terminus of the huIgG1 Fc sequence be altered to eliminate the GU invariant sequence, thereby breaking the resemblance to the splicing donor site. This will avoid the potential issue of Fc-extension completely while maintaining vector integrity and not negatively affecting protein production levels.
Potential impact of product quality of C-terminus extension caused by alternative splicing
Alternative splicing occurs as a normal, regulated process in eukaryotes to enable the production of multiple protein products from fewer genes.46 B cells use alternative splicing and other mechanisms to produce all five different antibody isotypes (IgM, IgD, IgG, IgE and IgA) from a single Ig heavy chain (Igh).47 On the other hand, aberrant alternative splicing has been implicated as one of the causes of many diseases,48 including hypercholesterolemia, frontotemporal lobar dementia, spinal muscular atrophy, tauopathies, familial dysautonomia, and Hutchinson-Gilford progeria syndrome.
The effect of alternative and unintended splicing on the production of biotherapeutics, and whether it qualifies as a critical quality attribute,49 is less frequently discussed in the literature. The alternatively spliced product such as that described in this paper introduces product heterogeneity and is obviously a product quality attribute (PQA) for the biotherapeutic. To assess the criticality of the (C-terminal) Fc-extension as a potential critical quality attribute, knowledge and experience from N-terminal signal peptide extensions 11,50,51 can be leveraged. These two PQAs share very similar characteristics: both are product-related impurities that are usually difficult to remove by purification (unless they alter protein charge), but neither of them is near the complementarity-determining region, hinge, or CH2 regions, and are therefore not expected to affect activity, effector function, or pharmacokinetics. The main concern is immunogenicity, as the extra amino acid sequences potentially can be recognized by the immune system as a foreign species, especially when the extension is long, and solvent exposed (evident by its clipping during storage).
If possible, it is most desirable to remove these terminal heterogeneity issues 52 through various engineering efforts in the early stages of the drug development process, such as we have achieved here. When terminal heterogeneity is present in final products due to technical or historic reasons, its impact on product quality, especially immunogenicity, should be evaluated experimentally.52 Since we have implemented the alternative glycine codon strategy, we are no longer producing proteins with this attribute for the affected projects. Previous productions were also at small scale, intended for discovery research. Nevertheless, other proteins produced with the standard GGU codon still may have this Fc-extension (potentially with a different amino acid sequence), depending on a number of factors including DNA context, cell line choice, and expression conditions as discussed previously. Via this report, we hope the scientific community gains awareness of this possible PQA and establishes its biological impact through further investigations.
Materials and methods
Materials and reagents
Pierce brand pre-weighed dithiothreitol (DTT) and iodoacetamide (IAM) were purchased from Thermo Fisher Scientific (Rockford, IL). Trypsin was obtained from Sigma-Aldrich (St. Louis, MO), while Lys-C was a product of Wako (Osaka, Japan). HPLC mobile phase buffers (0.1% formic acid/H2O, 0.1% formic acid/acetonitrile, 0.1% TFA/H2O, and 0.1% TFA/acetonitrile) were purchased from Fisher Chemical (Fair Lawn, NJ).
Protein expression and purification
Proteins were produced using a stable CHO cell expression system. The antibody heavy and light chain genes were cloned into a mammalian expression vector, containing a puromycin or hygromycin selection cassette, respectively. Vectors were transfected into CHOK1 cells with Lipofectamine LTX (Thermo Fisher Scientific, Waltham, MA). 48 hours post transfection, antibiotic selection was applied to select for a stable pool. For protein production, cells were seeded at 1 × 106 cells/mL in a proprietary production media for 7 days and harvested by centrifugation. For purification, protein was bound and eluted from a 5 mL HiTrap MabSelect SuRe column, desalted/buffer exchanged into 10 mM sodium acetate/150 mM NaCl, and finally passed over size-exclusion chromatography all using a GE Healthcare Life Sciences (Pittsburgh, PA) ÄKTA chromatography system.
Protein mass spectrometry by LC-MS
Protein MS was performed on an LC-MS system that we have described previously.1,4 Briefly, the protein samples (either reduced or non-reduced) were analyzed on an LC-MS system comprised of an Agilent 6230 ESI-TOF (Agilent Technologies, Santa Clara, CA) connected to an Agilent 1260 HPLC equipped with a Zorbax 2.1 X 50 mm 300SB-C8 column (Agilent Technologies, Santa Clara, CA) heated to 70°C. Buffer A: 0.1% TFA; Buffer B: 0.1% TFA/90% n-propanol. Raw mass spectrometric data was deconvoluted with the MaxEnt algorithm using the Agilent MassHunter software (version B7.0, Agilent Technologies, Santa Clara, CA).
Protein digestion
For de novo sequencing, 60 µg of the protein sample was dried with a Savant SpeedVac and re-dissolved in 60 µL of 150 mM Tris, pH 7.5/8 M urea/40 mM hydroxylamine. The sample was reduced with 25 mM DTT at 37°C for 1 hour, then alkylated with 50 mM IAM at room temperature for 1 hour in the dark. Excess IAM was quenched by the addition of 25 mM DTT. The reduced and alkylated protein was then diluted ~4x with water to reduce the urea concentration to 2 M, and then digested with 5 µg trypsin overnight at 37°C. The digestion was acidified by adding 0.1% TFA to dilute the sample to 0.2 mg/mL. 4 µg of the digestion (20 µL) is typically injected onto the LC-MS/MS system described below.
For later confirmation of the elimination of +1177 Da species, the digestion conditions were altered slightly, as the modification is on the C-terminus and does not involve disulfide linkages. 50 µg of the protein was dried and re-dissolved in 25 µL of 150 mM Tris, pH 7.5/8 M urea/40 mM hydroxylamine. The sample was then diluted 1:1 with water to reduce the urea concentration to 4 M, and then digested with 2.5 µg Lys-C overnight at 37°C. Digestion was stopped by the addition of 0.4% TFA and 5 µg of digest (10 µL) was injected onto the LC-MS/MS system described below. The mobile phase modifier was changed from 0.1% formic acid to 0.1% TFA, and the column temperature was changed from 30°C to 50°C. Both changes are aimed at more reproducible LC-MS/MS results for semi-quantitative work, while the formic acid conditions were optimized for discovery work where sensitivity is more important.
Peptide mapping by LC-MS/MS
Digests of the sample exhibiting the +1177 Da mass increase were analyzed using an Agilent 1260 Capillary HPLC linked to a Thermo Fisher Scientific (San Jose, CA) Q-Exactive Plus Hybrid Quadrupole-Orbitrap mass spectrometer equipped with a HESI II ionization source. For chromatographic separation, the HPLC uses an Agilent Zorbax 300SB-C18 5µm 1.0 mm X 250 mm column with formic acid buffers (buffer A: 0.1% formic acid/H2O; buffer B: 0.1% formic acid/acetonitrile) at a flow rate of 60 µL/min with a column temperature of 30°C. The gradient consists of isocratic at 1% B for 10 minutes, to 55% B over 85 minutes, to 97% B over 5 minutes, isocratic at 97% B for 5 minutes, and then to 1% B over 10 minutes. The column effluent was sprayed into the mass spectrometer using a spray voltage of 3.5 kV. The data-dependent MS method consists of Full MS (70K resolving power, m/z [300–2000]), followed by HCD (17.5K resolving power) of the top 10 most abundant precursors. AGC target values are 1E6 (full MS) and 5E4 (HCD). Other MS parameters include an isolation width of 2.0 Da and NCE of 27.
The targeted HCD MS method consists of a Full MS (70K resolving power, m/z [400–2000]), and six targeted HCD MS/MS experiments (2 charge states combined with 3 collision energy settings): m/z 919.43/NCE = 20, m/z 919.45/NCE = 27, m/z 919.47/NCE = 32, m/z 612.95/NCE = 20, m/z 612.97/NCE = 27, and m/z 612.99/NCE = 32. The slight differences in precursor mass were used to overcome a software issue where all HCD spectra with the same precursor ion m/z are automatically combined. This technique therefore enabled comparison of the results from the three NCE settings. The precursor ion mass difference (0.02 m/z units) is small enough not to cause any meaningful ion population difference, as the precursor ion isolation window of 2 m/z units is much larger. All other parameters remain unchanged. Deconvolution of the resulting targeted HCD spectra was performed using Xtract, an add-on program to Xcalibur. Averaged spectra from m/z 400–2000 were deconvoluted using a resolution of 100K (default) at m/z 400 and a S/N threshold of 2.0, then displayed as monoisotopic [M + H]+. Data from these LC-MS/MS runs were analyzed with the Amgen internal software MassAnalyzer,53 as well as the PEAKS software31 (Bioinformatics Solutions, Inc., Waterloo, ON, Canada).
De novo sequencing
For de novo sequencing of the modified peptide, the most complete and best-quality targeted HCD spectra for m/z 919.43 using NCE = 20 were averaged, then deconvoluted as described above. To establish linear peptide sequence, different fragment ions in the spectra were manually selected in the Thermo software (“label relative to selected mass”) to look for single amino acid mass shifts between neighboring fragment ions. If a single amino acid mass shift was observed, the process was repeated on the new fragment ion with the goal of looking for additional single amino acid extensions. Linking together enough amino acid mass shifts may be suitable for establishing peptide sequence. As the instrumental mass accuracy is 5ppm or greater, any mass discrepancies (precursor or fragment ion) significantly greater than that indicates an error somewhere in the de novo sequencing results. Leu/Ile cannot be discriminated with our current instrument and must be determined by other means including MS3 (ETD-HCD).34-36
Abbreviations
- LC-MS/MS
liquid chromatography tandem mass spectrometry
- HCD
higher-energy collision-induced dissociation
- NCE
normalized collision energy
- CHO
Chinese hamster ovary
- PTM
post-translational modification
Acknowledgments
The authors would like to thank Alison Castellano and Vivian Li for production of proteins utilizing alternate codons. Countless others in the Biologics Optimization Department also contributed to the production of the original proteins. We also thank Rick Jacobsen and Bram Estes for helpful discussions.
References
- 1.Spahr C, Shi SD-H, Lu HS.. O-glycosylation of glycine-serine linkers in recombinant Fc-fusion proteins: attachment of glycosaminoglycans and other intermediates with phosphorylation at the xylose sugar subunit. MAbs. 2014;6:904–914. doi: 10.4161/mabs.28763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spahr C, Kim JJ, Deng S, Kodama P, Xia Z, Tang J, Zhang R, Siu S, Nuanmanee N, Estes B, et al. Recombinant human lecithin-cholesterol acyltransferase Fc fusion: analysis of N- and O-linked glycans and identification and elimination of a xylose-based O-linked tetrasaccharide core in the linker region. Protein Sci. 2013;22:1739–1753. doi: 10.1002/pro.2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verrastro I, Pasha S, Jensen KT, Pitt AR, Spickett CM. Mass spectrometry-based methods for identifying oxidized proteins in disease: advances and challenges. Biomolecules. 2015;5:378–411. doi: 10.3390/biom5020378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spahr C, Gunasekaran K, Walker KW, Shi SD-H. High-resolution mass spectrometry confirms the presence of a hydroxyproline (Hyp) post-translational modification in the GGGGP linker of an Fc-fusion protein. MAbs. 2017;9:812–819. doi: 10.1080/19420862.2017.1325556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hao P, Adav SS, Gallart-Palau X, Sze SK. Recent advances in mass spectrometric analysis of protein deamidation. Mass Spectrom Rev. 2016. doi: 10.1002/mas.21491. [DOI] [PubMed] [Google Scholar]
- 6.Yang H, Zubarev RA. Mass spectrometric analysis of asparagine deamidation and aspartate isomerization in polypeptides. Electrophoresis. 2010;31:1764–1772. doi: 10.1002/elps.201000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brady LJ, Martinez T, Balland A. Characterization of nonenzymatic glycation on a monoclonal antibody. Anal Chem. 2007;79:9403–9413. doi: 10.1021/ac7017469. [DOI] [PubMed] [Google Scholar]
- 8.Tyshchuk O, Volger HR, Ferrara C, Bulau P, Koll H, Molhoj M. Detection of a phosphorylated glycine-serine linker in an IgG-based fusion protein. MAbs. 2017;9:94–103. doi: 10.1080/19420862.2016.1236165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Masuda K, Yamaguchi Y, Takahashi N, Jefferis R, Kato K. Mutational deglycosylation of the Fc portion of immunoglobulin G causes O-sulfation of tyrosine adjacently preceding the originally glycosylated site. FEBS Lett. 2010;584:3474–3479. doi: 10.1016/j.febslet.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 10.Gadgil HS, Bondarenko PV, Pipes GD, Dillon TM, Banks D, Abel J, Kleemann GR, Treuheit MJ. Identification of cysteinylation of a free cysteine in the Fab region of a recombinant monoclonal IgG1 antibody using Lys-C limited proteolysis coupled with LC/MS analysis. Anal Biochem. 2006;355:165–174. doi: 10.1016/j.ab.2006.05.037. [DOI] [PubMed] [Google Scholar]
- 11.Kotia RB, Raghani AR. Analysis of monoclonal antibody product heterogeneity resulting from alternate cleavage sites of signal peptide. Anal Biochem. 2010;399:190–195. doi: 10.1016/j.ab.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 12.Zhang T, Huang Y, Chamberlain S, Romeo T, Zhu-Shimoni J, Hewitt D, Zhu M, Katta V, Mauger B, Kao Y-H. Identification of a single base-pair mutation of TAA (Stop codon) –> GAA (Glu) that causes light chain extension in a CHO cell derived IgG1. MAbs. 2012;4:694–700. doi: 10.4161/mabs.22232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scott RA, Rogers R, Balland A, Brady LJ. Rapid identification of an antibody DNA construct rearrangement sequence variant by mass spectrometry. MAbs. 2014;6:1453–1463. doi: 10.4161/mabs.36222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ytterberg AJ, Dunsmore J, Lomeli SH, Thevis M, Xie Y, Loo RRO, Loo JA. The role of mass spectrometry for peptide, protein, and proteome characterization In: Cole RB, editor. Electrospray and MALDI mass spectrometry. John Wiley & Sons, Inc; 2010. p. 683–721. [Google Scholar]
- 15.Mirgorodskaya OA, Shevchenko AA, Chernushevich IV, Dodonov AF, Miroshnikov AI. Electrospray-ionization time-of-flight mass spectrometry in protein chemistry. Anal Chem. 1994;66:99–107. doi: 10.1021/ac00073a018. [DOI] [Google Scholar]
- 16.Zubarev RA, Makarov A. Orbitrap mass spectrometry. Anal Chem. 2013;85:5288–5296. doi: 10.1021/ac4001223. [DOI] [PubMed] [Google Scholar]
- 17.Marshall AG, Hendrickson CL, Jackson GS. Fourier transform ion cyclotron resonance mass spectrometry: A primer. Mass Spectrom Rev. 1998;17:1–35. doi: 10.1002/(SICI)1098-2787(1998)17:1<1::AID-MAS1>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 18.Qu M, An B, Shen S, Zhang M, Shen X, Duan X, Balthasar JP, Qu J. Qualitative and quantitative characterization of protein biotherapeutics with liquid chromatography mass spectrometry. Mass Spectrom Rev. 2017;36:734–754. doi: 10.1002/mas.21500. [DOI] [PubMed] [Google Scholar]
- 19.Rogstad S, Faustino A, Ruth A, Keire D, Boyne M, Park J, Retrospective A. Evaluation of the use of mass spectrometry in FDA biologics license applications. J Am Soc Mass Spectrom. 2017;28:786–794. doi: 10.1007/s13361-016-1531-9. [DOI] [PubMed] [Google Scholar]
- 20.Akesson P, Moritz L, Truedsson M, Christensson B, Von Pawel-Rammingen U. IdeS, a highly specific immunoglobulin G (IgG)-cleaving enzyme from Streptococcus pyogenes, is inhibited by specific IgG antibodies generated during infection. Infect Immun. 2006;74:497–503. doi: 10.1128/IAI.74.1.497-503.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.An Y, Zhang Y, Mueller H-M, Shameem M, Chen X. A new tool for monoclonal antibody analysis: application of IdeS proteolysis in IgG domain-specific characterization. MAbs. 2014;6:879–893. doi: 10.4161/mabs.28762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kleemann GR, Beierle J, Nichols AC, Dillon TM, Pipes GD, Bondarenko PV. Characterization of IgG1 immunoglobulins and peptide−Fc fusion proteins by limited proteolysis in conjunction with LC−MS. Anal Chem. 2008;80:2001–2009. doi: 10.1021/ac701629v. [DOI] [PubMed] [Google Scholar]
- 23.Zhang T, Zhang J, Hewitt D, Tran B, Gao X, Qiu ZJ, Tejada M, Gazzano-Santoro H, Kao Y-H. Identification and characterization of buried unpaired cysteines in a recombinant monoclonal IgG1 antibody. Anal Chem. 2012;84:7112–7123. doi: 10.1021/ac301426h. [DOI] [PubMed] [Google Scholar]
- 24.Kim J, Jones L, Taylor L, Kannan G, Jackson F, Lau H, Latypov RF, Bailey B. Characterization of a unique IgG1 mAb CEX profile by limited Lys-C proteolysis/CEX separation coupled with mass spectrometry and structural analysis. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878:1973–1981. doi: 10.1016/j.jchromb.2010.05.032. [DOI] [PubMed] [Google Scholar]
- 25.Bendtsen JD, Nielsen H, Von Heijne G, Brunak S. Improved prediction of signal peptides: signalP 3.0. J Mol Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 26.Yaefremova Y, Al-Majdoub M, Opuni KFM, Koy C, Cui W, Yan Y, Gross ML, Glocker MO. “De-novo” amino acid sequence elucidation of protein G′e by combined “Top-Down” and “Bottom-Up” mass spectrometry. J Am Soc Mass Spectrom. 2015;26:482–492. doi: 10.1007/s13361-014-1053-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harris RJ. Processing of C-terminal lysine and arginine residues of proteins isolated from mammalian cell culture. J Chrom A. 1995;705:129–134. doi: 10.1016/0021-9673(94)01255-D. [DOI] [PubMed] [Google Scholar]
- 28.Luo J, Zhang J, Ren D, Tsai W-L, Li F, Amanullah A, Hudson T. Probing of C-terminal lysine variation in a recombinant monoclonal antibody production using Chinese hamster ovary cells with chemically defined media. Biotechnol Bioeng. 2012;109:2306–2315. doi: 10.1002/bit.24510. [DOI] [PubMed] [Google Scholar]
- 29.Olsen JV, Macek B, Lange O, Makarov A, Horning S, Higher-Energy MM. C-trap dissociation for peptide modification analysis. Nat Methods. 2007;4:709–712. doi: 10.1038/nmeth1060. [DOI] [PubMed] [Google Scholar]
- 30.Horn DM, Zubarev RA, McLafferty FW. Automated de novo sequencing of proteins by tandem high-resolution mass spectrometry. Proc Natl Acad Sci U S A. 2000;97:10313–10317. doi: 10.1073/pnas.97.19.10313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma B, Zhang K, Hendrie C, Liang C, Li M, Doherty-Kirby A, Lajoie G. PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun Mass Spectrom. 2003;17:2337–2342. doi: 10.1002/rcm.1196. [DOI] [PubMed] [Google Scholar]
- 32.Medzihradszky KF, Chalkley RJ. Lessons in de novo peptide sequencing by tandem mass spectrometry. Mass Spectrom Rev. 2015;34:43–63. doi: 10.1002/mas.21406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perkins DN, Pappin DJC, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 34.Bagal D, Kast E, Cao P. Rapid distinction of leucine and isoleucine in monoclonal antibodies using nanoflow LCMSn. Anal Chem. 2017;89:720–727. doi: 10.1021/acs.analchem.6b03261. [DOI] [PubMed] [Google Scholar]
- 35.Xiao Y, Vecchi MM, Wen D. Distinguishing between leucine and isoleucine by integrated LC-MS analysis using an orbitrap fusion mass spectrometer. Anal Chem. 2016;88:10757–10766. doi: 10.1021/acs.analchem.6b03409. [DOI] [PubMed] [Google Scholar]
- 36.Johnson RS, Martin SA, Biemann K, Stults JT, Watson JT. Novel fragmentation process of peptides by collision-induced decomposition in a tandem mass spectrometer: differentiation of leucine and isoleucine. Anal Chem. 1987;59:2621–2625. doi: 10.1021/ac00148a019. [DOI] [PubMed] [Google Scholar]
- 37.Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular biology of the cell. 5th ed. New York (NY): Garland Science; 2007. p. 349. [Google Scholar]
- 38.Sharp PM, Li W-H. The codon adaptation index - a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15:1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakamura Y, CGojibori T, Ikemura T. Codon usage tabulated from international DNA sequence databases: status for the year 2000. Nucleic Acids Res. 2000;28:292. doi: 10.1093/nar/28.1.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Want EJ, Cravatt BF, Siuzdak G. The expanding role of mass spectrometry in metabolite profiling and characterization. ChemBioChem. 2005;6:1941–1951. doi: 10.1002/cbic.200500151. [DOI] [PubMed] [Google Scholar]
- 41.Lefranc M-P, Giudicelli V, Duroux P, Jabado-Michaloud J, Folch G, Aouinti S, Carillon E, Duvergey H, Houles A, Paysan-Lafosse T, et al. IMGT(R), the international ImMunoGeneTics information system(R) 25 years on. Nucleic Acids Res. 2015;43:D413–22. doi: 10.1093/nar/gku1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nott A, Meislin SH, Moore MJ. A quantitative analysis of intron effects on mammalian gene expression. RNA. 2003;9:607–617. doi: 10.1261/rna.5250403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Proudfoot NJ. How RNA polymerase II terminates transcription in higher eukaryotes. Trends Biochem Sci. 1989;14:105–110. doi: 10.1016/0968-0004(89)90132-1. [DOI] [PubMed] [Google Scholar]
- 44.Kuehner JN, Pearson EL, Moore C. Unravelling the means to an end: RNA polymerase II transcription termination. Nat Rev Mol Biol. 2011;12:283–294. doi: 10.1038/nrm3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Plant KE, Dye MJ, Lafaille C, Proudfoot NJ. Strong polyadenylation and weak pausing combine to cause efficient termination of transcription in the human Ggamma-globin gene. Mol Cell Biol. 2005;25:3276–3285. doi: 10.1128/MCB.25.8.3276-3285.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 47.Yabas M, Elliott H, Hoyne GF. The role of alternative splicing in the control of immune homeostasis and cellular differentiation. Int J Mol Sci. 2015:17. doi: 10.3390/ijms17010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tazi J, Bakkour N, Stamm S. Alternative splicing and disease. Biochim Biophys Acta. 2009;1792:14–26. doi: 10.1016/j.bbadis.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rathore A, Winkle H. Quality by design for biopharmaceuticals. Nat Biotechnol. 2009;27:26–34. doi: 10.1038/nbt0109-26. [DOI] [PubMed] [Google Scholar]
- 50.Huang Y, Fu J, Ludwig R, Tao L, Bongers J, Ma L, Yao M, Zhu M, Das T, Russell R. Identification and quantification of signal peptide variants in an IgG1 monoclonal antibody produced in mammalian cell lines. J Chromatogr B Analyt Technol Biomed Life Sci. 2017;1068-1069:193–200. doi: 10.1016/j.jchromb.2017.08.046. [DOI] [PubMed] [Google Scholar]
- 51.Ying H, Liu H. Identification of an alternative signal peptide cleavage site of mouse monoclonal antibodies by mass spectrometry. Immunol Lett. 2007;111:66–68. doi: 10.1016/j.imlet.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 52.Brorson K, Jia AY. Therapeutic monoclonal antibodies and consistent ends: terminal heterogeneity, detection, and impact on quality. Curr Opin Biotechnol. 2014;30:140–146. doi: 10.1016/j.copbio.2014.06.012. [DOI] [PubMed] [Google Scholar]
- 53.Zhang Z. Large-scale identification and quantification of covalent modifications in therapeutic proteins. Anal Chem. 2009;81:8354–8364. doi: 10.1021/ac901193n. [DOI] [PubMed] [Google Scholar]