

ABSTRACT
RNA binding proteins have emerged as critical oncogenic factors and potential targets in cancer therapy. In this study, we evaluated Musashi1 (Msi1) targeting as a strategy to treat glioblastoma (GBM); the most aggressive brain tumor type. Msi1 expression levels are often high in GBMs and other tumor types and correlate with poor clinical outcome. Moreover, Msi1 has been implicated in chemo- and radio-resistance. Msi1 modulates a range of cancer relevant processes and pathways and regulates the expression of stem cell markers and oncogenic factors via mRNA translation/stability. To identify Msi1 inhibitors capable of blocking its RNA binding function, we performed a ~ 25,000 compound fluorescence polarization screen. NMR and LSPR were used to confirm direct interaction between Msi1 and luteolin, the leading compound. Luteolin displayed strong interaction with Msi1 RNA binding domain 1 (RBD1). As a likely consequence of this interaction, we observed via western and luciferase assays that luteolin treatment diminished Msi1 positive impact on the expression of pro-oncogenic target genes. We tested the effect of luteolin treatment on GBM cells and showed that it reduced proliferation, cell viability, colony formation, migration and invasion of U251 and U343 GBM cells. Luteolin also decreased the proliferation of patient-derived glioma initiating cells (GICs) and tumor-organoids but did not affect normal astrocytes. Finally, we demonstrated the value of combined treatments with luteolin and olaparib (PARP inhibitor) or ionizing radiation (IR). Our results show that luteolin functions as an inhibitor of Msi1 and demonstrates its potential use in GBM therapy.
KEYWORDS: RNA binding protein, Musashi1, cancer therapy, glioblastoma, luteolin
Introduction
Glioblastoma (GBM), the most common and aggressive brain tumor type, is associated with an extremely poor prognosis [1]. Due to its diffusely infiltrative nature, tumor relapse is very frequent even after total surgical resection [2]. Temozolomide (TMZ), an alkylating agent, is the standard of care for GBM but only improves 2-year survival in 27% of patients [3]. TMZ- and radio-resistance are the greatest therapeutic challenges in GBM [4]. In recent years, many targeted anti-cancer drugs, such as small-molecule inhibitors and monoclonal antibodies have been evaluated in preclinical research or clinical trials. Unfortunately, only a single anti-angiogenic agent, bevacizumab, which disrupts the VEGF-VEGF-Receptor interaction, has been approved for the treatment of recurrent GBM [5]. Therefore, novel targeted drugs and efficient combination strategies are greatly needed to advance GBM therapy.
There are over 1,500 RNA binding proteins (RBPs) in the human genome [6] and these have a massive impact on gene expression. By regulating numerous processes including RNA splicing, poly-adenylation, mRNA decay and translation, RBPs contribute quantitatively and qualitatively to the protein profile of a cell. RBPs are particularly relevant in the nervous system where they function as key players in neurogenesis, brain development, synaptogenesis and memory. Additionally, a growing number of RBPs have been implicated in neurological disorders, neurodegenerative diseases and tumorigenesis [7,8]. Thanks to their broad regulatory nature, ‘onco-RBPs’ can contribute to cell transformation and to the acquisition of multiple phenotypes required for tumor growth [9]. The idea of targeting onco-RBPs is starting to be explored in cancer therapy. In contrast to strategies that focus on a specific signaling pathway, RBP targeting could potentially affect multiple oncogenic pathways simultaneously [10].
Musashi1 (Msi1) is an evolutionarily conserved RBP that regulates the balance between self-renewal and differentiation [11,12]. In cancer, Msi1 functions as a pro-tumorigenic factor in GBM, medulloblastomas, colon cancer, breast cancer, lung cancer and many more tumor types [13]. High Msi1 expression is prevalent in high-risk medulloblastoma subgroups 3 and 4 and correlates with poor prognosis [14]. A similar scenario is observed in gliomas [15]. Msi1 regulates translation, mRNA decay and poly adenylation by binding to specific motifs located mainly in the 3’UTR of its target transcripts [16–18]. Genomic analyses identified more than 1,000 Msi1 targets whose biological functions supports its involvement in apoptosis, cell cycle, proliferation, adhesion, invasion, migration, DNA-repair and glioma cell survival [14,17]. Msi1 levels also impact chemo- and radio-resistance in tumor cells [13]. Importantly, neurosphere cultures from Msi1 knockout mice are similar to the ones from wild type mice, suggesting that ‘stem-like cells’ in tumor tissue are much more sensitive to a decrease in Msi1 levels than normal cells [19].
We proposed to use Msi1 targeting as an alternative approach to treat GBM patients. To find compounds that interfere with Msi1-RNA binding capacities, we conducted a high-throughput screen (HTS) using biochemical fluorescence polarization. We identified luteolin as the leading candidate; its interaction with Msi1 RNA binding domain 1 was validated by NMR and LSPR and this interaction was shown to impact Msi1 regulatory activity. Finally, assays in GBM cells and organoids showed that luteolin has anti-tumorigenic capacity and could potentiate the effect of radiation and anti-cancer agents.
Results
Identification of luteolin as a Msi1-RNA interaction inhibitor
We conducted a small molecule HTS to identify compounds capable of blocking the RNA binding surface of Msi1, thereby inhibiting Msi1 function. A fluorescence polarization assay was used to monitor binding of a recombinant Msi1 RBD1 to a short RNA oligonucleotide containing a Msi1 binding sequence and labeled with a fluorescent tag (5Cy3-iSp9-rGrUrArGrUrArGrU). Upon binding to Msi1, polarization of the Cy3-RNA fluorescence signal increased from 30mP (free oligonucleotide) to 150mP (Msi1-RNA complex). The addition of excess unlabeled RNA (rGrUrArGrUrArGrU) displaced the fluorescent probe resulting in reduction of polarization back to the 30mP baseline. Cy3-RNA probe bound to purified Msi1 RBD1 with a Kd of 1.5nM – Figure S1(a). The assay performed very well in the high-throughput format with a Z’ factor of 0.72. The collected data were calculated as % activity with Msi1 plus RNA-probe set to 100% and RNA-probe alone set to 0% activity. 25,588 compounds from several libraries were screened- Figure 1(a-b). Using a cut-off of 50% inhibition, 55 compounds were identified and rescreened in a 7-point dose response with concentrations ranging from 8nM to 60μM. As a result, 32 compounds were reconfirmed to inhibit Msi1 binding to the Cy3-RNA probe with IC50 values ranging from 1.0μM to 20μM. Six of these compounds were confirmed to directly disrupt Msi1-RNA interaction. Association curves for these six compounds are displayed in Supplementary Table 1. Luteolin, morin, aurincarboxylic acid and myricetin were identified as strong interactors while tryphostin-51 and piceatannol were classified as weak interactors. Direct binding of the compounds to the Msi1 RBD1 was also confirmed by NMR analysis. Compound binding was observed to shift and/or broaden NMR signals of protein residues previously shown to be involved in RNA binding – Figure 1(c).
Figure 2.
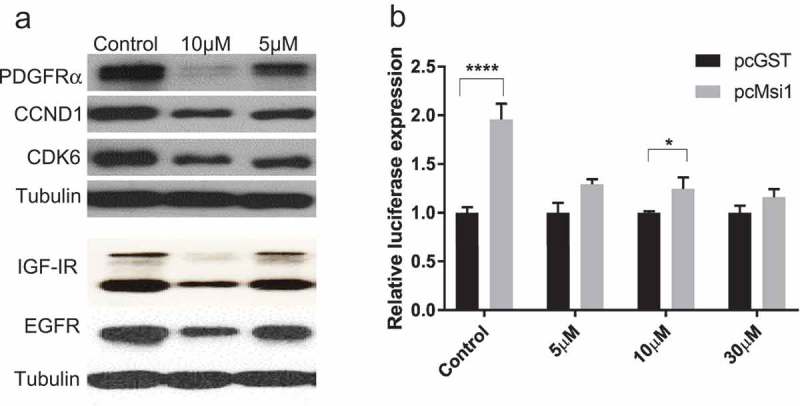
Luteolin impacts the expression of Musashi1 target genes. a) Western blot showing the impact of luteolin treatment on the expression of Msi1 target genes (PDGFRα, IGF-IR, EGFR, CCND1 and CDK6) 48 hours post-treatment. Msi1 functions as a positive regulator of these transcripts by increasing their translation or stability. Tubulin was used as loading control. b) We used a luciferase reporter containing the 3’UTR of PDGFRα to corroborate that luteolin treatment disrupts Msi1 regulatory functions. Msi1 is a positive regulator of PDGFRα translation. HT1080 cells were co-transfected with plasmids expressing either GST or Msi1. Msi1 expression affected the expression of the reporter, resulting increased luciferase activity. The effect was largely diminished when luteolin was added after transfection. Experiments were done in triplicate. A representative western is shown. Statistical significance was calculated by multiple t test. All data are shown as means ± s.d. (*P < 0.05, ***P < 0.005).
We chose luteolin for further studies as it showed strong interaction with Msi1 and the best overall results in preliminary studies with GBM cells. We further corroborated the interaction of Msi1 RBD1 with luteolin using LSPR. The LSPR data clearly showed that luteolin interacts with Msi1 to form a stable 1:1 complex with an equilibrium dissociation constant (Kd) of ~ 3.2 ± 0.02μM. The LSPR data also suggested high association (Kon ~ 6.16 X 103 ± 42 M−1s−1) and dissociation (Koff ~ 1.97 X 10−2 ± 2.93 X 10−7 s−1) rates for this interaction – Figure 1(d).
Luteolin inhibits Msi1 regulatory functions
We tested if luteolin treatment can disrupt Msi1 regulatory functions. U251 and U343 cells were treated with luteolin for 48 hours and the effect on the expression levels of Msi1 targets (PDGFRα, IGF-IR, EGFR, CCND1 and CDK6) was evaluated by western blot. CLIP analyses identified multiple Msi1 binding sites in the 3’UTR of these transcripts. Moreover, Msi1 knockdown was shown to reduce their expression levels [17,18]. We observed a dose-dependent decrease in protein levels of all Msi1 targets tested – Figures 2(a) and S1(c,f).
Figure 1.
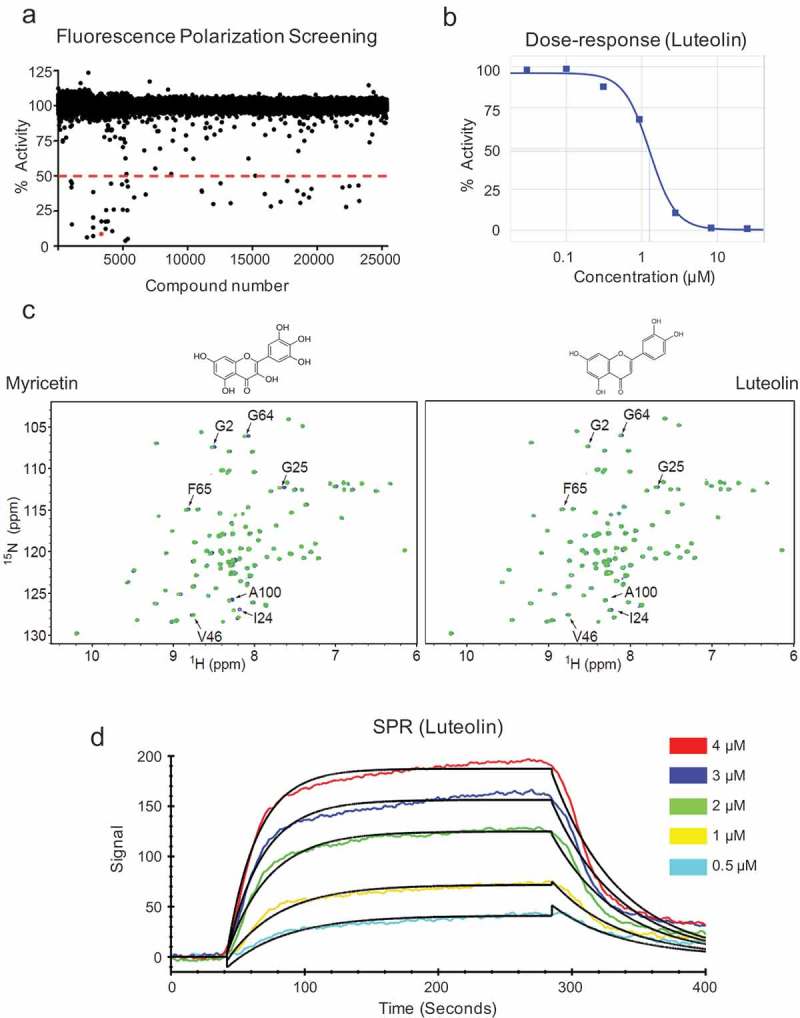
High throughput screen and identification of luteolin as an inhibitor of Msi1. a) Scatterplot displaying the screening results. 25,539 compounds were assayed in a high throughput screen targeting Msi1-RNA interaction. Compounds were tested for inhibition in 7-point dose response study with compounds ranging from 60μM to 8nM. The resulting data were expressed as % activity or the % of bound probe compared to untreated positive control (Msi1 plus Cy3-RNA probe). A 50% activity cutoff was employed to identify potential Msi1 inhibitors. b) Dose-response of luteolin. c) Validation of HTS hits by NMR spectroscopy. 15N HSQC spectra of free Msi1 (green) and Msi1 in the presence of 0.25mM myricetin or luteolin (blue). Chemical shift perturbations affect a subset of Msi1 residues previously shown to be involved in RNA binding. d) Msi1-luteolin interaction by localized surface plasmon resonance (LSPR). Representative sensograms for a series of luteolin concentrations (cyan, 0.5μM; yellow, 1μM; green, 2μM; blue, 3μM; red, 4μM) show both the association and dissociation phases of Msi1-luteolin interaction. The black lines represent the fitting of data to 1:1 binding model.
To corroborate luteolin’s impact on Msi1 regulatory activities, we checked if luteolin treatment impairs the positive effect Msi1 has on PDGFRa expression via its 3’UTR. CLIP results indicated that Msi1 has multiple binding sites on PDGFRa – Figure S1(g), and Msi1 knockdown decreases its translation [17]. Binding of Msi1 to PDGFR. 3’UTR was corroborated via RNA pulldown – Figure S1(g). We cloned the 3’UTR of PDGFRα into a luciferase reporter vector. The resulting luc-3’UTR PDGFRα clone and a Msi1 expression vector or control (GST) were transfected into HeLa or HT1080 cells. As expected based on Msi1 positive effect on PDGFRα translation [17], co-transfection with Msi1 produced an increase in luciferase activity. We then repeated this assay, this time adding luteolin or DMSO to the media 12 hours post-transfection. Luteolin treatment prevented Msi1-driven luc-3’UTR PDGFRα reporter expression – Figure 2(b) and S1(d). A control experiment performed with a luciferase reporter construct containing the 3’UTR of a non-Msi1 target (Prohibitin, PHB) shows that co-transfection of plasmids expressing Msi1 or GST have the similar impact on reporter activity in the presence or absence of luteolin – Figure S1(e).
Luteolin inhibits proliferation, colony formation, migration and invasion of glioblastoma cells
Msi1 silencing inhibits multiple cancer relevant phenotypes and abolishes growth of multiple tumor types including glioblastoma [14,15,17,20–22]. We evaluated if luteolin treatment could produce similar results in GBM cells. We selected U251 and U343 cells for analysis since these cells display high Msi1 expression and respond to its knockdown [17]. First, an Essen Bioscience IncuCyte automated microscope system was used to measure GBM proliferation over time and over a range of luteolin concentrations. Luteolin inhibited proliferation of both U251 and U343 cells – Figures 3(a) and S2(a). U343 cells appeared to be more resistant to treatment. This trend was observed in all assays we performed and may be expected since U343 cells display much higher levels of Msi1 expression than U251 – Figure S1(b). Next, we conducted a MTS assay to evaluate the impact of luteolin treatment on cell viability in GBM and normal astrocytes in parallel to evaluate any possible side effects of luteolin treatment. Differentiated cells display no or barely detectable levels of Msi1 expression [11]. Luteolin treatment decreased the viability of both U251 and U343 cells but not normal astrocytes at similar concentrations – Figures 3(b,c) and S2(b). In a colony formation assay, luteolin-treated cells also formed fewer and smaller colonies when compared to control – Figures 3(e) and S2(c). Since luteolin was identified straight from a screening, it is unrealistic to expect that it binds exclusively to Msi1. To determine how much Msi1 inhibition contributes to the impact luteolin has on glioblastoma cells, we treated U251 Msi1 knockout cells and conducted a cell viability assay. Once compared to the results obtained in U251 cells, we observed that Msi1 KO cells are a lot less sensitive to luteolin, supporting the idea that Msi1 inhibition is a critical component of luteolin’s effect on GBM cells – Figure 3(b,c).
Figure 3.
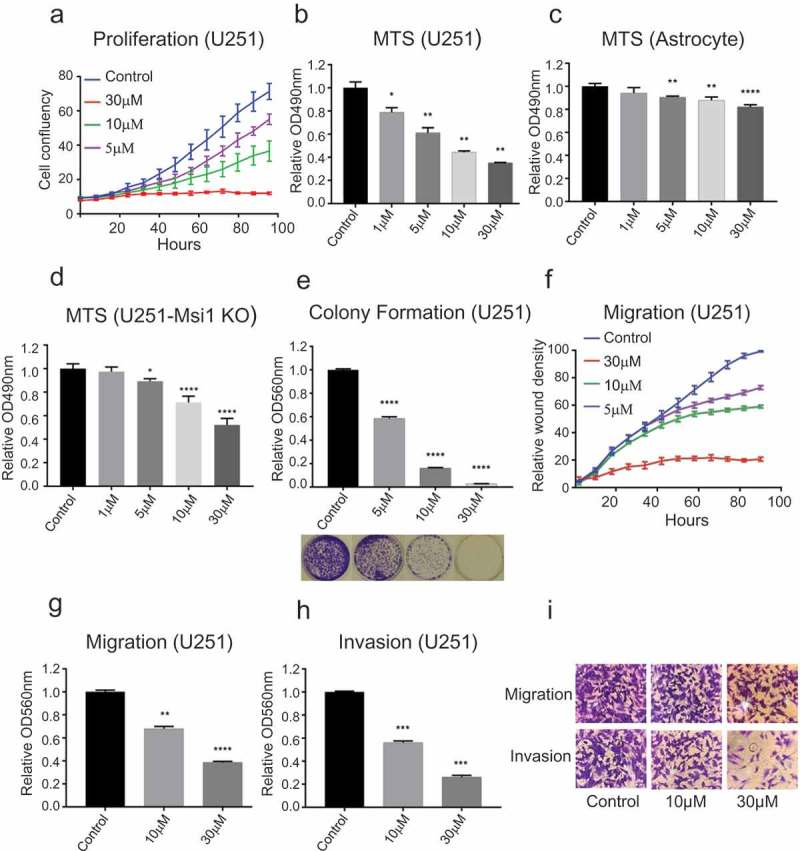
Luteolin impact on cancer relevant phenotypes. a) The Essen Bioscience IncuCyte automated microscope system was used to follow proliferation of U251 glioblastoma cells over a period of 96 hours. Treatment with luteolin produced a reduction in cell proliferation. b) MTS assay shows a decrease in viability of U251 cells after treated with luteolin. c) To determine possible side effects of luteolin treatment, we analyzed its impact on the viability of astrocytes by MTS assay. The results indicate that astrocytes tolerate luteolin even at high concentrations. d) To determine if Msi1 is a key target of luteolin in glioblastoma cells, we conducted an MTS assay with U251 Msi1 knockout cells. The results show that luteolin has stronger effects on viability of U251 cells (B) in comparison to U251 Msi1 knockout cells. e) After treated with luteolin or diluent (DMSO), U251 cells were seeded and their capacity to form colonies was evaluated. Cells exposed to luteolin formed less and smaller colonies compared to control. f) An in vitro scratch assay was used to evaluate the impact of luteolin on the migration of U251 cells. The Essen Bioscience IncuCyte automated microscope system recorded the cell density of the wound over a period of 96 hours. The graph shows an obvious decrease in wound density of the luteolin-treated cells. g-i) Two kinds of chambers from Corning were used to measure the effect of luteolin on migration and invasion of U251 cells. After treatment with luteolin for 48 hours, cells were plated onto the upper chamber (containing serum-free medium). 24 hours later the invaded or migrated cells in the lower chamber (containing 10% FBS medium) were stained and extracted with acetic acid. The relative OD560nm was used to quantify the relative cell invasion or migration. The graph shows that fewer cells migrated and invaded from the upper chamber to the lower chamber after treatment with luteolin; the relative OD560nm was proportional to the concentration of luteolin. DMSO was used as control in all biological assays. All experiments were performed in triplicate. Statistical significance was calculated by one-way ANOVA and t test. All data are shown as means ± s.d. (*P < 0.05, **P < 0.005, ***P < 0.0005, ****P < 0.0001).
Target analysis and subsequent biological assays in GBM cells have shown Msi1 mediated regulation of adhesion, migration and invasion [17]. We therefore asked if luteolin treatment inhibit GBM migration and invasion. We first performed in-vitro scratch assays with the IncuCyte system and determined that luteolin treatment impairs the ability of cells to close a wound – Figures 3(f) and S2(d). Next, transwell assays (Corning) were used to evaluate the effect of luteolin on migration and invasion. Optical density (OD560nm) was measured to determine cell density. We observed a dose-dependent decrease in the number of cells able to migrate or invade the basement membrane in response to luteolin – Figure 3(g-i) and S2(e-g).
Finally, we evaluated the effect of luteolin on patient-derived GIC cultures [23] and GBM organoids. Two GIC lines, 19NS and 84NS [24], which express high level of Msi1 (Figure S1(b)) were used. Luteolin treatment significantly decrease proliferation and viability of the two cell lines – Figures S3(a-d). Patient derived three-dimensional cultures (tumor organoids) recapitulate features of in vivo cell growth, differentiation and heterogeneity and serve as an excellent model to test anti-cancer drugs [25]. GBM528 and CCF1914 GBMs organoids were grown for > 2 months and then treated with DMSO or 30µM luteolin for 7 days. We found a dramatic reduction in 3-dimensional proliferation as measured by mitotic marker phospho-Histone H3 using immunohistochemistry in luteolin treated samples compared to control – Figure 4(a,b).
Figure 4.
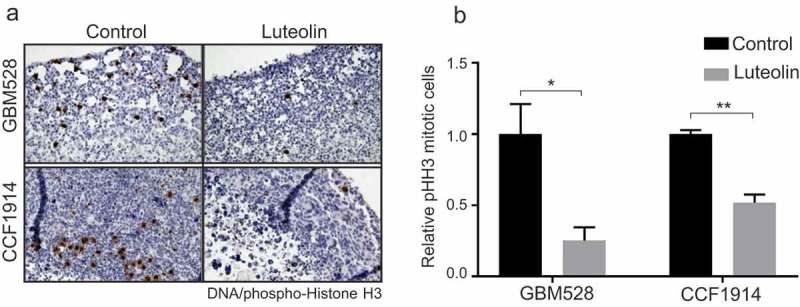
Luteolin inhibits proliferation of GBM organoids. Patient derived GBM528 or CCF1914 glioblastoma specimens were grown as 3-dimensional organoids for > 2 months to establish mature GBM tissue structures. Organoids were then treated with DMSO (control) or 30μM luteolin for 7 days. Treated organoids were probed for the mitotic marker phospho-Histone H3 by immunohistochemistry. Full digital slide scans were performed and 3–6 non-overlapping high power fields were extracted from each sample for automated quantification. Representative full 20x microscope fields are also shown. Statistical significance was calculated by one-way ANOVA and t test. All data are shown as means ± s.d. and performed as technical triplicates. (*P < 0.05, **P < 0.005).
Luteolin sensitizes glioblastoma cells to radiation and PARP inhibition
Glioblastoma is refractory to most treatments, suggesting that single agent therapies are unlikely to be effective alone. We have implicated Msi1 in radio-resistance and regulation of DNA replication and repair [11]. Similarly, we and others have shown that Msi1 levels influence chemo-sensitivity [13]. We therefore tested the value of luteolin in combined treatments.
We first asked if luteolin at low concentration could potentiate the effect of IR treatment. U251 and U343 cells were first treated with DMSO or 1.5 μM luteolin. At this concentration, luteolin produces a small effect on cell proliferation and viability. After 48 hours, cells were exposed to radiation from 0-10Gy. Subsequently, the treated cells were seeded for proliferation and colony formation assays. Dual treatment with luteolin clearly enhanced the effects of radiation on U251 proliferation compared to controls – Figure 5(a,b). To better illustrate the differences, we have plotted side by side the results of single and dual treatments at a 108-hour time point – Figure 5(c). Colony formation assay showed similar results – Figure 5(d,e). The benefit of dual treatment is particularly notable at high dosages of radiation. In U343 cells, the positive effects of dual treatment were also observed but less dependent on radiation dosage – Figure S4. To measure any synergistic effects of dual treatment, we calculated the combination index (CI) [26], in which CI < 1, = 1 or > 1, indicates that the treatments have synergistic effect respectively. Synergy was observed at all radiation doses, with the greatest synergistic effects at higher doses – Table S2.
Figure 5.
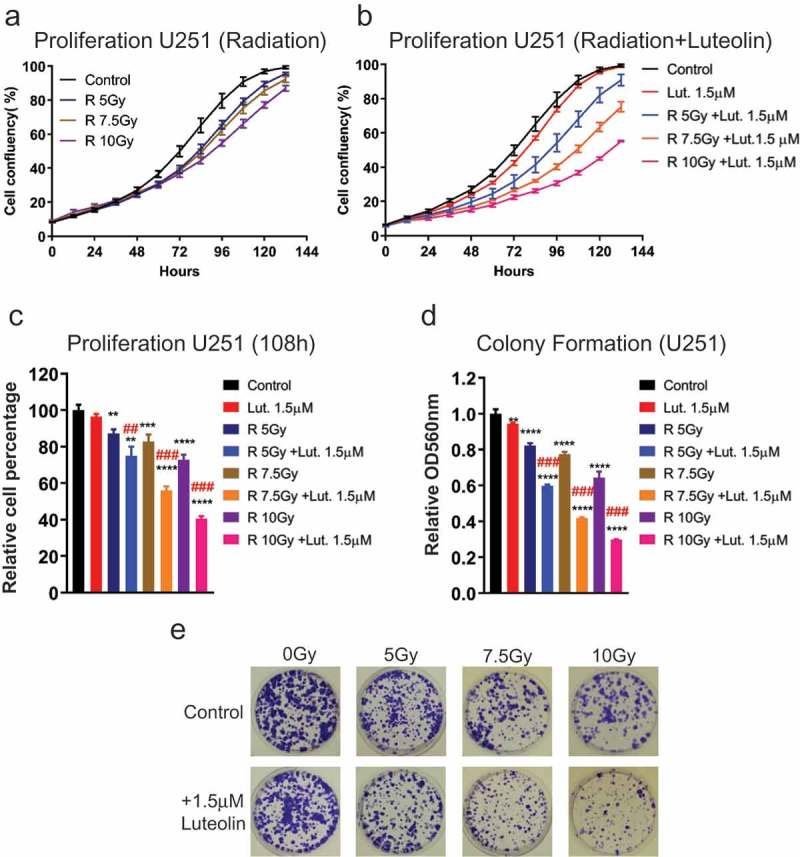
Luteolin sensitizes glioblastoma cells U251 to radiation. Glioblastoma cells were treated with luteolin or DMSO (control). 48 hours later, cells were exposed to radiation from 0-10Gy. Treated cells were harvested and seeded for proliferation and colony formation assays. a-b) The Essen Bioscience IncuCyte automated microscope system was used to follow proliferation of U251 glioblastoma cells over a period of 100 hours. Graphs show that comparatively, cells exposed to radiation and luteolin proliferate less than cells just exposed to radiation. c) The graph shows side-by-side differences in proliferation between single and combined treatment at 108 hours. d) Graph shows the result of colony formation assay. Colonies were extracted with acidic solution and relative absorbance was measured at 560nm. Combined treatment showed a more pronounced effect on colony formation. e) Colony formation plates were stained with violet blue. Cells treated with radiation and luteolin produce fewer and smaller colonies in comparison to the ones just exposed to radiation. All experiments were done in triplicate. The combination treatment of luteolin and radiation showed synergistic effect both in proliferation and colony formation, which was judged by the Combination Index (CI) (##CI< 0.9, ###CI< 0.7). Statistical significance was calculated by one-way ANOVA and multiple t test. All data are shown as means ± s.d. (**P < 0.005, ***P < 0.0005, ****P < 0.0001).
Olaparib, is a poly (adenosine diphosphate [ADP]-ribose) polymerase 1 (PARP1) inhibitor, capable of suppressing activities of PARP1 in single-strand break (SSB) repair [27]. PARP inhibitors have been shown in several studies to control glioblastoma growth in combination with different agents [28,29] and are currently being evaluated in six different clinical trials. Importantly, the catalytic subunit of ribonucleotide reductase M2 (RRM2) which we determined to be regulated by Msi1, protects GBM cells from replication stress, DNA damage and apoptosis. Treatment with RRM2 inhibitor increased radio-sensitivity, prevented tumor growth and sensitized GBM cells to PARP inhibition [30]. Therefore, we decided to test the potential use of combined luteolin-olaparib treatment in GBM therapy. We first tested different concentrations of olaparib on a cell proliferation assay and defined an effective concentration range from 0 to 20μM. As above, proliferation and colony formation assays were used to evaluate the benefit of combined treatment. Combined luteolin and olaparib-treatment of U251 cells displayed a stronger inhibition of proliferation than olaparib alone – Figure 6(a,b). To better illustrate the gains obtained with the addition of luteolin, we plot side-by-side the results of single and dual treatments at time point 90 hours – Figure 6(c). The results obtained in the colony formation assay show a similar trend – Figure 6(d,e). In U343 cells, the positive effects of dual treatment were also observed but less dependent on olaparib concentration – Figure S5. As above, we determined that luteolin and olaparib have synergistic effect – Table S3.
Figure 6.
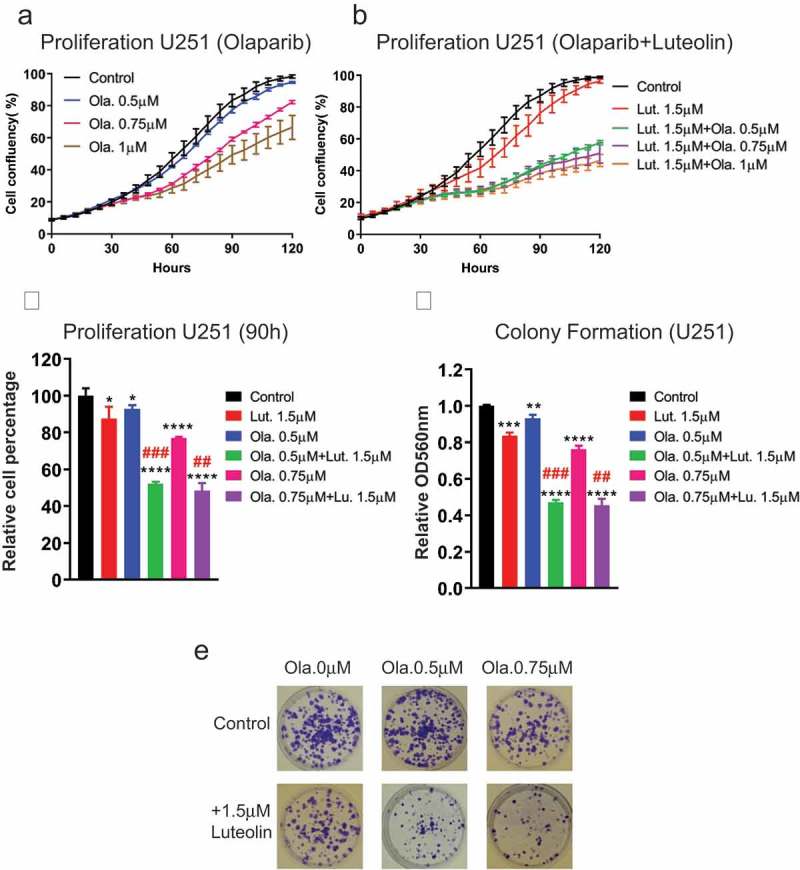
Luteolin sensitizes glioblastoma cells U251 to PARP inhibitor (olaparib). Glioblastoma cells were treated with compounds (luteolin, olaparib or combination of both) and DMSO (control). 48 hours later, the treated cells were harvested and seeded for proliferation and colony formation assays. a,b) The Essen Bioscience IncuCyte automated microscope system was used to follow proliferation of U251 glioblastoma cells over a period of 120 hours. Graphs show cells grew slower in response to combination treatment than olaparib single-treatment. c) The graph shows side by side differences in proliferation between single and combined treatment at 90 hours. d) Graph shows the result of colony formation assay. Colonies were extracted with acidic solution and relative absorbance was measured at 560nm. Combined treatment showed a more pronounced effect on colony formation. e) Colony formation plates were stained with violet blue. Cells treated with olaparib and luteolin produce fewer and smaller colonies in comparison to the ones just exposed to olaparib. All experiments were performed in triplicate. The combination treatment of luteolin and olaparib showed synergistic effect both in proliferation and colony formation, which was judged by the Combination Index (CI) (##CI< 0.9, ###CI< 0.7). Statistical significance was calculated by one-way ANOVA and t test. All data are shown as means ± s.d. (*P < 0.05, **P < 0.005, ***P < 0.0005, ****P < 0.0001).
Luteolin analogs and interaction with Msi1
Inhibitors identified in a screening require further analysis and rounds of modifications to prepare them for clinical applications, these include alterations to enhance binding, specificity and ‘drug-like’ properties. We examined nine luteolin analogs using our fluorescence polarization competition assay as a first attempt to identify molecular features required for interaction with Msi1 – Table S4. Only quercetin and myricetin interacted with Msi1 RBD1. We tested then their impact on the proliferation of U251 and U343 cells in comparison to luteolin. Although, both reduced proliferation of GBM cells, the effect was less dramatic compared to the results obtained with luteolin – Figure S6.
Discussion
Targeting RNA-binding proteins in cancer therapy
RNA-binding proteins modulate gene expression in multiple ways, from RNA processing to translation. Analysis of TCGA data revealed a large number of mutations in RBPs and dramatic alterations in their expression levels across tumor types [31,32]. These studies helped with the identification of a growing number of RBPs involved in tumor initiation and development and paved the way to explore them as targets in cancer therapy [10]. Since Msi1 has been implicated in the development of multiple tumor types [13], the use of luteolin could be expanded to treat other malignances besides glioblastoma.
Different approaches can be used to target onco-RBPs. Perhaps the most cost-effective one is the use of small molecule inhibitors as exemplified by our study. In this regard, splicing regulators have been the center of attention with multiple specific inhibitors developed in the past decade [33]. Similar to our study, an HTS identified several compounds able to block HuR-mRNA interaction [34]. Another common strategy is the use RNA aptamers or modified RNA oligos mimicking the consensus binding motif of the RBP to be targeted. The efficiency of these methods depends on the nature of the oligo and inclusion of RNA modifications that can increase stability and delivery. The last resource to target RBPs is to regulate their expression levels rather than function. For instance, intravenous injection of a specific antisense oligonucleotide against eIF4E prevented tumor growth by repressing the translation of oncogenic factors highly dependent on elF4E expression levels [35].
Musashi1 as oncogenic factor in multiple tumor types
Since Musashi1 has been implicated in the development of multiple tumor types [13], the use of luteolin could be expanded to treat other malignances. In medulloblastoma, we established that high Msi1 expression correlates with poor prognosis and therapy response and is prevalent in high risk sub-groups 3 and 4 [14]. Msi1 knockdown reduced neurosphere formation and expression of stem cell markers, proliferation and tumor growth and increased chemo-sensitivity [14,36]. Msi1 has been well characterized in the context cancers of the digestive system. In colorectal cancer, knockdown of Msi1 suppressed cell proliferation, colony formation, tumorsphere formation, G0/G1 arrest and tumor growth [37]. Msi gene deletion inhibited tumorigenesis in different mouse models of intestinal cancer [38]. Msi1 contributes to tumorigenesis in colon cancer by activating multiple pathways such as Notch and the PDK-Akt-mTORC1 axis [20,37,39]. Transgenic expression of Msi1 in the intestine increased cell proliferation and expansion of the progenitor layer. RNAseq analysis showed that genes affected by Msi1 induced expression are preferentially associated with cell cycle, DNA replication and drug metabolism [18]. Msi1 has been shown to be significantly upregulated in gastric cancer and to correlate with tumor size, tumor-node-metastasis (TNM) stage, Lauren classification, depth of invasion, vessel invasion, lymph node metastasis and distant metastasis. High levels of Msi1 expression also correlate with poor patient survival [40]. Msi1 modulates cell growth and cell cycle in hepatoma by activation of Wnt pathway via inhibition of APC and DKK1 [41]. In cervical cancer, silencing of Msi1 inhibited cell migration, invasion as well as epithelial-to-mesenchymal transition (EMT) through activation of the Wnt signaling pathway [42]. In breast and lung cancers, Notch and Wnt pathways are also Msi1’s preferential routes of action [22,43].
Luteolin as an anti-cancer therapy agent
Luteolin (3’,4’,5,7-tetrahydroxyflavone), a common dietary flavonoid, is well-known in Chinese traditional medicine and used in the treatment of cardiac diseases [44], inflammatory disorders [45] and cancer [46]. Recently, luteolin has been explored more formally as a potential therapeutic agent. In colon cancer, luteolin treatment inhibited tumorigenesis through the regulation of multiple events including antioxidant activity, EMT, COX-2 expression and modulation of MAPK signaling pathway [47,48]. In gastric cancer, luteolin treatment suppressed angiogenesis by blocking Notch1/VEGF signaling [49] and reversed EMT by inhibiting Notch signaling pathway [50]. In breast cancer, luteolin prevented lung metastasis, and decreased cell migration, viability and secretion of VEGF [51]. In prostate cancer, luteolin treatment inhibited proliferation and migration by downregulating the expression of Anoctamin 1 [52]. In glioblastoma, luteolin decreased EGFR-mediated proliferation through Akt and MAPK pathway [53], induced apoptosis by ROS/ER stress and mitochondrial dysfunction [54], and reduced migration via inhibition of p-IGF-1R/PI3K/AKT/mTOR signaling pathway and downregulation of CDC42 expression [55,56]. Moreover, luteolin and silibinin showed synergistic effect on migration, invasion and apoptosis in glioblastoma cells and overexpression of miR-7-1-3p amplified their effects on apoptosis and inhibition of autophagy [57,58].
Glioblastoma is a very aggressive tumor type and it is very unlikely that a single treatment agent would lead to major improvements in survival. In this regard, our results corroborate several studies showing luteolin’s potential in combined therapy. Various studies have shown that luteolin combined therapy decreased proliferation, apoptosis and autophagy; drugs used include kaempherol in lung adenocarcinoma and 5-fluorouracil in hepatocellular carcinoma [58–60].
Ionizing radiation therapy is part of GBM standard-of-care. DNA double-strand breaks (DSB) are the main cytotoxic lesions induced by radiations. Our previous study has shown that Msi1 plays an important role in DSB repair in glioblastoma via regulation of DNA-protein kinase catalytic subunit [16]. In fact, our target analysis indicates that Msi1 impact on DNA replication and repair is much more robust and it likely modulates other repair mechanisms, relevant targets include RRM2, GINS4, RBBP4, RBM14, Tipin, KIAA0101 and CDC6 [17]. As expected, Msi1 knockdown via siRNA increased GBM cells sensitivity to radiation [16]. Having these results in mind, we tested and demonstrated the positive effects of luteolin and radiation combined treatment. Similar to our results, luteolin and radiation displayed synergistic effects on invasiveness and clonogenicity of oral carcinoma cells via inactivation of IL-6/STAT3 signaling [61].
In conclusion, we identified luteolin as an inhibitor of Msi1. We confirmed the direct interaction between Msi1 RNA binding domain and luteolin and its ability to block Msi1 positive effect on the expression of a few target genes. Genomic analyses are still required to fully evaluate the impact of luteolin on Msi1 regulatory functions. Luteolin affected several cancer relevant phenotypes in GBM cells, GICs and tumor-organoids and worked well in combined treatments with PARP inhibitor and IR. We established Msi1 targeting as a potential treatment option and paved the way to explore other oncogenic RBPs as therapeutic targets.
Material and methods
Production of recombinant Musashi1 protein
pET-Msi1 plasmid encoding the sequence of RNA binding domain of Msi1 (RBD1), was transformed into BL21(DE3) competent cells. Colonies were cultured in M9 medium containing carbenicillin, 15N-NH4Cl and glucose. Cells were induced at OD600 ~ 0.6 with 0.084mM IPTG and incubated overnight at 37ºC. Cells pellets were re-suspended in 50mM Tris buffer (pH 8.0) with 1M NaCl and lysed by sonication. Supernatant was subjected to Ni2+ affinity chromatography and was further purified using Sephadex G75 gel filtration chromatography. The purity of Msi1 RBD1 was checked on SDS-PAGE gel.
Fluorescence polarization competition assay
We used a fluorescent RNA probe, 5Cy3-iSp9-rGrUrArGrUrArGrU, containing a Msi1 binding sequence to perform a HTS to identify compounds interacting with Msi1 RBD1 [62]. The equilibrium dissociation constant (Kd) value was calculated using a linear regression analysis for a one site binding isotherm. Through a series of optimization steps, a highly robust and reproducible assay with a Z’ factor score of 0.72 was developed. The change in polarization of the Cy3-RNA probe ranged from 30mP (free unbound) to 150mP (bound to Msi1). Excess unlabeled RNA (rGrUrArGrUrArGrU) displaced the probe, resulting in 100% free ligand and polarization of 30mP. The final assay conditions had the following components: 40nM recombinant Msi1 protein, 10nM RNA-probe in a 50mM Tris buffer, pH 7.5, containing 50mM NaCl. Each plate contained 320 distinct compounds at a final concentration of 20μM and the necessary controls. These controls included (1) Cy3-RNA-probe alone, (2) Msi1 with the Cy3-RNA-probe and (3) Ms1 with the Cy3-RNA-probe in an excess of unlabeled RNA. In total 25,588 compounds from the Prestwick FDA-approved (1,200 compounds), Library of Pharmacologically Active Compounds (LOPAC, 1,280 compounds), Cambridge NovaCORE (20,000 compounds) and the Life Chemical FSP3-enriched (3,108 compounds) libraries were screened. The collected data were then recalculated as % activity with Msi1 plus RNA-probe set to 100% and RNA-probe alone set to 0% activity. Potentially, active compounds were cherry picked and rescreened in a 7-point dose response with concentrations ranging from 8nM to 60μM. As a result, compounds were reconfirmed as inhibitors of Cy3-RNA probe binding with IC50 values ranging from 1.0μM to 20μM.
Nuclear magnetic resonance (NMR)
Direct physical interaction of HTS hits with Msi1 was evaluated using solution NMR spectroscopy. Msi1 RBD1 was expressed in M9 minimal media containing 15N-labeled ammonium chloride as the sole nitrogen source as described above. Msi1 NMR samples were prepared in 20mM MES pH 6.5, 100mM NaCl buffer at 0.1mM Msi1 concentration. 15N HSQC spectra of Msi1 with and without compounds were collected using a 500MHz Bruker spectrometer equipped with a 1.7mm cryoprobe. Compounds were added to Msi1 to yield 0.25mM final concentration. Spectra were processed using NMR Pipe software package.
Localized surface plasmon resonance (LSPR)
To determine the kinetics of Msi1-luteolin interaction, we used LSPR method (openSPR XT, Nicoya Lifesciences, ON, Canada). The purified Msi1 RBD1 was diluted to 3μM concentration into activation buffer. The protein was immobilized on COOH sensor chip. All experiments were performed using filtered and degassed HBS-EP buffer (10mM HEPES pH 7.5, 150mM NaCl, 3mM EDTA, 0.005% tween 20) at a continuous flow rate of 20μl/min at 20°C. The analyte (luteolin 0.5 – 4μM) was passed over the immobilized Msi1 protein. The sensor chip surface was regenerated by flowing the running buffer for longer time periods. The association and dissociation phases were recorded for 240 and 600 seconds, respectively. The dissociation curves up to 400 seconds were included in data analysis. We also passed the analyte over a blank COOH sensor chip to measure background responses upon luteolin binding to sensor chip. The background subtracted data were analyzed and fitted with 1:1 binding model using Trace Drawer software.
Cell culture
Human tumor cell lines HT-1080 and HeLa were obtained from ATCC. Glioblastoma lines U251 and U343 were obtained from Uppsala University (Uppsala, Sweden). U251 Msi1 KO lines was described in [16]. GIC lines 19NS and 84NS were obtained from Dr. Ichiro Nakano and are described in [24]. Human astrocytes were established as described in [63]. HT-1080 cells were cultured in EMEM medium (Lonza; Cat# 12662F). U251, U343, HeLa and astrocytes were cultured in DMEM medium (HyClone; Cat# SH30243.01). 19NS and 84NS cells were cultured in DME/F-12 medium (HyClone; Cat# SH30023.01) with 2% B27 (Invitrogen; Cat# 17504044), 32ku heparin (Sigma Aldrich; Cat# H3149110KU), 20ng/ml bFGF (Peprotech; Cat# 100-18B) and 20ng/ml EGF (Peprotech; Cat# AF-100-15). Media were supplemented with 10% FBS (Corning; Cat# 35015CV) and 1% penicillin/streptomycin (Gibco; Cat# 10378016).
Luteolin and Dimethyl sulfoxide (DMSO) were obtained from Sigma Aldrich (Cat# L9283; Cat# D8418).
Organoid culture
Primary glioblastoma samples were obtained in accordance with protocol #2559 (Cleveland Clinic Institutional Review Board) and were cultured as tumorspheres in NBM complete media – neurobasal medium supplemented with EGF (R&D Systems; Cat# P01133), bFGF (R&D Systems; Cat# P09038), B27 (Invitrogen), glutamine (CCF media core; Cat# CCFGB002), sodium pyruvate (Invitrogen; Cat# 11360070) and antibiotics (Invitrogen; Cat# 15240062). Organoids were formed and propagated as previously described [25]. Organoids were formed by suspending tumor cells in BD Matrigel and forming 20μl pearls on parafilm molds prior to solidification and culture. Then, organoids were cultured in bulk 10cm plates, shaking in NBM complete media. Individual organoids were arrayed into 12-well plates for luteolin treatment studies, shaking for 7 days. Treated or control organoids were fixed in 4% paraformaldehyde and embedding in paraffin. 7μm sections were stained with phospho-histone H3 (Ser10) (Cell Signaling Technology; Cat# 9701) and detected with chromogenic DAB staining. Full high-resolution slide scans were acquired using a Leica Aperio slide scanner and multiple non-overlapping high-power fields were extracted for quantification. Quantification of cell division rates was performed using ImageJ (FIJI package) software and color-threshold to define hematoxylin (marking total nuclei) and DAB-pHH3 (dividing cells) in 3–6 high power fields per sample.
Western blotting
U251 and U343 cells were treated with luteolin at different concentrations and incubated for 48 hours. Cells were harvested and lysed in Laemmli sample buffer. Cell extracts were separated on an 8% SDS-PAGE gel, and transferred to PVDF membranes, previously activated with methanol. Membranes were blocked in Tris-buffered saline with Tween 20 and 5% milk and probed with a collection of different antibodies, anti-IGF-IR (Santa Cruz Biotechnology; Cat# sc-713), anti-EGFR (Cell Signaling Technology; Cat# 2646), anti-PDGFRα (Cell Signaling Technology; Cat# 5241), anti-CCND1 (Millipore; Cat# EP272Y), anti-CDK6 (Cell Signaling Technology; Cat# 3136), and anti-α-tubulin (GeneTex; Cat# GTX12130). Horseradish peroxidase (HRP)-conjugated goat anti-rabbit antibody (Santa Cruz Biotechnology; Cat# sc-2030) or HRP-conjugated goat anti-mouse (Santa Cruz Biotechnology; Cat# sc-2005) were used as secondary antibodies. Immobilon Western chemo-luminescence substrate (Millipore; Cat# WBKLS0500) was used to detect selected proteins.
Luciferase reporter assay
Luciferase reporter assays were performed with Dual-Luciferase Reporter Assay System according to manufacturer’s instructions (Promega). HT-1080 cells were plated (96-well) at a density of 6,500 cells/well and HeLa 4,000 cells/well. GeneJammer Transfection Reagent (Agilent Technologies; Cat# 204130) was used to transfect cells. A luciferase reporter vector (pGLO-3’-UTR; Promega; Cat# E1330) containing the 3’UTR of PDGFRα (NM_001347830.1, 2987 nucleotides) was co-transfected with pcDNA expression vectors (Invitrogen; Cat# V79020) expressing either Msi1 or GST (negative control). Transfected cells were treated with either luteolin or DMSO. Forty-eight hours post transfection, cells were harvested and firefly luciferase reporter and Renilla luciferase activities were measured. Firefly luciferase reporter activity was expressed as a fold change following normalization to Renilla luciferase activity.
Cell proliferation assay
U251 and U343 cells were plated onto 96-well plates (800 cells/well for U251, 1,600 for U343). 19NS and 84NS cells were ressuspended in Gibco versene solution (Thermo Fisher Scientific; Cat# 15040066) and plated onto 96-well plates (5,000 cells/well) coated with Gibco GeltrexTM basement membrane matrix (Thermo Fisher Scientific; Cat# 12760013). 24 hours later, cells were treated with luteolin or DMSO. Plates were transferred to the IncuCyte automated microscope system (Essen BioScience) and cells were counted every 2 hours for 4–6 days.
MTS assay
U251, U343, 19NS, 84NS GSC and astrocytes were plated onto a 96-well plate (2,000 cells/well for U251 and 19NS, 4,000 cells for U343, 84NS and astrocytes), treated with luteolin or DMSO as described above and incubated at 37°C for 72 hours. Next, 20μl of MTS mixture (1,000μl MTS and 50μl PMS) were added to each well and samples were incubated at 37°C for an hour. Optical density was measured at absorbance 490nm with a Synergy HT microplate reader (BioTek).
Colony formation assay
U251 and U343 cells were plated onto 12-well plates (1,000 cells/well for U251, 2,000 for U343). Luteolin or DMSO were added at different concentrations the next day; medium containing 2% FBS with luteolin or DMSO was refreshed every three days. After 10–14 days incubation, colonies were fixed with 4% formaldehyde and then stained with 0.5% crystal violet. Stained cells were then extracted using 200μl of 33% acetic acid solution and the optical density was measured at 560nm.
Cell migration analysis using the Incucyte system
U251 and U343 cells were plated onto 96-well plates (5,000 cells/well for U251, 10,000 for U343). When cells reached 90–100% confluence, a 96-pin WoundMaker (Essen BioScience) was used to create precise and reproducible wounds. Then medium was replaced and luteolin or DMSO was added. The software was set to scan every 2 hours for 4 days. The data was analyzed by using the relative wound density integrated metric.
Cell migration and invasion by transwell assay
Cell invasion and migration were measured with the Corning BioCoat chamber (Corning; Cat# 354480 and Cat# 354578) – the basement membrane in the chamber for invasion contains the Matrigel matrix while migration do not – using 24-well plates. U251 and U343 cells were treated with luteolin or DMSO for three days, then harvested and counted. A total of 500μl of medium containing 10% fetal bovine serum was added to the lower chamber and 200μl serum-free medium with 5 × 104 cells were transferred to the upper chamber. Plate was incubated at 37°C for 24–48 hours. The chamber was fixed in 4% Formaldehyde, then stained with 4% crystal violet and washed with deionized water. Images of the migrated or invaded cells in the lower chamber was taken with a Nikon Eclipse TS2000 inverted microscope equipped with a DS-L2 camera control unit at × 20 magnification. The stained cells were then extracted using 200μl of 33% acetic acid and measured at absorbance 560nm with a microplate reader.
Combined treatment of luteolin with Ionizing Radiation (IR) or PARP inhibitor
For combined treatment with IR, U251 and U343 cells were first plated onto 35mm dish and 24 hours later treated with luteolin or DMSO. At 72 hours, cells were exposed to ionizing radiation (IR) at a dose ranging from 1-10Gy using the CP-160 Cabinet X-radiator (Faxitron X-ray Corporation). Next, treated cells were incubated at 37°C for 2 hours to stabilize, resuspended in fresh medium and plated onto 96-well plate for proliferation assay and 12-well plate for colony formation assay as described above.
For combined treatment with PARP inhibitor, U251 and U343 cells were plated onto 96-well plate for proliferation assay and 12-well plate for colony formation assay. 24 hours later, cells were treated with PARP inhibitor (olaparib) (Selleckchem; Cat# S1060) at several different concentrations combined with a low concentration of luteolin or DMSO. Proliferation and colony formation assays were done as described above.
Statistical analysis
Prism software (Prism, 7.0; GraphPad), one-way ANOVA and t-Test were used to analyze the data. A threshold of P < 0.05 was defined as statistically significant.
The synergistic effects of luteolin and radiation or PARP inhibitor (olaparib) were determined via the combination index (CI) [26]. CI = AB/ (A × B) where: AB = measured value for combined treatment/value for the control (DMSO), A and B = value for the single treatment/value for the control. Thus, CI < 1, = 1 or > 1, indicates that the combination treatments are synergistic, additive or antagonistic.
In vitro transcription assay
Nucleotides 716 to 1936 of PDGFRα 3’ UTR (both orientations) were inserted into the pTZ57R/T vector (ThermoFisher) and the resulting clones were digested and used as templates for in vitro transcription assays using T7 RNA Polymerase (Promega) according to manufacturer. Synthesized RNAs were incubated for 10 minutes with DNase TURBO and further precipitated with 3M ammonium acetate (pH 5.3) and 2.5 volumes of ethanol at −20°C for 24 hours. The tubes were then centrifuged and RNA samples were washed with cold 70% ethanol, dried and resuspended in water.
RNA pull down
175 pmol of transcribed RNA samples were Biotin-labeled using the Pierce RNA 3’ End Desthiobiotinylation Kit. Labeled RNAs were extracted with Chloroform:isoamyl alcohol (24:1) and precipitated for 24 hours, centrifuged, washed with cold 70% ethanol, dried and diluted in water. Pull down assays were carried out by incubating 30µg of fusion proteins GST-Msi1 or GST (negative control) with 50 pmol of labeled RNA (sense or antisense) for 1 hour at 4°C. RNA-protein complexes were pulled down with the Pierce Nucleic-Acid compatible streptavidin magnetic beads, resolved in SDS-PAGE and further analyzed by Western blotting by using an anti-GST antibody (sc-138 ; Santa Cruz).
Funding Statement
This work was supported by grants from Voices Against Brain Tumor and Greehey Children’s Cancer Research Institute. Caihong Yi was sponsored by the Second Xiangya Hospital of Central South University (CSU). The work performed in the Center for Innovative Drug Discovery was supported, in part, by the National Center for Advancing Translational Sciences, National Institutes of Health, under Grant UL1 TR001120. Christopher Hubert was supported by NIH under grant F32CA18964701 and by the Center for Transformational Nanoscience. The authors would also like to acknowledge the grant support (RP160884) from the Cancer Prevention and Research Institute of Texas. The Institutional NMR Core Facility at the UT Health San Antonio is supported in part as a Shared Resource of the NIH P30 CA054174 to the Mays Cancer Center - UT Health San Antonio.
Acknowledgments
The authors would like to thank Suzanne Burns for all her contributions over the years and the groundwork that put Musashi1 on the map. We would like to thank Ichiro Nakano and Jeremy Rich for GICs and organoids.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Ostrom QT, Gittleman H, Liao P, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro Oncol. 2017. November 6;19(suppl_5):v1–v88. PubMed PMID: 29117289; PubMed Central PMCID: PMCPmc5693142. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Weller M, Cloughesy T, Perry JR, et al. Standards of care for treatment of recurrent glioblastoma–are we there yet? Neuro Oncol. 2013. January;15(1):4–27. PubMed PMID: 23136223; PubMed Central PMCID: PMCPmc3534423. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Stupp R, Mason WP, van Den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005. March 10;352(10):987–996. PubMed PMID: 15758009; eng. [DOI] [PubMed] [Google Scholar]
- [4].Nam JY, de Groot JF.. Treatment of Glioblastoma. J Oncol Pract. 2017. October;13(10):629–638. PubMed PMID: 29020535; eng. [DOI] [PubMed] [Google Scholar]
- [5].Chinot OL, Wick W, Mason W, et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014. February 20;370(8):709–722. PubMed PMID: 24552318; eng. [DOI] [PubMed] [Google Scholar]
- [6].Gerstberger S, Hafner M, Tuschl T. A census of human RNA-binding proteins. Nat Reviews Genet. 2014. December;15(12):829–845. PubMed PMID: 25365966; eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lennox AL, Mao H, Silver DL. RNA on the brain: emerging layers of post-transcriptional regulation in cerebral cortex development. Wiley Interdiscip Rev Dev Biol. 2018. January;7(1):e290 PubMed PMID: 28837264; PubMed Central PMCID: PMCPmc5746464. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Brinegar AE, Cooper TA. Roles for RNA-binding proteins in development and disease. Brain Res. 2016. September 15;1647:1–8. PubMed PMID: 26972534; PubMed Central PMCID: PMCPmc5003702. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Pereira B, Billaud M, Almeida R. RNA-binding proteins in cancer: old players and new actors. Trends Cancer. 2017. July;3(7):506–528. PubMed PMID: 28718405; eng. [DOI] [PubMed] [Google Scholar]
- [10].Hong S. RNA binding protein as an emerging therapeutic target for cancer prevention and treatment. J Cancer Prev. 2017. December;22(4):203–210. PubMed PMID: 29302577; PubMed Central PMCID: PMCPmc5751837. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kaneko Y, Sakakibara S, Imai T, et al. Musashi1: an evolutionally conserved marker for CNS progenitor cells including neural stem cells. Dev Neurosci. 2000;22(1–2):139–153. PubMed PMID: 10657706; eng. [DOI] [PubMed] [Google Scholar]
- [12].MacNicol AM, Hardy LL, Spencer HJ, et al. Neural stem and progenitor cell fate transition requires regulation of Musashi1 function. BMC Dev Biol. 2015. March 18;15:15 PubMed PMID: 25888190; PubMed Central PMCID: PMCPmc4369890. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kudinov AE, Karanicolas J, Golemis EA, et al. Musashi RNA-binding proteins as cancer drivers and novel therapeutic targets. Clin Cancer Res. 2017. May 1;23(9):2143–2153. PubMed PMID: 28143872; PubMed Central PMCID: PMCPmc5413399. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Vo DT, Subramaniam D, Remke M, et al. The RNA-binding protein Musashi1 affects medulloblastoma growth via a network of cancer-related genes and is an indicator of poor prognosis. Am J Pathol. 2012. November;181(5):1762–1772. PubMed PMID: 22985791; PubMed Central PMCID: PMCPmc3761132. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kanemura Y, Mori K, Sakakibara S, et al. Musashi1, an evolutionarily conserved neural RNA-binding protein, is a versatile marker of human glioma cells in determining their cellular origin, malignancy, and proliferative activity. Differentiation. 2001. September;68(2–3):141–152. PubMed PMID: 11686236; eng. [DOI] [PubMed] [Google Scholar]
- [16].de Araujo PR, Gorthi A, Da Silva AE, et al. Musashi1 impacts radio-resistance in glioblastoma by controlling DNA-protein kinase catalytic subunit. Am J Pathol. 2016. September;186(9):2271–2278. PubMed PMID: 27470713; PubMed Central PMCID: PMCPmc5012509. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Uren PJ, Vo DT, de Araujo PR, et al. RNA-binding protein musashi1 is a central regulator of adhesion pathways in glioblastoma. Mol Cell Biol. 2015. September 1;35(17):2965–2978. PubMed PMID: 26100017; PubMed Central PMCID: PMCPmc4525321. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Cambuli FM, Correa BR, Rezza A, et al. A mouse model of targeted musashi1 expression in whole intestinal epithelium suggests regulatory roles in cell cycle and stemness. Stem Cells (Dayton, Ohio). 2015. December;33(12):3621–3634. PubMed PMID: 26303183; PubMed Central PMCID: PMCPmc4713339. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sakakibara S, Nakamura Y, Yoshida T, et al. RNA-binding protein Musashi family: roles for CNS stem cells and a subpopulation of ependymal cells revealed by targeted disruption and antisense ablation. Proc Natl Acad Sci USA. 2002. November 12;99(23):15194–15199. PubMed PMID: 12407178; PubMed Central PMCID: PMCPmc137566. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Plateroti M, de Araujo PR, Da Silva AE, et al. The RNA-binding protein Musashi1: a major player in intestinal epithelium renewal and colon cancer development. Curr Colorectal Cancer Rep. 2012;8(4):290–297. PubMed PMID: 23914149; PubMed Central PMCID: PMCPmc3728701. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lagadec C, Vlashi E, Frohnen P, et al. The RNA-binding protein Musashi-1 regulates proteasome subunit expression in breast cancer- and glioma-initiating cells. Stem Cells (Dayton, Ohio). 2014. January;32(1):135–144. PubMed PMID: 24022895; PubMed Central PMCID: PMCPmc3968686. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wang XY, Yu H, Linnoila RI, et al. Musashi1 as a potential therapeutic target and diagnostic marker for lung cancer. Oncotarget. 2013. May;4(5):739–750. PubMed PMID: 23715514; PubMed Central PMCID: PMCPmc3742834. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lathia JD, Mack SC, Mulkearns-Hubert EE, et al. Cancer stem cells in glioblastoma. Genes Dev. 2015. June 15;29(12):1203–1217. PubMed PMID: 26109046; PubMed Central PMCID: PMCPmc4495393. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mao P, Joshi K, Li J, et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc Natl Acad Sci USA. 2013. May 21;110(21):8644–8649. PubMed PMID: 23650391; PubMed Central PMCID: PMCPmc3666732. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hubert CG, Rivera M, Spangler LC, et al. A three-dimensional organoid culture system derived from human glioblastomas recapitulates the hypoxic gradients and cancer stem cell heterogeneity of tumors found in vivo. Cancer Res. 2016. April 15;76(8):2465–2477. PubMed PMID: 26896279; PubMed Central PMCID: PMCPmc4873351. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010. January 15;70(2):440–446. PubMed PMID: 20068163; eng. [DOI] [PubMed] [Google Scholar]
- [27].Deeks ED. Olaparib: first global approval. Drugs. 2015. February;75(2):231–240. PubMed PMID: 25616434; eng. [DOI] [PubMed] [Google Scholar]
- [28].Rasmussen RD, Gajjar MK, Jensen KE, et al. Enhanced efficacy of combined HDAC and PARP targeting in glioblastoma. Mol Oncol. 2016. May;10(5):751–763. PubMed PMID: 26794465; PubMed Central PMCID: PMCPmc5423160. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gupta SK, Mladek AC, Carlson BL, et al. Discordant in vitro and in vivo chemo-potentiating effects of the PARP inhibitor veliparib in temozolomide-sensitive versus -resistant glioblastoma multiforme xenografts. Clin Cancer Res. 2014. May 16;20(14):3730–3741. PubMed PMID: PMC4111895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Rasmussen RD, Gajjar MK, Tuckova L, et al. BRCA1-regulated RRM2 expression protects glioblastoma cells from endogenous replication stress and promotes tumorigenicity. Nat Commun. 2016. November 15;7:13398 PubMed PMID: 27845331; PubMed Central PMCID: PMCPmc5116074. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kechavarzi B, Janga SC. Dissecting the expression landscape of RNA-binding proteins in human cancers. Genome Biol. 2014. January 10;15(1):R14 PubMed PMID: 24410894; PubMed Central PMCID: PMCPmc4053825. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang J, Liu Q, Shyr Y. Dysregulated transcription across diverse cancer types reveals the importance of RNA-binding protein in carcinogenesis. BMC Genomics. 2015;16 Suppl 7:S5 PubMed PMID: 26100984; PubMed Central PMCID: PMCPmc4474540. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Choiniere M, Watt-Watson J, Victor JC, et al. Prevalence of and risk factors for persistent postoperative nonanginal pain after cardiac surgery: a 2-year prospective multicentre study. Cmaj. 2014. April 15;186(7):E213–23. PubMed PMID: 24566643; PubMed Central PMCID: PMCPmc3986330. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wang Z, Bhattacharya A, Ivanov DN. Identification of small-molecule inhibitors of the HuR/RNA interaction using a fluorescence polarization screening assay followed by NMR validation. PLoS One. 2015;10(9):e0138780 PubMed PMID: 26390015; PubMed Central PMCID: PMCPmc4577092. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Duffy AG, Makarova-Rusher OV, Ulahannan SV, et al. Modulation of tumor eIF4E by antisense inhibition: A phase I/II translational clinical trial of ISIS 183750-an antisense oligonucleotide against eIF4E-in combination with irinotecan in solid tumors and irinotecan-refractory colorectal cancer. Int J Cancer. 2016. October 1;139(7):1648–1657. PubMed PMID: 27194579; eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sanchez-Diaz PC, Burton TL, Burns SC, et al. Musashi1 modulates cell proliferation genes in the medulloblastoma cell line Daoy. BMC Cancer. 2008. September 30;8:280 PubMed PMID: 18826648; PubMed Central PMCID: PMCPmc2572071. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sureban SM, May R, George RJ, et al. Knockdown of RNA binding protein musashi-1 leads to tumor regression in vivo. Gastroenterology. 2008. May;134(5):1448–1458. PubMed PMID: 18471519; eng. [DOI] [PubMed] [Google Scholar]
- [38].Li N, Yousefi M, Nakauka-Ddamba A, et al. The Msi family of RNA-binding proteins function redundantly as intestinal oncoproteins. Cell Rep. 2015. December 22;13(11):2440–2455. PubMed PMID: 26673327; PubMed Central PMCID: PMCPmc4894540. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kharas MG, Lengner CJ. Stem cells, cancer, and MUSASHI in blood and guts. Trends Cancer. 2017. May;3(5):347–356. PubMed PMID: 28718412; PubMed Central PMCID: PMCPmc5546142. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Shou Z, Jin X, He X, et al. Overexpression of Musashi-1 protein is associated with progression and poor prognosis of gastric cancer. Oncol Lett. 2017. May;13(5):3556–3566. PubMed PMID: 28521458; PubMed Central PMCID: PMCPmc5431268. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chen K, Gao Q, Zhang W, et al. Musashi1 regulates survival of hepatoma cell lines by activation of Wnt signalling pathway. Liver Int. 2015. March;35(3):986–998. PubMed PMID: 24444033; eng. [DOI] [PubMed] [Google Scholar]
- [42].Gong P, Wang Y, Gao Y, et al. Msi1 promotes tumor progression by epithelial-to-mesenchymal transition in cervical cancer. Hum Pathol. 2017. July;65:53–61. PubMed PMID: 28088346; eng. [DOI] [PubMed] [Google Scholar]
- [43].Wang XY, Penalva LO, Yuan H, et al. Musashi1 regulates breast tumor cell proliferation and is a prognostic indicator of poor survival. Mol Cancer. 2010. August 21;9:221 PubMed PMID: 20727204; PubMed Central PMCID: PMCPmc2939568. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Luo Y, Shang P, Li D. Luteolin: a flavonoid that has multiple cardio-protective effects and its molecular mechanisms. Front Pharmacol. 2017;8:692 PubMed PMID: 29056912; PubMed Central PMCID: PMCPmc5635727. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Jeon IH, Kim HS, Kang HJ, et al. Anti-inflammatory and antipruritic effects of luteolin from Perilla (P. frutescens L.) leaves. Molecules. 2014. May 27;19(6):6941–6951. PubMed PMID: 24871572; eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lin Y, Shi R, Wang X, et al. Luteolin, a flavonoid with potential for cancer prevention and therapy. Curr Cancer Drug Targets. 2008. November;8(7):634–646. PubMed PMID: 18991571; PubMed Central PMCID: PMCPmc2615542. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kang KA, Piao MJ, Ryu YS, et al. Luteolin induces apoptotic cell death via antioxidant activity in human colon cancer cells. Int J Oncol. 2017. October;51(4):1169–1178. PubMed PMID: 28791416; eng. [DOI] [PubMed] [Google Scholar]
- [48].Liu Y, Lang T, Jin B, et al. Luteolin inhibits colorectal cancer cell epithelial-to-mesenchymal transition by suppressing CREB1 expression revealed by comparative proteomics study. J Proteomics. 2017. May 24;161:1–10. PubMed PMID: 28391045; eng. [DOI] [PubMed] [Google Scholar]
- [49].Zang M, Hu L, Zhang B, et al. Luteolin suppresses angiogenesis and vasculogenic mimicry formation through inhibiting Notch1-VEGF signaling in gastric cancer. Biochem Biophys Res Commun. 2017. August 26;490(3):913–919. PubMed PMID: 28655612; eng. [DOI] [PubMed] [Google Scholar]
- [50].Zang MD, Hu L, Fan ZY, et al. Luteolin suppresses gastric cancer progression by reversing epithelial-mesenchymal transition via suppression of the Notch signaling pathway. J Transl Med. 2017. February 27;15(1):52 PubMed PMID: 28241766; PubMed Central PMCID: PMCPmc5327575. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Cook MT, Liang Y, Besch-Williford C, et al. Luteolin inhibits lung metastasis, cell migration, and viability of triple-negative breast cancer cells. Breast Cancer (Dove Medical Press). 2017;9:9–19. PubMed PMID: 28096694; PubMed Central PMCID: PMCPmc5207335. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Seo Y, Ryu K, Park J, et al. Inhibition of ANO1 by luteolin and its cytotoxicity in human prostate cancer PC-3 cells. PLoS One. 2017;12(3):e0174935 PubMed PMID: 28362855; PubMed Central PMCID: PMCPmc5376326. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Anson DM, Wilcox RM, Huseman ED, et al. Luteolin decreases epidermal growth factor receptor-mediated cell proliferation and induces apoptosis in glioblastoma cell lines. Basic Clin Pharmacol Toxicol. 2018. June 23 PubMed PMID: 29935053; eng DOI: 10.1111/bcpt.13077 [DOI] [PubMed] [Google Scholar]
- [54].Wang Q, Wang H, Jia Y, et al. Luteolin induces apoptosis by ROS/ER stress and mitochondrial dysfunction in gliomablastoma. Cancer Chemother Pharmacol. 2017. May;79(5):1031–1041. PubMed PMID: 28393257; eng. [DOI] [PubMed] [Google Scholar]
- [55].Wang Q, Wang H, Jia Y, et al. Luteolin reduces migration of human glioblastoma cell lines via inhibition of the p-IGF-1R/PI3K/AKT/mTOR signaling pathway. Oncol Lett. 2017. September;14(3):3545–3551. PubMed PMID: 28927111; PubMed Central PMCID: PMCPmc5588063. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Cheng WY, Chiao MT, Liang YJ, et al. Luteolin inhibits migration of human glioblastoma U-87 MG and T98G cells through downregulation of Cdc42 expression and PI3K/AKT activity. Mol Biol Rep. 2013. September;40(9):5315–5326. PubMed PMID: 23677714; PubMed Central PMCID: PMCPmc3751389. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Chakrabarti M, Ray SK. Synergistic anti-tumor actions of luteolin and silibinin prevented cell migration and invasion and induced apoptosis in glioblastoma SNB19 cells and glioblastoma stem cells. Brain Res. 2015. December 10;1629:85–93. PubMed PMID: 26471408; eng. [DOI] [PubMed] [Google Scholar]
- [58].Chakrabarti M, Ray SK. Anti-tumor activities of luteolin and silibinin in glioblastoma cells: overexpression of miR-7-1-3p augmented luteolin and silibinin to inhibit autophagy and induce apoptosis in glioblastoma in vivo. Apoptosis. 2016. March;21(3):312–328. PubMed PMID: 26573275; eng. [DOI] [PubMed] [Google Scholar]
- [59].Sonoki H, Tanimae A, Endo S, et al. Kaempherol and luteolin decrease claudin-2 expression mediated by inhibition of STAT3 in lung adenocarcinoma A549 cells. Nutrients. 2017. June 13;9(6):597 PubMed PMID: 28608828; PubMed Central PMCID: PMCPmc5490576. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Xu H, Yang T, Liu X, et al. Luteolin synergizes the antitumor effects of 5-fluorouracil against human hepatocellular carcinoma cells through apoptosis induction and metabolism. Life Sci. 2016. January 1;144:138–147. PubMed PMID: 26656468; eng. [DOI] [PubMed] [Google Scholar]
- [61].Tu DG, Lin WT, Yu CC, et al. Chemotherapeutic effects of luteolin on radio-sensitivity enhancement and interleukin-6/signal transducer and activator of transcription 3 signaling repression of oral cancer stem cells. J Formos Med Assoc. 2016. December;115(12):1032–1038. PubMed PMID: 27742160; eng. [DOI] [PubMed] [Google Scholar]
- [62].Ohyama T, Nagata T, Tsuda K, et al. Structure of Musashi1 in a complex with target RNA: the role of aromatic stacking interactions. Nucleic Acids Res. 2012. April;40(7):3218–3231. PubMed PMID: 22140116; PubMed Central PMCID: PMCPmc3326303. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Sayre NL, Sifuentes M, Holstein D, et al. Stimulation of astrocyte fatty acid oxidation by thyroid hormone is protective against ischemic stroke-induced damage. J Cereb Blood Flow Metab. 2017. February;37(2):514–527. PubMed PMID: 26873887; PubMed Central PMCID: PMCPmc5381439. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.