

ABSTRACT
Datasets reporting microRNA expression profiles in normal and cancer cells show that miR-216b is aberrantly downregulated in pancreatic ductal adenocarcinoma (PDAC). We found that KRAS, whose mutant G12D allele drives the pathogenesis of PDAC, is a target of miR-216b. To suppress oncogenic KRAS in PDAC cells, we designed single-stranded (ss) miR-216b mimics with unlocked nucleic acid (UNA) modifications to enhance their nuclease resistance. We prepared variants of ss-miR-216b mimics with and without a 5ʹ phosphate group. Both variants strongly suppressed oncogenic KRAS in PDAC cells and inhibited colony formation in pancreatic cancer cells. We observed that the designed ss-miR-216b mimics engaged AGO2 to promote the silencing of KRAS. We also tested a new delivery strategy based on the use of palmityl-oleyl-phosphatidylcholine (POPC) liposomes functionalized with ss-miR-216b conjugated with two palmityl chains and a lipid-modified cell penetrating peptide (TAT). These versatile nanoparticles suppressed oncogenic KRAS in PDAC cells.
KEYWORDS: KRAS, PDAC cells, miR-216b, AGO2, POPC liposome
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the major causes of death in western countries [1,2]. As current treatments are not effective, there is an urgent need to develop new therapies [3]. The main genetic lesion present in > 90% of PDAC patients is a mutation in the KRAS proto-oncogene, mainly in exon 1 at codon 12, G12D (Gly→Asp) or G12C (Gly→Cys) [4]. It has been demonstrated that mutant KRAS is the major driver of PDAC and that the expression of KRAS G12D in transgenic mice pancreas causes intraepithelial neoplastic lesions progressing into full malignancy [4–6]. The expression of KRAS G12D is necessary for tumour maintenance and its extinction leads to a rapid tumor regression [6]. Recent work has demonstrated that pancreatic cancer cells are ‘addicted’ to mutant KRAS, as this oncogene reprograms the metabolism of tumor cells, in particular the glucose and glutamine pathways, in order to fuel a higher proliferation rate [7,8].
It is known that several properties of cancer cells, including proliferation, migration, invasion and gene expression, are regulated by small noncoding microRNAs [9–14]. These molecules are synthesized as long RNA strands folding into hairpin-loop structures processed by Drosha and Dicer into mature duplexes ranging from 17 to 26 nt in length [9–14]. The guide strand of the mature RNA duplexes forms a complex with argonaute proteins that bind to a 3ʹ-untraslated region (3ʹ-UTR) mRNA target. This mediates two modes of gene silencing: translation repression and/or RNA decay [12–15]. Synthetic double-stranded (ds) miRNAs have given encouraging results as antigene molecules [16] and recent studies have demonstrated that single-stranded (ss) RNAs, mimicking the guide strand of miRNAs, can also mediate an Ago-dependent inhibition of the target gene [17–21]. These findings provided new perspectives on the use of synthetic miRNAs as therapeutic agents.
In an attempt to inhibit KRAS in pancreatic cancer cells, we started from the observation that certain small noncoding RNAs are aberrantly expressed in cancer tissues [22]. In cancer, miRNAs inhibiting the expression of tumor suppressor genes are often upregulated, and this favors the development of the tumor. Instead, miRNAs behaving as tumor suppressors are downregulated and inhibit cancer growth [22–27]. Two miRNA-based therapeutic approaches have been developed: miRNA antagonists and miRNA mimics. MiRNA antagonists are single-stranded oligonucleotides that bind to oncogenic miRNAs and ablate their function. MiRNA mimics instead are used to restore a miRNA that is downregulated in the tumor, normally behaving as a tumor suppressor (replacement strategy) [23].
In our study we focused on a miRNA aberrantly down-regulated in PDAC, miR-216b, in order to design therapeutic agents suppressing KRAS in these tumor cells [28]. We designed single-stranded (ss) miR-216b mimics with unlocked nucleic-acid modifications, with or without a 5ʹ phosphate, and found that they strongly suppress oncogenic KRAS in PDAC cells. We also tested the activity of miR-216b conjugated to two palmityl chains and fixed on the surface of palmityl-oleyl-phosphatidylcholine (POPC) liposomes, functionalized with the trans-activator of transcription of the human immune-deficiency virus (TAT) cell penetrating peptide [29–32]. The results of our study may have relevance in cancer therapy, for designing single-stranded UNA-modified miRNA mimics against therapeutically important genes.
Results and discussion
We consulted miRNA expression profiles relative to PDAC and adjacent non-tumor tissues, deposited in the Array Express Archive of Functional Genomics. The three datasets analyzed, whose accession number are E-MTAB-753, GSE43796 and GSE41372 [28], showed that several miRNAs are differently expressed when the tumor PDAC tissue was compared with adjacent non-tumor tissue. Among the abnormally downregulated miRNAs, miR-216b showed an expression fold change of 27.95 (GSE43796) (Figure 1). Similar data were observed with E-MTAB-753 and GSE41372 (Fig. S1). In keeping with these data, Liu et al [33] have recently reported that the level of miR-216b in PDAC cells (Panc-1, BxPC3 and SW 1990) is 3- to 4-fold lower than in non-cancer cells. The targets of miR-216b in PDAC cells include TPT1, a gene encoding for the translationally-controlled tumor protein [34], and ROCK1, the ρ‐associated coiled-coil containing protein kinase 1 [33]. In addition, miR-216b targets the KRAS oncogene in nasopharyngeal tumor cells [35]. MiR-216b plays a critical role in PDAC, as in a transgenic mouse model this miRNA is downregulated in all steps of tumorigenesis, suggesting that it behaves as a tumor suppressor [36]. Considering that PDAC cells are addicted to KRAS, we asked if KRAS is a target of miR-216b even in this lethal cancer and if miR-216b mimics, properly modified, may be a valuable therapeutic tool to suppress mutant KRAS in PDAC cells.
Figure 1.
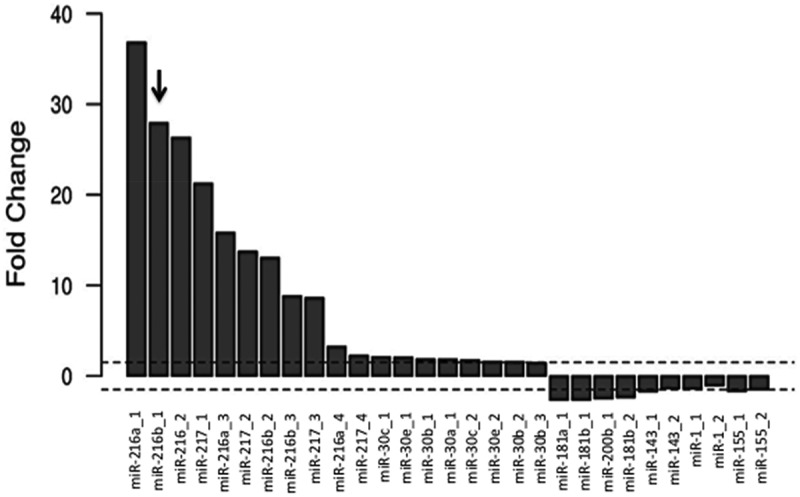
Fold change [FC = log2(Fnon-tumour/Ftumour)] of miRNAs in normal tissue over cancer PDAC tissue. Data have been obtained from GSE43796.
Design of single-stranded miRNA mimics specific for oncogenic KRAS
Synthetic miRNA mimics are normally double-stranded RNA molecules imitating mature microRNA duplexes [16]. Synthetic double-stranded miRNA mimics are incorporated into the miRNA-induced silencing complex (miRISC) that directs miRNA to its mRNA target in a sequence-specific manner for translation inhibition or mRNA degradation. Interestingly, previous studies have showed that both double- and ss-siRNAs act through the RNAi pathway and silence gene expression [20,21]. This led to the hypothesis that ss-miRNAs might also suppress gene expression. Indeed, ss-miRNAs are loaded into miRISC and inhibit gene expression [18]. Against this background, we designed synthetic ss-miRNA mimics to attempt KRAS suppression in pancreatic cancer cells. One might wonder why to use ss-miRNAs, considering that ds-miRNAs are potent tools for gene silencing. Two reasons have been put forward [18]: (i) ds-miRNAs, being more complex molecules than ss-miRNAs, are expected to be transported into the cells less efficiently than the single-stranded analogues [37]; (ii) ds-miRNAs are composed of the guide and passenger strands, and the latter may be a source of undesired off-target effects [38]. Figure 2(a) shows the primary sequence of the designed miR-216b mimics and their target in the 3ʹ-UTR of KRAS mRNA. At the 5ʹ-end the miR-216b sequence, UCUCUAAA-5ʹ, is perfectly complementary to mRNA and represents the ‘seed region’. Some complementarity with the target is also present at the 3ʹ-end of miR-216b, where 5ʹ-UAAAC is base-paired with mRNA. This should improve the interactions between miRNA and the target gene [39,40]. To increase their nuclease stability, we designed miR-216b mimics with one or two unlocked nucleic acid (UNA) modifications [41,42]. The key feature of UNA is the loss of C2ʹ-C3ʹ bond of the ribose, a modification that increases the flexibility of the RNA strand [43] (Figure 2(b)). A single UNA modification in the middle of a RNA/RNA duplex can lower the Tm by 5–10 °C, but when the UNA modification is placed near the duplex end, it causes a rather modest drop in Tm of 1–3 °C [43,44].
Figure 2.
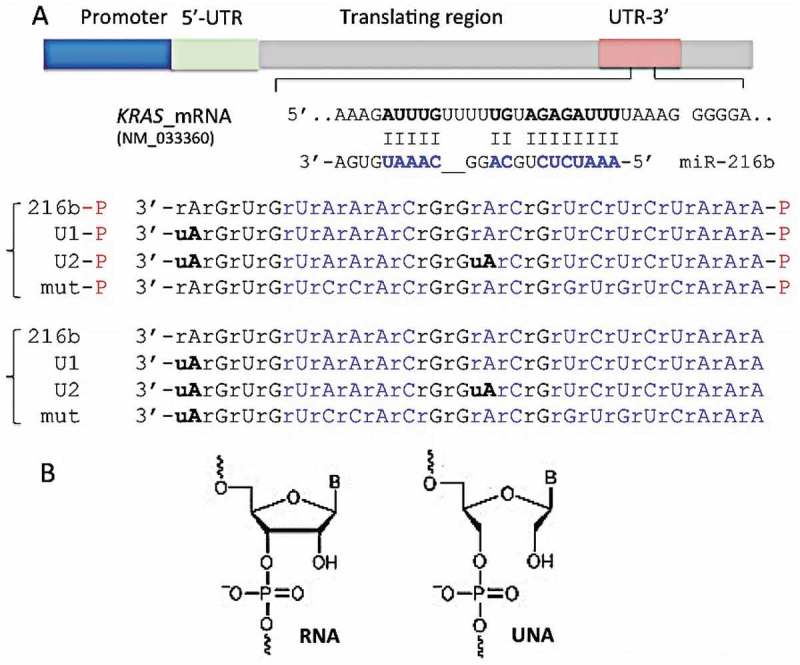
(a) Sequence of the KRAS 3ʹ-UTR recognized by miR-216b. We designed single-stranded UNA-modified miR-216b mimics with and without a 5ʹ phosphate: wild-type miR-216b and miR-216b-P; miR-216b with one UNA (U1 and U1-P) or two UNAs (U2 and U2-P). Mutated miRNAs with and without a 5ʹ phosphate (mut-P, mut) were used as a control (Supplementary S3); (b) The structures of RNA and UNA, lacking the covalent bond between C2ʹ and C3ʹ of the ribose, are shown.
We designed two UNA-modified miR-216b mimics: compound U1 with one modified adenine (uA) at the 3ʹ-end; compound U2 with two uAs, at the 3ʹ-end and in the middle of the oligoribonucleotide, but outside the seed sequence (Figure 2(a)). The uA at the 3ʹ-end is also located in a portion of miR-216b that does not pair with the mRNA target. Therefore, these modifications should not affect the hybridization of the UNA-modified mimics to the RNA target. Previous studies have reported that: (i) ss-miRNAs with 2ʹ-methyl, 2ʹ-fluoro, 2ʹ-O-methoxyethyl and with a phosphorothioate backbone are fairly active [17,45]; (ii) the binding of Argonaute 2 (AGO2) to modified siRNAs (including UNA-modified siRNAs) is not affected by chemical modifications [18,21]. Another important element in designing synthetic ss-miRNAs is the presence of a phosphate at the 5ʹ-end. Lima et al. found that a 5ʹ phosphate is a critical determinant for ss-siRNAs [17,18,21]. Yet, ss-siRNAs function through the RNAi pathway and require protein AGO2 [21]. Recently, Chorn et al. have compared the activity of ss-2ʹF-miRNAs against ss-2ʹOH-miRNAs [17]. They observed that the former had a higher capacity to suppress CD164 in HCT-116 cells than the latter. Moreover, with both types of miRNA the phosphorylation at the 5ʹ end was irrelevant. In contrast, when the ss-miRNAs contained both 2ʹ-F and 2ʹ-OH riboses, the 5ʹ phosphorylated analogues showed higher activity [17]. This suggests that the 5ʹ phosphorylation has a complex effect on miRNA activity. We thus designed UNA-modified ss-miRNAs with and without a 5ʹ phosphate group (Figure 2(a)). To investigate the gain in stability obtained with the UNA insertions, we treated the wild-type and UNA-modified miRNA mimics with cellular nucleases from a total Panc-1 extract. Figure 3(a) shows the integrity of the designed ss-miRNA mimics with a 5ʹ phosphate after incubation with a total Panc-1 extract for 1, 2 and 4 h, at 37°C. After 4 h, only ~ 10% of miR-216b-P was still intact. This percentage increased to ~ 20 and ~ 65%, respectively, when one or two UNA modifications were introduced in the oligonucleotide. Figure 3(b) shows that the non-phosphorylated analogues are slightly more resistant: after 4 h, ~ 27% of miR-216b was intact, while the percentage of undigested U1 and U2 was ~ 30 and ~ 80%, respectively. This proves that UNA insertions at the 3ʹ end and in the middle of the sequence boost the resistance of the designed mimics to PDAC cellular nucleases.
Figure 3.
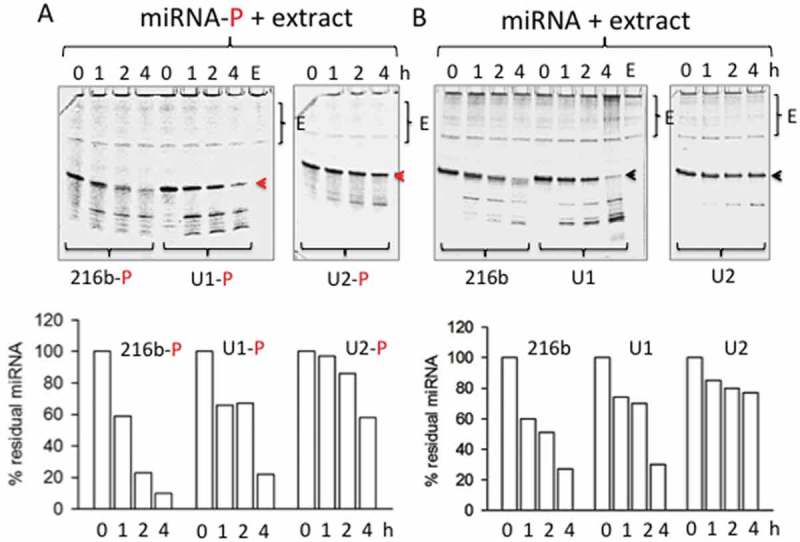
Denaturing (7 M urea) polyacrylamide gel showing the integrity of wild-type and UNA-modified mimics with (a) and without (b) a 5ʹ phosphate, after incubation for 1, 2 and 4 h with 2 μg Panc-1 cellular extract (E) at 37 °C. The mobility of E is shown in each panel. The arrow shows the undegraded miRNA. The % of undegraded miRNA at the various times is shown in the histograms.
Anti KRAS activity of UNA-modified miR-216b mimics in PDAC cells
To suppress oncogenic KRAS in Panc-1 cells we employed a replacement strategy. A previous study reported that miR-216b down-regulates KRAS in nasopharyngeal cancer cells [35]. Although miR-216b is aberrantly downregulated in PDAC, its therapeutic potential in pancreatic cancer cells has not yet been investigated. Our first step was to demonstrate that miR-216b is indeed KRAS-specific in PDAC. To this end, we hybridized wild-type and UNA-modified miR-216b mimics to their complementary sequence and obtained 22-mer ds-mimics with or without UNA insertions and 5ʹ phosphate in the guide strand (ds-216b, ds-U1, ds-U2 and ds-U2-P) (Figure 4). We also obtained a mutated ds-UNA-modified miRNA that was used as a control. Panc-1 cells were transfected two times with the duplexes and after an incubation of 48 h, a Western blot was performed to measure the level of KRAS protein (Figure 4). Duplexes ds-U1 and ds-U2 dramatically reduced the level of KRAS protein to ~ 10% of the control (ds-mut). Instead, duplex wild-type (ds-216b) produced a weaker protein suppression (~ 35% of the control). Duplex ds-U2-P, in which the guide strand bears a 5ʹ phosphate, reduced protein KRAS to ~ 40% of the control. This experiment clearly demonstrates that in PDAC cells oncogenic KRAS is a target of miR-216b.
Figure 4.
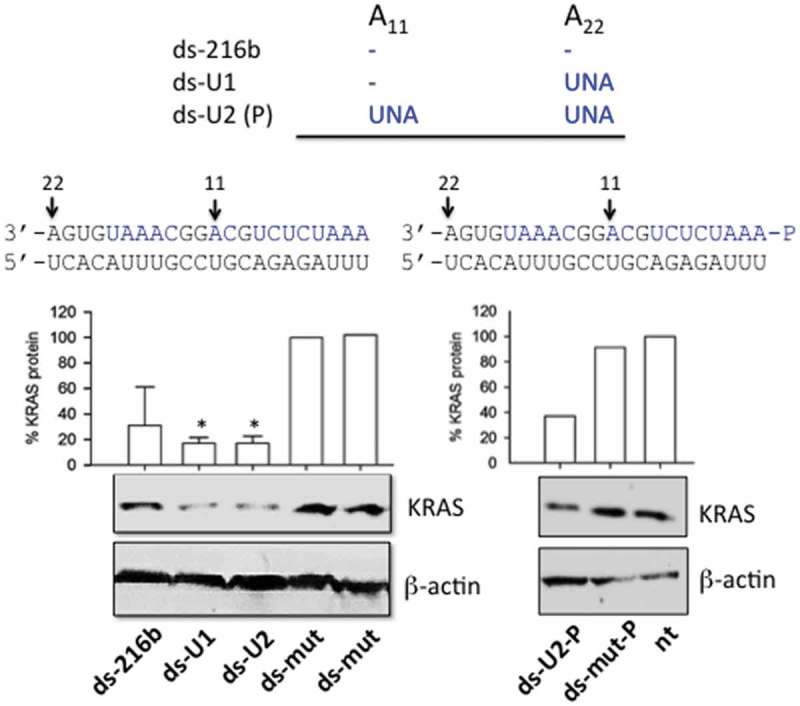
Effect of double-stranded UNA-modified mimics (ds-216b, ds-U1, ds-U2, ds-mut, ds-U2-P, ds-mut-P) on KRAS expression in Panc-1 cells. MiR-216b, U1, U2 and mut were hybridized to the complementary strand; Western blots were performed 48 h after the second transfection. The histogram shows the % KRAS protein determined as T/C x 100 where T = (KRAS/β-actin) in cells treated with ds-216b, ds-U1, ds-U2 and ds-U2-P, C = (KRAS/β-actin) in cells treated with ds-mut. The values are the average of three independent Western blot experiments, error bars represent ± SE. A Student’s t-test was performed, the asterisk (*) means P < 0.05 (n = 3).
Next, we designed ss-miR-216b mimics, with and without a 5ʹ phosphate, and examined their capacity to suppress KRAS in Panc-1 cells. As miRNAs inhibit gene expression either by translation repression and/or by mRNA decay [12–14], we measured by quantitative RT-PCR the level of KRAS transcript in Panc-1 cells treated with the designed ss-miR-216b mimics. As in the case of nasopharyngeal cells [35], we did not notice any significant reduction of KRAS mRNA at 16, 40 or 72 h (Fig. S2). Instead, when we measured by Western blot the level of the KRAS protein in Panc-1 cells treated the ss-miRNAs, we observed a dramatic suppression of the protein (Figure 5(a,b)). In keeping with the nuclease stability data, the compound showing the highest capacity to suppress protein KRAS was U2, the ss-miRNA with two UNA modifications. We found that both 5ʹ phosphorylated and non-phosphorylated single-stranded mimics caused a strong suppression of translation. UNA modifications turned out to be essential for the activity of the ss-miRNA mimics, which brought the protein down to < 10% of the control. The phosphorylation at the 5ʹ end did not seem to be an essential determinant, at least with these miRNAs containing UNA modifications. As a next issue, we wondered what the specificity of miR-216b for KRAS is, since this oncogene is highly homologous to HRAS and NRAS. As illustrated in Figure 5(c,d) none of the designed miR-216b mimics had any impact on HRAS and NRAS proteins. This was expected, as the matching between the seed region of the designed mimics and the 3ʹ-UTR sequence in these genes is suboptimal. In these cases, the base pairing in the miRNA 3ʹ half would have importance for stabilizing the interaction [12–14]. The lack of complementarity at the 3ʹ end makes the mimics ineffective against HRAS and NRAS. We can therefore conclude that, at least in pancreatic cancer cells, miR-216b regulates only the KRAS member of the ras family.
Figure 5.
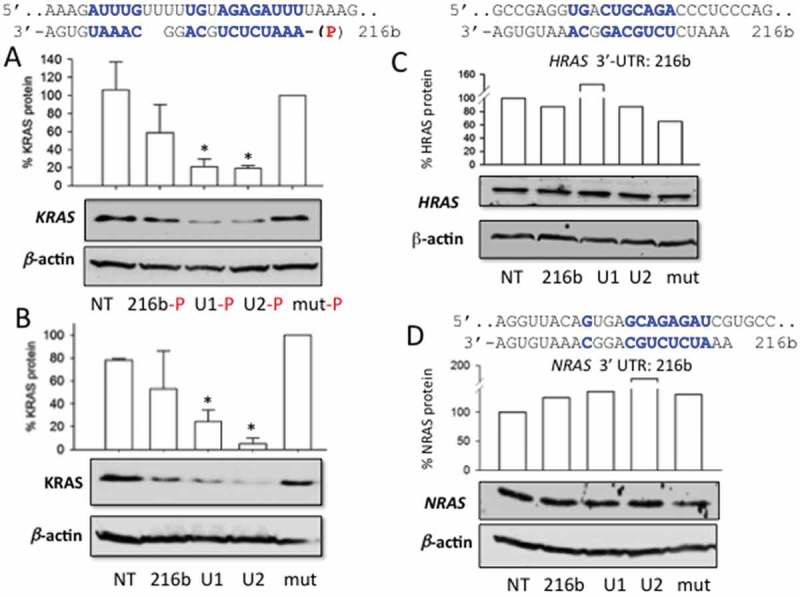
(a, b) Effect of single-stranded miRNA mimics with and without a 5ʹ phosphate on the expression of KRAS in Panc-1 cells. The cells, 24 h after seeding, were treated with the mimics, using Interferin. A second treatment was carried out 24 h after the first treatment. A Western blot was carried out 48 h after the second treatment; (a) Base-pairing between KRAS 3ʹ-UTR mRNA and miR-216b. The Western blot shows the impact on protein KRAS of the designed ss-miR-216b mimics with a 5ʹ phosphate. Histograms show the % KRAS protein in miRNA-treated cells compared to mut-treated cells is reported; (b) as in a but with single-stranded mimics without a 5ʹ phosphate; (c) and (d) match between HRAS and NRAS 3ʹ-UTR mRNA and miR-216b. The Western blots show the levels of HRAS and NRAS proteins in Panc-1 cells treated with the designed ss-miR-216b mimics. Results obtained from three independent Western blot experiments, error bars represent ± SE. A Student’s t-test was performed, the asterisk (*) means P < 0.05 (n = 3).
Single-stranded miR-216b without 5ʹ phosphate is AGO-dependent
Previous studies have demonstrated that AGO2 is a protein involved in RNAi gene silencing activated by ss-siRNAs [46] or ss-miRNA mimics with a 5ʹ phosphate [18]. On this basis, we asked if the activity of UNA-modified ss-miR-216b without a 5ʹ phosphate is also mediated by AGO2. To address this point, we silenced in Panc-1 cells AGO2 by siRNA and one day after we treated the cells with the designed ss-miR-216b mimics. The levels of proteins AGO2 and KRAS were measured by Western blot (Figure 6(a,b)). The results show that when protein AGO2 is suppressed, the designed miR-216b mimics are unable to knock out KRAS. In contrast, when the designed ss-miRNA mimics suppressed KRAS, AGO2 was expressed in the cells, as expected (Figure 6(c)). Together these results suggest that the silencing mechanism promoted by the ss-miR-216b mimics lacking a 5ʹ phosphate is clearly AGO2 dependent. The crystal between AGO2 and miR-20a showed that besides the 5ʹ phosphate, also the phosphates and ribose 2ʹ-OHs of the seed sequence promote contacts with the protein [46]. This suggests that in the absence of a 5ʹ phosphate, ss-miR-216b will only have more freedom to adopt a position within the binding site of AGO2.
Figure 6.
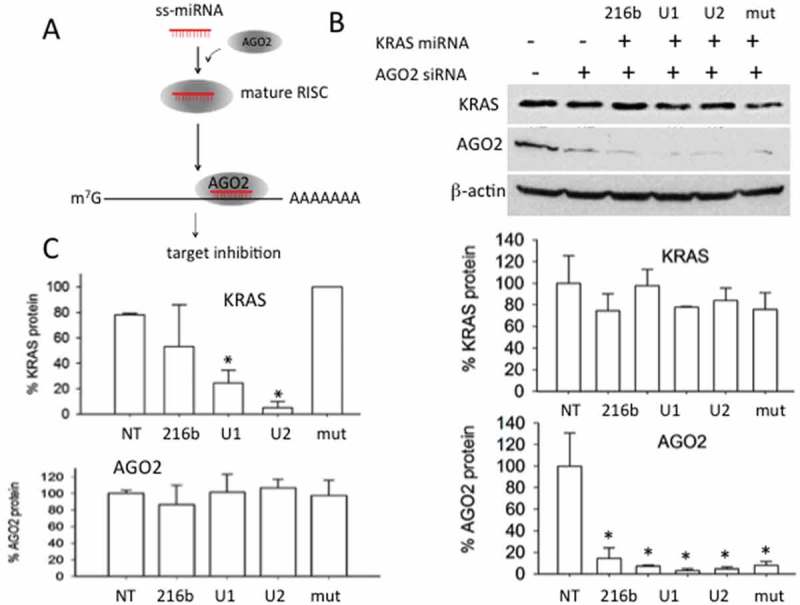
(a) Scheme showing the function of AGO2 in the silencing mechanism mediated by ss-miRNA; (b) Western blot showing that the silencing of AGO2 by a specific siRNA, results in the inactivation of the designed ss-miR-216b mimics. The histograms show the % of KRAS and AGO2 proteins in the cells treated with the designed ss-miR-216b mimics lacking a 5ʹ phosphate and with AGO2 siRNA as indicated; (c) histograms show the levels of AGO2 and KRAS proteins in Panc-1 cells treated with the designed ss-miR-216b mimic lacking a 5ʹ phosphate. Results obtained from two independent Western blot experiments, error bars represent ± SE.
Luciferase and clonogenic assays
To obtain further experimental evidence that the designed UNA-modified ss-miR-216b mimics lacking a 5ʹ phosphate are active and specific for KRAS, we tested them in a luciferase assay (Figure 7(a,b)). We used plasmid pGL4.75-KRAS LCS6m, carrying Renilla luciferase driven by the CMV promoter and the human KRAS 3ʹ UTR (3200 bp) containing downstream of luciferase the KRAS 3ʹ UTR. Panc-1 cells were treated with 10 nM miRNA mimics (complexed with Interferin), with 30 ng pGL4.75-KRAS LCS6m and 70 ng pGL3-Control Vector carrying Firefly luciferase driven by the SV40 promoter. When the cells were transfected only once with the miRNA mimics, the reduction of luciferase was proportional to the modifications introduced in ss-miR-216b. The molecule with the strongest anti-KRAS activity was U2, which decreased luciferase to ~ 60% of the control (luciferase expressed in mut-treated Panc-1 cells). Instead, when the cells were transfected twice with the single-stranded miR-216b, U1 and U2 (one treatment 24 h after the other) the suppression of luciferase was stronger: it was reduced to ~ 60, ~ 50 and ~ 40% by miR-216b, U1 and U2, respectively. A roughly similar result was obtained with the 5ʹ phosphorylated mimics (not shown). The luciferase experiment unambiguously proved that oncogenic KRAS can be downregulated by miR-216b. The luciferase assay gave a weaker effect than the western blot assay, probably because CMV is a stronger promoter than the KRAS promoter, and also because in the luciferase assay the target is not in its natural context.
Figure 7.
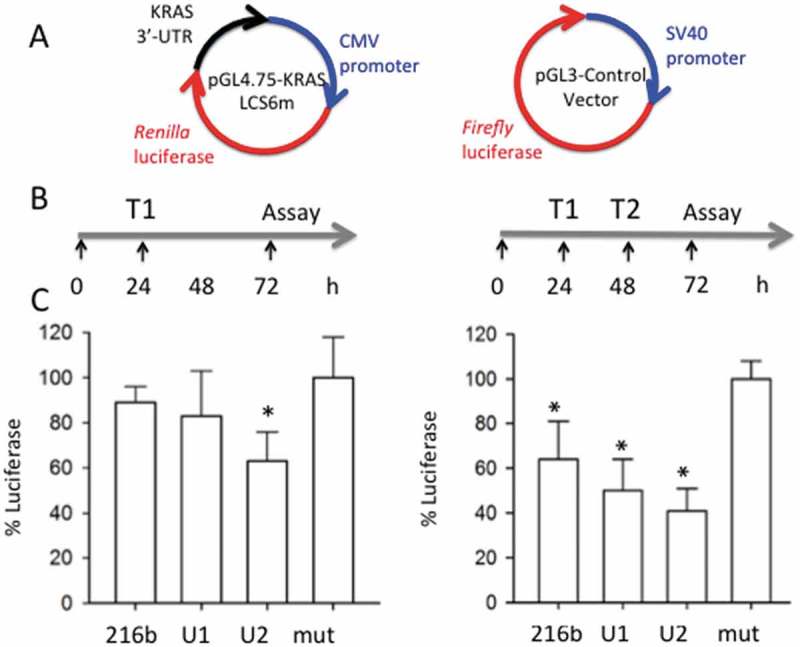
(a) Plasmids used for the luciferase experiments; (b) Outline of the luciferase experiments. T1: plasmids transfection and first miRNA treatment; T2: second miRNA treatment; (c) Histograms show the levels of Renilla luciferase in Panc-1 cells treated with miRNAs mut, 216-b, U1 and U2. The histograms report the % luciferase in Panc-1 cells treated with the designed ss-miRNAs (% luciferase = T/C x 100 where T = Renilla/Firefly in miRNA treated cells, C = Renilla/Firefly luciferase in controlled miRNA treated cells (mut)). Error bar is obtained from three independent experiments. A Student’s t-test was performed, the asterisk (*) means P < 0.05 (n = 3).
Since KRAS stimulates the pathway controlling cell growth, the suppression of protein KRAS should result in the inhibition of proliferation. To assess the effect of miR-216b, U1 and U2 on the growth of PDAC cells, we carried out a colony formation assay with Panc-1 and MIA PaCa-2 cells, carrying the KRAS mutations G12D and G12C, respectively. The PDAC cells were seeded in a medium after being diluted in a way that a single colony could be formed by each cell. After 10 days of growth, the colonies of at least 50 cells were counted and the results plotted in a histogram. Figure 8(a,b), show the data of a typical colony-formation assay obtained with Panc-1 and MIA PaCa-2 cells. The number of colonies on the untreated plate shows to be similar to the number on the plate treated with the control single-stranded mut sequence, which does not suppress oncogenic KRAS. In contrast, miR-216b and the UNA-analogues strongly reduced the number of colonies with both types of cells. U2 reduced the number of MIA PaCa-2 colonies to ~ 40% of the control. As KRAS controls cell adhesion via the integrin-linked kinase (ILK) [47–49], its suppression in Panc-1 cells resulted in cell aggregation and reduction of the number of colonies by ~ 50%.
Figure 8.
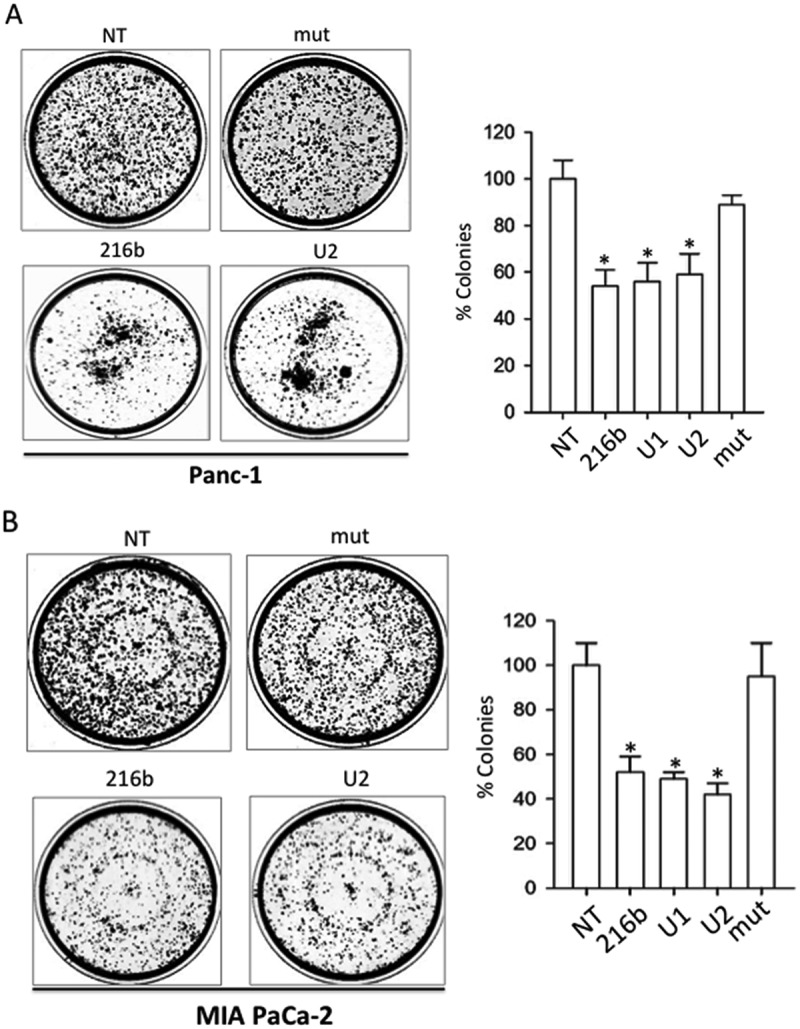
Clonogenic assays. MiRNAs 216-b, U1 and U2 decrease the % of colonies in Panc-1 (a) and Mia PaCa-2 (b) pancreatic cancer cells, while miRNA mut did not. NT = untreated cells. Histogram shows the % colonies in miRNA-treated cells compared to NT. Error bar is obtained from three independent experiments. A Student’s t-test was performed, the asterisk (*) means P < 0.05 (n = 3).
Lipid-modified miR-216d and POPC liposomes
In the experiments described above, the miRNA mimics were delivered to the cells complexed with Interferin, a commercial polyethyleneimine-based transfectant agent. To improve miRNA delivery, we used as a transporter palmityl-oleyl-phosphatidylcholine (POPC) liposomes in combination with surface attached functionalities [29,30]. POPC liposomes were functionalized with a cell-penetrating peptide, the trans-activator of transcription of the human immune-deficiency virus (TAT), and with miR-216b, as previously reported [29,30,50,51]. Both miR-216b and TAT were chemically modified with a palmityl membrane anchor to allow their rapid attachment to the liposome surface. This strategy, illustrated in Figure 9(a), has been recently adopted to suppress KRAS in Panc-1 cells with POPC liposomes functionalized with G4-decoy oligonucleotides and TAT [29]. In the present work, bioactive nanoparticles were obtained by treating POPC liposomes with lipid-modified miR-216b (216b-Pal) and TAT (TAT-Pal). The effector molecules spontaneously anchored to the liposome surface. Oligonucleotide 216b-Pal being not covalently attached to the liposomes can move freely on the lipid surface and interact efficiently with the mRNA target. The membrane anchor of miR-216b consists of a 3-amino-1,2-propanediol unit with two saturated palmityl chains (membrane anchor X) [29,30]. We treated two pancreatic cancer cells (Panc-1 and MIA PaCa-2) twice with POPC liposomes functionalized with TAT-Pal and 216b-Pal and then measured the level of KRAS protein by Western blot. It was observed that KRAS protein is reduced to about ~ 30–40% of the control (cells treated with empty liposomes or with 216b-mut) (Figure 9(b)). This supports the conclusion that POPC liposomes are an attractive vehicle to deliver miR-26b. Lastly, the bioactivity of 216b-Pal delivered via POPC liposomes was tested in a colony formation assay. The lipid-modified 216b-Pal fixed on POPC liposomes reduced Panc-1 and MIA PaCa-2 colony formation to about 60% of the control (Figure 10).
Figure 9.
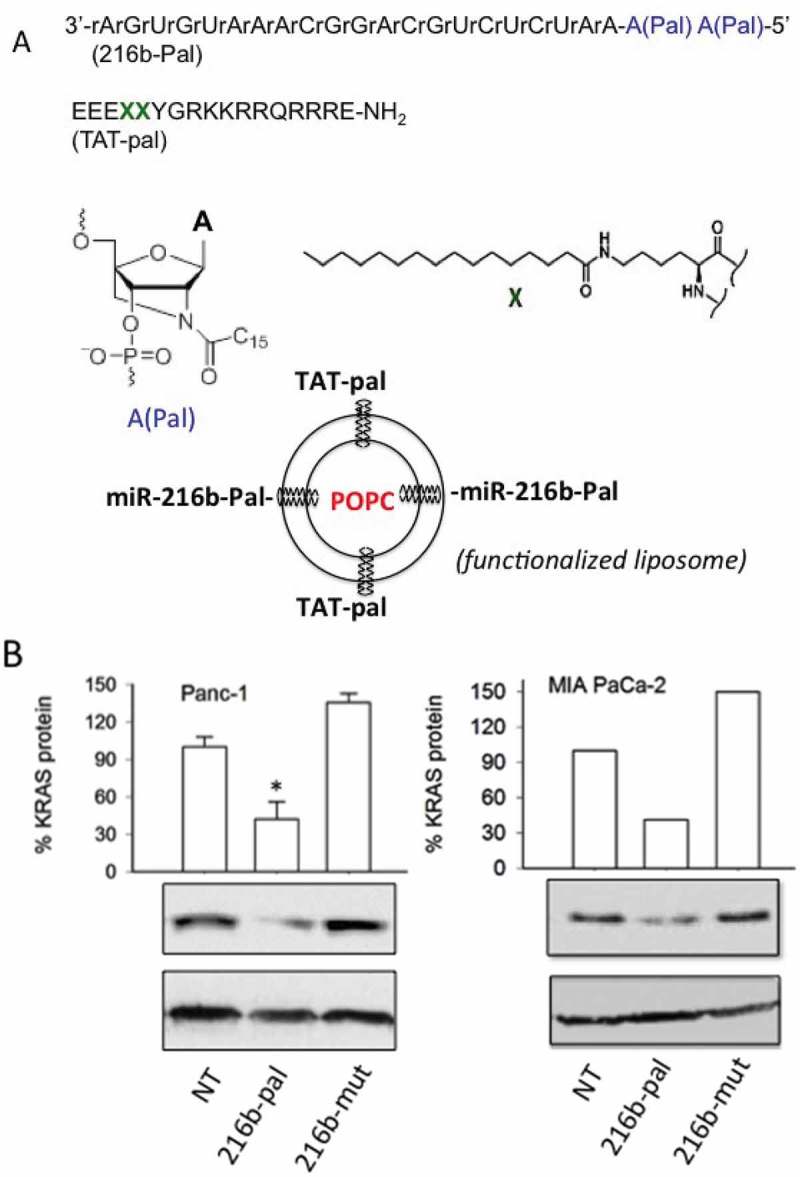
(a) Sequence of miR-216b with two palmityl chains (216b-pal) and of TAT peptide with two X insertions containing two saturated palmityl chains (TAT-pal). MiRNA 216b-pal and TAT-pal attached through their lipid modifications to the surface of POPC liposomes; (b) Western blot showing the reduction of KRAS protein in Panc-1 and MIA PaCa-2 cells treated with the POPC liposomes functionalized with TAT-pal and miR-216b-pal.
Figure 10.
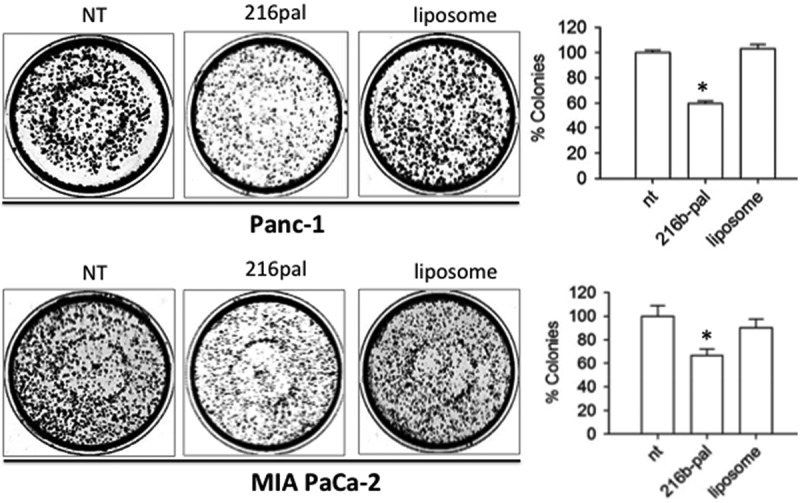
Clonogenic assays. Effect of functionalized POPC liposomes on colony formation in Panc-1 and Mia PaCa-2 pancreatic cancer cells. NT = untreated cells, 216-pal = cells treated with POPC liposomes functionalized with 216b-pal and TAT-pal; Liposome = cells treated with POPC non-functionalized with the effector molecules. Histogram shows the % colonies in miRNA-treated, liposome-treated and non-treated (NT) cells. Error bar is obtained from three independent experiments. A Student’s t-test was performed, the asterisk (*) means P < 0.05 (n = 3).
Conclusion
The aim of this study was to design oligoribonucleotides combining the potency of the RNAi pathway with the versatility of single-stranded RNA, in order to prepare antigene molecules specific for the KRAS oncogene, the main causative agent for pancreatic cancer. To this purpose we focused on miR-216b, a miRNA that is aberrantly downregulated in PDAC. We found that double–stranded miR-26b mimics, with UNA-modifications in the guide strand, specifically down regulate the KRAS gene but not the NRAS and HRAS analogues. We also observed that UNA-modified single-stranded miR-216b mimics exhibit a formidable inhibitory activity against oncogenic KRAS in pancreatic cancer cells. The designed ss-miRNAs acted through an AGO2-dependent mechanism, in keeping with the results of a recent study [18]. There are two earlier studies showing that ss-miRNAs may act as gene silencing agents [17,18]. Our study provides further evidence in support of this conclusion. We designed ss-miRNAs with and without a 5ʹ phosphate: in both cases the effector molecules suppressed the KRAS target gene with similar strength. Western blots showed that the designed ss-miR-216b mimic with two UNA modifications reduced dramatically (by 90%) the KRAS protein in Panc-1 cells as well as the capacity of colony formation (by 50%). To our knowledge, this is the first report showing that miR-216b targets oncogenic KRAS in PDAC cells. As a proof-of-principle we conjugated miR-216b with two palmityl chains and fixed it on the surface of POPC liposomes, functionalized with the TAT cell-penetrating peptide, which was also conjugated with two palmityl chains. The results obtained with two PDAC cell lines showed that the nanoparticle functionalized with TAT-Pal and 216b-Pal caused ~ 70% decrease of protein KRAS and ~ 40% inhibition of colony formation. This delivery strategy may have a interesting potential for in vivo studies
Experimental
Synthesis of UNA-modified miRNAs
The synthesis of the UNA-modified miRNA miR-216b mimics (216b, U1, U2, mut, U1-P, U2-P) was performed on an automated nucleic acid synthesizer as previously reported. The oligonucleotides were purified by HPLC by using a C18 3 µm 300 Å reversed phase column. Maldi MS analyses were carry out to confirm the structure of the designed UNA-modified miRNAs and evaluate their purity. Calculated and experimental masses were almost identical for all the oligonucleotides (Fig. S3 and S4).
Synthesis of lipid-conjugated TAT and miR-216b
The oligonucleotide was synthesized on an ExpediteTM 8900 nucleic acid synthesis system (Perceptive Biosystems Inc.). The synthesis was performed on a 1.0 µmol scale on GE Healthcare Custom Primer SupportTM T40s using standard conditions for automated synthesis with DCI as activator. However, the lipid modified phosphor amidite was dissolved in 2:1 DCE:MeCN at a concentration of 0.1 M, 42® was used as activator (Proligo reagent/Sigma-Aldrich) and the coupling time was increased to 20 min. The DMT protecting group on the last nucleotide in the sequence was removed. After deprotection and cleavage from the solid support using standard conditions (conc. NH3(aq.) over night at 55 ºC), the oligonucleotides were purified by HPLC.
TAT was synthesized on a Liberty 1 Microwave Peptide Synthesizer using a Rink Amide resin and an amino acid concentration of 0.2 M for unmodified amino acids and 0.1 M for the lipid-modified amino acids. After synthesis, the peptide was cleaved from the resin, deprotected and purified by HPLC using a C18 5 µm 10 × 150 mm column.
Cell cultures
The cells used in this study are the human pancreatic adenocarcinoma cells Panc-1 with a KRAS G12D (12 Gly→Asp) mutation and MIA PaCa- 2 with a KRAS G12C (12 Gly→Cys) mutation. The cell lines have been genotyped by Microsynth (CH) to verify their identity. As expected, they matched 100% to the DNA-profiles of the cell line of Panc-1 (ATCC® CRL-1469TM) and MIA PaCa-2 (ATCC® CRM-CRL-1420TM).
Preparation of POPC liposomes
Liposomes were prepared as previously described [31]. They were extruded on a LIPIX Extruder, Northern Lipids. 114 mg 1-palmityl-2-oleyl-sn-glycero-3-phosphocholine (POPC) was suspended in 1.5 mL 10 mM phosphate buffer (10 mM NaH2PO4. 2 H2O, 5 mM Na2HPO4, 140 mM Na+, pH 7.4) (100 mM POPC) and the resulting solution was extruded 10 times through two stacked polycarbonate filters with a pore size of 50 nm using compressed N2 (~ 30 bar). The size of the liposomes, determined using Nanoparticle Tracking Analysis, was about 80 nm.
Transfection experiments
The cells were transfected twice with the designed ds- or ss-miRNAs. When the cells were about 65% confluent, 10 nM ss- or ds-miRNAs were transfected in the cells in the presence of Interferin (Polyplus, France), following the manufacturing instructions. After 24 h of incubation a second transfection was performed. The double-stranded miRNA mimics have been prepared in DEPC water containing 100 mM NaCl, 50 mM Tris-HCl pH 7.4, heating the solution for 5 min at 80 °C and let the molecules to anneal overnight at room temperature.
Transfections with POPC liposomes were carried out as previously reported [29]. Briefly, Liposomes (2.8 μl, liposome stock solution ~ 10−12 moles/L) were mixed with 110 pmol ss/ds-miRNA in 200 μl phosphate buffer (10 mM NaH2PO4, 5 mM Na2HPO4, 140 mM NaCl pH 7.4). After overnight incubation at room temperature, 2.3 μg of peptide were added to the solution and let to incubate for 30 minutes (liposome mix). For all liposome formulations a statistic distribution of miRNA and CPPs during membrane anchoring to the surface of the liposomes was assumed. Finally, the liposome mix (200 μl) was added to the cells in 2 ml DMEM.
As for the clonogenic assays, the liposome mix was obtained by adding 100 pmol miRNA in 50 μl buffer (liposome and peptide were scaled down accordingly). Then 50 μl of liposome mix were in this case added to 120 μl cell medium. A second transfection was performed 24 h following the first. Then, 48 h after the second transfection the cells in each well were divided in 3 parts and seeded again in 3 wells.
Anti AGO2 siRNA (10 nM, OriGene, USA) were transfected in Panc-1 cells using Interferin (PolyPlus, France) according to manufacturing instructions.
Stability of miRNAs in cellular environment
To determine their nuclease resistance, the designed miRNAs (3 µM) have been incubated for 1, 2 and 4 h in a total extract from Panc-1 cancer cells (2 μg). After incubation the oligonucleotides have been run in a denaturing 20% polyacrylamide gel (7 M urea, 1 x TBE), which was stained with ‘stains all’.
Dual luciferase assays
Panc-1 cells were plated (15 x 103) in 96-well plate and after one day transfected with miR-216b or UNA-analogues and with plasmids pGL4.75-KRAS LCS6m (a gift from Frank Slack, Plasmid # 44,571, Addgene) and pGL3 Control Vector (Promega, USA). Transfection was performed by mixing 70 ng of pGL3 Control Vector (Firefly luciferase) with 30 ng of pGL4.75-KRAS LCS6m (Renilla luciferase), by using jet-PEI (Polyplus) as a transfectant agent. A second miRNA transfection was performed 24 h after the first transfection and the luciferase assays were performed 48 h after the second transfection. A Dual-Glo Luciferase Assay System (Promega, USA) was used. Samples were read on a Turner Luminometer and the relative luminescence expressed as T/C x 100, where T = Renilla luciferase/Firefly luciferase in miRNA treated cells and C = Renilla luciferase/Firefly luciferase in mut-treated cells.
Western blots
Total cell protein lysates (15–20μg) extracted from Panc-1 cells were sonicated for 10 minutes and the lysates were electrophoresed on 12% SDS-PAGE and transferred into a nitrocellulose membrane, at 70 V for 2 h. The filter was blocked for 1 h with 5% nonfat dry milk solution in PBS 0.05% Tween (Sigma-Aldrich, Italy) at room temperature. Membranes were incubated overnight at 4 °C with the primary antibodies: monoclonal anti-KRAS (clone 3B10-2F2, IgG1 mouse, 2.5 μg/mL, Sigma-Aldrich, USA), polyclonal anti-HRAS (IgG rabbit, diluted 1:300, Santa Cruz Biotechnology Inc, USA), monoclonal anti-NRAS (clone F155-227, IgG1 mouse, 2.5 μg/mL) and monoclonal anti-actin (clone JLA20, IgM mouse, 1x10−4 μg/mL, Calbiochem, Merck Millipore, Germany). The membranes were washed with a 0.05% Tween in PBS and then incubated 1 h with the secondary antibodies horseradish peroxidase conjugated: anti-mouse IgG (diluted 1:5000) and anti-mouse IgM (diluted 1:2000) (Calbiochem, Merck Millipore, Germany) and anti-rabbit IgG (diluted 1:5000) (Calbiochem, Merck Millipore, Germany). To detect the protein we used Super signal ® West PICO and FEMTO (ThermoFisher Scientific Pierce, USA). The exposure time depended on the antibody used and was usually between 30 s and 5 min. The protein levels were quantified by the Image Quant TL Version 2003 software (Amersham).
Colony forming assay
To assess the effect of miR-216b, U1 and U2 on the growth of PDAC cells we carried out colony formation assays with Panc-1 and MIA PaCa-2 cells, carrying the KRAS mutations G12D and G12C, respectively. The PDAC cells, treated with the designed miRNAs, were seeded in a medium after being diluted in a way that a single colony could be formed from each cell. After 7–13 days of growth, the colonies of at least 50 cells were counted and the results plotted in a histogram. NT = untreated cells. Histogram shows the % colonies in miRNA-treated cells compared to NT. Values obtained from three independent experiments. A Student’s t-test was performed, (*) = P < 0.05.
RNA Extraction and Real-time PCR (see Fig. S2).
Funding Statement
This work has been carried out with the financial support of AIRC (Italian association for cancer research, IG 2017, Project Code 19898).
Abbreviations
PDAC pancreatic ductal adenocarcinoma
KRAS Kirsten ras
UTR untranslated region
POPC palmityl-oleyl-phosphatydilcholine
TAT trans-activator of transcription
Author Contributions
The manuscript was written by LEX, through the contributions of all authors. All authors have given approval to the final version of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Supporting Information
The following information is available free of charge:
(i) Array Express Archive of Functional genomics E-MTAB-753, GSE41372; (ii) Levels of KRAS mRNA in miRNA treated Panc-1 cells measured by quantitative real-time PCR; (iii) Molecular masses of UNA-modified RNAs by MALDI MS and Maldi spectra; (iv) Further Materials and methods information.
References
- [1].Jemal A, Siegel R, Ward E, et al. Cancer statistics. Cancer J Clin 2007; 57:43–66. [DOI] [PubMed] [Google Scholar]
- [2].Jemal A, Siegel R, Ward E, et al. Cancer statistics. Cancer J Clin 2010; 60:277–300. [DOI] [PubMed] [Google Scholar]
- [3].Kleeff J, Reiser C, Hinz U, et al. Surgery for recurrent pancreatic ductal adenocarcinoma. Ann Surg 2007, 245:566–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bryant KL, Mancias JD, Kimmelman AC, et al. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci. 2014; 39:91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Collins MA, Bednar MA, Zhang F, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest 2012;122:639–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kong B, Michalski CW, Erkan M, et al, From tissue turnover to the cell of origin for pancreatic cancer Nat Rev Gastroenterol Hepatol 2011; 8: 467–472. [DOI] [PubMed] [Google Scholar]
- [7].Ying H, Kimmelman AC, Lyssiotis CA, et al, Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012; 149: 656–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Son J, Lyssiotis CA, Ying H, et al, Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013; 496: 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet 2015; 16: 421–433. [DOI] [PubMed] [Google Scholar]
- [10].Stefani G, Slack FJ. Small non-coding RNAs in animal development. Nat Rev Mol Cell Biol 2008; 9: 219–230. [DOI] [PubMed] [Google Scholar]
- [11].Krol J, Loedige I, Filipowicz W, The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet 2010; 11: 597–610. [DOI] [PubMed] [Google Scholar]
- [12].Filipowicz W, Bhattacharyya SN, Sonenberg N, Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat. Rev Genet 2008; 9: 102–114. [DOI] [PubMed] [Google Scholar]
- [13].Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet 2011; 12:99–110. [DOI] [PubMed] [Google Scholar]
- [14].Izaurralde E. Gene regulation. Breakers blockers-miRNAs Sci 2015; 349: 380–382. [DOI] [PubMed] [Google Scholar]
- [15].Iwakawa HO, Tomari Y, The functions of microRNAs: mRNA decay and translational repression. Trends Cell Biol. 2015; 25:651–665. [DOI] [PubMed] [Google Scholar]
- [16].Goldgraben MA, Russell R, Rueda OM, et al. A double-stranded microRNA mimics can induce length- and passenger strand–dependent effects in a cell type–specific manner. Rna 2016; 22:193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chorn G, Klein-McDowell M, Zhao L, et al. Single-stranded microRNA mimics. Rna 2012; 18:1796–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Matsui M, Prakash TP, Corey DR, Argonaute 2-dependent regulation of gene expression by single-stranded miRNA mimics. Mol Ther 2016; 24:946–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Schirle NT, Kinberger GA, Murray HF, et al. Structural analysis of human Argonaute-2 bound to a modified siR-NA guide. J Am Chem Soc 2016;138:8694–8697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yu D, Pendergraff H, Liu J, et al. Single-stranded RNAs use RNAi to potently and allele-selectively inhibit mutant huntingtin expression. Cell 2012; 150: 895–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lima WF, Prakash TP, Murray HM, et al. Single-stranded siRNAs activate RNAi in animals, Cell 2012;150:883–894. [DOI] [PubMed] [Google Scholar]
- [22].Di Leva G, Croce C, miRNA profiling of cancer. Curr Opin Genet Dev 2013; 23: 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ling H, Fabbri M, Ga C, MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat Rev Drug Discov 2013; 12: 847–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lin S, Gregory RI, MicroRNA biogenesis pathways in cancer. Nat Rev Cancer 2015; 15: 321–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Matsui M, Corey DR, Non-coding RNAs as drug targets. Nat Rev Drug Discov 2017; 16: 167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Seux M, Iovanna J, Dagorn JC et al. MicroRNAs in pancreatic ductal adenocarcinoma: new diagnostic and therapeutic clues. Pancreatology 2009; 9: 66–72. [DOI] [PubMed] [Google Scholar]
- [27].Ling H. Non-coding RNAs: therapeutic strategies and delivery systems.Adv Exp Med Biol. 2016; 937:229–237. [DOI] [PubMed] [Google Scholar]
- [28].Barrett T, Troup DB, Wilhite SE, et al, NCBI GEO: archive for functional genomics data sets—10 years on. Nucleic Acids Res. 2011; 39: 1005–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Cogoi S, Jakobsen U, Pedersen EB, et al, Lipid-modified G4-decoy oligonucleotide anchored to nanoparticles: delivery and bioactivity in pancreatic cancer cells. Sci Rep. 2016. 6:38468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jakobsen U, Vogel S. Assembly of liposomes controlled by triple helix formation. Bioconjug 2013; 24, 1485–1495. [DOI] [PubMed] [Google Scholar]
- [31].Jakobsen U, Vogel S, DNA-controlled assembly of liposomes in diagnostics. Methods Enzymol 2009; 464: 233–248. [DOI] [PubMed] [Google Scholar]
- [32].Jakobsen U, Simonsen AC, Vogel S, DNA-controlled assembly of soft nanoparticles. J Am Chem Soc 2008; 130: 10462–10463. [DOI] [PubMed] [Google Scholar]
- [33].Liu Y-A, Zhang Y, Zheng Z, et al, MicroRNA-216b reduces growth, migration and invasion of pancreatic ductal adenocarcinoma cells by directly targeting β–associated coiled-coil protein kinase 1. Oncol 2018; 15:6745–6751. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [34].You Y, Tan J, Gong Y, et al, MicroRNA-216b-5p functions as a tumor-suppressive RNA by targeting TPT1 in pancreatic cancer cells. J Cancer 2017; 8: 2854–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Deng M, Tang H, Zhou Y, et al, MiR-216b suppresses tumor growth and invasion by targeting KRAS in nasopharyngeal carcinoma. J Cell Sci 2011; 124: 2997–3005. [DOI] [PubMed] [Google Scholar]
- [36].Azevedo-Pouly AC, Sutaria DC, Jiang J, et al, MiR-216 and miR-217 expression is reduced in transgenic mouse models of pancreatic adenocarcinoma, knockout of miR-216/miR-217 host gene is embryonic lethal. Funct Integr Genomics 2017; 17: 203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Koller E, Vincent TM, Chappell A, Mechanisms of single-stranded phosphorothioate modified antisense oligonucleotide accumulation in hepatocytes. Nucleic Acids Res. 2011; 39: 4795–4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Jackson AL, Bartz SR, Schelter J, et al, Expression profiling reveals off-target gene regulation by RNAi. Nat Biotecnol 2003; 21: 635–637. [DOI] [PubMed] [Google Scholar]
- [39].Brennecke J, Stark A, Russell RA, et al, Principles of microRNA–Target recognition. Plos Biol. 2005; 3 e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Grimson A, Farh KK, Johnston KW, MicroRNA targeting specificity in mammals: determinants beyond Seed Pairing. Mol Cell 2007; 27: 91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bramsen JB, Laursen MB, Nielsen AF, et al, A large-scale chemical modification screen identifies design rules to generate siRNAs with high activity, high stability and low toxicity. Nucleic Acids Res. 2009; 37: 2867–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Snead NM, Escamilla-Powers JR, Rossi JJ, et al, 5′-unlocked nucleic acid modification improves siRNA targeting. Mol Ther Nucleic Acids 2013; 2 e103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Langkjaer N, Pasternak A, Wengel J, UNA (unlocked nucleic acid): a flexible RNA mimic that allows engineering of nucleic acid duplex stability. Bioorg Med Chem 2009; 17: 5420–5425. [DOI] [PubMed] [Google Scholar]
- [44].Pasternak A, Wengel J, Unlocked nucleic acid – an RNA modification with broad potential. Bioorg Med Chem 2011; 9: 3591–3597. [DOI] [PubMed] [Google Scholar]
- [45].Watts JK, Yu D, Charisse K et al., Effect of chemical modifications on modulation of gene expression by duplex antigene RNAs that are complementary to non-coding transcripts at gene promoters. Nucleic Acids Res. 2010, 38: 5242–5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Elad Elkayam E, Kuhn CD, Tocilj A, et al, The structure of human argonaute-2 in complex with miR-20. Cell 2012; 150: 100–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Novak A, Hsu S, Leung-Hagesteijn C et al., Cell adhesion and the integrin-linked kinase regulate the LEF-1 and β-catenin signaling pathways. Proc Natl Acad Sci U S A 1998; 95: 4374–4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Cogoi S, Rapozzi V, Cauci S et al., Critical role of hnRNP A1 in activating KRAS transcription in pancreatic cancer cells: a molecular mechanism involving G4 DNA. Biochim Biophys Acta 2017; 1861: 1389–1398. [DOI] [PubMed] [Google Scholar]
- [49].Chu PC, Yang MC, Kulp SK, et al, Regulation of oncogenic KRAS signaling via a novel KRAS-integrin-linked kinase-hnRNPA1 regulatory loop in human pancreatic cancer cells. Oncogene 2016; 35: 3897–3908. [DOI] [PubMed] [Google Scholar]
- [50].Green M, Loewenstein PM, Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell 1988; 55: 1179–1188. [DOI] [PubMed] [Google Scholar]
- [51].Frankel AD, Pabo CO, Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988; 55: 1189–1193. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.