

ABSTRACT
MicroRNA (miRNA)-mediated crosstalk between coding and non-coding RNAs of various types is known as the competing endogenous RNA (ceRNA) concept. Here, we propose that there is a specific variant of the ceRNA language that takes advantage of simple sequence repeat (SSR) wording. We applied bioinformatics tools to identify human transcripts that may be regarded as repeat-associated ceRNAs (raceRNAs). Multiple protein-coding transcripts, transcribed pseudogenes, long non-coding RNAs (lncRNAs) and circular RNAs (circRNAs) showing this potential were identified, and numerous miRNAs were predicted to bind to SSRs. We propose that simple repeats expanded in various hereditary neurological diseases may act as sponges for miRNAs containing complementary repeats that would affect raceRNA crosstalk. Based on the representation of specific SSRs in transcripts, expression data for SSR-binding miRNAs and expression profiling data from patients, we determined that raceRNA crosstalk is most likely to be perturbed in the case of myotonic dystrophy type 1 (DM1) and type 2 (DM2).
KEYWORDS: ceRNA hypothesis, non-coding RNAs, microsatellite repeats, miRNA sponge, miRNA cooperativity, repeat expansion diseases, myotonic dystrophy
Introduction
Simple sequence repeats (SSRs), known also as microsatellites, are tandemly reiterated 1–6 base pair-long DNA motifs that occur in coding and non-coding regions and are ubiquitous in genomes [1]. High instability of these sequences, i.e., expansion or contraction of the repeated tract, mainly results from DNA slippage during replication and from unequal DNA recombination. With mutation rates several orders of magnitude higher than rates of point mutations, the SSRs are the most variable regions in genomes. The high variability of SSRs facilitates evolutionary changes and adaptations to new environments, making these sequences rich sources of phenotypic variation [2].
In contrast, abnormally expanded SSRs are harmful genetic elements that cause a number of hereditary neurological disorders in humans via various gain-of-function and loss-of-function mechanisms [3] (Supplementary Table I). The trinucleotide repeat expansion diseases (TREDs) constitute the largest subgroup of these disorders. They are triggered by an expansion of trinucleotide repeats that occur both in coding and non-coding regions of human genes. Specifically, Huntington’s disease (HD) is triggered by the expansion of a CAG repeat in the translated region of the HTT gene, both fragile X syndrome (FXS) and fragile X-associated tremor/ataxia syndrome (FXTAS) are caused by a CGG repeat expansion in the 5ʹ untranslated region (UTR) of the FMR1 gene, and Friedreich ataxia (FRDA) is caused by a GAA repeat expansion in the first intron of the FXN gene. The TREDs subgroup also includes a neuromuscular disorder, myotonic dystrophy type 1 (DM1), whose source is a CTG repeat expansion in the 3ʹ UTR of the DMPK gene. Furthermore, 4- and 5-nt repeat tracts of CCTG, ATTCT and TGGAA are implicated in myotonic dystrophy type 2 (DM2), spinocerebellar ataxias (SCAs) type 10 and type 31, respectively. In addition, expansion of a hexanucleotide GGCCTG repeat causes SCA36, while expansion of GGGGCC leads to the amyotrophic lateral sclerosis (ALS)/frontotemporal dementia (FTD) pathology. Disease-causing SSRs are present in different regions of protein-coding transcripts, including 5ʹ UTRs, open reading frames (ORFs), introns and 3ʹ UTRs, and their pathogenic repeat thresholds also differ [4] (Supplementary Table I).
As a major part of the human genome undergoes transcription, a plethora of RNAs (large and small, sense and antisense, linear and circular, and coding and non-coding) are formed and function in cells, including transcripts harboring SSRs. Most protein-coding genes are subject to negative posttranscriptional regulation by microRNAs (miRNAs). The latter bind to miRNA response elements (MREs) located within the 3ʹ UTRs of transcripts, which results in transcript degradation and/or translational repression [5–7]. However, individual miRNA-MRE associations should not be considered in isolation because miRNAs form complex regulatory networks. According to the competing endogenous RNA (ceRNA) concept, multiple cellular transcripts communicate with and co-regulate each other by competing for binding to a shared pool of miRNAs [8]. These transcripts include not only protein-coding RNAs but also non-coding RNAs (ncRNAs), such as long ncRNAs (lncRNAs), transcribed pseudogenes and circular RNAs (circRNAs) [9]. In this context, the question arises of whether the SSRs themselves might be directly involved in the regulation of mRNA translation by miRNAs and whether they are potentially involved in a wider miRNA-mediated crosstalk between coding and non-coding transcripts.
In this study, we examined the scale of putative crosstalk between coding and non-coding transcripts via miRNAs binding to corresponding SSRs (Supplementary Fig. S1). We present the results of a computational survey of potentially crosstalking SSRs among all human trinucleotide repeat-containing transcripts and disease-relevant tetra-, penta-, and hexanucleotide repeats in all major types of human transcripts. First, we estimated the representation, number and length distribution of selected SSRs in coding and non-coding transcripts. Next, we identified miRNAs with the potential to bind to SSRs and assessed the number of MREs and their density in various types of transcripts. Finally, we addressed the relevance of postulated repeat-associated ceRNA (raceRNA) crosstalk to pathology using RNA expression data. We found that expanded CUG and CCUG tracts, present in DM1 and DM2 patients, are most potent among all of pathogenic SSRs in influencing raceRNA crosstalk.
Results
Representation of triplet repeat tracts varies between protein-coding and non-coding RNAs
Here, we analyzed the occurrence of all possible triplet repeat tracts composed of at least 5 consecutive repeats in human protein-coding transcripts, lncRNAs, circRNAs and pseudogenes and compared the occurrence of these repeats with their frequency in the genome (Fig. 1A). Comparison of the representation rates of triplet repeats in mRNAs showed a clear overrepresentation of CGA, CGG, CAG, CCG, CUG and AGG repeats forming stable RNA structures, such as hairpins or quadruplexes, consistent with results of previous studies [10]. Interestingly, similar but milder enrichment of these repeat tracts was found in lncRNAs, except for CGA tracts. CGA tracts were the most enriched repeat tracts in mRNAs (showing a 22-fold difference), but their representation in lncRNAs was comparable with their occurrence in the genome (only a 1.1-fold difference). In contrast, CGU repeats, another hairpin-forming repeat, were strongly enriched in lncRNAs (14-fold), while their enrichment in protein-coding transcripts was quite moderate (1.5-fold). Additionally, repeats that are unable to form stable RNA structures were mildly underrepresented by 2–7-fold, in both protein-coding RNAs and lncRNAs. Triplet repeat tracts were generally rather underrepresented in pseudogenes, with a greater underrepresentation of repeats not forming stable structures, such as UUG, CAA, AUU and UAA tracts, which were underrepresented 11-, 12-, 39- and 69-fold, respectively. The representation rates of triplet repeats in circRNAs did not significantly differ from their genomic occurrence. It can be speculated that there is a mechanism for retention of certain SSR tracts in lncRNA sequences. While such a phenomenon is well described for mRNAs, providing advantageous effects on protein levels [11,12], the functionality of hairpin-forming triplet repeats in ncRNAs must manifest at the DNA/RNA level. Such repeats could contribute to the regulation of the expression of lncRNAs, shape their structure or mediate interactions with other RNAs and RNA-binding proteins (RBPs) [13].
Figure 1.
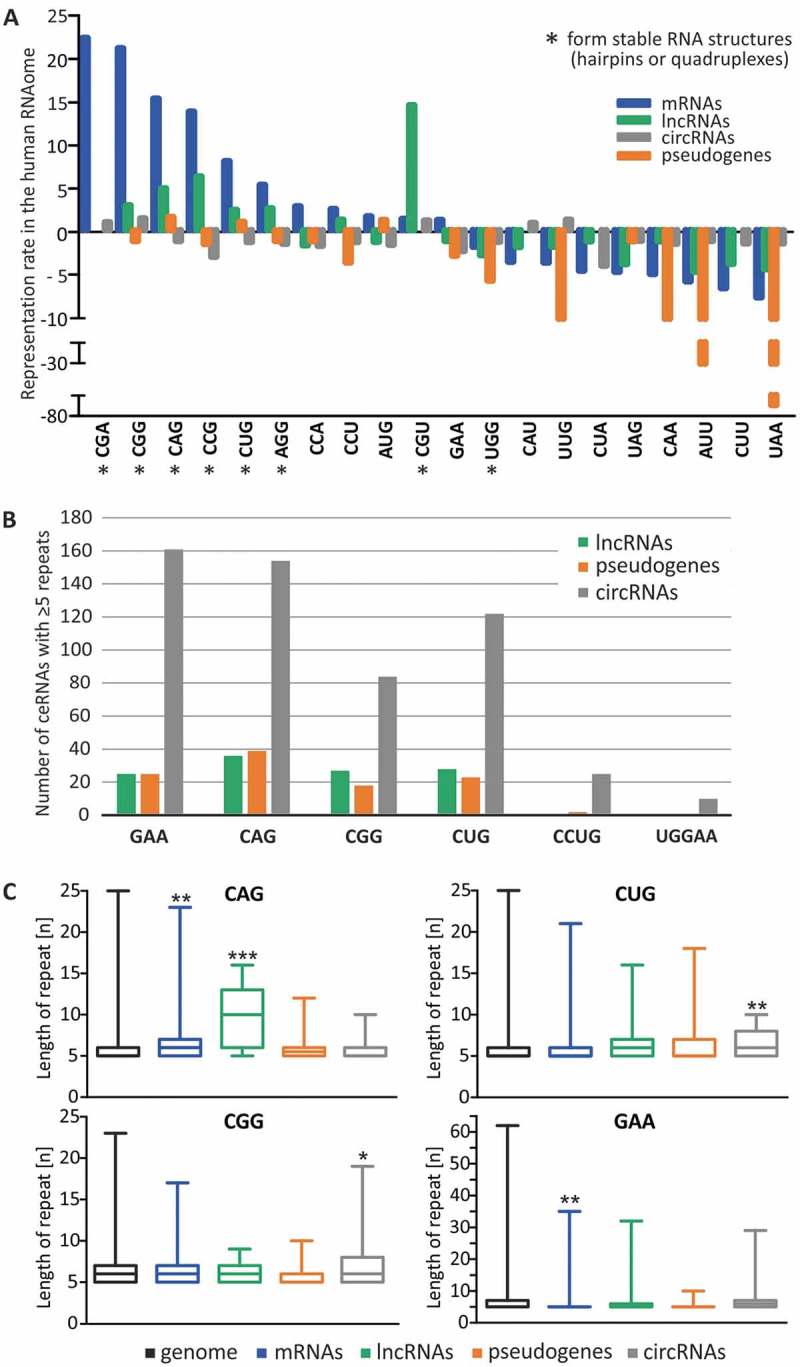
Characterization of SSRs found in human coding and non-coding transcripts. (A) Representation rate of all triplet repeats in the human RNAome compared with their occurrence in the human genome. Tracts of at least 5 consecutive triplet repeats were analyzed in the genome, protein-coding transcripts, lncRNAs, pseudogenes and circRNAs. Positive and negative values represent the fold over- and under-representation, respectively. Repeats capable of forming stable RNA structures are marked with an asterisk. (B) Number of non-coding transcripts containing at least 5 consecutive disease-relevant SSRs. No RNAs were found for AUUCU, GGCCUG and GGGGCC repeat tracts. (C) Distribution of the lengths of tracts containing at least 5 consecutive disease-relevant triplet repeats in the genome and RNAome, * − P-value < 0.05; ** − P-value < 0.01, *** − P-value < 0.001. Boxes and whiskers represent the minimum and maximum values.
Non-coding transcripts contain a substantial number of SSR tracts
The existence of SSR tracts in lncRNAs, pseudogenes and circRNAs has never been examined previously. We addressed this issue by analyzing the occurrence of tracts of at least 5 consecutive repeats for all possible triplet repeats, including those triggering TREDs as well as disease-relevant tetra-, penta- and hexanucleotide sequence motifs. A number of ncRNAs were identified for each triplet repeat tract, ranging from 15 to 160 sequences, while fewer were found for CCUG and UGGAA repeats (Fig. 1B and Supplementary Data). Because some circRNAs are very long, these were the type of ncRNA with the greatest number of sequences bearing SSRs. Comparative analysis of the lengths of these tracts in the genome, mRNAs and ncRNAs revealed that most of the identified tracts among the analyzed RNA classes were of moderate length (less than 10 repeats) (Fig. 1C). The notable exception was CAG tracts present in lncRNAs, which were significantly longer than in the other analyzed groups (median of 10 vs. 6 repeats) but were still shorter than pathogenically expanded CAG tracts, which typically range from approximately 30 to 100 repeats (Supplementary Table I). The length of SSR tracts varies in population and higher heterozygosity is found for non-protein-coding genomic regions [14,15]. In general, the variability in the number of potential MREs for SSR-binding miRNAs may result in slight differences in functioning of raceRNA network between the individuals in population. In the case of disease, the degree of raceRNA network deregulation is expected to depend on the length of the SSR tract in the mutant gene, which is known to be highly variable in patient groups (Supplementary Table I).
Examples of structures formed by SSRs tracts found in ncRNAs
Some of the transcribed SSR tracts are predicted to form specific secondary structures such as hairpins and quadruplexes [16]. In case of diseases, expanded tracts of CAG, CUG, CGG, CCUG repeats are predicted to form long stable hairpins which are considered factors responsible for gain-of-function RNA toxicity [17,18]. One of the interesting examples that we found is circRNA hsa_circ_0055538 which has closely located 20 CGG repeat tracts (composed of 5, 13, 10, 10, 5, 7, 12, 11, 9, 19, 10, 10, 7, 8, 9, 11, 6, 12, 13 and 7 repeats) separated by short GC-rich sequences that are predicted to form multiple hairpins (Supplementary Fig. S2A). The second example is a family of 9 circRNAs originating from a pericentrosomal region of chromosome 2q12-q13, including hsa_circ_0003581, which bears tracts of 33 and 68 consecutive TGGAA repeats that are separated by a 19-nt bridging sequence consisting of TGGAA-like sequences. The first repeat tract is also predicted to form a long bulged hairpin structure, whereas the second is expected to be less structured (Supplementary Fig. S2B). Two aspects should be mentioned regarding secondary structures formed by SSR tracts in ncRNAs: (I) some RNAs can gain a toxic function if these repeat tracts undergo expansion and may be a cause of rare diseases, (II) the SSR tract region may be accessible or not for interacting miRNAs, which would determine their function in raceRNA network.
Numerous miRNAs have the potential to bind to SSR tracts, including disease-relevant tracts
To examine whether miRNAs can bind to SSR tracts, including disease relevant tracts, we searched for miRNAs exhibiting either 6 or 7 continuous matches to the repeat sequence within their 7-nt seed region. For all triplet repeats and the selected longer repeated motifs, up to 25 potentially binding miRNAs were identified per repeat tract (Fig. 2A and Supplementary Table II). The greatest number of miRNAs was predicted to bind to CCUG and CUG tracts, suggesting that in particular the expansion of these repeats could influence miRNA-mediated regulation of gene expression. Secondary structure models of complexes between miRNAs and repeat tracts show that these complexes represent rather classical miRNA-mRNA interactions, with full or nearly full complementarity within the miRNA seed and additional matching within the miRNA 3ʹ region (Figs. 2B and S3).
Figure 2.
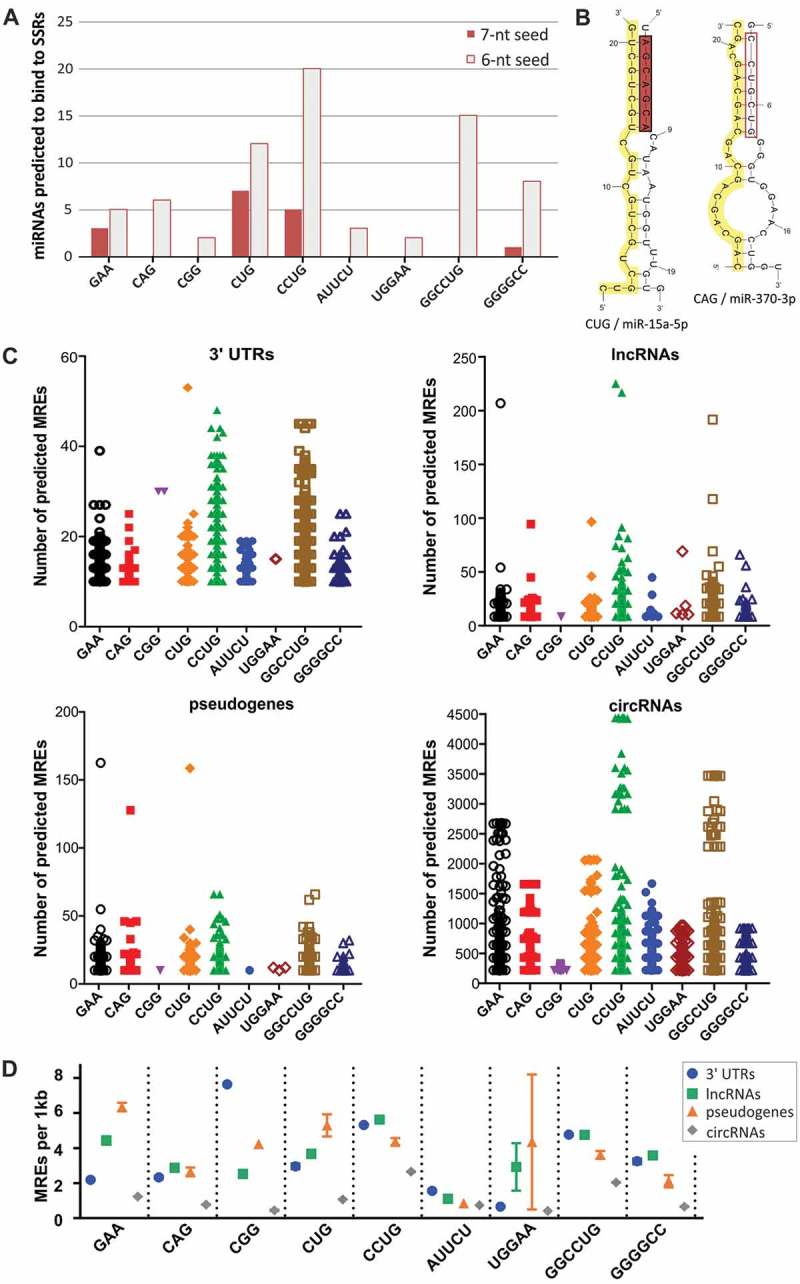
Identification of miRNAs binding to SSR tracts. (A) Number of miRNAs predicted to bind to disease-relevant SSRs showing 7-nt and 6-nt complementarity to SSRs within their seed regions. (B) Secondary structure representations of complexes between CUG repeats and miR-15a-5p and between CAG repeats and miR-370-3p. SSR tracts are highlighted in yellow; miRNA seed regions are highlighted in red. (C) Distribution of the number of predicted MREs for miRNAs interacting with SSRs in the 3ʹ UTRs of protein-coding transcripts and in ncRNAs (lncRNAs, pseudogenes and circRNAs). Each point represents a single RNA; only RNAs with a minimum of 10 MREs are shown. (D) The density of predicted MREs was calculated as the number of MREs per 1 kb of the analyzed RNAs with multiple (minimum of 10) MREs and is depicted as the mean value with the SEM (symbols with bars).
To determine which RNAs can be regulated by these subsets of miRNAs, we identified possible MREs in the 3ʹ UTRs of protein-coding transcripts and in ncRNAs: lncRNAs, pseudogenes and circRNAs. For almost all repeat-binding miRNAs, we found many RNAs with multiple (10 and more) interaction sites (Fig. 2C). In general, more MREs were identified for subsets containing the greatest numbers of repeat-associating miRNAs, such as CCUG-, GGCCUG- and CUG-binding miRNAs. Because some circRNAs are of considerable length, they were predicted to host up to a few thousand potential binding sites for these miRNAs. To compare the frequency of the occurrence of these sites across different classes of RNAs that vary in their average length, we calculated the number of putative MREs per 1 kb for RNAs with multiple MREs (Fig. 2D). The density varied from 0 to 8 sites per 1 kb for different repeats, following trends observed in the raw number of binding sites (Fig. 2C). The MRE frequency was very similar in 3ʹ UTRs, lncRNAs and pseudogenes and considerably lower in circRNAs, which clearly shows that lncRNAs and transcribed pseudogenes can constitute a potent reservoir of SSR-associated MREs and play an important role in the miRNA-mediated regulation of protein-coding genes.
RaceRNA crosstalk may be disrupted in myotonic dystrophies
RNA crosstalk mediated by miRNAs depends mostly on the concentrations of miRNAs and target RNAs as well as a number of MREs and their accessibility [19–22]. We examined the expression levels of miRNAs predicted to bind to selected SSR tracts in tissues that are primarily affected in individuals with repeat expansion diseases. We reasoned that these miRNAs should be expressed at relatively high levels, together with their target RNAs, to be able to effectively regulate gene expression under physiological conditions. We employed human miRNA atlas data [23] to examine the miRNA expression in the muscle for CUG- and CCUG- repeats causing DM1 and DM2 and in the brain for selected repeats resulting in central nervous system pathologies. We found that there was a subset of CUG- and CCUG-binding miRNAs that were highly expressed in muscle (Fig. 3A), while repeat-binding miRNAs showed considerably lower expression in the brain (Figs. 3A and S4). Therefore, we decided to investigate how the expansion of CUG and CCUG tracts in patients with DM1 and DM2 could influence miRNA crosstalk in myoblasts.
Figure 3.
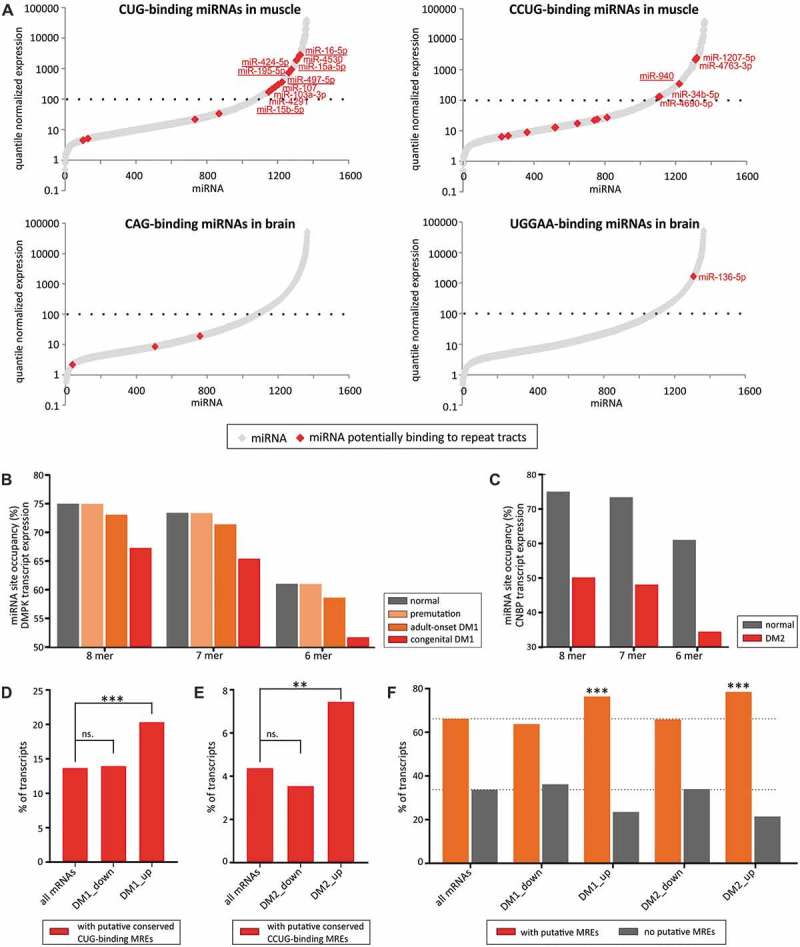
SSRs mediate raceRNA crosstalk. (A) Expression levels of CUG- and CCUG repeat-binding miRNAs in muscle (upper graphs) and expression of CAG and UGGAA repeat-binding miRNAs in the brain (lower graphs). Expressed miRNAs are depicted with gray diamond-shaped symbols; miRNAs binding to repeats are shown in red. MiRNAs showing 7-nt matching within the seed region are underscored. (B) Modeling of miRNA-mediated crosstalk between CUG repeats and a group of CUG-binding miRNAs (with a 6-nt AGCAGC seed region (hsa-miR-15a-5p, hsa-miR-15b-5p, hsa-miR-16-5p, hsa-miR-195-5p, hsa-miR-424-5p, hsa-miR-497-5p, hsa-miR-503-5p, hsa-miR-646 and hsa-miR-6838-5p)) in human myoblasts. The graph depicts the effects on miRNA site occupancy upon DMPK transcript expression (normal (~2 MREs), with premutation (~5 MREs), in adult-onset DM1 (60 MREs) or congenital DM1 (240 MREs)). (C) Modeling of miRNA crosstalk between CCUG repeats and a group of CCUG-binding miRNAs (with a 6-nt AGGCAG seed region (hsa-miR-34b-5p, hsa-miR-449c-5p, hsa-miR-940, hsa-miR-1910-3p, hsa-miR-2682-5p, hsa-miR-6808-5p, hsa-miR-6893-5p and hsa-miR-6511a-5p)) in human myoblasts. The graph depicts the effects on miRNA site occupancy upon of CNBP transcript expression (normal (3 MREs) or present in DM2 (800 MREs)). (D) The graph depicts the percentage of mRNAs bearing putative conserved MREs for conserved CUG-binding miRNAs in their 3ʹ UTRs in all human mRNAs and mRNAs downregulated or upregulated in DM1 patients (Supplementary Table III), *** − P-value < 0.001. (E) The graph depicts the percentage of mRNAs bearing putative conserved MREs for conserved CCUG-binding miRNAs in their 3ʹ UTRs in all human mRNAs and mRNAs downregulated or upregulated in DM2 patients (Supplementary Table III), ** − P-value < 0.01. (F) The graph depicts the percentage of mRNAs bearing putative conserved MREs for all conserved miRNAs in their 3ʹ UTRs in in all human mRNAs, mRNAs downregulated or upregulated in DM1 patients and mRNAs downregulated or upregulated in DM2 patients, *** − P-value < 0.001.
We used a quantitative steady-state model to study the effects of changes in MRE concentrations on target site occupancy [21]. The combination of single-cell transcriptome and miRNAome data from human myoblasts [24] with the identified putative MREs for miRNAs binding to SSR tracts allowed us to simulate the effect of abnormally elongated mutant transcript expression on endogenous MRE occupancy (Fig. 3B, C). Based on experimentally verified transcript copy numbers in DM1 patient myoblasts [25,26], we assumed the existence of 15 copies per cell of a mutant DMPK transcript with the CUG tract expansion. In healthy individuals, a DMPK transcript exhibits up to ~20 CUG repeats, which equates to ~2 MREs, whereas approximately 40 repeats (~5 MREs) are present in healthy carriers of premutation alleles that are unstable and can lead to large expansions in progeny. Our modeling showed that premutation of DMPK transcripts has a negligible effect on MRE site occupancy (Fig. 3B). In contrast, the 500 and 2000 repeats found in patients with classical adult-onset DM1 and congenital DM1, respectively, are predicted to have a greater impact on MRE site occupancy (~10% increase in unbound MREs in the case of congenital DM1 patients compared with the initial presumed state). It is worth to point out that number of copies of DMPK transcript is estimated to be higher in muscle cells than in myoblasts or fibroblasts.
The CNBP transcript in healthy individuals bears approximately 20 CCUG repeats, constituting 3 MREs, while the mean length of the CCUG tract in DM2 patients is 5000 repeats [27], which forms 800 MREs. We assumed that the copy number of the mutant CNBP is comparable to the number of copies of the DMPK transcript (i.e., 15 copies per cell) [28] and we examined the influence of MRE site occupancy on raceRNA crosstalk (Fig. 3C). We discovered that even a few copies of the mutant RNA can alter MRE site occupancy and potentially exceed the effects triggered by the CUG repeat expansion.
If the predicted effects of the CUG and CCUG tract expansions on raceRNA crosstalk occur in DM1 and DM2, they should cause deregulation of the expression of genes that exhibit MREs for miRNAs interacting with these repeats. Consequently, this would lead to elevated levels of transcripts containing these MREs. To verify this point, we employed expression data from muscle biopsies of DM1 and DM2 patients (Supplementary Table III). We observed a statistically significant increase of the number of protein-coding genes with conserved MREs for conserved CUG-binding miRNAs among genes upregulated in DM1 patients compared with all human genes. Such an enrichment was not observed for genes downregulated in DM1 patients (20% vs. 14%, p < 0.0001 and 14% vs. 14%, p = 0.77, respectively; Fig. 3D). Similar trend was observed for the increase of the number of protein-coding genes with conserved MREs for conserved CCUG-binding miRNAs among genes upregulated in DM2 patients compared with all human genes but not for genes downregulated in DM2 patients (7% vs. 4%, p = 0.0034 and 4% vs. 4%, p = 0.59, respectively; Fig. 3E). Such enrichments support the notion that abnormally elongated SSR tracts can lead to observable changes in miRNA-mediated gene regulation. Additionally, we noticed that 3ʹ UTRs of transcripts upregulated in DM1, as well as DM2 samples, are enriched in conservative MREs compared to all human transcripts (76% vs. 66%, p < 0.0001 and 78% vs. 66%, p < 0.0001, respectively; Fig. 3F). It suggests that expression of elongated CUG and CCUG tracts can lead to more global de-repression of miRNA-mediated gene regulation. Moreover, we employed a dataset of genes that are deregulated in the muscle tissue of DM2 patients [29] and observed a mild but statistically significant enrichment of MREs for miRNAs predicted to bind CCUG tracts. This could be observed among genes that are upregulated in DM2 muscle samples compared with all human genes containing any MREs (74% vs. 68%, p = 0.0005). This trend was consistent for putative MREs for most of subsets of CCUG-binding miRNAs grouped by common 6-mer sequences in the seed regions (10 groups) and when examining only miRNAs with 7-mer sites for CCUG tracts (40% vs. 37%, p = 0.04) (Supplementary Table IV).
Discussion
In this study, we aimed to examine the potential perturbation of the crosstalk between raceRNAs in simple repeat expansion diseases. For this purpose, we extracted a fraction of human cellular transcripts that could use the wording of SSRs in RNA crosstalk from existing nucleotide sequence databases. We searched for all possible trinucleotide repeats and longer repeated motifs known to be implicated in human genetic diseases and identified hundreds of ceRNA candidates belonging to the human ‘repeatome’. Our survey comprised all major classes of non-coding transcripts, most of which are as yet of unknown function. Although we succeeded in identifying SSR-bearing non-coding transcripts, without performing dedicated studies, we cannot state whether their repeat tracts are beneficial, neutral or potentially harmful to cells. To date, none of the known repeat expansion diseases has been linked to expanded repeats located in lncRNAs, circRNAs or transcribed pseudogenes. However, we identified nearly 90 miRNAs potentially interacting with various disease-relevant SSRs. It appears to us that raceRNA crosstalk and engagement of at least some of these transcripts in such crosstalk are very likely to occur in cells.
A unique feature of raceRNA crosstalk is that SSRs form naturally occurring reiterated MREs in which miRNA regulation may more freely realize its potential for cooperative action. In earlier work, we observed an increase of miRNA regulatory activity with the length of CUG tracts, which we explained as the effect of miRNA cooperativity [30]. This finding is in agreement with several reporter assay-based studies conducted with non SSR-MREs [31–34]. These studies have demonstrated that multiple neighboring MREs often exhibit greater regulatory effects than are predicted from the cumulative action of individual MREs and from the observed effects of the same number of MREs when they are distantly spaced. It can be then speculated that the affinity of SSR tracts for miRNA-loaded RNA-induced silencing complexes (RISCs) is higher than that of typical MREs; thus, the modeled effects of expanded SSR tracts on raceRNA crosstalk proposed in this study could be likely underestimated. The cooperative action of RISCs programmed with CUG repeat small interfering RNAs (siRNAs) that interact with expanded CAG repeat tracts was demonstrated previously in the reverse situation where exogenous miRNA-like siRNAs were tested as potential therapeutics for several polyglutamine (polyQ) diseases [35–38]. These siRNAs form base mismatches with CAG repeats and function more like miRNAs preferentially inhibiting mutant mRNA translation.
The ceRNA concept postulates the existence of a miRNA-mediated network that regulates the expression levels of the transcriptome, where coding and ncRNAs compete for a limited pool of miRNAs [8]. As a result, changes in the expression of RNAs with many MREs, acting as miRNA sponges, have been shown to influence mRNA expression [9]. However, transcriptome-wide studies suggest that changes in the abundance of a single RNA are often not sufficient to have a global effect on miRNA activity and that ceRNA crosstalk may be not as widespread phenomenon as previously hypothesized (reviewed in: [22,39]). The biological relevance of ceRNA activity has been extensively debated [19–21,40] and it was proposed that competitor RNAs should be among the most abundant RNAs in the cell, or that they should contain dozens of binding sites for a single miRNA species. Moreover, it was speculated that miR-15/16, which is also our top candidate in DM1-related analyses, is especially prone to ceRNA perturbations but would likely require unphysiological target increases to affect repression [20]. The substantial evidence supporting the ceRNA hypothesis is still missing; to date there is only one study reporting functional circRNA and of a physiologically relevant ceRNA mechanism in mammals [41]). We believe that SSR-containing transcripts are ideal candidates for a physiological ceRNA network, which can be altered by pathological repeat expansions (Fig. 4). SSR tracts have the potential to fulfill stringent requirements for serving as modulators of miRNA-mediated crosstalk due to their (1) ability to create many MREs that are located next to each other, which may increase their affinity for miRNA-loaded RISCs and makes transcripts harboring SSRs efficient ceRNAs, even when present at low abundance; (2) capacity for efficient miRNA sequestration and functional depletion; and (3) ability to associate with more than one miRNA family, extending the possibility of miRNA crosstalk in various tissues showing differences in miRNA expression.
Figure 4.
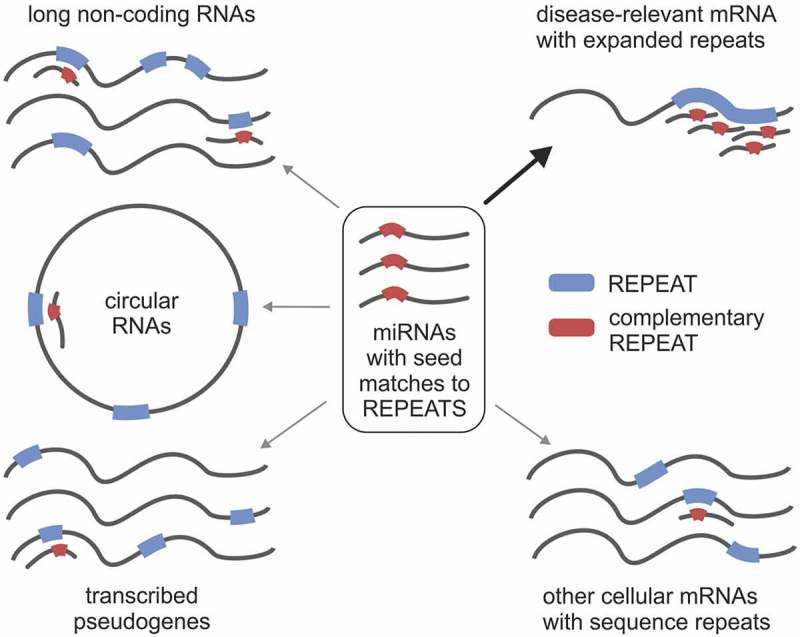
The concept of miRNA-mediated crosstalk between coding and non-coding transcripts using SSR tracts. Different types of RNAs containing SSR tracts (protein-coding RNAs and ncRNAs, including pseudogenes, lncRNAs and circRNAs) can function as ceRNAs. SSR-binding miRNAs play a central role in the crosstalk between various cellular RNAs. Aberrant expansion of SSRs may lead to disruption of this network.
The binding of repeat-containing miRNAs to MREs composed of complementary repeats may engage RISCs to different extents. Upon the binding of an miRNA-loaded RISC to SSR tracts, the RISC may remain associated with raceRNAs, which in the case of pathologically expanded SSR tracts, may lead to increased RISC sequestration, affecting global miRNA-mediated gene regulation by lowering the availability of Ago proteins for other miRNAs. We also anticipate that structures formed by some SSR-MREs [16] or high-affinity protein binding [13,21] will affect the accessibility of these sequences for raceRNA crosstalk.
Another important aspect to be considered is the dynamics of interactions between expanded repeats and miRNAs. Some RNAs containing expanded disease-relevant SSR tracts are known to form cellular foci composed of mutant transcripts and repeat-binding proteins that are implicated in pathogenesis [42,43] and we have recently shown that CUG RNA foci also contain selected CUG-binding miRNAs [30]. Although these RNA foci predominantly localize to the nucleus while miRNAs mature and function in the cytoplasm, interactions can occur between them. First, the CUG and CCUG RNA foci can form and exist (at least transiently) in the cytoplasm [44,45]. Second, several studies have demonstrated that many miRNAs and other components of RNAi machinery are also present and active in the nucleus [46,47]. In addition, a shift in the subcellular distribution of several miRNAs to a more nuclear localization was observed in DM1 skeletal muscles [48], strongly suggesting that expanded transcripts are able to alter miRNA localization. These findings illustrate an additional mechanism by which expanded SSRs may influence the miRNA-mediated crosstalk: by altering the subcellular localization of miRNAs and effectively perturbing their accessibility to other MREs.
Our analyses show that mutant CUG and CCUG tracts, observed in DM1 and DM2, are likely to perturb naturally occurring raceRNA crosstalk due to (1) the length of expanded repeats observed in affected individuals, (2) the large number of miRNAs predicted to associate with these tracts and (3) the relatively high abundance of these miRNAs in muscle. Such perturbation may contribute to the phenotypic changes observed in patients, and the influence of expanded SSRs on miRNA-mediated crosstalk is predicted to be stronger with an increase in mutant repeat length. Our modeling predicts a relatively small impact of repeat tracts in individuals with classical adult-onset DM1 but a considerably greater influence of CUG repeat tracts present in congenital DM1. As the expanded CCUG tracts in DM2 patients are typically longer than the CUGs observed in DM1, most of the mutant repeats associated with DM2 are predicted to be sufficient to perturb miRNA-mediated crosstalk. This prediction is in agreement with the low correlation found between expanded CCUG tract length and the severity of symptoms in DM2 [27].
Taken together, the results of this work indicate that cellular transcripts that harbor stuttering CUG and CCUG sequences and miRNAs whose seed sequences are complementary to these repeats have a unique potential to participate in the raceRNA dialogue, and their voice is predicted to become louder in DM1 and DM2 due to cooperative action of multiple RISCs.
Materials and methods
Prediction of miRNAs binding to SSR tracts
An in-house script written in Python was employed to identify human miRNAs that are potentially sequestered by SSR tracts. The program entitled miRNAfinder.py is available at the laboratory’s GitHub repository (https://github.com/krzyzosiak-lab/). Mature miRNA sequences were obtained from miRBase release 21 (http://www.mirbase.org/). MiRNAs with binding potential were defined as those bearing either 6 or 7 continuous complementary matches within their 7-nt seed region to the analyzed repeat sequence. The analyzed SSRs consisted of all possible triplet repeats, including those known to trigger triplet repeat expansion diseases, as well as the selected tetra-, penta- and hexanucleotide sequence motifs CCUG, AUUCU, GGCCUG and GGGGCC, which are implicated in myotonic dystrophy type 2 (DM2), spinocerebellar ataxia type 10 (SCA10), spinocerebellar ataxia 36 (SCA36) and amyotrophic lateral sclerosis (ALS)/frontotemporal dementia (FTD) pathology, respectively.
Analysis of SSRs in coding and ncRNAs
An in-house script written in Python was employed to identify ceRNAs containing SSRs among transcripts, lncRNAs, circRNAs and transcribed pseudogenes. The program entitled repeatfinder.py is available at the laboratory’s GitHub repository (https://github.com/krzyzosiak-lab/). Coding transcripts and lncRNAs were retrieved directly from GENCODE version 19 (https://www.gencodegenes.org/releases/19.html). The pseudogene transcript dataset, including all annotated pseudogenes except for polymorphic pseudogenes, was obtained from GENCODE version 19 using the UCSC Table Browser (https://genome.ucsc.edu/cgi-bin/hgTables). Human circular RNA datasets [49–52] were retrieved from circBase, an on-line repository of public circRNA datasets (http://www.circbase.org). A threshold of a minimum of 5 consecutive SSRs was established. The identified ncRNAs with SSR tracts can be found in the Supplementary Data. The calculation of repeat tract sizes allowed us to examine their distribution. The representation rate of triplet repeats in the human RNAome was calculated as the ratio of the density of SSR tracts, calculated as their summary length per 1 Mbp, found in coding and ncRNAs, compared with their density in the entire human genome. Version GRCh37.p13 of the human genome was obtained from the Genome Research Consortium (http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/).
Prediction of MREs in coding and ncRNAs
Computational identification of putative MREs in ncRNAs and the 3ʹ UTRs of coding transcripts was conducted with a TargetScan 6.0 Perl script (http://www.targetscan.org/vert_60/) using mature human miRNA sequences (obtained from miRBase release 21 (http://www.mirbase.org/) and the 3ʹ UTRs of protein-coding genes (obtained from TargetScan 6.0 (http://www.targetscan.org/vert_60/)) and consisted of all human RefSeq transcripts following NCBI annotation of the human genome (hg19)) or ncRNA sequences, where the sources were as stated above. The prediction results were further processed with our in-house Python script to identify RNAs with multiple (minimum 10) 7- and 8-mer MREs for miRNAs predicted to bind to and be potentially sequestered by SSRs. The program entitled RNAwithmultipleMREfinder.py is available at the laboratory’s GitHub repository (https://github.com/krzyzosiak-lab/).
SSR expansion influence on ceRNA crosstalk
The modeling of the influence of additional MREs, due to the expansion of disease-triggering SSRs on miRNA-mediated regulatory crosstalk was performed using a quantitative model developed by Jens and Rajewsky (http://dorina.mdc-berlin.de/public/rajewsky/rna_competition/). Single-cell mRNA and miRNA expression data from myoblasts were obtained from Zeng W et al. [24]. The 8-mer and 7-mer binding sites for CUG- and CCUG-binding miRNAs were predicted with a TargetScan 6.0 Perl script (http://www.targetscan.org/vert_60/) using mature human miRNA sequences and the 3ʹ UTRs of protein-coding genes, where the sources were as indicated above. Six-mer sites were additionally identified using an in-house Python script. The combination of expression data with predicted MREs allowed us to assess the total number of possible 6-, 7- and 8-mer MREs in all transcripts expressed in myoblasts and the expression-weighted concentrations of CUG- and CCUG-binding miRNAs. Based on high expression levels, we modeled the effects of CUG tract expansion on a group of miRNAs with a 6-nt AGCAGC seed region (hsa-miR-15a-5p, hsa-miR-15b-5p, hsa-miR-16-5p, hsa-miR-195-5p, hsa-miR-424-5p, hsa-miR-497-5p, hsa-miR-503-5p, hsa-miR-646 and hsa-miR-6838-5p) and the effects of CCUG tract expansion on a group of miRNAs with a 6-nt AGGCAG seed region (hsa-miR-34b-5p, hsa-miR-449c-5p, hsa-miR-940, hsa-miR-1910-3p, hsa-miR-2682-5p, hsa-miR-6808-5p, hsa-miR-6893-5p and hsa-miR-6511a-5p). Taking into account the false discovery rates of miRNA-target prediction algorithms and 50% MRE accessibility, approximately 23,000 to 25,000 active MREs were predicted for each group of the abovementioned miRNAs per cell. The main parameters for examining MRE occupancy by miRNAs were the same as those used by Jens and Rajewsky [21] and were inferred from experimental data. Namely, we assumed 250,000 protein-coding RNAs per cell and 150,000 Ago complexes per cell; the Kd values for 6-, 7- and 8-mer MREs were set as 61 pM, 67 pM and 118 pM, respectively, for 37°C; and an initial 75% site occupancy was assumed for 8-mer MREs. For the calculation of MRE numbers in DMPK and CNBP transcripts, we defined the length of a single MRE as 25 nucleotides, which is a region sufficient to harbor an miRNA molecule.
Analysis of gene dysregulation in DM1 and DM2
Gene expression analysis data from skeletal muscle biopsies of DM1 patients (n = 7), DM2 patients (n = 7) and healthy individuals (n = 8) were obtained from [53]. Among transcripts with statistically significant altered expression, only transcripts with high expression levels (PLIER (probe logarithmic intensity error) value of 200 and above) were subjected to further analysis. Prediction of conserved MREs of all conserved miRNA families and conserved miRNA families with CUG and CCUG repeat binding miRNAs for all human mRNAs and for mRNA with altered expression in DM1 and DM2 samples was performed using TargetScan 6.0 [54].
Gene expression analysis data from muscle biopsies of DM2 patients and healthy individuals were obtained from [29] (GEO accession number: GSE45331). A group of genes that are upregulated in DM2 samples vs. controls was obtained using the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo, one-tailed t-test with cut-off level p < 0.01). All possible 7- and 8-mer sites in the 3ʹ UTRs of mRNAs with altered expression and all human mRNAs for CCUG repeat-binding miRNAs were predicted using a TargetScan 6.0 Perl script (http://www.targetscan.org/vert_60/).
Secondary structure prediction
The secondary structures of complexes between ncRNAs and miRNAs were calculated using UNAfold (http://unafold.rna.albany.edu/) with default parameters for RNA folding.
Quantification and statistical analysis
All statistical analyses were performed and graphs generated using GraphPad Prism 6 (GraphPad Software). In Fig. 1C, the statistical significance of differences in the length distribution of triplet repeats was assessed using the Kruskal-Wallis test, followed by Dunn’s test for multiple comparisons. The comparison of the number of genes containing MREs for CUG tract-binding miRNAs between genes with altered expression in DM1 and DM2 patients, and all human genes (Fig. 3D, E and F) were performed using Fisher’s exact test. The comparison of the number of genes containing MREs for CCUG tract-binding miRNAs between genes that are upregulated in DM2 patients and all human genes was performed using Fisher’s exact test; please see Supplementary Table IV for the number of genes in each category and for the calculated p-values. The statistical significance cut-offs in all tests were set as follows: * − P-value < 0.05; ** − P-value < 0.01, *** − P-value < 0.001.
Data and software availability
The IDs and sequences of the identified human lncRNAs, circRNAs and transcribed pseudogenes with SSR tracts can be found in the Supplementary Data. The identified human miRNAs with potential to bind to SSRs can be found in Supplementary Table II. The list of genes with altered expression in DM1 and DM2 patients can be found in Supplementary Table III.
The scripts created in-house have been deposited to the laboratory’s GitHub repository (https://github.com/krzyzosiak-lab). Please refer to Wiki guide for detailed information about how to use these programs, input file requirements and example files (https://github.com/krzyzosiak-lab/raceRNA/wiki).
Key points
Multiple non-coding and protein-coding transcripts harbor simple sequence repeats (SSRs)
Some SSRs in human transcripts participate in miRNA-mediated cross-regulation
Repeat-associated ceRNAs crosstalk is most likely altered in two myotonic dystrophies (DM1 and DM2), associated with extended SSRs.
Funding Statement
This work was supported by the National Science Centre [2014/15/B/NZ1/01880 to W.J.K. and 2015/17/D/NZ5/03443 to A.F.] and the Polish Ministry of Science and Higher Education [under the KNOW program and a scholarship to A.F.].
Acknowledgments
We are grateful to Witold Filipowicz and Krzysztof Sobczak for critical reading of the manuscript and helpful suggestions.
Disclosure statement
No potential conflict of interest was reported by the authors.
Author contributions
WJK, TMW and EK devised the project and the main conceptual ideas. TMW performed bioinformatics analyses, while EK designed figures. WJK and AF received funding. All authors analyzed the data, discussed the results and contributed to the final manuscript.
Supplementary material
Supplementary data for this article can be acceseed here.
References
- [1].Toth G, Gaspari Z, Jurka J.. Microsatellites in different eukaryotic genomes: survey and analysis. Genome Res. 2000;10:967–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Gemayel R, Vinces MD, Legendre M, et al. Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu Rev Genet. 2010;44:445–477. [DOI] [PubMed] [Google Scholar]
- [3].Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30:575–621. [DOI] [PubMed] [Google Scholar]
- [4].Rohilla KJ, Gagnon KT. RNA biology of disease-associated microsatellite repeat expansions. Acta Neuropathol Commun. 2017;5:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. 2010;79:351–379. [DOI] [PubMed] [Google Scholar]
- [7].Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 2015;16:421–433. [DOI] [PubMed] [Google Scholar]
- [8].Salmena L, Poliseno L, Tay Y, et al. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505:344–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kozlowski P, de Mezer M, Krzyzosiak WJ. Trinucleotide repeats in human genome and exome. Nucleic Acids Res. 2010;38:4027–4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Faux NG, Bottomley SP, Lesk AM, et al. Functional insights from the distribution and role of homopeptide repeat-containing proteins. Genome Res. 2005;15:537–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Willadsen K, Cao MD, Wiles J, et al. Repeat-encoded poly-Q tracts show statistical commonalities across species. BMC Genomics. 2013;14:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Morriss GR, Cooper TA. Protein sequestration as a normal function of long noncoding RNAs and a pathogenic mechanism of RNAs containing nucleotide repeat expansions. Hum Genet. 2017;136:1247–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Duitama J, Zablotskaya A, Gemayel R, et al. Large-scale analysis of tandem repeat variability in the human genome. Nucleic Acids Res. 2014;42:5728–5741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Willems T, Gymrek M, Highnam G, et al. The landscape of human STR variation. Genome Res. 2014;24:1894–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sobczak K, Michlewski G, de Mezer M, et al. Structural diversity of triplet repeat RNAs. J Biol Chem. 2010;285:12755–12764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Blaszczyk L, Rypniewski W, Kiliszek A. Structures of RNA repeats associated with neurological diseases. Wiley Interdiscip Rev RNA. 2017;8:e1412. [DOI] [PubMed] [Google Scholar]
- [18].Krzyzosiak WJ, Sobczak K, Wojciechowska M, et al. Triplet repeat RNA structure and its role as pathogenic agent and therapeutic target. Nucleic Acids Res. 2012;40:11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Denzler R, Agarwal V, Stefano J, et al. Assessing the ceRNA hypothesis with quantitative measurements of miRNA and target abundance. Mol Cell. 2014;54:766–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bosson AD, Zamudio JR, Sharp PA. Endogenous miRNA and target concentrations determine susceptibility to potential ceRNA competition. Mol Cell. 2014;56:347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Jens M, Rajewsky N. Competition between target sites of regulators shapes post-transcriptional gene regulation. Nat Rev Genet. 2015;16:113–126. [DOI] [PubMed] [Google Scholar]
- [22].Thomson DW, Dinger ME. Endogenous microRNA sponges: evidence and controversy. Nat Rev Genet. 2016;17:272–283. [DOI] [PubMed] [Google Scholar]
- [23].Ludwig N, Leidinger P, Becker K, et al. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016;44:3865–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zeng W, Jiang S, Kong X, et al. Single-nucleus RNA-seq of differentiating human myoblasts reveals the extent of fate heterogeneity. Nucleic Acids Res. 2016;44:e158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gudde AE, Gonzalez-Barriga A, van den Broek WJ, et al. A low absolute number of expanded transcripts is involved in myotonic dystrophy type 1 manifestation in muscle. Hum Mol Genet. 2016;25:1648–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wojciechowska M, Sobczak K, Kozlowski P, et al. Quantitative methods to monitor RNA biomarkers in myotonic dystrophy. Sci Rep. 2018;8:5885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Liquori CL, Ricker K, Moseley ML, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293:864–867. [DOI] [PubMed] [Google Scholar]
- [28].Thomas JD, Sznajder LJ, Bardhi O, et al. Disrupted prenatal RNA processing and myogenesis in congenital myotonic dystrophy. Genes Dev. 2017;31:1122–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Screen M, Jonson PH, Raheem O, et al. Abnormal splicing of NEDD4 in myotonic dystrophy type 2: possible link to statin adverse reactions. Am J Pathol. 2014;184:2322–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Koscianska E, Witkos TM, Kozlowska E, et al. Cooperation meets competition in microRNA-mediated DMPK transcript regulation. Nucleic Acids Res. 2015;43:9500–9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Saetrom P, Heale BS, Snove O Jr., et al. Distance constraints between microRNA target sites dictate efficacy and cooperativity. Nucleic Acids Res. 2007;35:2333–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Grimson A, Farh KK, Johnston WK, et al. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27:91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Broderick JA, Salomon WE, Ryder SP, et al. Argonaute protein identity and pairing geometry determine cooperativity in mammalian RNA silencing. Rna. 2011;17:1858–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Doench JG, Sharp PA. Specificity of microRNA target selection in translational repression. Genes Dev. 2004;18:504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hu J, Liu J, Corey DR. Allele-selective inhibition of huntingtin expression by switching to an miRNA-like RNAi mechanism. Chem Biol. 2010;17:1183–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Fiszer A, Mykowska A, Krzyzosiak WJ. Inhibition of mutant huntingtin expression by RNA duplex targeting expanded CAG repeats. Nucleic Acids Res. 2011;39:5578–5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hu J, Liu J, Yu D, et al. Mechanism of allele-selective inhibition of huntingtin expression by duplex RNAs that target CAG repeats: function through the RNAi pathway. Nucleic Acids Res. 2012;40:11270–11280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Fiszer A, Olejniczak M, Galka-Marciniak P, et al. Self-duplexing CUG repeats selectively inhibit mutant huntingtin expression. Nucleic Acids Res. 2013;41:10426–10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Smillie CL, Sirey T, Ponting CP. Complexities of post-transcriptional regulation and the modeling of ceRNA crosstalk. Crit Rev Biochem Mol Biol. 2018;53:231–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Denzler R, McGeary SE, Title AC, et al. Impact of MicroRNA levels, target-site complementarity, and cooperativity on competing endogenous RNA-regulated gene expression. Mol Cell. 2016;64:565–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Piwecka M, Glazar P, Hernandez-Miranda LR, et al. Loss of a mammalian circular RNA locus causes miRNA deregulation and affects brain function. Science. 2017;357. [DOI] [PubMed] [Google Scholar]
- [42].Pettersson OJ, Aagaard L, Jensen TG, et al. Molecular mechanisms in DM1 - a focus on foci. Nucleic Acids Res. 2015;43:2433–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhang N, Ashizawa T. RNA toxicity and foci formation in microsatellite expansion diseases. Curr Opin Genet Dev. 2017;44:17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Xia G, Ashizawa T. Dynamic changes of nuclear RNA foci in proliferating DM1 cells. Histochem Cell Biol. 2015;143:557–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Giagnacovo M, Malatesta M, Cardani R, et al. Nuclear ribonucleoprotein-containing foci increase in size in non-dividing cells from patients with myotonic dystrophy type 2. Histochem Cell Biol. 2012;138:699–707. [DOI] [PubMed] [Google Scholar]
- [46].Gagnon KT, Li L, Chu Y, et al. RNAi factors are present and active in human cell nuclei. Cell Rep. 2014;6:211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Roberts TC. The MicroRNA biology of the mammalian nucleus. Mol Ther Nucleic Acids. 2014;3:e188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Perbellini R, Greco S, Sarra-Ferraris G, et al. Dysregulation and cellular mislocalization of specific miRNAs in myotonic dystrophy type 1. Neuromuscul Disord. 2011;21:81–88. [DOI] [PubMed] [Google Scholar]
- [49].Jeck WR, Sorrentino JA, Wang K, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. Rna. 2013;19:141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Memczak S, Jens M, Elefsinioti A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–338. [DOI] [PubMed] [Google Scholar]
- [51].Salzman J, Chen RE, Olsen MN, et al. Cell-type specific features of circular RNA expression. PLoS Genet. 2013;9:e1003777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Zhang Y, Zhang XO, Chen T, et al. Circular intronic long noncoding RNAs. Mol Cell. 2013;51:792–806. [DOI] [PubMed] [Google Scholar]
- [53].Nakamori M, Sobczak K, Puwanant A, et al. Splicing biomarkers of disease severity in myotonic dystrophy. Ann Neurol. 2013;74:862–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Garcia DM, Baek D, Shin C, et al. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat Struct Mol Biol. 2011;18:1139–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.