

ABSTRACT
Epstein-Barr virus (EBV) expresses an abundant nuclear noncoding RNA called EBER2, which interacts with and acts as a guide RNA for the host transcription factor PAX5. This ribonucleoprotein complex localizes to the terminal repeat (TR) regions of the EBV genome via RNA-RNA interactions between EBER2 and nascent transcripts originating from these target sites. Given the fact that EBER2 base pairs with a viral RNA, we developed a protocol to identify EBER2-interacting RNAs in a transcriptome-wide manner. Our approach entails psoralen-mediated crosslinking, selection with antisense oligonucleotides targeting EBER2, and RNase V1 digestion coupled to next-generation sequencing. The use of RNase V1 circumvents the need of extensive computational analysis post data acquisition to search for predicted RNA hybrids, as the RNase V1 cleavage site marks the region of RNA duplex formation. As proof of principle, we show that our approach correctly identifies the known EBER2 interaction with TR RNAs. Moreover, we identify the host functional noncoding RNAs MRP, H1, and 7SL RNAs as well as three putative enhancer RNAs as candidate EBER2-interacting RNAs. As all of these gene loci exhibit PAX5 occupancy, we propose that EBER2 is recruited to these sites through its binding partner PAX5 and forms RNA-RNA interactions with nascent transcripts on chromatin. Thus, our novel approach facilitates the identification of targeted RNA-RNA-interactions and minimizes the need of downstream computational analyses to predict RNA duplexes.
KEYWORDS: Epstein-Barr virus, noncoding RNA, RNA-RNA interaction, next-generation sequencing
Introduction
Epstein-Barr virus expresses two noncoding RNAs called EBV-encoded RNA 1 (EBER1) and EBER2. Both RNAs are transcribed in infected cells to high levels (>2.5 × 105 copies per cell) and localize exclusively to the nucleus [1–3]. While the function of EBER1 remains poorly understood [4], much insight has been gained recently about the molecular mode of action of EBER2. By studying potential chromatin and protein interactions, EBER2 was found to form a ribonucleoprotein (RNP) complex with the host transcription factor PAX5 (Paired box-containing protein 5) and localize to the so-called terminal repeat (TR) regions of the EBV genome; EBER2-PAX5 binding to the TR regions is required for efficient viral lytic replication [5,6]. Intriguingly, this RNP complex is recruited to its binding sites on the EBV genome not through the DNA-binding domain of PAX5, but via RNA-RNA interactions between one of the two accessible regions of EBER2 and nascent RNA from the TR regions. This RNA duplex formation is evolutionarily conserved in a related primate gamma-herpesvirus expressing an EBER2 homolog [5]. Thus, EBER2 acts as a guide RNA for PAX5 recruitment to its viral target sites.
The estimated copy number of EBER2 (~2.5 × 105 copies) by far surpasses the number of EBV episomes, which are present at 5–800 copies per infected cell during latent infection [7]. Given the apparent stoichiometric imbalance, the question arises as to whether EBER2 may be executing other physiologically relevant functions than base paring with viral nascent transcripts. One possibility would be to form as-yet unidentified RNA-RNA interactions with host RNA molecules. Recently, several methods have been described for elucidating RNA-RNA interactions at a transcriptome-wide level. These conceptually related and targeted approaches include CLASH (crosslinking ligation and sequencing of hybrids), hiCLIP (RNA hybrid and individual-nucleotide resolution UV crosslinking and immunoprecipitation), RAP (RNA Antisense Pulldown), and RICC (RNA–RNA interaction identification by crosslinking and capture) [8–11]. All of these techniques combine covalent crosslinking, either via UV light irradiation or a crosslinking agent, with deep sequencing technology, but differ in how the RNA of interest is enriched. Notably, none of these approaches provide information about the exact site of RNA duplex formation and subsequent extensive computational analyses are required to predict regions of putative RNA-RNA interactions. Three recent publications have even described a shotgun approach to infer global RNA interaction maps [12–14], which require massive computational prowess to analyze the resulting next-generation sequencing data sets.
In this study, we adapted steps from existing protocols and extended them by leveraging the enzymatic activity of RNase V1 to identify EBER2-interacting host RNAs. This ribonuclease isolated from the venom of the Caspian cobra (Naja oxiana) has preferred specificity towards double-stranded RNA substrates and has thus been instrumental in the past in uncovering the secondary structures of many RNAs [15]. Given the compatibility of RNase V1 digestion with subsequent ligation using T4 RNA ligase, as cleavage produces a 5′ phosphate and 3′ hydroxyl group at the cut site, it can be readily used in combination with adapter ligation in the preparation of next-generation sequencing libraries [16]. As this valued reagent in RNA biology is currently commercially unavailable, we partially purified this enzyme from lyophilized cobra venom. After RNA selection and RNase V1 cleavage, a linker is ligated to the digested RNA duplex site. The junction between the linker and the cut site marks the region engaging in RNA-RNA interaction and can be identified bioinformatically following deep sequencing, thus providing a biochemical, rather than bioinformatical solution for identifying RNA-RNA interaction sites. We observed that base pairing of EBER2 is not limited to viral RNA, and several cellular RNAs putatively base pair with EBER2. Our approach can be applied to other target RNAs and identify their interacting RNA partners while minimizing the need of computational analyses.
Results
Developing a transcriptome-wide method to identify EBER2-interacting RNAs
Given the RNA-RNA interaction of EBER2 with viral RNA, we asked whether the available region for hybridization in EBER2 also potentially interacts with host RNAs. We devised a strategy entailing crosslinking of intact cells with the cell-permeable crosslinking agent aminomethyl trioxsalen (AMT), a psoralen derivative, to preserve in vivo RNA duplex formation [17], followed by DNA antisense oligonucleotide (ASO)-mediated selection of EBER2 from crosslinked total RNA, as previously performed for EBER2 to identify chromatin binding sites and interacting proteins [5,6]; this ASO targets the bottom loop region of EBER2 (nts 101–124). In addition, the selected EBER2 hybrids were subjected to digestion with the double-strand specific RNase V1 enzyme, which would result in cleavage at the RNA-RNA interaction sites between EBER2 and interacting cellular RNAs (Fig. 1A), to which a linker was subsequently ligated with T4 RNA ligase. RNase V1 digestion results in the generation of a 5′ phosphate group and 3′ hydroxyl group at the cut site and is therefore compatible with subsequent ligation with T4 RNA ligase without any further enzymatic treatment. As the starting material was phosphatase-treated prior to EBER2 selection, the linker will be ligated only to the newly generated 5′ phosphate group at the RNase V1 cut site. Following deep sequencing of the selected RNAs, the junction between the linker and ligated RNA can be identified computationally and indicates the site of RNA-RNA interaction between EBER2 and the co-selected RNA. Thus, this additional enzymatic step circumvents the need for extensive computational analyses to predict putative RNA-RNA interaction sites.
Figure 1.
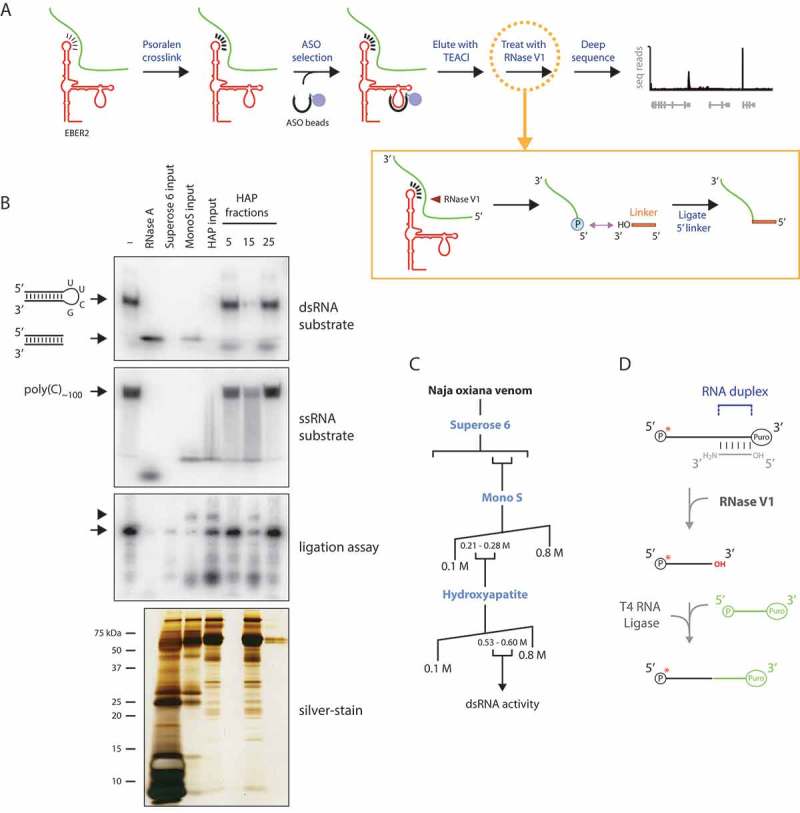
Identifying EBER2-interacting RNAs by combining psoralen crosslinking, ASO-mediated selection, and RNase V1 treatment. (A) The psoralen derivative AMT is used to crosslink RNA duplexes in intact cells to preserve in vivo RNA-RNA interactions. An EBER2-targeting ASO is then used to select EBER2 together with crosslinked interacting RNAs. These duplexes are eluted from the ASO beads using TEACl-containing buffer and are subjected to RNase V1 digestion. Following cleavage of double-stranded regions, a linker is ligated to the newly-generated 5′ phosphate group at the cut site using T4 RNA ligase (inset). Only one possible cleavage event is depicted for simplicity. After deep sequencing, not only can the interacting RNAs be identified, but also the site of RNA-RNA interactions can be deduced, which are specified by the junction of the linker and interacting RNA. (B) Cobra venom fractions were examined for activity towards doubled-stranded and single-stranded substrates. The double-stranded substrate consists of a shRNA with a pyrimidine-rich loop, which can be digested by single-strand specific RNases, such as RNase A. The trimmed RNA duplex with no loop region migrates faster in a native polyacrylamide gel. Digestion within the stem region by a double-strand specific RNase results in the disappearance of radioactive signal, as observed after digestion with all input material as well as hydroxyapatite (HAP) fraction 15; note that the weak activity of the MonoS input sample is due to the great dilution of protein concentration following size exclusion chromatography. Indicated fractions were also used in a ligation assay (outlined in D) to verify the compatibility of RNase V1 digest with T4 RNA ligase reaction. A silver-stained gel of the purified fractions is shown in the bottom panel, revealing the partial purification only of RNase V1; many other proteins are present in our sample preparation, which, importantly, do not interfere with RNase V1 activity. (C) Purification scheme of RNase V1 from Naja oxiana venom. (D) Outline of ligation reaction after RNase V1 digest. An oligonucleotide blocked at the 3′ end with puromycin was 5′ end-labeled (arrow in B, third panel from top) and annealed to a partially complementary oligonucleotide with a 3′ amino modifier. A free 3′ OH group is created only after RNase V1 digest, to which a 5′ phosphorylated linker blocked at the 3′ end with puromycin can be ligated using T4 RNA ligase. This ligation product is the only one that can be visualized by autoradiography as shown in B (arrowhead, third panel from top).
An essential reagent in our strategy is RNase V1, which is currently commercially unavailable. RNase V1 has been isolated from the venom of the Caspian cobra by a dual protein fractionation step [15]. We obtained lyophilized venom and used conventional protein chromatography following the original protocol to partially purify RNase V1 (Fig. 1B+C). We devised an assay depending on cleavage of a radiolabeled short hairpin RNA (shRNA) substrate to monitor for activity towards double-stranded RNA (Fig. 1B, top panel). This shRNA substrate contains a pyrimidine-rich loop region that can be targeted by enzymes with activity towards single-stranded RNA, such as RNase A. Following digestion of the loop region, the trimmed RNA duplex would migrate faster in polyacrylamide gel electrophoresis, while digestion within the stem region by a double-strand specific RNase would result in no resolved bands and disappearance of radioactive signal. We also used a single-stranded substrate consisting of poly(C) oligonucleotides to verify preferential double-stranded specificity (Fig. 1B, second panel from top). In addition to size exclusion and ion exchange chromatography described in the original publication, we included a subsequent separation over a hydroxyapatite column to further remove residual single-stranded RNase activity (compare ‘HAP input’ and ‘HAP fraction 15ʹ lanes in ssRNA substrate autoradiograph in Fig. 1B). Finally, we verified that the purified double-strand specific activity is compatible with subsequent ligation with T4 RNA ligase by producing a 5′ phosphate group and 3′ hydroxyl group at the cut site. For this, we used the purified double-strand specific RNase to digest a partial RNA duplex, which was 5′ end-labeled on one strand and blocked at both 3′ ends to prevent chain elongation. A free 3′ hydroxyl group would only be generated following digestion with RNase V1, to which a linker was ligated with T4 RNA ligase (Fig. 1B, third panel from top; Fig. 1D). Based on these results that match the known enzymatic properties of RNase V1, we hereafter refer to the double-strand specific RNase we purified from cobra venom as RNase V1.
The known EBER2-TR interaction is correctly detected
As proof-of-principle, we examined whether our method would correctly identify the known RNA-RNA interaction between EBER2 and nascent transcripts from the TR regions (Fig. 2A) [5]. Using EBER2-targeting ASO we selected EBER2 from total RNA purified from DMSO-treated (-AMT) or AMT-crosslinked (+AMT) EBV-positive cells. RNAs selected from the latter sample were split into two aliquots of which one was further subjected to RNase V1 digest followed by linker ligation (+AMT; +RNase V1). All three samples were converted into Illumina-compatible libraries and deep sequenced. As expected, all three selections robustly enriched for EBER2 (Fig. 2B+C). Notably, no reads were recovered aligning to the even more abundant EBER1 viral noncoding RNA, demonstrating that our approach generates a minimal number of contaminating reads. Based on our experimental setup, the 5′ end of each sequence read from the RNase V1-treated sample represents regions of RNA duplex formation. Accordingly, we observed that the profile of the EBER2 peak differed for the RNase V1-treated sample as compared to the other two in that sharp edges were present in the peak profile (Fig. 2C, red arrows in bottom panel), indicating preferential RNase V1 cut sites in EBER2 through a noticeably elevated number of sequence reads whose 5′ ends start at these positions. Interestingly, these sites coincided with the previously predicted RNA-RNA interaction site with TR RNA within EBER2 (Fig. 2A, compare highlighted nucleotides and red arrows). While the deep sequencing coverage throughout the EBER2 locus is not uniform and underrepresented at its 3′ end, possibly due to internal secondary structures interfering with the reverse transcriptase reaction as part of the library preparation, the reported sharp edges observed following RNase V1 treatment have not been observed in other RNA-seq experiments covering the EBER locus conducted thus far (Fig. 2E).
Figure 2.
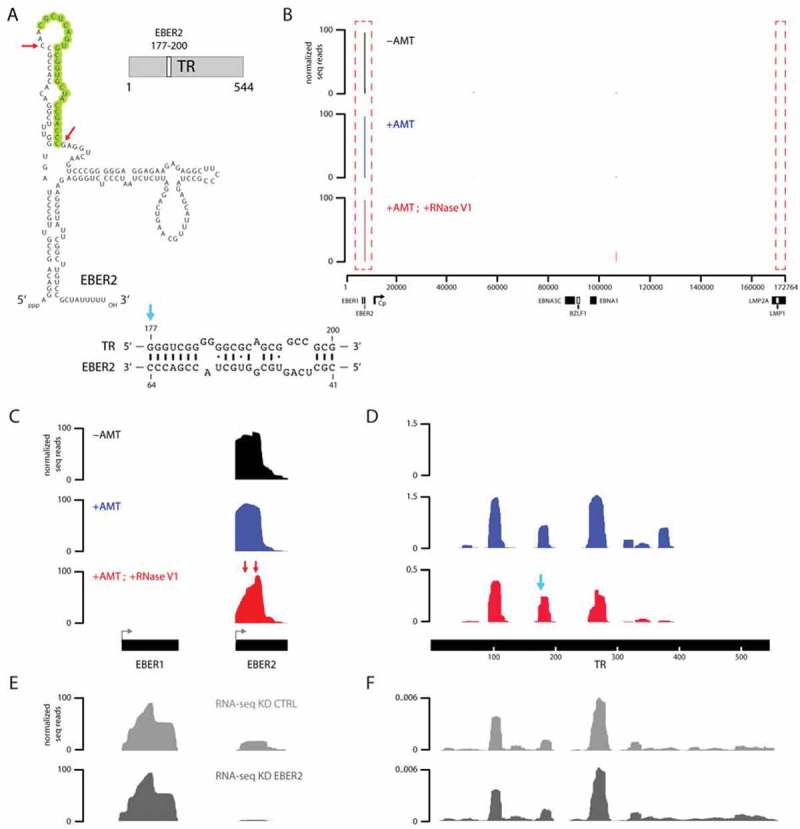
Verification of the previously predicted RNA-RNA interaction between EBER2 and nascent transcript from the TR region. (A) Secondary structure of EBER2 and previously predicted evolutionarily conserved RNA-RNA interaction between EBER2 and TR RNA is shown. (B) Tracks of ASO-selected RNAs from untreated (-AMT), AMT-crosslinked (+AMT), as well as AMT-crosslinked and RNase V1 digested (+AMT; +RNase V1) samples. X-axis shows the entire EBV genome; y-axis shows the number of sequence reads normalized to total mapped reads. (C) Zoomed-in view to the EBER locus of the tracks shown in B. Note that no reads for the highly abundant EBER1 are observed. The EBER2 profile for the AMT-crosslinked and RNase V1-digested sample differs from the other two in that abrupt edges are observed, which are indicated by red arrows. The positions within EBER2 of these sites are also denoted by red arrows in A. (D) Zoomed-in view to the TR repeat region of the tracks shown in B. No read aligning to the TR region is recovered from the -AMT sample; coverage is observed only after AMT-crosslinking. Blue arrow indicates a distinct peak shape for the ‘+AMT; +RNase V1ʹ sample compared to the +AMT sample. The location of this abrupt peak is also indicated by a blue arrow in A. (E) RNA-seq tracks of input RNA from control knockdown (KD CTRL) or EBER2 knockdown (KD EBER2) cells. While EBER1 levels are not affected, the knockdown efficiency of EBER2 to 14% of wildtype levels is evident in the sequencing tracks. (F) RNA-seq tracks of input RNA for the EBV TR region.
We next examined the TR locus for deep sequencing coverage after EBER2 ASO selection. Consistent with the fact that ASO selection was performed under denaturing conditions in the presence of urea, no reads were recovered for the TR region in the absence of AMT crosslinking; inclusion of AMT-treatment was necessary to co-purify nascent RNAs from this region (Fig. 2D). We noticed that the coverage over the TR regions was not uniform, but fragmentary and limited to several peaks. This observation may possibly be due to the high GC content of 78% of the TR region, which may create a bias and processivity issue for reverse transcriptase during library preparation, resulting in the underrepresentation of GC-rich regions that form strong secondary structures. Of note, we observed a peak in the RNase V1-treated sample whose profile differed from the one in the AMT-crosslinked sample by exhibiting a sharp edge, indicating a preferred RNase V1 cut site (Fig. 2D, blue arrow). Again, this sharp edge in the RNA sequencing profile is not observed in the deep sequencing tracks of other samples, such as input RNA (Fig. 2F). Notably, the position of this nucleotide (nt) coincided with nt 177 of the TR RNA (Fig. 2A, blue arrow), which is the first nucleotide of the predicted EBER2-TR RNA duplex. A second peak around nucleotide 280 exhibited an altered peak as well. However, as this region contains an extended GC-stretch with no apparent complementarity to EBER2, the RNase V1 digestion here may be caused not by RNA duplex formation with EBER2 but with other regions within the TR RNA. In summary, our approach using psoralen crosslinking combined with ASO selection and subsequent RNase V1 digest correctly identifies at nucleotide resolution known RNA-RNA interactions between EBER2 and nascent transcript from the TR regions.
Host RNAs that interact with EBER2
We next searched for candidate cellular RNAs that interact with EBER2. As we utilized an ASO for selection that targets the bottom loop region of EBER2, the putative base pairing with cellular RNAs would have to be formed by the other accessible region for hybridization in EBER2 (nts 41–64, highlighted in Fig. 2A). The two criteria to qualify as a candidate interacting partner were i) enrichment after EBER2 selection in the AMT-crosslinked (+AMT) sample over control (-AMT) and ii) sequencing coverage over the candidate gene region also in the RNase V1-treated sample. We used the software Cuffdiff of the Tuxedo Suite [18] to filter candidates with a more than twofold enrichment based on the calculated FPKM values of annotated genes in the hg38 human genome assembly and retrieved eleven candidates. Subsequently, we manually examined the eleven candidates for sequencing coverage also in the RNase V1-treated sample and found only three candidates with coverage over their exons. The three cellular RNAs that met both criteria were MRP RNA (RNA subunit of the RNase mitochondrial RNA processing), H1 RNA (Ribonuclease P RNA Component H1), and 7SL RNA (Fig. 3A). Interestingly, all three RNAs are functional noncoding RNAs transcribed by RNA polymerase III [19–21]. MRP RNA plays a role in processing precursor ribosomal RNA in the nucleolus and RNA in mitochondria to generate primers for DNA replication [22–24]. H1 RNA as part of RNase P is best known as the endoribonuclease that generates the 5′ ends of transfer RNAs [25], but other substrates have also been described [26,27]. 7SL RNA is an integral component of the signal recognition particle (SRP) that mediates protein translocation into the ER lumen during translation by ER-associated ribosomes [28]. For these RNAs, we were able to locate preferential RNase V1 cleavage sites in the deep sequencing tracks (Fig. 3A, arrows) and searched for potential RNA duplex formation between these regions and the accessible region in EBER2 using the program RNAhybrid [29]. We were able to retrieve predicted RNA hybrids where the RNase V1 cut site coincided with the end of a helix (Fig. 3C–E, arrows), which are the preferred cleavage sites of this enzyme. We also observed a prominent RNase V1 cleavage site within 7SL RNA (Fig. 3A, arrowhead). However, we were unable to predict an RNA duplex consisting of the surrounding region and EBER2. As the cut site is at nucleotide 151 of 7SL, which is located at the end of helix 6 of its predicted secondary structure [30], RNase V1 cleavage at this site is likely caused by intramolecular duplex formation, similar to our observation at the TR region. To confirm the EBER2 interaction with MRP, H1, and 7SL RNA observed by our deep-sequencing approach, we examined the co-selection of these cellular RNAs by qRT-PCR following AMT-treatment and ASO-selection. As a negative control, we used ASO against EBER1 and verified that both EBER1 and EBER2 were precipitated at comparable levels in the absence or following AMT-treatment (Fig. 3F, left panel). MRP, H1, and 7SL RNA were indeed enriched after EBER2 selection, when the cells were treated with AMT, while no such enrichment was observed for EBER1 selection (Fig. 3F, right panel). This observation supports the notion that our deep-sequencing based approach can be used to identify EBER2-interacting RNAs.
Figure 3.
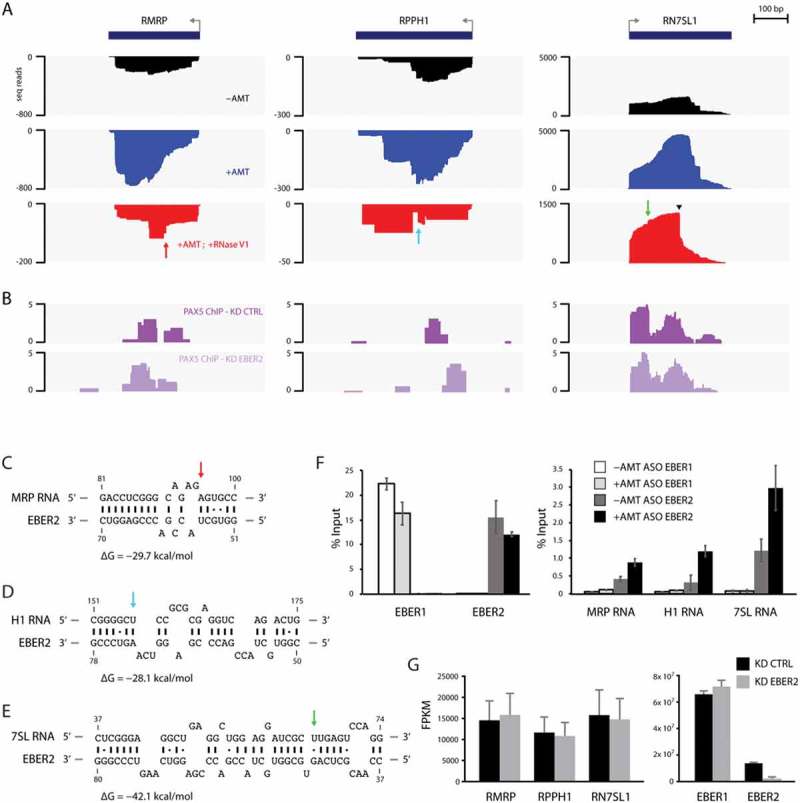
Candidate host EBER2-interacting RNAs. (A) Tracks for the gene loci RMRP, RPPH1 and RN7SL1 are shown after ASO-selection. All three functional noncoding RNAs exhibit increased coverage after AMT-crosslinking compared to ASO-selection of untreated total RNA. They also exhibit a distinct alignment profile when comparing the AMT-crosslinked sample to the one including RNase V1-digest. The latter shows abrupt edges in their profiles, indicative of RNA duplex formation. (B) PAX5 ChIP tracks of control (PAX5 ChIP–KD CTRL) and EBER2 knockdown (PAX5 ChIP–KD EBER2) BJAB-B1 cells. (C-E) Predicted RNA duplexes generated by the program RNAhybrid between the top stem-loop region of EBER2 and MRP (C), H1 (D), and 7SL RNAs (E). The calculated free energies of these duplexes are shown. Arrows indicate the abrupt edges shown in the sequencing tracks. (F) Enrichment of candidate host RNAs after AMT-crosslinking and EBER2 selection determined by quantitative RT-PCR. ASO targeting EBER1 was used as a negative control. While both EBER1 and EBER2 are precipitated at comparable levels, co-selection for MRP, H1, and 7SL RNA is only observed for ASO EBER2 and enhanced following AMT-crosslinking. Error bars indicate standard deviation of three independent experiments. (G) EBER2 depletion does not affect steady-state RNA levels of MRP, H1, and 7SL RNAs. FPKM values of the functional noncoding RNAs in control (KD CTRL) and EBER2 knockdown (KD EBER2) BJAB-B1 cells as determined by Cufflinks. Error bars indicate confidence intervals calculated by Cufflinks.
We serendipitously noticed that all three gene loci are bound by the host transcription factor PAX5 (Fig. 3B; the PAX5 ChIP-seq data will be published elsewhere), which is an interaction partner of EBER2 [5,6]. As the recruitment of PAX5 to the TR regions of the EBV genome is guided by EBER2, we asked whether the same EBER2-mediated recruitment is occurring for PAX5 localization at the candidate gene loci. To this end, we performed PAX5 ChIP-seq analysis after EBER2 knockdown and observed that PAX5 localization is not quantitatively altered under conditions of EBER2 depletion. As shown in Figs. 2E and 3G, EBER2 was efficiently knocked down to 14% of wildtype levels in our experiment. Thus, unlike PAX5 recruitment to the TR regions, its binding to the RMRP, RPPH1, and RN7SL1 gene loci is independent of EBER2 and likely not mediated by RNA-RNA interactions. Furthermore, the fact that PAX5 localizes to these gene loci suggests that EBER2 is targeted to these chromatin sites as a component of the PAX5 complex and likely interacts with nascent transcripts on chromatin rather than the mature RNAs within their respective RNP complexes. Consistent with the notion that EBER2 has a negligible role in PAX5 recruitment, knockdown of EBER2 did not have an apparent effect on the steady-state levels of these functional noncoding RNAs (Fig. 3G). Taken together, we propose that EBER2 interacts with the nascent transcripts of MRP, H1, and 7SL RNAs, but interaction with EBER2 does not impact their overall RNA levels.
EBER2 interacts with putative enhancer RNAs
To identify EBER2-interacting candidate RNAs that lie outside of annotated exons, we focused on the raw read counts rather than FPKM values output by Cuffdiff to find regions that show enhanced read coverage in the AMT-crosslinked sample compared to control. We identified three loci in apparently intronic regions of the genes HFM1, SLCO5A1, and PDE3A, which show enhanced selection after AMT-crosslinking as well as read coverage in the RNase V1-treated sample (Fig. 4A–C; gene coordinates shown in Table 1). As it is unlikely that unstable, random intronic sequences would interact with EBER2, we tested the possibility that these regions are not mere intronic sequences by consulting other available track information on the UCSC genome browser, such as RNA-seq and ChIP-seq data provided by the ENCODE project. All of these intronic regions showed robust transcription compared to adjacent genomic regions as well as strong H3K27Ac ChIP signals (Fig. 4D–F), one of the hallmark histone modifications of active enhancer regions [31], suggesting that these transcripts are putative enhancer RNAs.
Figure 4.
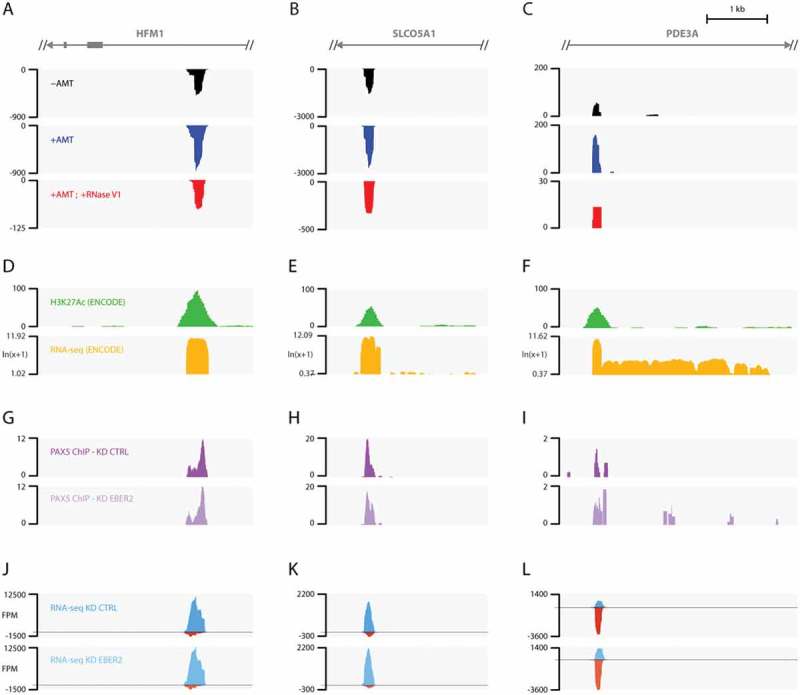
EBER2 interacts with putative host enhancer RNAs. (A-C) Tracks after ASO-selection are shown for intronic regions of HFM1 (A), SLCO5A1 (B), and PDE3A (C) genes. Please see Table 1 for genome coordinates. (D-F) H3K27Ac and RNA-seq tracks provided by the ENCODE project on the UCSC genome browser. The EBER2-selection coverages overlap with H3K27Ac marks, a hallmark of active enhancers. RNA-seq tracks indicate robust transcription at these loci compared to surrounding regions. (G-L) PAX5 ChIP tracks (G-I) and RNA-seq data (J-L) of control (KD CTRL) and EBER2 knockdown (KD EBER2) BJAB-B1 cells. Detailed analyses of these tracks will be published elsewhere. These putative enhancer regions are bound by PAX5; their localization is not altered by EBER2 depletion. These loci are bi-directionally transcribed (blue and red tracks indicate transcription of Watson and Crick strand, respectively). Levels of these enhancer RNAs do not markedly change upon EBER2 knockdown. Note that EBER2 interacts with the less abundantly transcribed strand.
Table 1.
EBER2-interacting host RNAs. Gene ID, genome coordinates, and raw read count at maximum peak height are shown.
| Gene | Locus | Raw read count at max peak height −AMT | Raw read count at max peak height +AMT |
|---|---|---|---|
| RMRP | chr9: 35657750-35658018 | 249 | 748 |
| RPPH1 | chr14: 20343071-20343411 | 128 | 271 |
| RN7SL1 | chr14: 49586580-49586878 | 1494 | 4643 |
| HFM1 (intron) | chr1: 91387198-91387598 | 607 | 853 |
| SLCO5A1 (intron) | chr8: 69690050-69690385 | 1954 | 2619 |
| PDE3A (intron) | chr12: 20551271-20551741 | 55 | 156 |
To examine the mechanism by which EBER2 initiates interaction with these enhancer RNAs, we probed for PAX5 localization at these sites. ChIP-seq data revealed PAX5 peaks at all three enhancers, which did not change in peak intensity when EBER2 was knocked down (Fig. 4G–I). Given the fact that PAX5 is found at these sites and that enhancer RNAs are chromatin-associated, EBER2 likely interacts with the nascent transcripts here as well. We next asked whether EBER2 association with the enhancer RNAs regulates their abundance, but RNA-seq data of control and EBER2 knockdown cells showed no effect on enhancer RNA levels under conditions of EBER2 depletion (Fig. 4J–L). According to the RNA-seq data, these enhancers are bi-directionally expressed, yet interestingly EBER2 interacts exclusively with the less abundant strand in all cases. In summary, EBER2 interacts with three putative host enhancer RNAs likely on chromatin, but its interaction does not markedly affect enhancer RNA levels.
Discussion
We developed an approach for identifying cellular EBER2-interacting RNAs using ASO selection coupled to deep sequencing that requires minimal bioinformatic analysis to predict the regions engaging in RNA-RNA interactions (Fig. 1A). We provide proof-of-principle for our approach by correctly identifying the known RNA-RNA interaction between EBER2 and nascent RNA from the TR regions of the EBV genome (Fig. 2). Furthermore, we extend our analysis to identify host RNAs that base pair with EBER2. Interestingly, three candidates are functional noncoding RNAs expressed by RNA polymerase III; the second category of EBER2-interacting RNAs comprises putative enhancer RNAs (Figs. 3 and 4).
Many ribonucleases with distinct enzymatic activities are widely used in molecular biology [32], of which RNase V1 is valued due to its specificity preference towards double-stranded RNA substrates. Even though it is an essential reagent in RNA biology, RNase V1 is currently not available commercially, which prompted us to isolate this enzyme de novo from lyophilized cobra venom. Our purification scheme was based on a previously reported chromatography strategy [15], but entails a modified assay to monitor for enzymatic activity (Fig. 1B). Silver-staining of our purified fractions indicated that we only partially purified RNase V1. Nevertheless, the contaminating proteins did not interfere with its enzymatic activity, and we have thus abstained from additional chromatography steps for further protein separation.
In our approach, the use of RNase V1 and its digestion at RNA duplexes circumvents the need for extensive downstream computational analyses. This achievement relies on linker ligation at the RNase V1 cut site, as the resulting junction marks the site of RNA duplex formation, and linker-adjacent sequences are determined by subsequent deep sequencing (Fig. 1A, inset). The generation of a 5′ phosphate group after RNase V1 digestion enables direct linker ligation by T4 RNA ligase without the need of any prior enzymatic treatment. As the starting RNA material is phosphatase-treated before ASO selection, the newly generated 5′ phosphate group after RNase V1-mediated digestion is the only available site to which the linker can be ligated. After deep sequencing, we therefore filtered for sequence reads that contain the linker sequence and only mapped those to the reference genome after trimming the linker sequence at their 5′ ends. Thus, the first nucleotide following the linker sequence, i.e. the first nucleotide of each mapped read, in theory marks the region of an RNA duplex between EBER2 and its interacting RNA. These preferential RNase V1 cut sites present themselves in the deep sequencing tracks as sharp edges in the peak profile (Figs. 2 and 3, arrows). Subsequent searches for RNA duplexes with EBER2 and their interacting RNAs using the RNAhybrid tool indicated these preferred cut sites to coincide with ends of helices, which are preferential cleavage sites of RNase V1.
Based on the criteria that candidate EBER2-interacting RNAs are enriched after ASO selection in the AMT-crosslinked sample and exhibit deep sequencing coverage also in the RNase V1-treated sample, we identified three functional noncoding RNAs and three putative enhancer RNAs as candidate interactors. Following reasons argue against the possibility that these RNAs are false-positives. First, as shown by the lack of sequence coverage over the TR regions of the EBV genome in the non-crosslinked control sample (Fig. 2D, -AMT), our experimental outline generates minimal amount of background. The low-level coverage in the non-crosslinked sample over the candidate gene loci are likely remnants of EBER2-interaction that persist under the denaturing conditions of ASO selection. Second, using the RNAhybrid program we were able to retrieve RNA duplexes containing regions surrounding the preferred RNase V1 cut sites, and those are found at the end of a helix adjacent to bulges within these duplexes. We did not observe clear preferential RNase V1 cleavage sites within the enhancer RNAs, which may partially be due to the low coverage over these regions. Third, the putative enhancers are expressed bi-directionally (Fig. 4J–L), yet EBER2 interacts exclusively with the less abundant strand, indicating a specific interaction rather than non-specific background. Fourth, a common denominator for all candidates is the fact that PAX5 binding is observed at these gene loci, suggesting the mechanism by which EBER2 interacts with these candidate RNAs. As EBER2 forms a complex with PAX5, we propose that EBER2 is recruited to these candidate genes by piggybacking on PAX5 and interacts with nascent, chromatin-associated transcripts. Of note, unlike the recruitment of PAX5 to the TR regions of the EBV genome, PAX5 binding at these host loci is independent of EBER2 and not mediated by its guide RNA function, as EBER2 knockdown does not affect PAX5 occupancy at these loci. Further support for interaction with nascent RNA comes from the fact that some of the RNAs localize to different subcellular compartments than the nuclear EBER2, such as 7SL, which is part of the SRP that associates with the ER-associated translocon [28]. Moreover, as the known secondary structures of MRP, H1, and 7SL RNAs are incompatible with the predicted RNA duplexes with EBER2 [25,30,33] and the nucleotides predicted to interact with EBER2 would be unavailable due to intramolecular secondary structure formation, it is likely that interaction occurs with nascent transcripts before the known conformations in the fully functional RNPs are adopted.
The steady-state levels of the EBER2-interacting RNAs are not affected by EBER2 depletion. Future studies will have to address the physiological relevance of EBER2 interaction with these cellular RNAs, and it remains to be seen whether these interactions affect one of the diverse functions of RMRP, RNase P, or SRP. Furthermore, it is possible that EBER2 interacts with additional RNAs that have escaped our detection. EBER2 contains two accessible regions for hybridization, and we only searched for RNA-RNA interactions formed by the region that also interacts with TR RNA. The second accessible site (bottom loop region) may interact with a different set of RNAs. Our approach can thus be applied not only to EBER2 using a second ASO, but also to uncover RNA-RNA interactions formed by other noncoding RNAs.
Materials and methods
Purification of RNase V1 from cobra venom
Lyophilized Naja oxiana (Caspian cobra) venom was purchased from Miami Serpentarium Laboratories. Venom was resuspended in TS0 buffer (50 mM Tris-succinate pH 5.6, 20% glycerol, 0.2 mM EDTA, 0.5 mM DTT; 0 M KCl) and fractionated using column chromatography as outlined in Fig. 1C. Based on a previously described purification scheme [15], cobra venom was purified by size-exclusion and ion-exchange chromatography: Total venom was first separated over a Superose 6 column by eluting with 20 ml TS buffer containing 200 mM KCl and collecting 0.25 ml fractions. These were assayed for double-strand RNA-specific activity (see below), which indicated that RNase V1 eluted between 16.75 ml and 18.0 ml elution volume. Based on the molecular weight protein standards, this elution volume corresponds to a molecular weight of 22–46 kDa. Subsequently, active fractions were applied to a MonoS column, eluting fractions by applying a linear gradient from 0.1 M to 0.8 M KCl. Double-strand specific activity eluted with 0.21–0.28 M salt. An additional purification step using hydroxyapatite column was included to further separate enzymatic activity towards double-stranded RNA from single-stranded RNA specificity. For this, MonoS fractions harboring double-strand RNA-specific activity were pooled and dialysed in BP10 buffer (5 mM HEPES pH 5.6, 20% glycerol, 0.04 M KCl, 0.01 mM CaCl2, 0.01% Triton X-100, 1 mM DTT; 10 mM potassium phosphate), applied to a hydroxyapatite column, and eluted with a linear gradient from 0.1 to 0.8 M potassium phosphate. Double-strand specific activity eluted with 0.53–0.60 M phosphate. Further fractionation using heparin or phenyl column did not result in improved separation of double-strand from single-strand RNA-specific activity.
To assay for double-strand RNA-specific activity, a shRNA (5′- GGGCUUGUCGGGAGCGCCACCCUCUGCUUCGGCAGAGGGUGGCGCUCCCGACAAGCCC-3′; the 4 nt-loop is underlined and contains pyrimidines that can be cleaved by RNase A) was in vitro transcribed with T7 polymerase, using as a template two DNA oligonucleotides that were annealed to each other (sequences were: 5′-GTAGCTAATACGACTCACTATAGGGCTTGTCGGGAGCGCCACCCTCTGCTTCGGCAGAGGGTGGCGCTCCCGACAAGCCC-3′ and 5′- GGGCTTGTCGGGAGCGCCACCCTCTGCCGAAGCAGAGGGTGGCGCTCCCGACAAGCCCTATAGTGAGTCGTATTAGCTAC-3′; T7 promoter sequence is underlined). The shRNA was CIP-treated, purified by phenol-chloroform extraction, and then PNK-treated in the presence of γ-32P-ATP. Instead of 5′-end labeling, body-labeled shRNA substrates were also generated using Ambion’s MAXIscript T7 Transcription Kit in the presence of α-32P-CTP to distinguish phosphatase activity from double-strand RNA-specific activity. Both substrates yielded identical results. Digestions were set up in a 20 µl-reaction containing 50 ng of radiolabeled shRNA, 5 µl of each fraction dialysed against TS100, and 2 µl of 10x V1 Digestion Buffer (100 mM Tris pH 7.2, 50 mM MgCl2, 1 M NaCl, 50 mM DTT). Reactions were incubated for 15 min at 37°C, followed by phenol-chloroform extraction of RNA, and subsequently resolved on a 12% native polyacrylamide gel at 200 V for 45 min. Gels were dried and exposed to a phosphor imager screen.
To generate a substrate for single-strand RNA-specific activity, poly(C) (Sigma P4903) was resuspended in 40 mM Tris-acetate pH 8.3, 100 mM potassium acetate, 30 mM magnesium acetate, and incubated 10 min at 95°C to fragment the polymer. The reaction was resolved on a denaturing polyacrylamide gel and poly(C) chains of ~100 nucleotides were isolated by gel-purification and radiolabeled by PNK-treatment. The same digestion conditions as described above were used for single-strand RNA substrate.
To confirm the generation of a phosphate group at the 5′ end and a hydroxyl group at the 3′ end after cleavage with purified RNase V1, oligonucleotide RL3 (5′-OH-GUGUCAGUCACUUCCAGCGG-Puromycin-3′)was radiolabeled by PNK-treatment and annealed to oligonucleotide V1_Anneal containing a 3′ amino modifier (5′-GCUGGA -/3AmMO-3′; complementary region in RL3 is underlined). This partially complementary/doubled-stranded substrate (1 nmol) was digested with indicated RNase V1 fractions in a 20 µl-reaction as stated above, and RNA was isolated by phenol-chloroform extraction. RNA was resuspended in 14.5 µl H2O and subjected to T4 RNA Ligase reaction by adding 1 µl of 20 µM 5′-phosporylated RL3 (5′-P -GUGUCAGUCACUUCCAGCGG-Puromycin-3′), 2 µl 10× T4 Ligase Buffer, 2 µl BSA, 0.5 µl T4 RNA Ligase (ThermoFisher), and incubated overnight at 16°C. Reactions were ethanol precipitated and resolved on a 12% denaturing polyacrylamide gel.
Selection of EBER2-interacting RNAs using ASO
Intact EBV-positive BJAB-B1 lymphoma cells (5× 106 cells) were incubated in 1 ml culture medium containing 50 µg/ml AMT (aminomethyl trioxsalen) or DMSO (control) for 5 min at 37°C. Cells were transferred on ice and irradiated with 365 nm UV light for 30 min using a hand-held UV illuminator from a distance of 15 cm and protected by a 2 mm-thick glass plate. Cells were washed once with ice-cold PBS and resuspended in 100 µl of PBS containing 0.5% SDS, 10 mM EDTA, 20 µg Proteinase K, and incubated 30 min at 50°C, followed by RNA isolation using Trizol and calf intestinal phosphatase-treatment.
Biotinylated DNA ASO against EBER2 (5′-CCTGACTTGCAAATGCTCTAGGCG/3BioTEG−3′, complementary to nucleotides 101–124) were complexed with MyOne Streptavidin C1 Dynabeads (ThermoFisher). 50 µg of total RNA (with or without AMT-mediated crosslinking) were resuspended in 100 µl TE buffer, incubated 3 min at 95°C to disrupt secondary structures, and transferred on ice. 100 µl H2O, 100 µl Denaturant buffer (100 mM HEPES pH 7.5, 8 M urea, 200 mM NaCl, 2% SDS), and 300 µl 2x Hybridization buffer (1.12 M urea, 1.5 M NaCl, 10x Denhardt’s solution, 10 mM EDTA) including 5 µl RNasin Plus (Promega) were added together with 50 µl of EBER2 ASO-beads and incubated for 4 h at RT on a rotator. Beads were washed twice with Wash buffer (10 mM HEPES pH 7.5, 250 mM NaCl, 2 mM EDTA, 1 mM EGTA, 0.1% N-Lauroylsarcosine, 0.2% SDS), twice with PNK buffer (50 mM Tris pH 7.4, 10 mM MgCl2, 0.5% NP40), followed by on-bead CIP-treatment for 20 min at 37°C. Beads were washed once with Wash buffer, and RNA was eluted by adding 200 µl TEACl buffer (10 mM Tris pH 7.4, 2.4 M TEACl, 0.05% Tween-20) and incubating the beads for 5 min at 40°C. Supernatant was collected, RNA was isolated using Trizol, resuspended in 150 µl of Reverse Crosslink Buffer (10 mM Tris pH 7.0, 50% formamide, 150 mM NaCl) and irradiated for 10 min with 254 nm UV light on ice, followed by phenol-chloroform extraction of RNA. Selected EBER2-interacting RNAs from the -AMT and +AMT-sample were then converted into an Illumina-compatible deep sequencing library using the SMARTer Stranded RNA-Seq Kit (Clontech).
To generate a deep sequencing library from the ‘+AMT; +RNase V1’ sample, ASO-selected RNAs from AMT-crosslinked total RNA were resuspended in 15 µl H2O and RNase V1-digested by incubating for 15 min at 37°C after adding 5 µl 10x V1 Digestion Buffer, 25 µl 4 M urea, and 5 µl of RNase V1-containing hydroxyapatite fraction. RNase V1 activity was active in the presence of 2 M urea, which was added to disrupt weak RNA duplex formation and foster the preferential digestion of AMT-crosslinked duplexes. 150 µl of Reverse Crosslink Buffer was added, and reaction was irradiated for 10 min with 254 nm UV light on ice, followed by phenol-chloroform extraction of RNA. Precipitated RNA was resuspended in 6.5 µl H2O and incubated overnight at 16°C after adding 1 µl 10× T4 RNA Ligase Buffer, 1 µl BSA, 1 µl of 20 µM RL5 linker (5′-OH-AGGGAGGACGAUGCGG-OH-3′), and 0.5 µl T4 RNA Ligase. The junction of RL5 to the 5′ end of the ligated RNA indicates the RNase V1 cleavage site and thus the region of RNA-RNA interaction. Reaction was then DNase-treated by adding 79 µl H2O, 11 µl 10× DNase Buffer, 5 µl RNasin Plus, 5 µl RQ1 DNase (Promega) and incubating for 20 min at 37°C. Non-ligated RL5 was removed by adding 198 µl Agencourt RNAClean XP beads (Beckman Coulter) to the reaction following manufacturer’s instructions. The cleaned-up RNA was converted into an Illumina-compatible deep sequencing library using the SMARTer Stranded RNA-Seq Kit. Deep sequencing was performed on a NextSeq 500 system with a 300 cycle paired-end run. For the ‘-AMT’, ‘+AMT’, and ‘+AMT; RNase V1’ samples 3.0, 6.3, and 8.5 million reads, respectively, were obtained, of which 23.1%, 30.9%, and 1.92%, respectively, mapped to both the EBV and human genome.
To confirm enrichment of EBER2-interacting candidate RNAs after AMT-treatment and ASO selection by qRT-PCR, following primer pairs were used: RMRP 5′-AGGACTCTGTTCCTCCCCTTTCCGCCTA-3′ and 5′-TGGAGTGGGAAGCGGGGAATGTCTA-3′; RPPH1 5′-GCCGGAGCTTGGAACAGA-3′ and 5′-AATGGGCGGAGGAGAGTAGTCT-3′; RN7SL 5′-ATCGGGTGTCCGCACTAAGTT-3′ and 5′-CAGCACGGGAGTTTTGACCT-3′ [34–36]. Primer pairs for EBER1 and EBER2 have been described [5]. Selected RNA after reverse crosslinking with UV irradiation as described above was subjected to cDNA synthesis followed by real-time PCR analysis.
Identification of EBER2-interacting host RNAs
Deep sequencing data were deposited in the Sequence Read Archive under accession no. SRP136709. Sequence reads were paired using PEAR software [37] and then mapped to the EBV reference genome (NCBI accession number DQ279927.1) or the hg38 human reference genome using Novoalign or TopHat2 of the Tuxedo Suite [38]. The Cufflinks program of the Tuxedo Suite was then used to calculate FPKM values and identify RNAs that are enriched in the AMT-crosslinked sample over control [18]. To locate transcripts enriched in the AMT-crosslinked sample that lie outside of exons, such as enhancer RNAs, we sorted by the raw read count over the maximum peak height to identify genomic regions where sequencing coverage is higher in one sample over the other.
To map the RNase V1 cut sites of the AMT-crosslinked sample, the deep sequencing reads were collapsed for PCR duplicates and scanned for the presence of RL5 sequence. Only those reads were retained for genome alignment that contained the linker sequence in their sequence read, which was trimmed prior to mapping. Thus, the start site of each read indicates a duplex forming region.
To predict RNA duplexes between EBER2 and putative interacting RNAs, the RNAhybrid tool [29] was used to search for RNA-RNA interactions between the top stem-loop region of EBER2 and the functional noncoding host RNAs MRP, H1, and 7SL RNAs.
PAX5 ChIP-seq and RNA-seq following EBER2 knockdown
Knockdown of EBER2 and PAX5 ChIP were performed as described [5]. Immunoprecipitated material from PAX5 ChIP experiments and isolated RNAs from control and EBER2-depleted cells were subjected to Illumina next-generation sequencing; detailed analyses of these data will be published elsewhere.
Acknowledgments
We would like to thank Morgan Coren and Sarah Haralam for technical assistance.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Fok V, Friend K, Steitz JA.. Epstein-Barr virus noncoding RNAs are confined to the nucleus, whereas their partner, the human La protein, undergoes nucleocytoplasmic shuttling. J Cell Biol. 2006. May 8;173(3):319–325. PubMed PMID: 16682524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Howe JG, Steitz JA. Localization of Epstein-Barr virus-encoded small RNAs by in situ hybridization. Proc Natl Acad Sci USA. 1986. December;83(23):9006–9010. PubMed PMID: 3024161; PubMed Central PMCID: PMCPMC387063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Moss WN, Steitz JA. Genome-wide analyses of Epstein-Barr virus reveal conserved RNA structures and a novel stable intronic sequence RNA. BMC Genomics. 2013. August 09;14:543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lee N, Pimienta G, Steitz JA. AUF1/hnRNP D is a novel protein partner of the EBER1 noncoding RNA of Epstein-Barr virus. RNA. 2012. November;18(11):2073–2082. PubMed PMID: 23012480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lee N, Moss WN, Yario TA, et al. EBV noncoding RNA binds nascent RNA to drive host PAX5 to viral DNA. Cell. 2015. February 12;160(4):607–618. PubMed PMID: 25662012; PubMed Central PMCID: PMCPMC4329084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lee N, Yario TA, Gao JS, et al. EBV noncoding RNA EBER2 interacts with host RNA-binding proteins to regulate viral gene expression. Proc Natl Acad Sci U S A. 2016. March 7 PubMed PMID: 26951683 DOI: 10.1073/pnas.1601773113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sugden B, Phelps M, Domoradzki J. Epstein-Barr virus DNA is amplified in transformed lymphocytes. J Virol. 1979. September;31(3):590–595. PubMed PMID: 229241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Engreitz J, Lander ES, Guttman M. RNA antisense purification (RAP) for mapping RNA interactions with chromatin. Methods Mol Biol. 2015;1262:183–197. PubMed PMID: 25555582. [DOI] [PubMed] [Google Scholar]
- [9].Helwak A, Kudla G, Dudnakova T, et al. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell. 2013. April 25;153(3):654–665. PubMed PMID: 23622248; PubMed Central PMCID: PMCPMC3650559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sugimoto Y, Vigilante A, Darbo E, et al. hiCLIP reveals the in vivo atlas of mRNA secondary structures recognized by Staufen 1. Nature. 2015. March 26;519(7544):491–494. PubMed PMID: 25799984; PubMed Central PMCID: PMCPMC4376666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gorbea C, Mosbruger T, Cazalla D. A viral Sm-class RNA base-pairs with mRNAs and recruits microRNAs to inhibit apoptosis. Nature. 2017. October 12;550(7675):275–279. . PubMed PMID: 28976967; PubMed Central PMCID: PMCPMC5864290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lu Z, Zhang QC, Lee B, et al. RNA duplex map in living cells reveals higher-order transcriptome structure. Cell. 2016. May 19;165(5):1267–1279. PubMed PMID: 27180905; PubMed Central PMCID: PMCPMC5029792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sharma E, Sterne-Weiler T, O’Hanlon D, et al. Global mapping of human RNA-RNA interactions. Mol Cell. 2016. May 19;62(4):618–626. PubMed PMID: 27184080. [DOI] [PubMed] [Google Scholar]
- [14].Aw JG, Shen Y, Wilm A, et al. In vivo mapping of eukaryotic RNA interactomes reveals principles of higher-order organization and regulation. Mol Cell. 2016. May 19;62(4):603–617. PubMed PMID: 27184079. [DOI] [PubMed] [Google Scholar]
- [15].Lockard RE, Kumar A. Mapping tRNA structure in solution using double-strand-specific ribonuclease V1 from cobra venom. Nucleic Acids Res. 1981. October 10;9(19):5125–5140. PubMed PMID: 7031604; PubMed Central PMCID: PMCPMC327503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kertesz M, Wan Y, Mazor E, et al. Genome-wide measurement of RNA secondary structure in yeast. Nature. 2010. September 2;467(7311):103–107. PubMed PMID: 20811459; PubMed Central PMCID: PMCPMC3847670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cimino GD, Gamper HB, Isaacs ST, et al. Psoralens as photoactive probes of nucleic acid structure and function: organic chemistry, photochemistry, and biochemistry. Annu Rev Biochem. 1985;54:1151–1193. PubMed PMID: 2411210. [DOI] [PubMed] [Google Scholar]
- [18].Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012. March 1;7(3):562–578. PubMed PMID: 22383036; PubMed Central PMCID: PMCPMC3334321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Baer M, Nilsen TW, Costigan C, et al. Structure and transcription of a human gene for H1 RNA, the RNA component of human RNase P. Nucleic Acids Res. 1990. January 11;18(1):97–103. PubMed PMID: 2308839; PubMed Central PMCID: PMCPMC330208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Elder JT, Pan J, Duncan CH, et al. Transcriptional analysis of interspersed repetitive polymerase III transcription units in human DNA. Nucleic Acids Res. 1981. March 11;9(5):1171–1189. PubMed PMID: 7232214; PubMed Central PMCID: PMCPMC326744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yuan Y, Reddy R. 5ʹ flanking sequences of human MRP/7-2 RNA gene are required and sufficient for the transcription by RNA polymerase III. Biochim Biophys Acta. 1991. May 2;1089(1):33–39. PubMed PMID: 1709054. [DOI] [PubMed] [Google Scholar]
- [22].Schmitt ME, Clayton DA. Nuclear RNase MRP is required for correct processing of pre-5.8S rRNA in saccharomyces cerevisiae. Mol Cell Biol. 1993. December;13(12):7935–7941. PubMed PMID: 8247008; PubMed Central PMCID: PMCPMC364865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Stohl LL, Clayton DA. Saccharomyces cerevisiae contains an RNase MRP that cleaves at a conserved mitochondrial RNA sequence implicated in replication priming. Mol Cell Biol. 1992. June;12(6):2561–2569. PubMed PMID: 1588958; PubMed Central PMCID: PMCPMC364449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chang DD, Clayton DA. A mammalian mitochondrial RNA processing activity contains nucleus-encoded RNA. Science. 1987. March 6;235(4793):1178–1184. PubMed PMID: 2434997. [DOI] [PubMed] [Google Scholar]
- [25].Kikovska E, Svard SG, Kirsebom LA. Eukaryotic RNase P RNA mediates cleavage in the absence of protein. Proc Natl Acad Sci U S A. 2007. February 13;104(7):2062–2067. . PubMed PMID: 17284611; PubMed Central PMCID: PMCPMC1892975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Altman S. Ribonuclease P. Philos Trans R Soc Lond B Biol Sci. 2011. October 27;366(1580):2936–2941. PubMed PMID: 21930585; PubMed Central PMCID: PMCPMC3158923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wilusz JE, Freier SM, Spector DL. 3ʹ End processing of a long nuclear-retained noncoding RNA yields a tRNA-like cytoplasmic RNA. Cell. 2008. November 28;135(5):919–932. . PubMed PMID: 19041754; PubMed Central PMCID: PMCPMC2722846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Walter P, Blobel G. Signal recognition particle contains a 7S RNA essential for protein translocation across the endoplasmic reticulum. Nature. 1982. October 21;299(5885):691–698. PubMed PMID: 6181418. [DOI] [PubMed] [Google Scholar]
- [29].Rehmsmeier M, Steffen P, Hochsmann M, et al. Fast and effective prediction of microRNA/target duplexes. RNA. 2004. October;10(10):1507–1517. PubMed PMID: 15383676; PubMed Central PMCID: PMCPMC1370637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zwieb C, van Nues RW, Rosenblad MA, et al. A nomenclature for all signal recognition particle RNAs. RNA. 2005. January;11(1):7–13. PubMed PMID: 15611297; PubMed Central PMCID: PMCPMC1370686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Creyghton MP, Cheng AW, Welstead GG, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010. December 14;107(50):21931–21936. PubMed PMID: 21106759; PubMed Central PMCID: PMCPMC3003124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Rittie L, Perbal B. Enzymes used in molecular biology: a useful guide. J Cell Commun Signal. 2008. June;2(1–2):25–45. . PubMed PMID: 18766469; PubMed Central PMCID: PMCPMC2570007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Esakova O, Krasilnikov AS. Of proteins and RNA: the RNase P/MRP family. RNA. 2010. September;16(9):1725–1747. . PubMed PMID: 20627997; PubMed Central PMCID: PMCPMC2924533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Glazov EA, Zankl A, Donskoi M, et al. Whole-exome re-sequencing in a family quartet identifies POP1 mutations as the cause of a novel skeletal dysplasia. PLoS Genet. 2011. March;7(3):e1002027 PubMed PMID: 21455487; PubMed Central PMCID: PMCPMC3063761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kuramitsu M, Okuma K, Yamagishi M, et al. Identification of TL-Om1, an adult T-cell leukemia (ATL) cell line, as reference material for quantitative PCR for human T-lymphotropic virus 1. J Clin Microbiol. 2015. February;53(2):587–596. PubMed PMID: 25502533; PubMed Central PMCID: PMCPMC4298509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Galiveti CR, Rozhdestvensky TS, Brosius J, et al. Application of housekeeping npcRNAs for quantitative expression analysis of human transcriptome by real-time PCR. RNA. 2010. February;16(2):450–461. PubMed PMID: 20040593; PubMed Central PMCID: PMCPMC2811673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zhang J, Kobert K, Flouri T, et al. PEAR: a fast and accurate illumina paired-end reAd mergeR. Bioinformatics. 2014. March 1;30(5):614–620. PubMed PMID: 24142950; PubMed Central PMCID: PMCPMC3933873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kim D, Pertea G, Trapnell C, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013. April 25;14(4):R36 PubMed PMID: 23618408; PubMed Central PMCID: PMCPMC4053844. [DOI] [PMC free article] [PubMed] [Google Scholar]