

ABSTRACT
The advantages of site-specific over stochastic bioconjugation technologies include homogeneity of product, minimal perturbation of protein structure/function, and – increasingly – the ability to perform structure activity relationship studies at the conjugate level. When selecting the optimal location for site-specific payload placement, many researchers turn to in silico modeling of protein structure to identify regions predicted to offer solvent-exposed conjugatable sites while conserving protein function. Here, using the aldehyde tag as our site-specific technology platform and human IgG1 antibody as our target protein, we demonstrate the power of taking an unbiased scanning approach instead. Scanning insertion of the human formylglycine generating enzyme (FGE) recognition sequence, LCTPSR, at each of the 436 positions in the light and heavy chain antibody constant regions followed by co-expression with FGE yielded a library of antibodies bearing an aldehyde functional group ready for conjugation. Each of the variants was expressed, purified, and conjugated to a cytotoxic payload using the Hydrazinyl Iso-Pictet-Spengler ligation to generate an antibody-drug conjugate (ADC), which was analyzed in terms of conjugatability (assessed by drug-to-antibody ratio, DAR) and percent aggregate. We searched for insertion sites that could generate manufacturable ADCs, defined as those variants yielding reasonable antibody titers, DARs of ≥ 1.3, and ≥ 95% monomeric species. Through this process, we discovered 58 tag insertion sites that met these metrics, including 14 sites in the light chain, a location that had proved refractory to the placement of manufacturable tag sites using in silico modeling/rational approaches.
Keywords: Aldehyde tag, formylglycine, fGly, formylglycine generating enzyme, FGE, antibody-drug conjugate, ADC, site-specific, bioconjugation
Introduction
Since its introduction more than 10 years ago,1 the aldehyde tag has become accepted as a convenient, facile method for the site-specific introduction of a bioorthogonal chemical handle into a protein of interest. The chemical handle can be selectively ligated to a compatible linker/payload to yield a defined protein conjugate. Specifically, the aldehyde tag comprises a six amino acid sequence (LCTPSR) that serves as a recognition sequence for formylglycine generating enzyme (FGE). FGE oxidizes the cysteine (Cys) in the context of the recognition sequence to a formylglycine (fGly) residue containing an aldehyde, which serves as the bioorthogonally-reactive group. Compatible conjugation chemistries include the Hydrazinyl Iso-Pictet-Spengler (HIPS) ligation and the trapped-Knoevenagel ligation, both of which result in stable C-C bonds.2,3
The LCTPSR sequence can be introduced genetically into a protein of interest, followed by coexpression of the protein along with FGE. The conversion of Cys to fGly occurs cotranslationally, and the protein is ready for conjugation to a payload following standard protein purification methods. A number of applications have emerged for this approach, ranging from the development of therapeutic drug candidates (e.g., antibody-drug conjugates (ADCs)),4 through industrial applications (e.g., enzyme immobilization for continuous use),5–7 to the enablement of novel molecular geometries (e.g., DNA-mediated protein assembly)8 and sophisticated analytical techniques (e.g., single molecule force spectroscopy).9 The technology is scalable and robust. For example, antibodies bearing the aldehyde tag have been expressed as high as 5 g/L with fGly conversion levels of ≥ 95%.10 Furthermore, multiple aldehyde tags can be inserted into a single antibody while maintaining high titers and fGly conversion levels.
Aldehyde tag technology has been used to install an fGly chemical handle on proteins expressed in both E. coli and mammalian systems.11–13 While the technology has demonstrated utility in a wide range of specific contexts, the generality of the aldehyde tag in terms of placement within a protein has yet to be explored. The LCTPSR sequence, while short, contains features that could modify protein structure (proline) and local charge (arginine), potentially limiting the contexts for tag insertion. Payload placement can also affect biophysical properties of a conjugated protein, such as hydrophobicity, aggregation, payload stability, and in vivo pharmacodynamics/pharmacokinetics. The effect of payload placement has been most extensively explored in the context of ADCs, where a number of research groups have shown that the pharmacokinetics, efficacy, and toxicity of a therapeutic drug can vary widely depending on conjugation site.12,14–17 Therefore, we elected to use ADCs as the context for an aldehyde tag insertion screen testing the generalizability of the conjugation platform across a protein sequence.
Here, we sequentially inserted the LCTPSR sequence next to each amino acid residue in the kappa light chain and heavy chain human IgG1 constant regions. This aldehyde tag scanning project covered 106 locations in the light chain and 331 locations in the heavy chain. The tagged antibodies were expressed, purified, and conjugated to produce ADCs. We assessed each tag insertion in terms of manufacturability based on antibody titers, conjugatability, and percent monomer; a subset of conjugates was also tested for retention of antigen binding. 13% of the tag insertions produced manufacturable ADCs, demonstrating the robust and customizable nature of this technology.
Results
We began this work by scanning insertion of the aldehyde tag in the kappa light chain constant region, which contains 106 residues. The aldehyde tag, LCTPSR, was directly inserted after each residue (Figure 1). Antibodies were expressed in 6 mL cultures by transfecting three plasmids into ExpiCHO-S cells: vectors expressing the antibody light chain, the antibody heavy chain, and the human FGE. As a control for titers, antibody tagged at the C-terminus of the heavy chain (CT-tagged) was also produced. The CT-tag has been successfully paired with multiple antibody variable regions and titers up to 5 g/L have been achieved with it.10 Therefore, CT-tagged titers were used as a benchmark for acceptable expression levels in this small-scale, unoptimized transfection system.
Figure 1.

Illustration of aldehyde tag scanning insertion.
In order to carry out an unbiased scan of aldehyde tag placement throughout the antibody constant regions, we designed a scheme in which the FGE recognition sequence, LCTPSR, would be inserted next to each amino acid residue in the constant regions of the light and heavy chains. Our numbering system is sequential based on the first N-terminal residue in the constant region of each antibody chain.
All 106 light chain-tagged antibodies were transfected and purified at the same time to avoid day-to-day cell variability. IgG expression levels were quantified by ForteBio on Day 8 post-transfection. Titers ranged from 0 to 65 mg/L, with CT-tagged antibody expression at 24 mg/L. Eighty antibodies (75%) had titers above 10 mg/L (Figure 2). Seventy-two antibodies, including 70 of those with titers ≥ 10 mg/L, were purified by Protein A followed by conjugation to RED-106, a HIPS-functionalized non-cleavable linker bearing a cytotoxic maytansine payload.4 Of the antibodies that were tested in this step, 36 (50%) yielded recoverable ADCs with drug-to-antibody ratios (DARs) above 0.5. Sixteen antibodies showed DAR values of ≥ 1.3 (Figure 2). Furthermore, 14 of the 16 light chain conjugates with DARs of ≥ 1.3 were highly monomeric, ranging from < 1.0 to 5.2% high molecular weight. Complete details of the light chain results can be found in Supplemental Table 1.
Figure 2.
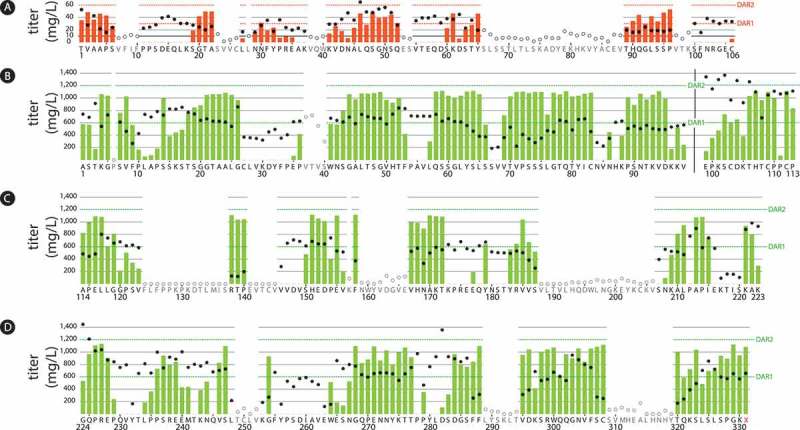
Titers and conjugation results from an aldehyde tag scan of the human kappa light chain and IgG1 heavy chain human IgG1 constant regions.
The aldehyde tag was inserted either after (light chain, Panel A) or before (heavy chain, Panels B-D) each amino acid residue in the constant regions. Tagged antibodies were expressed using a transient system, purified, and conjugated to a cytotoxic payload using a HIPS-functionalized non-cleavable linker. Titers (circles corresponding to the left y-axes) were measured by Protein A on Day 8 post-transfection. Conjugation, reported as drug-to-antibody ratio (DAR) was determined by chromatography. The maximum possible DAR value for each conjugate was 2 (one payload per tagged light or heavy chain). DAR values are indicated as red (light chain) or green (heavy chain) bars corresponding to the right y-axes as follows: Panel A, light chain constant region; Panel B, heavy chain CH1 and hinge domains to the left and right of the black vertical bar, respectively; Panel C, heavy chain CH2 domain; Panel D, heavy chain CH3 domain. In the CH3 region, the heavy chain stop codon is indicated by 331X. Antibodies that expressed poorly or did not bind well to Protein A were not purified or conjugated (open circles denote titers, no DAR values were obtained). Distinct transfection protocols were used to produce the light chain-tagged antibodies (Panel A) and the heavy chain-tagged antibodies (Panels B-D), which accounts for the marked difference in antibody titers and DAR values between the light and heavy chain datasets.
Table 1.
Summary of results from aldehyde tag scanning insertions across 436 sites in antibody constant regions.
| Antibody domain | Number of Sites Scanned | % Scanned Sites Expressed as mAbsa | % of Expressed mAbs Conjugatedb | % of High DAR Conjugatesc | % of Highly Monomeric High DAR ADCsd | Total Number of Manufacturable Sitese | % of Manufacturable Sites Per Sites Scanned |
|---|---|---|---|---|---|---|---|
| Light chain constant region | 106 | 75 | 50 | 22 | 88 | 14 | 13 |
| Heavy chain CH1 | 98 | 99 | 70 | 46 | 51 | 23 | 23 |
| Heavy chain Hinge | 15 | 100 | 87 | 40 | 0 | 0 | 0 |
| Heavy chain CH2 | 110 | 57 | 60 | 35 | 27 | 6 | 5 |
| Heavy chain CH3 | 107 | 80 | 72 | 37 | 44 | 14 | 13 |
| All constant regions (light and heavy chain) | 436 | 76 | 68 | 37 | 49 | 58 | 13 |
aTiters ≥ 10 mg/L (light chain) or ≥ 80 mg/L (heavy chain) and purifiable by Protein A.
bAnalyzable ADCs with DARs ≥ 0.5.
cADCs with DARs above 1.3 (light chain) or 1.5 (heavy chain)
dOf High DAR conjugated antibodies, % monomer above 95.
eNumber of highly monomeric, high DAR ADCs produced per number of sites scanned.
Antibody conjugatability depends on both high conversion of Cys to fGly and a favorable steric environment around the tag site. As a preliminary check to determine whether one or the other of these factors was the primary driver of our results, we used a mass spectrometric-based method18 to assess fGly conversion at five sites representing DAR values ranging from 0 to 1.5. The results showed that conjugation tracked with fGly conversion for four of the five sites (Supplemental Table 2). Only one site demonstrated appreciable conversion (56%) with little to no conjugation (DAR 0).
The effects of payload placement or linker technology on the overall hydrophobicity of ADCs are of substantial interest to the ADC field. These characteristics are thought to inform on the solvent accessibility of the linker/payload, which can influence stability. Together, ADC stability and hydrophobicity are linked to in vivo pharmacokinetic and pharmacodynamic outcomes, including ADC half-life, efficacy, and tolerability.19–21 Therefore, we determined the relative retention time (RRT) of the ADCs in our panel as measured by hydrophobic interaction chromatography (HIC), a typical measure of hydrophobicity. The RRT is calculated by dividing the retention time of the DAR 2 ADC by that of the unconjugated antibody.21 The HIC RRTs of the light chain-tagged ADCs ranged from 1.3 to 1.8, suggesting the possibility of selecting improved ADC configurations as a result of this tag screen (Supplemental Table 1).
As an additional assessment of the effects of aldehyde tag insertion and conjugation on antibody structure and function, the 14 manufacturable ADCs (defined for light chain-tagged antibodies as titers ≥ 10 mg/L, DARs ≥ 1.3, and % monomer ≥ 95%) that emerged from the light chain tag screen were tested for antigen binding. An indirect ELISA-based assay was used to compare antigen binding of the ADCs compared to the untagged, unconjugated parental antibody. In general, the binding curves were similar among the ADCs and antibody control (Supplemental Figure 1), indicating that insertion of the aldehyde tag and conjugation to the RED-106 linker/payload did not abrogate antigen recognition.
The success of the light chain scanning project encouraged us to continue scanning through the heavy chain constant regions. For the heavy chain scan, we used the same strategies and methods applied to the light chain scan with the following exception. By this point, our laboratory had developed a new transient transfection approach using a clonal Expi-CHO-S cell line stably-expressing human FGE, obviating the need for a 3-plasmid transfection and delivering improved fGly conversion efficiency compared to our previous transient system. Use of this production cell line allowed us to obtain generally higher antibody titers and better DARs in this portion of the project compared to those obtained during the light chain scanning work. Therefore, our definition of a manufacturable ADC for the heavy chain-tagged antibodies was titers ≥ 80 mg/L, DARs ≥ 1.5, and % monomer ≥ 95%. Due to the size of the heavy chain, we conducted the scanning process of the constant region in four stages N-terminus to C-terminus by domain: CH1, hinge, CH2, and CH3. For each stage, the heavy chain-tagged antibodies within a domain were transfected and purified at the same time to avoid day-to-day cell variability. IgG expression levels were quantified by ForteBio on Day 8 post-transfection. As a control for titers, untagged antibody was also produced using this new transfection method, achieving titers in the range of 600–750 mg/L.
There are 98 residues in the CH1 region. We were able to place the aldehyde tag at every position. One tagged antibody (6P) did not produce. The other 97 antibodies were produced in high titers, ranging from 188 to 913 mg/L (Figure 2). These were purified and conjugated to the RED-106 linker/payload. Of the 97 antibodies that were tested in this step, 68 (70%) yielded recoverable ADCs with DARs above 0.5. Forty-eight antibodies showed promising DAR values of ≥ 1.5 (Figure 2), with 29 of those displaying DARs of ≥ 1.75 (range 1.75 to 1.86). Of the CH1-tagged conjugates with DARs of ≥ 1.5, 25 (52%) were highly monomeric, with ≤ 5.0% high molecular weight species (Figure 3). Complete details of the heavy chain CH1 results can be found in Supplemental Table 3.
Figure 3.
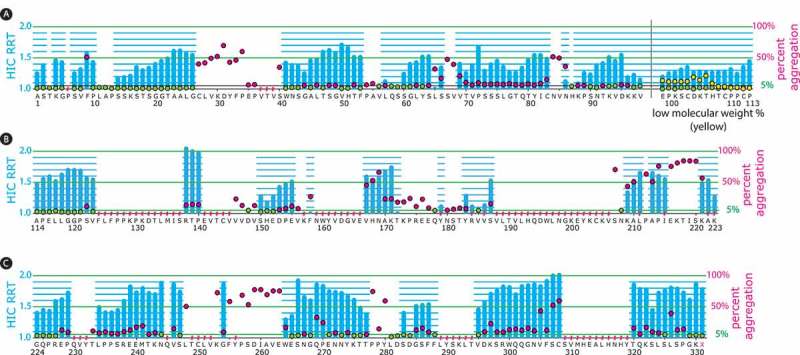
Percent ADC aggregate and HIC relative retention times from an aldehyde tag scan of the heavy chain human IgG1 constant regions.
The aldehyde tag was inserted before each amino acid residue in the heavy chain constant regions. Tagged antibodies were expressed using a transient system, purified, and ADCs were generated through conjugation to a cytotoxic payload using a HIPS-functionalized non-cleavable linker. The ADC HIC relative retention time (RRT) values, calculated by dividing the HIC retention time for the DAR 2 species by that of the unconjugated antibody, are depicted with blue bars corresponding to the left y-axes as follows: Panel A, heavy chain CH1 and hinge domains to the left and right of the black vertical bar, respectively; Panel B, heavy chain CH2 domain; Panel C, heavy chain CH3 domain. The percent aggregate (high-molecular weight species) in the ADC preparations was determined by size-exclusion chromatography (SEC) and is depicted with green or red circles corresponding to the right y-axes. Green circles indicate ADCs with ≤ 5% aggregate, which was the cut-off that we set for manufacturability. Red circles indicate ADCs with > 5% aggregate. When the aldehyde tag was placed in the hinge region, low-molecular weight species were detected (depicted with yellow circles corresponding to the right y-axis). At some tag insertion sites, we could not obtain data for the corresponding ADCs (indicated with a red hash mark above the amino acid). This typically occurred because antibody titers (as detected by Protein A) at that position were insufficient (SEC) or because the ADC resolved poorly on HIC.
There are 15 residues in the hinge region. All 15 hinge-tagged antibodies expressed well, with titers ranging from 488 to 943 mg/L (Figure 2). All but two tag sites yielded recoverable ADCs with DARs of at least 0.5, with six variants showing DARs of ≥ 1.5 (Figure 2). However, it appeared that inserting the aldehyde tag in this region disrupted the association between the antibody light and heavy chains. We found a significant amount of low-molecular weight material in the six highly-conjugated ADCs (Figure 3), rendering the constructs unmanufacturable. Complete details of the heavy chain hinge results can be found in Supplemental Table 4.
The CH2 region contains 110 residues. We were able to place the aldehyde tag at every position in this region. As assessed by Protein A binding, antibody titers ranged from 0 to 982 mg/L. However, because the CH2 and CH3 domains form the binding interface with Protein A,22 we anticipated that some antibody variants carrying aldehyde tags in these regions would be expressed but undetected by our Protein A-coated ForteBio sensor used for titer measurements. Accordingly, scanned regions of the CH2 domain where antibody titers were low as measured by Protein A (e.g., 188V through 206V) were tested for antibody titers by ForteBio using a Protein L-coated sensor. Protein L recognizes the kappa light chain,23 and thus binding would likely not be affected by tag insertions into the CH2 region. As anticipated, the Protein L ForteBio data indicated that CH2-tagged antibodies undetectable with Protein A were expressed in reasonable concentrations (e.g., the titers of variants 188V through 206V ranged from 146 to 253 mg/L). In fact, pairing Protein A and Protein L titer assessments showed that all CH2-tagged antibodies were expressed at levels potentially high enough for development (data not shown). However, acknowledging the prevalent use of Protein A purification in antibody manufacturing, we elected to classify those antibodies that did not bind to Protein A as unmanufacturable.
There were 63 antibodies from the CH2 region (57% of scanned sites) with Protein A-measured titers ≥ 90 mg/L (Figure 2); these were purified and conjugated to RED-106. Of the 63 antibodies carried forward, 38 (60%) yielded recoverable ADCs with DARs above 0.5. Twenty-two of the antibodies showed promising conjugation, with DARs of ≥ 1.5 (Figure 2), with 13 of those displaying crude DARs of ≥ 1.75 (range 1.75 to 1.86). Of the CH2-tagged conjugates with DARs of ≥ 1.5, 6 (27%) were highly monomeric, with ≤ 5.0% high molecular weight species (Figure 3). Complete details of the heavy chain CH2 results can be found in Supplemental Table 5.
There are 108 residues in the heavy chain CH3 region. We were able to place the aldehyde tag at every position except 251L. The other 107 constructs were transfected and yielded antibody titers ranging from 100 to 1448 mg/L as measured by Protein A or Protein L. Ninety tagged antibodies were produced with Protein A titers higher than 80 mg/L (Figure 2). In four cases, the protein recoveries were too low for subsequent conjugation. The remaining 86 were conjugated to RED-106. Sixty-two antibodies (72%) yielded recoverable ADCs with DARs above 0.5. Thirty-four antibodies conjugated well, with DARs of ≥ 1.5 (Figure 2), with 14 of those displaying crude DARs of ≥ 1.75 (range 1.75 to 1.89). Of CH3-tagged conjugates with DARs of ≥ 1.5, 15 ADCs (44%) were highly-monomeric, with ≤ 5.0% high molecular weight species (Figure 3). Complete details of the heavy chain CH3 results can be found in Supplemental Table 6.
Regarding the effect of tag placement in the heavy chain as it relates to ADC hydrophobicity, we assessed the HIC RRTs of 182 conjugatable variants (Figure 3). The values ranged from a low of 1 (where no difference in HIC retention was observed in the DAR 2 species relative to the unconjugated antibody) to a high of 2 (where the HIC retention time was doubled for the DAR 2 species relative to the unconjugated antibody). In general, current trends in the ADC field suggest that constructs with low RRTs offer superior performance compared to more hydrophobic constructs.19–21 The aldehyde tag scan approach allowed us to identify 43 heavy chain-tagged RED-106 ADC variants (23% of conjugatable tag insertions) displaying RRT values of ≤ 1.3. Of these, 21 had RRT values of ≤ 1.2, and 12 had values of 1, where no change in retention time was detected upon conjugation. Our tagged-antibody panels thus represent a resource for the future exploration of best-in-class ADC compositions.
Although this tag scanning work grew out of our initial failures using rational design to produce light chain-tagged ADCs, we recognized that the large dataset generated here provided an opportunity to revisit the possibility of structure-based predictions. Thus, we performed a bioinformatic analysis of our dataset to look for trends relating tag site with bioconjugatability. Conjugation results were plotted according to tag insertion site near each of the 20 amino acid residues (Supplemental Figure 2). Then, the effects of amino acid proximity on bioconjugation were assessed with GraphPad Prism software by using a one sample t-test comparing the mean bioconjugation levels to 50% as the null hypothesis. Only insertions next to glycine and threonine residues gave statistically significant differences, in both cases improvements, in conjugatability (P < 0.05, two tailed).
Discussion
In our previous work,12 we described a panel of antibodies containing the aldehyde tag at eight different locations within the IgG1 heavy and light chain constant regions. These sites were selected based on their predicted solvent accessibility, which we expected would improve conjugation. Our goal was to generate high titer antibodies with efficient conjugation. Although we achieved this goal with two heavy chain tags, at that time our best conjugating light chain-tagged antibody returned low titers compared to untagged antibodies and was deemed unmanufacturable as a result. However, because other groups reported improved pharmacokinetics or in vivo efficacy by using ADCs conjugated to the light chain,15,16 we refocused our efforts on identifying useful tag insertions in that region. As a next step, we tried computer-modeling approaches to predict optimal light chain tag site locations based on solvent accessibility as assessed by IgG crystal structures. During that project, we generated > 30 additional light chain-tagged antibodies; however, we failed to find a variant that yielded both high titers and high DARs. It was at that point that we decided to change tactics and apply an unbiased, systematic approach to the problem by inserting the aldehyde tag sequence after every residue of the light chain constant region. The stunning success of this approach, which returned 14 manufacturable ADC variants in just a few months, prompted us to continue scanning placement of the aldehyde tag through the entire IgG1 constant region. Overall, we created 58 new antibody variants that support manufacturable ADC production. These tagged variants comprise a high-potential library that in future studies can be mined for constructs displaying specific features, e.g., stability with a particular linker system or payload; minimized ADC hydrophobicity.
With respect to the utility of predictive vs. empirical payload placement, our experience appears to dovetail with that of other groups, including groups from Genentech and Pfizer, who applied distinct approaches to generating site-specifically conjugated ADCs. This probably reflects both the difficulties of predicting the effects of mutations – especially insertions and deletions – on protein folding, structure, and function,24–27 as well as the fact that there are not yet sufficient data relating ADC biophysical characteristics with functional outcomes.
Early work from Genentech that employed modeling to rationally select three engineered cysteine conjugation sites with differing local structural environments revealed that ADCs derived from the three variants conjugated similarly and performed similarly in vitro, but yielded very different in vivo efficacies.15,24–27 The distinct outcomes were not predicted by the structural modeling, but rather were discovered empirically. More recent work from Genentech used a scanning approach similar to ours, whereby a cysteine was substituted at every residue in an anti-HER2 IgG1.28 Once conjugated, these constructs generated a large ADC library that was interrogated in order to identify those with improved stability relative to conventional cysteine conjugates. Then, the team used structural information to evaluate whether solvent accessibility around the conjugation sites contributed to stability; no significant correlation was found between the two features.
Pfizer took an intermediate approach between unbiased scanning and rational modeling when investigating conjugation sites using transglutaminase and a four amino acid “glutamine tag” target sequence.16 They tested the glutamine tag at ~90 sites identified as surface accessible within the constant domains of an IgG1. Of these sites, 12 yielded highly conjugated ADCs with good biophysical properties, translating to a success rate of ~13%, similar to that of our own unbiased scanning method (Table 1). Thus, it appears that, for the moment, unbiased scanning offers faster and higher returns on investment compared to targeted insertions based on protein modeling information. As the ADC field progresses, our collective ability to rationally predict function from structure may improve.
Materials and Methods
Light and heavy chain expression vector backbone preparation
Catalent plasmid pRW449 (encoding human antibody #1 kappa light chain) was used as the starting material for generating the light chain vector backbone. pRW449 was digested with BamH1-HF (NEB) and DraIII-HF (NEB) to remove the wild type human kappa light chain constant region. The digested plasmid DNA was purified by 1% agarose gel electrophoresis and QIAquick gel extraction kit (QIAGEN). The purified DNA was used as a vector backbone for cloning the human IgG1 kappa light chain constant region containing the aldehyde tag at different positions.
Catalent plasmid pRW1064 (encoding human antibody #1 IgG1 heavy chain) was used as the starting material for the generation of the heavy chain vector backbone. pRW1064 was digested with KpnI-HF (NEB) and DraIII-HF to remove the wild-type human IgG1 heavy chain constant region. The digested plasmid DNA was purified by 1% agarose gel electrophoresis and QIAquick gel extraction kit. The purified DNA was used as a vector backbone for cloning human IgG1 heavy chain constant regions containing the aldehyde tag at different positions.
Primer design for generating antibody constant regions bearing the aldehyde tag at different positions
Two PCR fragments (5’ and 3’), which introduced the aldehyde tag, were generated for each position along the light chain constant region. The 5’ part of the kappa light chain constant region was generated by using a forward PCR primer based on the forward vector backbone sequence with a BamH1 site (5’-TCAAACGTGAGTAGAATTTAAACTT-3’) and variant reverse aldehyde tag PCR primers (reverse aldehyde tag DNA sequence + reverse vector backbone sequence). The 3’ part of the kappa light chain constant region was generated by using variant forward aldehyde tag PCR primers (forward aldehyde tag DNA sequence + forward vector backbone sequence) and a reverse PCR primer based on the reverse vector backbone sequence with a DraIII site (5’-AAAGGGCGAAAAACCGTCTATCAGG-3’).
The approach used to design the light chain primers was also used to design the heavy chain primers. The 5’ part of the IgG1 heavy chain constant region was generated by using a forward PCR primer based on the forward vector backbone sequence with a KpnI site (5’-GGGTCGCATACATTAGTAGTGGTGGTG-3’) and variant reverse PCR primers (reverse aldehyde tag DNA sequence + reverse vector backbone sequence). The 3’ part of IgG1 heavy chain constant region was generated by using variant forward primers (forward aldehyde tag DNA sequence + forward vector backbone sequence) and a reverse PCR primer based on the reverse vector backbone sequence with a DraIII site (5’-AAAACCGTCTATCAGGGCGATGGCCCA-3’).
Integrated DNA Technologies (IDT) synthesized all of the primers.
PCR amplification
Plasmid pRW449 was used as a PCR template for kappa light chain constant region amplification. Plasmid pRW1064 was used as a PCR template for IgG1 heavy chain constant region amplification.
Q5 High-Fidelity DNA Polymerase (New England BioLabs) was used as DNA polymerase for PCR amplification. All PCR fragments were amplified by pre-heating at 98°C for 1 minute followed by 30 cycles of 98°C for 10 seconds, 60°C for 10 seconds, 72°C for 20 seconds concluding with a 1 minute final extension step at 72°C.
DNA assembly for making light chain and heavy chain plasmid DNA
To assemble the two PCR fragments and vector backbone, we added 100 ng vector backbone, 0.5 μL of the 5’ part of PCR fragment, 0.5 μL of the 3’ part of PCR fragment, 5 μL of 2X Gibson Assembly Master Mix (New England BioLabs) and water to a final volume of 10 μL in a 96-well PCR plate. We incubated the plate at 50°C for 1 hour and transformed 2 μL of the assembled product into Top10 chemically competent cells. After plating, we picked a single colony and sent it for sequencing verification (Sequetech Corporation, Mountain View, CA).
Plasmid DNA isolation and IgG expression
After sequence verification, a QIAprep Spin Miniprep kit (QIAGEN) was used to isolate the light chain and heavy chain plasmid DNA.
An ExpiFectamine TM CHO Transfection Kit (Thermo Fisher) was used for transfection following the manufacturer’s protocol. Six mL of cells were aliquotted into a 50 mL mini bioreactor (Corning) and 100 mM CuSO4 (Sigma) was added to a final concentration of 100 µM. For the light chain scanning, 3 plasmids were cotransfected encoding the antibody heavy chain: antibody light chain: and human FGE. The ratio of these plasmids was 1.2: 1.8: 1, respectively. ExiCHO-S cells (Thermo Fisher) were used as a host cell for expression of the light chain-tagged antibodies.
For the heavy chain scanning, 2 plasmids were cotransfected encoding the antibody heavy chain: and antibody light chain. The ratio of these plasmids was 2: 3, respectively. An in-house derived ExpiCHO-S cell line stably-expressing human FGE was used as a host cell for expression of the heavy chain-tagged antibodies.
Titer assessment
Expression cultures were harvested by centrifugation at day 8 post-transfection. IgG was quantified using a BLItz system (ForteBio) with a Protein A Biosensor (ForteBio). The expression of some CH2- or CH3-tagged antibodies was also assessed using the same system equipped with a Protein L Biosensor.
IgG purification
Expression culture supernatant was incubated with preequilibrated MabSelect SuRe Protein A resin (Millipore Sigma) at room temperature with rocking for 1–2 h. The resin was washed with 5 mL of 20 mM sodium citrate pH 7.2, 150 mM NaCl. IgG was eluted with 4 mL 20 mM sodium acetate pH 3.5, 50 mM NaCl and was neutralized with 444 µL of 250 mM triethanolamine. Purified IgG was concentrated by using an Amicon Ultra-4 Centrifugal Filter Ultracel-30K (Millipore).
Conjugation and analysis of conjugates
Aldehyde-tagged antibodies were conjugated using HIPS chemistry to the RED-106 non-cleavable maytansine payload4 for 24 h at 37°C in 50 mM sodium citrate, pH 5.5, 50 mM NaCl in the presence of 2.5% DMA. Residual free payload was removed using Amicon centrifugal filters (EMD Millipore). Maytansine ADCs were analyzed by hydrophobic interaction chromatography (HIC) to determine DAR and retention time (Tosoh column 14,947; mobile phase A: 1.5 M ammonium sulfate, 25 mM sodium phosphate, pH 7.0; mobile phase B: 25% isopropanol, 18.75 mM sodium phosphate, pH 7.0). When the separation between DAR 0, DAR 1, and DAR 2 peaks was insufficient to calculate DAR, samples were analyzed by reversed-phase chromatography of dithiothreitol-reduced samples (Agilent PLRP-S #PL1912-1802; mobile phase A: 0.1% trifluoroacetic acid (TFA) and mobile phase B: 0.1% TFA in CH3CN). All ADCs were also analyzed by size exclusion chromatography to determine the % aggregate of the final product (Tosoh column 08541; mobile phase: 300 mM Nacl, 25 mM NaPO4, pH 6.8).
ELISA-based detection of antigen binding
Eleven 1:2 serial dilutions (ranging from 200 to 0.2 ng/mL) of untagged or aldehyde tagged antibodies were captured with HIS-tagged recombinant human antigen (Sino Biological; coated at 1 µg/mL) and detected with a horseradish peroxidase-conjugated goat anti-human IgG Fcg specific secondary antibody (Jackson ImmunoResearch #709–035-098). Bound secondary antibody was detected using Ultra TMB One-Step ELISA substrate (Thermo Fisher). After quenching the reaction with sulfuric acid, signal absorbances at 450 nm were read on a Molecular Devices Spectra Max M5 plate reader equipped with SoftMax Pro software. Data were analyzed using GraphPad Prism and Microsoft Excel software.
Disclosure statement
All authors are employees of Catalent Biologics.
Abbreviations
- ADC
antibody-drug conjugate
- CT
C-terminal
- Cys
cysteine
- DAR
drug-to-antibody ratio
- ELISA
enzyme-linked immunosorbent assay
- FGE
formylglycine-generating enzyme
- fGly
formylglycine
- HIC
hydrophobic interaction chromatography
- HIPS
Hydrazinyl Iso-Pictet-Spengler
- IgG1
immunoglobulin G1
- RRT
relative retention time
Supplemental Material
Supplemental data for this article can be access on the publisher’s website.
References
- 1.Carrico IS, Carlson BL, Bertozzi CR.. Introducing genetically encoded aldehydes into proteins. Nat Chem Biol. 2007;3:321–322. doi: 10.1038/nchembio878. [DOI] [PubMed] [Google Scholar]
- 2.Agarwal P, Kudirka R, Albers AE, Barfield RM, de Hart GW, Drake PM, Jones LC, Hydrazino-Pictet-Spengler RD. Ligation as a biocompatible method for the generation of stable protein conjugates. Bioconjug. 2013;24:846–851. doi: 10.1021/bc400042a. [DOI] [PubMed] [Google Scholar]
- 3.Kudirka R, Barfield RM, McFarland J, Albers AE, de Hart GW, Drake PM, Holder PG, Banas S, Jones LC, Garofalo AW, et al. Generating site-specifically modified proteins via a versatile and stable nucleophilic carbon ligation. Chem Biol. 2015;22:293–298. doi: 10.1016/j.chembiol.2014.11.019. [DOI] [PubMed] [Google Scholar]
- 4.Drake PM, Carlson A, McFarland JM, Banas S, Barfield RM, Zmolek W, Kim YC, Huang BCB, Kudirka R, Rabuka D. CAT-02-106, a site-specifically conjugated anti-CD22 antibody bearing an MDR1-resistant maytansine payload yields excellent efficacy and safety in preclinical models. Mol Cancer Ther. 2018;17:161–168. doi: 10.1158/1535-7163.MCT-17-0776. [DOI] [PubMed] [Google Scholar]
- 5.Jian H, Wang Y, Bai Y, Li R, Site-Specific GR. Covalent immobilization of dehalogenase ST2570 catalyzed by formylglycine-generating enzymes and its application in batch and semi-continuous flow reactors. Molecules. 2016;21:895. doi: 10.3390/molecules21070895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zang B, Ren J, Xu L, Jia L. Direct site-specific immobilization of protein A via aldehyde-hydrazide conjugation. J Chromatogr B Analyt Technol Biomed Life Sci. 2016;1008:132–138. doi: 10.1016/j.jchromb.2015.11.016. [DOI] [PubMed] [Google Scholar]
- 7.Wang A, Du F, Wang F, Shen Y, Gao W, Zhang P. Convenient one-step purification and immobilization of lipase using a genetically encoded aldehyde tag. Biochem Eng J. 2013;73:86–92. doi: 10.1016/j.bej.2013.02.003. [DOI] [Google Scholar]
- 8.Liang SI, McFarland JM, Rabuka D, Gartner ZJ, Modular A. Approach for assembling aldehyde-tagged proteins on DNA scaffolds. J Am Chem Soc. 2014;136:10850–10853. doi: 10.1021/ja504711n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walder R, M-A L, van Patten WJ, Edwards DT, Greenberg JA, Adhikari A, Okoniewski SR, Sullan RMA, Rabuka D, Sousa MC, et al. Rapid characterization of a mechanically labile α-helical protein enabled by efficient site-specific bioconjugation. J Am Chem Soc. 2017;139:9867–9875. doi: 10.1021/jacs.7b02958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.York D, Baker J, Holder PG, Jones LC, Drake PM, Barfield RM, Bleck GT, Rabuka D. Generating aldehyde-tagged antibodies with high titers and high formylglycine yields by supplementing culture media with copper(II) In: BMC Biotechnology. 2016;16:23. doi: 10.1186/s12896-016-0254-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dickgiesser S, Rasche N, Nasu D, Middel S, Hörner S, Avrutina O, Diederichsen U, Kolmar H. Self-assembled hybrid aptamer-fc conjugates for targeted delivery: A modular chemoenzymatic approach. ACS Chem Biol. 2015;10:2158–2165. doi: 10.1021/acschembio.5b00315. [DOI] [PubMed] [Google Scholar]
- 12.Drake PM, Albers AE, Baker J, Banas S, Barfield RM, Bhat AS, de Hart GW, Garofalo AW, Holder P, Jones LC, et al. Aldehyde tag coupled with HIPS chemistry enables the production of ADCs conjugated site-specifically to different antibody regions with distinct in vivo efficacy and PK outcomes. Bioconjug. 2014;25:1331–1341. doi: 10.1021/bc500189z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hudak JE, Yu HH, Bertozzi CR. Protein glycoengineering enabled by the versatile synthesis of aminooxy glycans and the genetically encoded aldehyde tag. J Am Chem Soc. 2011;133:16127–16135. doi: 10.1021/ja206023e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ra K, Rm B, Jm M, Pm D, Carlson A, Banas S, Zmolek W, Aw G, Rabuka D. Site-specific tandem knoevenagel condensation–michael addition to generate antibody–drug conjugates. ACS Med Chem Lett. 2016;7:994–998. doi: 10.1021/acsmedchemlett.6b00253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen B-Q, Xu K, Liu L, Raab H, Bhakta S, Kenrick M, Parsons-Reponte KL, Tien J, Yu S-F, Mai E, et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat Biotechnol. 2012;30:184–189. doi: 10.1038/nbt.2108. [DOI] [PubMed] [Google Scholar]
- 16.Strop P, Liu S-H, Dorywalska M, Delaria K, Dushin RG, Tran -T-T, Ho W-H, Farias S, Casas MG, Abdiche Y, et al. Location matters: site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem Biol. 2013;20:161–167. doi: 10.1016/j.chembiol.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 17.Dorywalska M, Strop P, Melton-Witt JA, Hasa-Moreno A, Farias SE, Galindo Casas M, Delaria K, Lui V, Poulsen K, Loo C, et al. Effect of attachment site on stability of cleavable antibody drug conjugates In: Bioconjugate Chem. 2015;26:650–659. 150220133437002. [DOI] [PubMed] [Google Scholar]
- 18.Zmolek W, Banas S, Barfield RM, Rabuka D, Drake PM. A simple LC/MRM-MS-based method to quantify free linker-payload in antibody-drug conjugate preparations. J Chromatogr B Analyt Technol Biomed Life Sci. 2016;1032:144–148. doi: 10.1016/j.jchromb.2016.05.055. [DOI] [PubMed] [Google Scholar]
- 19.Lyon RP, Bovee TD, Doronina SO, Burke PJ, Hunter JH, Neff-LaFord HD, Jonas M, Anderson ME, Setter JR, Senter PD. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat Biotechnol. 2015;33:733–735. doi: 10.1038/nbt.3212. [DOI] [PubMed] [Google Scholar]
- 20.Puthenveetil S, He H, Loganzo F, Musto S, Teske J, Green M, Tan X, Hosselet C, Lucas J, Tumey LN, et al. Multivalent peptidic linker enables identification of preferred sites of conjugation for a potent thialanstatin antibody drug conjugate. PLoS ONE. 2017;12:e0178452. doi: 10.1371/journal.pone.0178452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tumey LN, Leverett CA, Vetelino B, Li F, Rago B, Han X, Loganzo F, Musto S, Bai G, Sukuru SCK, et al. Optimization of tubulysin antibody–drug conjugates: a case study in addressing ADC metabolism. ACS Med Chem Lett. 2016;7:977–982. doi: 10.1021/acsmedchemlett.6b00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deisenhofer J. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at. 2.9- And 2.8-A Resolution. Biochemistry. 20;1981:2361–2370. [PubMed] [Google Scholar]
- 23.Nilson BH, Solomon A, Björck L, Protein AB. L from peptostreptococcus magnus binds to the kappa light chain variable domain. J Biol Chem. 267;1992:2234–2239. [PubMed] [Google Scholar]
- 24.Ng PC, Henikoff S. Predicting the effects of amino acid substitutions on protein function. Annu Rev Genom Hum Genet. 2006;7:61–80. doi: 10.1146/annurev.genom.7.080505.115630. [DOI] [PubMed] [Google Scholar]
- 25.Kim R, Guo J-T. Systematic analysis of short internal indels and their impact on protein folding. BMC Struct Biol. 2010;10:24. doi: 10.1186/1472-6807-10-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feyfant E, Sali A, Fiser A. Modeling mutations in protein structures. Protein Sci. 2007;16:2030–2041. doi: 10.1110/(ISSN)1469-896X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi Y, Ge S, Murphy S, Jr M, Ap C. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE. 2012;7:e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohri R, Bhakta S, Fourie-O’Donohue A, Cruz-Chuh Dela J, Sp T, Cook R, Wei B, Ng C, Aw W, Ab B, et al. High-throughput cysteine scanning to identify stable antibody conjugation sites for maleimide- and disulfide-based linkers. Bioconjug. 2018;29:473–485. doi: 10.1021/acs.bioconjchem.7b00791. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.