

ABSTRACT
Animal models used to evaluate efficacies of immune checkpoint inhibitors are insufficient or inaccurate. We thus examined two xenograft models used for this purpose, with the aim of optimizing them. One method involves the use of peripheral blood mononuclear cells and cell line-derived xenografts (PBMCs-CDX model). For this model, we implanted human lung cancer cells into NOD-scid-IL2Rg−/− (NSI) mice, followed by injection of human PBMCs. The second method involves the use of hematopoietic stem and progenitor cells and CDX (HSPCs-CDX model). For this model, we first reconstituted the human immune system by transferring human CD34+ hematopoietic stem and progenitor cells (HSPCs-derived humanized model) and then transplanted human lung cancer cells. We found that the PBMCs-CDX model was more accurate in evaluating PD-L1/PD-1 targeted immunotherapies. In addition, it took only four weeks with the PBMCs-CDX model for efficacy evaluation, compared to 10–14 weeks with the HSPCs-CDX model. We then further established PBMCs-derived patient-derived xenografts (PDX) models, including an auto-PBMCs-PDX model using cancer and T cells from the same tumor, and applied them to assess the antitumor efficacies of anti-PD-L1 antibodies. We demonstrated that this PBMCs-derived PDX model was an invaluable tool to study the efficacies of PD-L1/PD-1 targeted cancer immunotherapies. Overall, we found our PBMCs-derived models to be excellent preclinical models for studying immune checkpoint inhibitors.
KEYWORDS: Non-small-cell-lung cancer, humanized mouse model, patient-derived-xenograft, anti-PD-L1/PD-1 monoclonal antibody, immunotherapy
Introduction
Lung cancer is the leading cause of cancer-related mortality worldwide. 1 Two types of lung cancer have been identified clinically and pathologically: small cell lung cancer (SCLC) and non–small cell lung cancer (NSCLC). 2 The latter category, which includes large cell carcinoma, squamous cell carcinoma, and adenocarcinoma, accounts for more than 80% of lung cancer deaths. 3
Traditional lung cancer treatments include surgery, radiation, and chemotherapy. 4 Molecularly targeted small molecule drugs such as epidermal growth factor receptor tyrosine kinase inhibitors (e.g., gefitinib and erlotinib), 5,6 and anaplastic lymphoma kinase (ALK) tyrosine kinase inhibitors (e.g., crizotinib) have been developed as therapeutic options. 7 More recently, immunotherapies have emerged as one of the most promising approaches to cancer treatment. 8,9 The discovery of immune checkpoint inhibitors, such as programmed cell death protein 1 (PD-1) and programmed death-ligand 1 (PD-L1, also known as B7-H1) axis, has further revolutionized the treatment of multiple cancers, including lung cancer. 10–15 PD-1 is expressed on double-negative αβ and γδ T cells in thymus and activated T cells, including CD4+ Th cells, CD8+ CTL cells and memory T cells, and binds to its ligands, which are expressed on tumor cells or tumor-associated stromal cells, to inhibit T-cell activation and induce T-cell exhaustion. 16,17 Numerous clinical trials have indicated that cancer patients experienced positive clinical response to PD-L1/PD-1 signaling targeted therapies. 11,13,18 In recent years, several monoclonal antibodies that target the PD-L1/PD-1 axis, such as nivolumab, pembrolizumab, atezolizumab, and durvalumab, have yielded excellent benefits on prolonging progression-free survival and overall survival for both second-line and first-line NSCLC patients, and have received US Food and Drug Administration approval. 19–21 However, responses typically occur only in a subset of patients (20–30%) with given tumor histologic profile, despite the demonstrated success of PD-1/PD-L1 blockade in a variety of tumors. 22 Additionally, such treatment is expensive and associated with immune-mediated adverse events. 23,24 Therefore, determining which patients will derive clinical benefit from immunotherapy is a compelling clinical question.
Cell line-derived xenografts (CDX), in which cultured cancer cell lines are injected into immunodeficient mice, are widely used to examine the antitumor effects of drug candidates. 25,26 However, CDX models cannot recapitulate complex human cancer components such as the heterogeneity of tumor cells and the tumor microenvironment. 27,28 Patient-derived xenografts (PDX) generated by implanting tumor samples from patients into immunodeficient mice have therefore become a favored preclinical model to study tumor biology. 29–31 In addition, the immune-PDX model, which is based on the PDX model but contains both human tumor cells and immune cells, is emerging as a promising translational platform for evaluating efficacies of new immunotherapeutic agents. 32–34 The reconstitution of the human immune system in mice based on delivery of human CD34+ hematopoietic stem and progenitor cells (HSPCs) to immunodeficient mice has been the focus of most previous studies;35–38 few studies have established humanized mice using peripheral blood mononuclear cells (PBMCs).39,40 In this study, we systematically optimized the methods to generate immune-PDX models. We showed that the accuracies and resolutions of PBMCs-CDX for evaluating the antitumor efficacies of anti-PD-1/PD-L1 antibodies were higher than that of HSPCs-CDX models, while the time costs of PBMCs-CDX were lower. We also generated allogenic PBMCs-PDX and autogenic PBMCs-PDX models and used them to assess the efficacies of three anti-PD-L1 antibodies.
Results
Establishing humanized mice by transplanting human CD34+ HSPCS or PBMCS
To optimize the xenograft models to assess the efficacies of anti-PD-L1/PD-1 antibodies, we first established immune-humanized tumor-bearing mice. Experiments conducted in tumor-bearing mice typically use human CD34+ HSPCs or PBMCs as sources of T cells. Therefore, we established humanized mice by engrafting CD34+ HSPCs or PBMCs and compared the advantages of CD34+ HSPCs and PBMCs in reconstituting human T cells in NOD-scid-IL2Rg−/− (NSI) mice. The CD34+ HSPCs transplanted mice exhibited increased peripheral blood levels of human T cells over time during a 12-week period (Figure 1A). Compared with CD34+ HSPC-derived T cells, human T cells proliferated more rapidly when NSI mice were transplanted with PBMCs (Figure 1B), and these mice reconstituted higher levels of human T cells (Figure 1(A-B)). In summary, transplantation of either hCD34+ HSPCs or PBMCs into NSI mice led to human T cells reconstitution, but PBMCs-implanted mice reconstituted more T cells.
Figure 1.
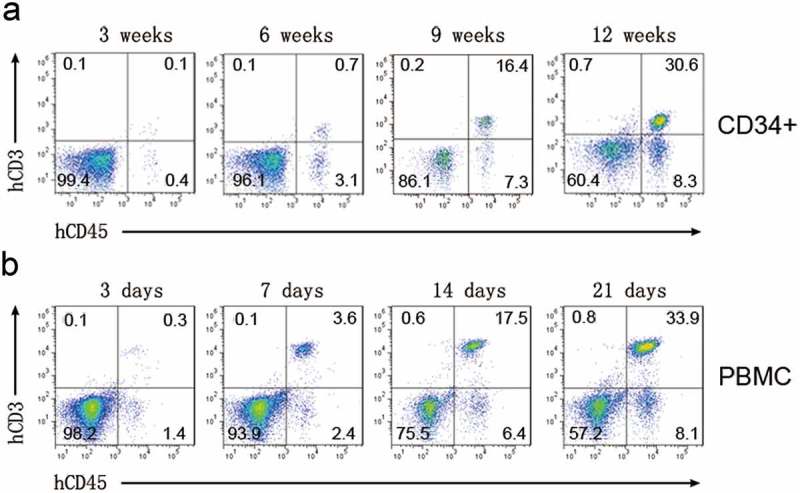
Human T cells reconstitution in NSI mice. Representative FACS profiles of human T cell reconstitution in NSI mice transplanted with CD34+ HSPCs (A) and PBMCs (B) at indicated time points.
Tumor cell line selection
To generate the CDX mouse model to test the efficacy of PD-L1- and PD-1-targeted monoclonal antibodies, we selected tumor cell lines based on the expression of PD-L1. We evaluated the expression levels of PD-L1 in H460, A549, H23, H441, and H522 lung cancer cell lines and found that H23, H460, and H522 expressed high levels of PD-L1 and A549 expressed low levels of PD-L1 (Figure 2A). Flow cytometry confirmed these findings, and identified H460 and A549 as exhibiting the strongest and weakest expression levels of PD-L1, respectively (Figure 2B). Therefore, we chose H460 (high PD-L1 expression) and A549 (low PD-L1 expression) for subsequent studies. As described below, we then compared two T cell sources for assessing the antitumor effects of anti-PD-L1/PD-1 antibodies in our mouse model.
Figure 2.
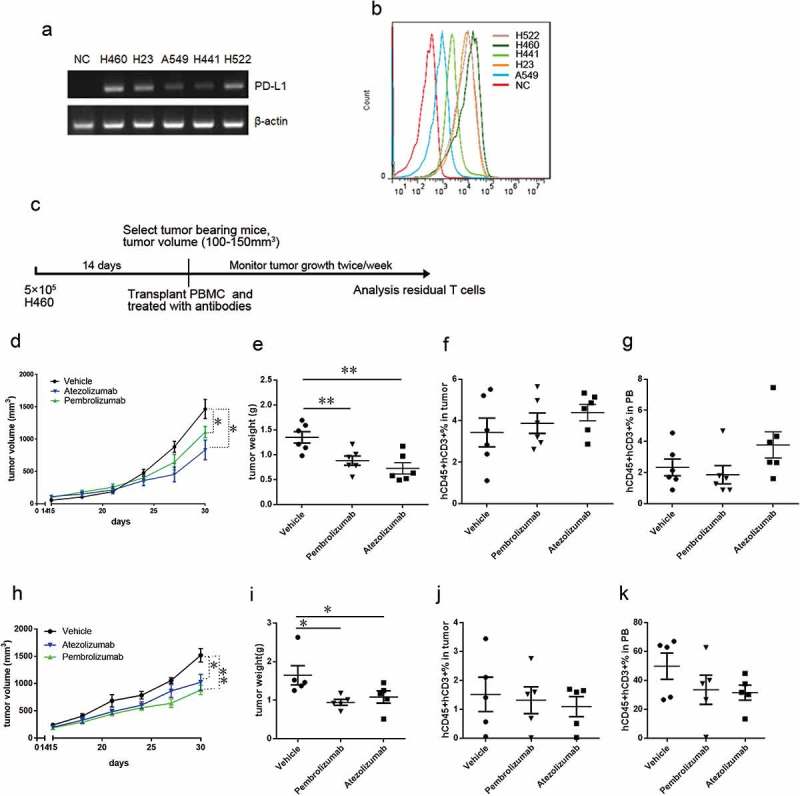
Evaluating the antitumor effect of anti-PD-L1/PD-1 monoclonal antibodies in PBMCs-CDX mouse model. (A) Real-time PCR analyzes PD-L1 (top) and ACTIN (bottom) in indicated lung cancer cell lines. (B) Representative FACS profiles of PD-L1 expression in indicated cell lines. (C) Experimental design to evaluate the antitumor effects of anti-PD-L1/PD-1 monoclonal antibodies in PBMCs-CDX mouse model. (D) Tumor growth curve showing reduced tumor growth in H460-bearing PBMCS-CDX mice by treating with atezolizumab (blue), and pembrolizumab (green); n = 6. (E) Tumor weight showing reduced tumor growth in PBMCS-CDX mice by treating with atezolizumab, and pembrolizumab; n = 6. Summary of percentages of human T cells in tumor tissues (F) and peripheral blood (G) from H460-bearing PBMCs-CDX mice treated with atezolizumab, and pembrolizumab. (H) Tumor growth curve showing reduced tumor growth in A549-bearing PBMCS-CDX mice by treating with atezolizumab (blue) and pembrolizumab (green); n = 5. (I) Tumor weight showing reduced tumor growth in A549-bearing PBMCS-CDX mice by treating with atezolizumab and pembrolizumab; n = 5. Summary of percentages of human T cells in tumor tissues (J) and peripheral blood (K) from A549-bearing PBMCs-CDX mice treated with atezolizumab and pembrolizumab. Means ± SEM are shown in graphs. *p < 0.05, **p < 0.01, and ***p < 0.001; One-way ANOVA. Statistics on tumor growth curve (d, h), p-values are significance for measurements of tumor volume before sacrificing mice.
Evaluating the antitumor effect of anti-PD-L1/PD-1 monoclonal antibody in PBMCS-CDX mouse model
We established PBMCs-CDX mice by transplanting human PBMCs (PBMCs-CDX mice) as illustrated in Figure 2C. We implanted H460 cells in NSI mice; treatment was started 2 weeks after tumor implantation, followed by twice-weekly anti-PD-L1/PD-1 antibody injections at high dose (10 mg/kg). We tested two antibodies: atezolizumab (MPDL3280a, a high-affinity engineered anti-PD-L1 monoclonal IgG1-kappa antibody that blocks the interaction of PD-L1 with PD-1 and B7.1 (CD80)) and pembrolizumab (a high-affinity engineered anti-PD-1 monoclonal IgG4-kappa antibody that binds to the PD-1 receptor and blocks its interaction with PD-L1 and PD-L2). Treatment with atezolizumab (MPDL3280a) and pembrolizumab significantly inhibited tumor growth in PBMCs-CDX mice (Figure 2(D-E)). Atezolizumab exhibited better tumor inhibitory effect compared to pembrolizumab (Figure 2(D-E)). At the end of antibody treatment, we collected peripheral blood and tumor tissues form these PBMCs-CDX mice, and the T cell population was analyzed by flow cytometry. No significant difference of T cell frequencies was observed in PBMCs-CDX mice treated with or without anti-PD-L1 or anti-PD-1 antibody (Figure 2(F-G)). In addition, we re-examined the efficacy of anti-PD-L1 or anti-PD-1 antibody in A549 implanted PBMCs-CDX mice with the experimental protocol illustrated in Figure 2C. Both atezolizumab and pembrolizumab significantly inhibited tumor growth (Figure 2(H-I)), and T cells from the atezolizumab group or pembrolizumab group remained at levels similar to the control group (Figure 2(J-K)).
We further quantified the exhausted status of T cells in PBMCs-CDX mice by flow cytometry. Pembrolizumab treatment significantly reduced the PD-1+ cell population in human T cells (Supplementary Figure 1a-b), indicating that pembrolizumab bound to human T cells and competed with anti-PD-1 staining by antibody detection, and also suggesting that pembrolizumab prevented T cells from entering an exhausted state by blocking of the PD-L1/PD-1 signaling pathway. In addition, both atezolizumab and pembrolizumab treatment significantly decreased LAG3+ TIM3+ cells detected in the human T-cell population in tumor tissues (Supplementary Figure 1c), but no significant difference in the frequencies of exhausted T cells was observed in peripheral blood (Supplementary Figure 1d), suggesting that atezolizumab and pembrolizumab treatment suppressed T-cell exhaustion inside tumors. Collectively, these results demonstrated that PBMCs-CDX humanized mice were an invaluable tool for assessing the antitumor effects of anti-PD-L1/PD-1 antibodies.
Evaluating the antitumor effect of anti-PD-L1/PD-1 monoclonal antibody in HSPCS-CDX mouse model
As human T cell reconstitution from hCD34+ HSPCs occurs more slowly, we transplanted hCD34+ HSPCs into NSI mice (HSPCs-CDX mice) before tumor cell line engraftment and anti-PD-L1/PD-1 antibody treatment (Figure 3A). Twelve weeks after the CD34+ cells transplantation, mice containing similar frequencies of hCD45+ hCD3+ cells (15–25%) (Figure 3B) were selected for antibody assessment. We implanted the H460 cells in humanized mice; treatment was started immediately after tumor implantation, followed by twice-weekly anti-PD-L1/PD-1 antibody injections. Although tumor growth exhibited a declining tendency after antibody treatment, no significant decreases in tumor burdens were apparent among HSPCs-CDX mice treated with anti-PD-L1/PD-1 antibodies when the mice were sacrificed (Figure 3(C-D)). At the end of antibody therapy, we detected residual human T cells in the tumor tissues and peripheral blood by flow cytometry. The percentage of T cells was not affected by anti-PD-L1/PD-1 antibody treatment (Figure 3(E-F)). We further reexamined the efficacy of anti-PD-L1/PD-1 antibody in A549-implanted HSPCs-CDX mice; mice contained similar frequencies of human T cells before therapy assessment (Figure 3G). Both atezolizumab and pembrolizumab treatment resulted in similar tumor growth rate as control mice (Figure 3(H-I)). At the end of antibody therapy, residual T cells from the atezolizumab group or pembrolizumab group remained at levels similar to the control group (Figure 3(J-K)). In addition, although we barely detected PD-1 + T cells in tumor tissue and peripheral blood from the pembrolizumab group (Supplementary Figure 2a-b), which was similar to the results in PBMCs-CDX model, LAG3+ TIM3 + T cells from the pembrolizumab group remained at levels similar to the control group (Supplementary Figure 2c-d). Notably, both the percentage of PD-1 + T cells and LAG3+ TIM3 + T cells from atezolizumab group were increased (Supplementary Figure 2a-d), indicating an exhausted T cell status, which also explained the rapid tumor growth in the atezolizumab group. These results indicated that anti-PD-L1/PD-1 antibodies could not rescue exhausted T cells, and the T cells in HSPCs-CDX mice might be mostly functional incompetent.
Figure 3.
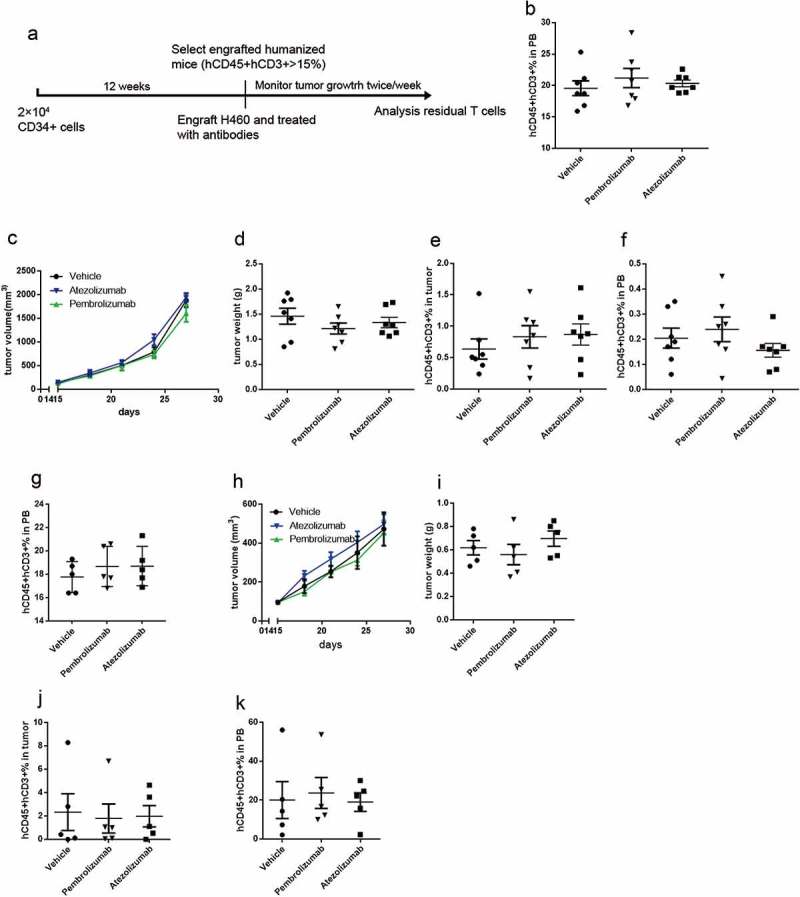
Evaluating the antitumor effect of anti-PD-L1/PD-1 monoclonal antibodies in HSPCs-CDX mouse model. (A) Experimental design to evaluate the antitumor effects of anti-PD-L1/PD-1 monoclonal antibodies in HSPCs-CDX mouse model. (B) Summary of percentages of human T cells in peripheral blood (PB) of H460 tumor-bearing mice before treated with atezolizumab or pembrolizumab. (C) Tumor growth curve and tumor weight (D) showing no reduced tumor growth in H460-bearing HSPCs-CDX mice by treating with atezolizumab or pembrolizumab; n = 7. Summary of percentages of human T cells in tumor tissues (E) and peripheral blood (F) from H460-bearing HSPCs-CDX mice treated with atezolizumab or pembrolizumab. (G) Summary of percentages of human T cells in peripheral blood (PB) of A549 tumor-bearing mice before treated with atezolizumab or pembrolizumab. (H) Tumor growth curve and tumor weight (I) showing no reduced tumor growth in A549-bearing HSPCs-CDX mice by treating with atezolizumab or pembrolizumab; n = 5. Summary of percentages of human T cells in tumor tissues (J) and peripheral blood (k) from A549-bearing HSPCs-CDX mice treated with atezolizumab or pembrolizumab.
Accordingly, we determined that the PBMCs-derived humanized model was superior for assessing the antitumor efficacy of anti-PD-L1/PD-1 antibodies, given the reduced time requirement, availability of T cells, and accuracies for evaluating the antitumor efficacies.
Evaluating the antitumor effects of anti-PD-L1 monoclonal antibodies in PDX mouse model
PDX models have been used in many applications, such as cancer biomarker identification, drug evaluation, and guiding clinical decisions, due to its high fidelity of reconstituting human tumors. We have established lung cancer PDX models for studying tumor biology and cancer immunotherapy, as previous described. 41,42 Here, we established the PBMCs-PDX model to further test whether our PBMC-derived T cell mouse model could be used to evaluate anti-PD-L1 antibodies. We selected two PDXs (Table 1), LC2 and LC23, that expressed high levels of PD-L1 (Figure 4A), and transplanted human PBMCs into mice bearing these tumors, followed by anti-PD-L1 antibody treatment. We evaluated three anti-PD-L1 antibodies in this model: atezolizumab no mutation, atezolizumab N298A mutation and MSB2311 N298A mutation. MSB2311 is an anti-PD-L1 antibody developed by MabSpace Biosciences. MSB2311 binds PD-L1 in a pH-dependent manner, and may thus be able to recycle from the PD-L1-expressing tumor cells and penetrate into the tumor more effectively. The N298A mutation was introduced into the Fc region of human IgG1 to remove the antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) activity of MSB2311 or atezolizumab. Significant tumor growth inhibition was observed after treatment with anti-PD-L1 antibodies, suggesting there are enhanced cytotoxic T-cell responses in PBMCs-PDX-LC2 (Figure 4B and supplementary Figure 3a) and PBMCs-PDX-LC23 (Figure 4C and supplementary Figure 3b). No significant differences in antitumor effects were observed between anti-PD-L1 antibodies without and with the N298A mutation, but MSB2311 was more effective in PDX-LC23 (Figure 4(B-C) and supplementary Figure 3a-b). At the end of PD-L1 therapy, we detected residual human T cells in both the tumors and peripheral blood of PBMCs-PDX-LC2 (Figure 4(D-E)) and PBMCs-PDX-LC23 mice (Figure 4(F-G)), and found more tumor-infiltrating T cells in MSB2311-treated mice These results indicated that PBMCs-PDX could be a useful tool to evaluate anti-PD-L1 antibodies.
Table 1.
Information of patients whose samples were used to establish patient-derived xenograft (PDX) models.
| Sample | Gender | Age | Histology |
|---|---|---|---|
| LC2 | Male | 65 | Lung adenocarcinoma |
| LC23 | Male | 62 | Lung squamous carcinoma |
| LC50 | Male | 63 | Lung squamous carcinoma |
Figure 4.
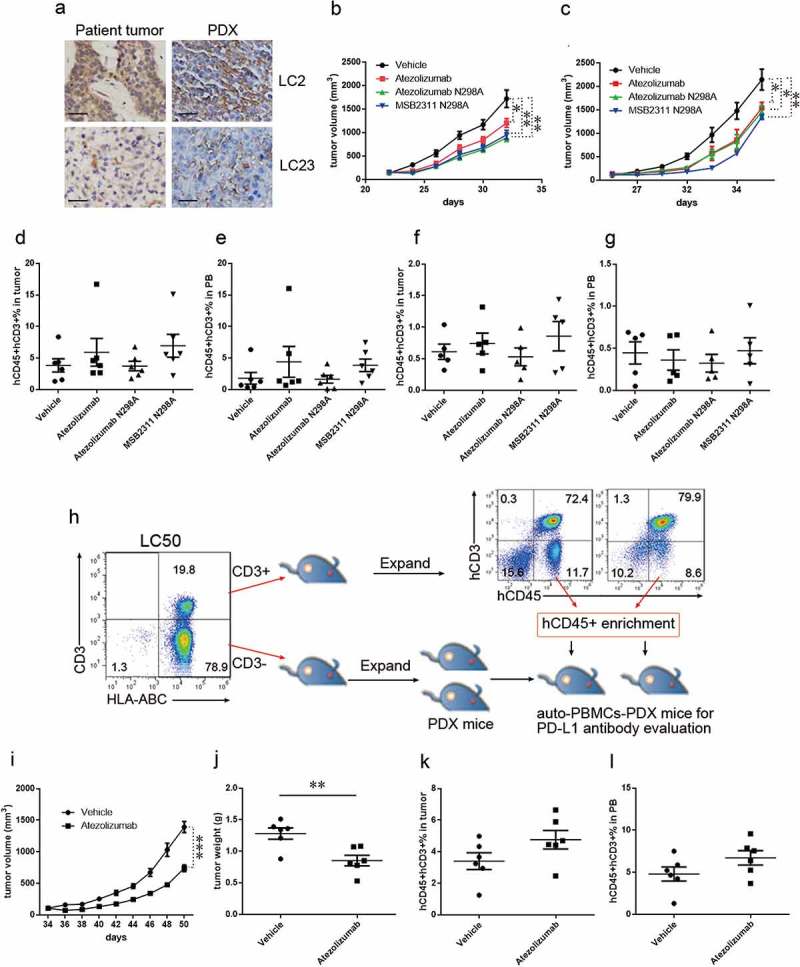
Evaluating the antitumor effect of anti-PD-L1 monoclonal antibodies in PBMCs-PDX mouse model. (A) Immunochemical analyzes PD-L1 in LC2 and LC23 and matched PDX; Scale bar: 100 μm. Tumor growth curve showing reduced tumor growth in PBMCS-PDX2 (B) and PBMCS-PDX23 (C) mice by treating with atezolizumab (red), atezolizumab (N298A mutation, green), and MSB2311 (N298A mutation, blue). Summary of percentages of human T cells in tumor tissues (D) and peripheral blood (E) from PBMCS-PDX2 mice treated with atezolizumab, atezolizumab (N298A mutation), and MSB2311 (N298A mutation). Summary of percentages of human T cells in tumor tissues (F) and peripheral blood (G) from PBMCS-PDX23 mice treated with atezolizumab, atezolizumab (N298A mutation), and MSB2311 (N298A mutation). (H) Experimental design to evaluate the antitumor effects of anti-PD-L1 monoclonal antibody in auto-PBMCs-PDX50 mouse model. Tumor growth curve (I) and tumor weight (J) showing reduced tumor growth in auto-PBMCs-PDX50 mice by treating with atezolizumab; n = 6. Summary of percentages of human T cells in tumor tissues (K) and peripheral blood (L) of auto-PBMCs-PDX50 mice treated with atezolizumab. Means ± SEM are shown in graphs. *p < 0.05, **p < 0.01, and ***p < 0.001; One-way ANOVA (b, c), and unpaired two-tailed t-test (i, j). Statistics on tumor growth curve (b, c and i), p-values are significance for measurements of tumor volume before sacrificing mice.
The mechanism for tumor rejection in PBMCs-CDX mice and PBMCs-PDX mice treated with anti-PD-1/anti-PD-L1 is mostly mediated by alloreactivity, which is does not fit with the tumor biology of most cancer patients. We further established auto-PBMCs-PDX mice that are engrafted with cancer cells and T cells from the same tumor. We detected considerable CD3 + T cell infiltration in the resected primary tumor LC50 (Table 1, Figure 4H). We isolated CD3+ and CD3- cells from this tumor and transplanted two types of the cells into two groups of NSI mice (Figure 4H). CD3- cells formed a tumor that could be serially passaged in NSI mice. CD3+ cells proliferated vigorously in vivo, and some transplanted mice developed graft vs. host disease. We detected human T cell frequencies exceeding 70% in the peripheral blood and spleens of these CD3+ cell-transplanted mice (Figure 4H). Next, we isolated hCD45+ cells from the CD3+ transplanted mice and transplanted these cells into CD3- derived PDX transplanted mice, followed by twice-weekly anti-PD-L1 antibody treatment with atezolizumab (Figure 4H). Antibody treatment was effective against the tumors (Figure 4(I-J)). At the end of PD-L1 therapy, we could detect residual human T cells in both the tumors and peripheral blood of auto-PBMCs-PDX-LC50 mice (Figure 4(K-L)). These results indicate that PD-L1/PD-1 was the major inhibitory signaling axis involved in immune surveillance and the auto-PBMCs-PDX mice could be valuable tools for evaluating antitumor effects of anti-PD-L1 antibodies.
Discussion
In this study, we established PBMCs-derived humanized mouse models and HSPCs-derived humanized models and assessed their fitness for the evaluation of antibody-based immunotherapies targeting PD-L1 or PD-1. Either human CD34+ HSPCs or PBMCs could be engrafted in CDX or PDX mouse models as a source of human T cells. Although a HSPCs-derived humanized model has been reported as a useful tool to investigate the efficacy of anti-PD-1 antibodies, 33 no previous reports have described the advantages or disadvantages of either source of human T cells when evaluating PD-L1/PD-1 targeted therapy. Here, we show that a PBMCs-derived humanized model was superior for assessing the antitumor efficacy of anti-PD-L1/PD-1 antibodies. We demonstrated that T cell reconstitution took less time when PBMCs were used; PBMCs-implanted mice reconstituted more human T cells and showed higher veracity of antitumor effects.
Anti-PD-L1/PD-1 antibodies block the interactions of PD-1 molecules on T cells with PD-L1 molecules on tumor cells or antigen-presenting cells. We observed the PD-L1/PD-1 axis was blockaded by anti-PD-L1/PD-1 antibody treatments, and T cell exhaustion was rescued in PBMCs-CDX mice. T cell exhaustion occurs during human cancer and chronic infections 43–45 due to the persistence of high levels of antigen. 46 Recently, researchers have made substantial efforts to establish strategies to restore the exhausted T cells in tumor therapy. The important role of PD-1 in T cell exhaustion has been demonstrated in basic research and preclinical studies. 47–49 These findings drove the clinical trials of PD-L1/PD-1-targeted immunotherapy for cancer patients (e.g., NCT02304458, NCT02301039, NCT02500797).50 However, some patients do not benefit from PD-L1/PD-1-targeted immunotherapy,51 in part because there are an insufficient number of tumor infiltrating T cells.52,53 Our study showed that MSB2311, an anti-PD-L1 antibody with unique pH-dependent binding to PD-L1, showed more efficacy in preserving tumor infiltrating T cells and suppressed tumor growth in mice. Thus, modifying anti-PD-L1/PD-1 antibodies to fit the tumor microenvironment is a promising way to improve the efficacy of cancer immunotherapy. The N298A mutation was designed to reduce ADCC and CDC activity of anti-PD-L1 antibodies to protect immune cells from autoimmunity, but human natural killer cells or complement were not well restored in mice; thus, N298A mutation may not affect the efficacy of anti-PD-L1 antibodies in mouse models. In addition, T cell exhaustion was not affected by anti-PD-L1/PD-1 antibodies in HSPCs-derived humanized mice. The mechanisms for the non-sensitive T cells in HSPCs-derived humanized mice remain to be elucidated.
PDX mouse models are used to assess the clinical relevance of cancer therapy. In addition, autologous immune-humanized mouse models have become an emerging platform to study cancer immunotherapy. 54,55 In this study, we demonstrated that auto-PBMCs-PDX mice can also be used to evaluate the efficacy of anti-PD-L1/PD-1 antibody therapies. Thus, this model may help to guide clinical decisions, such as advising if patients may derive clinical benefit from PD-L1/PD-1-targeted immunotherapy. This promising model may also represent a novel platform to predict clinical efficacy of other immunotherapies.
In conclusion, our findings show that PBMCs-CDXs, PBMCs-PDX, and auto-PBMCs-PDX models are excellent preclinical models with which to study inhibitory signaling pathways necessary to immunological surveillance and immune therapy. These models may provide promising platform for drug development, evaluating immunotherapies as well as guiding clinical decisions.
Materials and methods
Mice
All animal experiments were performed at the Laboratory Animal Center of the Guangzhou Institutes of Biomedicine and Health (GIBH), and all procedures were approved by the Animal Welfare Committee of GIBH. NOD-scid-IL2Rg−/− (NSI) mice were derived at GIBH.56 All mice were maintained in specific pathogen-free cages and provided autoclaved food and water. Animal protocols were approved by the institutional animal care and use committee (IACUC).
Cell lines
The human non-small cell lung carcinoma cell line H23, A549, H441 H460 and H522 were purchased from American Type Culture Collection (ATCC) and cultured in RPMI-1640 (Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco). Cells were passaged upon reaching 80% confluence.
Anti-PD-L1 and anti-PD-1 antibodies
The PD-L1-specific antibodies atezolizumab, atezolizumab (N298A mutation), MSB2311-N298A (a novel humanized anti-PD-L1 antibody with pH-dependent PD-L1 binding), and the PD-1-specific antibodies pembrolizumab, were generous gifts from MabSpace Biosciences.
Cord blood and peripheral blood
All procedures were approved by the Research Ethics Board of GIBH. Cord blood samples were collected at the South China Medical University (SCMU) Department of Gynecology and Obstetrics under informed consent for research purposes only, and this process was monitored by the Institutional Review Boards of SCMU. Human whole peripheral blood mononuclear cells were isolated using Lymphoprep (StemCell Technologies) according to the manufacturer’s instructions. Human umbilical cord blood CD34+ cells were enriched via magnetic cell sorting (Miltenyi Biotec) according to the manufacturer’s instructions.
T cell reconstitution
For CD34+ HSPCs-based reconstitution, 2 × 104 CD34+ cells were intravenously transplanted into each sub-lethally irradiated (0.5 Gy) 4-week-old NSI mouse. Peripheral blood from all mice was monitored for T cell (hCD45+ hCD3+) reconstitution every 2 weeks. For PBMC-based reconstitution, 5 × 106 human PBMCs were transplanted into sub-lethally irradiated (0.5 Gy) 4-week-old NSI mice. Peripheral blood from all mice was monitored for T cell (hCD45+ hCD3+) reconstitution at the indicated time.
Establishment of immune-CDX and immune-PDX models
PBMCs-CDX mouse model: H460 human lung cancer cells (5 × 105) were injected into the hind flanks of 6–8-week-old NSI mice. The mice were subjected to tumor growth monitoring, and tumor volumes were calculated from caliper measurements using the following formula: (length × width2)/2. Mice were sub-lethally irradiated (0.5 Gy) when tumors reached 120–180 mm3 in volume, and intravenously transplanted with 5 × 106 human PBMCs after a 4-hour interval. Antibodies were administered via intravenous injection at a dosage of 10 mg/kg twice weekly. Tumor growth was monitored every 3 days.
HSPCs-CDX mouse model: Four-week-old NSI mice were sub-lethally irradiated with 0.5 Gy. Four hours after irradiation, each mouse was intravenously transplanted with 2 × 104 CD34+ cells and monitored for T cell (hCD45+ hCD3+) reconstitution every 2 weeks. Once the hCD45+ hCD3+ frequency in NSI mice exceeded 15% (12 weeks post-transplantation), each mouse was engrafted with 5 × 105 H460 cells and subsequently received intravenous injections of anti-PD-L1/PD-1 antibody (10 mg/kg) twice weekly. Tumor growth was monitored every 3 days.
PDX mouse model: All human lung cancer tissues were obtained from Sun Yat-Sen University Cancer Center (SYSUCC), Guangzhou, China. Written informed consent was obtained from each patient. Tissues used in this study were approved by the committee for the ethical review of research involving human subjects at SYSUCC. Fresh tissues were immediately dissected following collection, minced into tissue blocks (diameter: ~ 3 mm) and placed in Matrigel Matrix (Corning) containing antibiotics. These blocks were then subcutaneously transplanted into 6–8-week-old NSI mice within 2 hours after resection. Mice were monitored daily for tumor growth. Tumors were removed and passaged once they reached 1.5 cm3 in diameter.
PBMCs-PDX mouse model: All PDX tumor-bearing mice were sub-lethally irradiated (0.5 Gy) once tumor volumes had reached 120–180 mm3 (measured by calipers). Four hours later, 5 × 106 human PBMCs were transplanted into each mouse. Antibodies were administered via intravenous injection at a dosage of 10 mg/kg twice weekly. Tumor growth was monitored every 3 days.
Auto-PBMCs-PDX mouse model in which T cells and tumor cells were derived from the same patient: Human lung cancer tissues were obtained from SYSUCC after written informed consent was provided by each patient. Tissues used in this study were approved by the committee for the ethical review of research involving human subjects at SYSUCC. Fresh tissues were immediately dissected upon collection and minced into tissue blocks (diameter: ~ 3 mm). The blocks were incubated for 45 minutes at 37°C in serum-free L-15 Leibovitz media (HyClone, Lakewood, NJ, USA) containing collagenase type I and IV (Sigma, St. Louis, MO, USA; 170 mg/l = 45–60 U/ml), collagenase type II (Sigma, 56 mg/l = 15–20 U/ml), DNase I (Sigma, 25 mg/l), elastase (Sigma, 25 mg/l), and 1% penicillin-streptomycin (Life Technologies, Carlsbad, CA, USA). Following this incubation, any visible tumor pieces were vigorously pipetted against the side of a 50-mL tube to enhance disaggregation, followed by an additional 30–50-minute incubation under the same conditions during which larger pieces of tumor tissue were permitted to settle to the bottom of the tube. The supernatant was passed through a 70-μm nylon cell strainer (BD Falcon, Franklin Lakes, NJ, USA), and subjected to red blood cell lysis with 1x Red Blood Cell (RBC) Lysis Buffer (eBioscience). The remaining cells were washed twice in RPMI supplemented with 2% FBS and re-suspended in cell culture media.
CD3+ cells were enriched via magnetic cell sorting (Miltenyi Biotec) according to the manufacturer’s instructions, after which 2 × 104 to 2 × 105 CD3+ cells were intravenously transplanted into each sub-lethally irradiated (0.5 Gy) mouse. Mice were monitored weekly for T cell (hCD45+ hCD3+) reconstitution and were sacrificed when the hCD45+ hCD3+ frequency exceeded 50%. PBMCs and splenocytes were isolated and re-injected (1–5x106 cells/mouse) to generate secondary immunized mice. Subsequently, hCD45+ hCD3+ cells were isolated from these secondary immunized mice to establish immune-PDX models or for cryopreservation in liquid nitrogen. For the CD3- compartment, 5 × 104 to 2 × 105 cells were resuspended in Matrigel Matrix (Corning) with antibiotics and injected under the renal capsules of 6–8-week-old NSI mice as previously described. 57 These mice were palpated weekly to monitor tumor growth, and were sacrificed once tumors could be readily identified by palpation (generally, these tumors were equivalent to 5–10% of body weight). Fresh tumor tissues were collected and immediately dissected, minced into tissue blocks (diameter: ~ 3 mm), and placed in Matrigel Matrix (Corning) with antibiotics. These PDX blocks were subcutaneously transplanted into 6–8-week-old NSI mice to generate secondary tumors, which were removed and passaged once they had reached 1.5 cm in diameter. For auto-PBMCs-PDX mouse model establishment, all tumor-bearing mice were sub-lethally irradiated (0.5 Gy) once the PDX tumor volumes reached 120–180 mm3 (measured by calipers). Four hours later, 5 × 106 hCD45+ hCD3+ cells derived from parallel CD3+ engrafted mice were transplanted into each mouse. Antibodies were administered via intravenous injection at a dosage of 10 mg/kg twice weekly. Tumor growth was monitored every 3 days.
Flow cytometry
Tumor tissues were mechanically chopped with scalpels and placed in culture medium (DMEM with 5% FBS, 0.5 mg/mL collagenase A, 0.2 mg/mL hyaluronidase V, and 0.02 mg/mL DNase I). They were digested for 45 min at 37°C. The resulting suspensions were resuspended in phosphate-buffered saline, and the cells were pelleted at 300 r.c.f. for 3 min. Venous blood were collected, red blood cells were lysed by Red Blood Cell Lysis Buffer (Sigma), the white blood cells were pelleted at 300 r.c.f. for 3 min.
All antibodies were purchased from eBioscience unless otherwise noted. Flow cytometric analysis was performed using an Accuri C6 or LSR FORTESSA device (BD Biosciences, San Jose, CA, USA). Anti-human CD274 (PD-L1, MIH1), anti-human CD45 APC (HI30), anti-human HLA-ABC PE (W6/32), anti-human CD3 FITC (OKT3), anti-human PD-1 APC (NAT103), anti-human LAG3 PerCP-cyanine5.5 (11C3C65), and anti-human TIM3 PE-CY7 (F38-2E2) were used in these analyses.
For fluorescence activated cell sorting (FACS) separation, tumor cells from PDX mice were labeled with anti-human HLA-ABC PE (W6/32) and anti-human CD3 FITC (OKT3) for 20 min at 4°C and were sorted with a MoFlo Astrios cell sorter (Beckman Coulter). Post-sort analysis usually indicated purities of >95%.
Immunohistochemical staining and analyses
Paraffin-embedded tumor sections were deparaffinized, stained with a rabbit anti-human PD-L1 (ab205921, Abcam) as the primary antibodies. After heat-mediated antigen retrieval, sections were incubated with the anti-PD-L1 antibody (1:200) overnight at 4°C. Sections incubated with peroxidase-labeled anti-rabbit antibody at 37°C for 60 minutes. Slides were then stained with DAB and counterstained with hematoxylin. All slides were imaged with a microscope (DMI6000B; Leica Microsystems).
Statistical analyses
Statistical analyses used are detailed in the figure legends. One-way ANOVA or unpaired two tailed Student’s t-test was used to establish the statistical significance using GraphPad Prism (GraphPad Software). Statistical significance set at p < 0.05.
Biography
SL, GH and LC contributed to the conception and design, collection and/or assembly of data, data analysis and interpretation, and manuscript writing. ZL, YX, QD, and YJ contributed to the provision of study material or patients, collection and/or assembly of data. BL, SL, SW, and QW provided animal care and administrative supports. YL, PL, WW, DP, YY, ZW, XZ, YW, ZZ, and SC contributed to the provision of study material and manuscript revising. HY and SC contributed to revise the dosing regimens for in vivo experiments. XS contributed to revise design of the HSPCs-CDX mouse model. XQ and PL contributed to the conception and design, data analysis and interpretation, manuscript writing, and final approval of manuscript and provided financial support. All authors read and approved the final manuscript.
Funding Statement
This study was supported by National Natural Science Foundation of China (NSFC) - 81522002, 81773301; The Strategic Priority Research Program of the Chinese Academy of Sciences, Grant No. XDB19030205, No. XDA12050305; The Natural Science Fund of Guangdong Province: Distinguished Young Scholars (Grant No.: 2014A030306028), Doctoral Foundation (Grant No.: 2017A030310381); The National Major Scientific and Technological Special Project for “Significant New Drugs Development” (Grant No.: SQ2018ZX090201); The Guangdong Provincial Applied Science and Technology Research& Development Program (Grant No.: 2016B020237006); The Frontier and key technology innovation special grant from the Department of Science and Technology of Guangdong province, (2015B020227003, 2014B020225005, 2016B030229006).
Disclosure of potential conflicts of interest
No potential conflict of interest was reported by the authors.
Abbreviations
- CDX
Cell line-derived xenografts
- FACS
Fluorescence-activated Cell Sorting
- HSPC
Hematopoietic stem and progenitor cells
- PBMC
Peripheral blood mononuclear cells
- PD-1
Programmed cell death protein 1
- PD-L1
Programmed death-ligand 1
- PDX
Patient-derived xenografts
Supplementary materials
Supplementary data for this article can be accessed here.
References
- 1.Dholaria B, Hammond W, Shreders A, Lou YY.. Emerging therapeutic agents for lung cancer. J Hematol Oncol. 2016;9(1):138. doi: 10.1186/s13045-016-0365-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Farago AF, Snyder EL, Jacks T. 2012. SnapShot: lung cancer models. Cell. 149:246-+. doi: 10.1016/j.cell.2012.03.015. [DOI] [PubMed] [Google Scholar]
- 3.Kim HS, Mendiratta S, Kim J, Pecot CV, Larsen JE, Zubovych I, Seo BY, Kim J, Eskiocak B, Chung H, et al. Systematic identification of molecular subtype-selective vulnerabilities in non-small-cell lung cancer. Cell. 2013;155:552–566. doi: 10.1016/j.cell.2013.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hirsch FR, Suda K, Wiens J, Bunn PA Jr.. 2016. New and emerging targeted treatments in advanced non-small-cell lung cancer. Lancet. 388:1012–1024. doi: 10.1016/S0140-6736(16)31473-8. [DOI] [PubMed] [Google Scholar]
- 5.Piotrowska Z, Sequist LV. 2016. Treatment of EGFR-mutant lung cancers after progression in patients receiving first-line EGFR tyrosine kinase inhibitors: a review. JAMA Oncology. 2:948–954. doi: 10.1001/jamaoncol.2016.0333. [DOI] [PubMed] [Google Scholar]
- 6.Wu SG, Liu YN, Yu CJ, Yang PC, Shih JY. 2016. Association of BIM deletion polymorphism with intrinsic resistance to EGFR tyrosine kinase inhibitors in patients with lung adenocarcinoma. JAMA Oncology. 2:826–828. doi: 10.1001/jamaoncol.2016.0016. [DOI] [PubMed] [Google Scholar]
- 7.Larkins E, Blumenthal GM, Chen H, He K, Agarwal R, Gieser G, Stephens O, Zahalka E, Ringgold K, Helms W, et al. FDA approval: alectinib for the treatment of metastatic, ALK-positive non-small cell lung cancer following crizotinib. Clinical Cancer Res: Off J Am Assoc Cancer Res. 2016;22:5171–5176. doi: 10.1158/1078-0432.CCR-16-1293. [DOI] [PubMed] [Google Scholar]
- 8.Lin JJ, Shaw AT. 2017. Raising the bar on first-line immunotherapy in lung cancer. Lancet Oncol. 18:2–3. doi: 10.1016/S1470-2045(16)30594-0. [DOI] [PubMed] [Google Scholar]
- 9.Mitchell PL, John T. 2016. Lung cancer in 2016: immunotherapy comes of age. Lancet Respir Med. 4:947–949. doi: 10.1016/S2213-2600(16)30379-4. [DOI] [PubMed] [Google Scholar]
- 10.Shien K, Papadimitrakopoulou VA, Wistuba II. 2016. Predictive biomarkers of response to PD-1/PD-L1 immune checkpoint inhibitors in non-small cell lung cancer. Lung Cancer. 99:79–87. doi: 10.1016/j.lungcan.2016.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garon EB, Rizvi NA, Hui RN, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, et al. 2015. Pembrolizumab for the treatment of non-small-cell lung cancer. New Engl J Med. 372:2018–2028. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 12.Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, Weber JS, Joshua AM, Hwu W-J, Gangadhar TC, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014;384:1109–1117. doi: 10.1016/S0140-6736(14)60958-2. [DOI] [PubMed] [Google Scholar]
- 13.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xue S, Hu M, Iyer V, Yu JM. Blocking the PD-1/PD-L1 pathway in glioma: a potential new treatment strategy. J Hematol Oncol. 2017;10(1):81. doi: 10.1186/s13045-017-0455-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu SY, Wu YL. 2017. Ongoing clinical trials of PD-1 and PD-L1 inhibitors for lung cancer in China. J Hematol Oncol. 10:136. doi: 10.1186/s13045-017-0506-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petrovas C, Price DA, Mattapallil J, Ambrozak DR, Geldmacher C, Cecchinato V, Vaccari M, Tryniszewska E, Gostick E, Roederer M, et al. SIV-specific CD8(+) T cells express high levels of PD1 and cytokines but have impaired proliferative capacity in acute and chronic SIVmac251 infection. Blood. 2007;110:928–936. doi: 10.1182/blood-2007-01-069112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blackburn SD, Shin H, Freeman GJ, Wherry EJ. 2008. Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc Natl Acad Sci U S A. 105:15016–15021. doi: 10.1073/pnas.0801497105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chakravarti N, Prieto VG. 2015. Predictive factors of activity of anti-programmed death-1/programmed death ligand-1 drugs: immunohistochemistry analysis. Transl Lung Cancer Res. 4:743–751. doi: 10.3978/j.issn.2218-6751.2015.12.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hashimoto M, Kamphorst AO, Im SJ, Kissick HT, Pillai RN, Ramalingam SS, Araki K, Ahmed R. 2018. CD8 T cell exhaustion in chronic infection and cancer: opportunities for interventions. Annu Rev Med. 69:301–318. doi: 10.1146/annurev-med-012017-043208. [DOI] [PubMed] [Google Scholar]
- 21.Liu BS, Song YP, Liu DL. 2017. Recent development in clinical applications of PD-1 and PD-L1 antibodies for cancer immunotherapy. J Hematol Oncol. 10:174. doi: 10.1186/s13045-017-0541-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McDermott DF, Sosman JA, Sznol M, Massard C, Gordon MS, Hamid O, Powderly JD, Infante JR, Fassò M, Wang YV, et al. Atezolizumab, an anti-programmed death-ligand 1 antibody, in metastatic renal cell carcinoma: long-term safety, clinical activity, and immune correlates from a phase ia study. J Clinical Oncology: Official Journal Am Soc Clin Oncol. 2016;34:833–842. doi: 10.1200/JCO.2015.63.7421. [DOI] [PubMed] [Google Scholar]
- 23.Fujita Y, Yagishita S, Hagiwara K, Yoshioka Y, Kosaka N, Takeshita F, Fujiwara T, Tsuta K, Nokihara H, Tamura T, et al. The clinical relevance of the miR-197/CKS1B/STAT3-mediated PD-L1 network in chemoresistant non-small-cell lung cancer. Mol Therapy: J Am Soc Gene Ther. 2015;23:717–727. doi: 10.1038/mt.2015.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Powles T, Jp E, Gd F, Fs B, Loriot Y, Cruz C, Bellmunt J, Burris HA, Petrylak DP, Teng S-L, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515:558–562. doi: 10.1038/nature13904. [DOI] [PubMed] [Google Scholar]
- 25.Gong Z, Xu H, Su Y, Wu W, Hao L, Han C. 2015. Establishment of a novel bladder cancer xenograft model in humanized immunodeficient mice. Cellular Physiol Biochem: Int J Experimental Cellular Physiol, Biochem, Pharmacology. 37:1355–1368. doi: 10.1159/000430401. [DOI] [PubMed] [Google Scholar]
- 26.Zhou Q, Facciponte J, Jin M, Shen Q, Lin Q. 2014. Humanized NOD-SCID IL2rg-/- mice as a preclinical model for cancer research and its potential use for individualized cancer therapies. Cancer Letters. 344:13–19. doi: 10.1016/j.canlet.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 27.Herter-Sprie GS, Kung AL, Wong KK. 2013. New cast for a new era: preclinical cancer drug development revisited. J Clin Invest. 123:3639–3645. doi: 10.1172/JCI68340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Siolas D, Hannon GJ. 2013. Patient-derived tumor xenografts: transforming clinical samples into mouse models. Cancer Res. 73:5315–5319. doi: 10.1158/0008-5472.CAN-13-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hidalgo M, Amant F, Biankin AV, Budinska E, Byrne AT, Caldas C, Clarke RB, De Jong S, Jonkers J, Mælandsmo GM, et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discovery. 2014;4:998–1013. doi: 10.1158/2159-8290.CD-14-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, Arcaroli JJ, Messersmith WA, Eckhardt SG. 2012. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol. 9:338–350. doi: 10.1038/nrclinonc.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lai YX, Wei XR, Lin SH, Qin L, Cheng L, Li P. 2017. Current status and perspectives of patient-derived xenograft models in cancer research. J Hematol Oncol. 10:106. doi: 10.1186/s13045-017-0470-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kawaguchi T, Foster BA, Young J, Takabe K. 2017. Current update of patient-derived xenograft model for translational breast cancer research. J Mammary Gland Biol. 22:131–139. doi: 10.1007/s10911-017-9378-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang M, Yao LC, Cheng M, Cai D, Martinek J, Pan CX, Shi W, Ma A-H, De Vere White RW, Airhart S, et al. Humanized mice in studying efficacy and mechanisms of PD-1-targeted cancer immunotherapy. FASEB Journal: off Publ Federation Am Societies Experimental Biol. 2018;32:1537–1549. doi: 10.1096/fj.201700740R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao Y, Shuen TWH, Toh TB, Chan XY, Liu M, Tan SY, Fan Y, Yang H, Lyer SG, Bonney GK, et al. Development of a new patient-derived xenograft humanised mouse model to study human-specific tumour microenvironment and immunotherapy. Gut. 2018;67:1845–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Werner-Klein M, Proske J, Werno C, Schneider K, Hofmann HS, Rack B, Buchholz S, Ganzer R, Blana A, Seelbach-Göbel B, et al. Immune humanization of immunodeficient mice using diagnostic bone marrow aspirates from carcinoma patients. PloS One. 2014;9(5):e97860. doi: 10.1371/journal.pone.0097860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee EK, Joo EH, Song KA, Choi B, Kim M, Kim SH, Kim SJ, Kang MS. 2016. Effects of lymphocyte profile on development of EBV-induced lymphoma subtypes in humanized mice (vol 112, pg 13081, 2015). Proc Natl Acad Sci U S A. 113:E2470–E. doi: 10.1073/pnas.1605284113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Skirecki T, Kawiak J, Machaj E, Pojda Z, Wasilewska D, Czubak J, Hoser G. Early severe impairment of hematopoietic stem and progenitor cells from the bone marrow caused by CLP sepsis and endotoxemia in a humanized mice model. Stem Cell Res Ther. 2015;6(1):1–14. doi: 10.1186/scrt535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma SD, Xu XQ, Jones R, Delecluse HJ, Zumwalde NA, Sharma A, Gumperz JE, Kenney SC, Ling PD. 2016. PD-1/CTLA-4 blockade inhibits epstein-barr virus-induced lymphoma growth in a cord blood humanized-mouse model. Plos Pathog. 12:5. doi: 10.1371/journal.ppat.1005642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yi SN, Ji M, Wu JJ, Ma XQ, Phillips P, Hawthorne WJ, O’Connell PJ. 2012. Adoptive transfer with in vitro expanded human regulatory T cells protects against porcine islet xenograft rejection via interleukin-10 in humanized mice. Diabetes. 61:1180–1191. doi: 10.2337/db11-1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lm T, Me H, English K, Bp M. 2013. Human mesenchymal stem cells suppress donor CD4+T cell proliferation and reduce pathology in a humanized mouse model of acute graft-versus-host disease. Clin Exp Immunol. 172:333–348. doi: 10.1111/cei.12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wei XR, Lai YX, Li J, Qin L, Xu YD, Zhao RC, Li B, Lin S, Wang S, Wu Q, et al. PSCA and MUC1 in non-small-cell lung cancer as targets of chimeric antigen receptor T cells. Oncoimmunology. 2017;6(3). doi: 10.1080/2162402X.2017.1284722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin SH, Huang GH, Xiao YR, Sun W, Jiang YC, Deng QH, Peng M, Wei X, Ye W, Li B, et al. CD215+myeloid cells respond to interleukin 15 stimulation and promote tumor progression. Front Immunol. 2017;8:1713. doi: 10.3389/fimmu.2017.01713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wherry EJ, Kurachi M. 2015. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 15:486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cl D, De K, Kiepiela P, Ja B, Es M, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 45.Baitsch L, Baumgaertner P, Devevre E, Raghav SK, Legat A, Barba L, Wieckowski S, Bouzourene H, Deplancke B, Romero P, et al. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. J Clin Invest. 2011;121:2350–2360. doi: 10.1172/JCI46102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mueller SN, Ahmed R. 2009. High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A. 106:8623–8628. doi: 10.1073/pnas.0809818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dong HD, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat Med. 2002;8(8):793. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 49.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. 2002. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 99:12293–12297. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pianko MJ, Liu Y, Bagchi S, Lesokhin AM. 2017. Immune checkpoint blockade for hematologic malignancies: a review. Stem Cell Investig. 4:32. doi: 10.21037/sci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Diggs LP, Hsueh EC. 2017. Utility of PD-L1 immunohistochemistry assays for predicting PD-1/PD-L1 inhibitor response. Biomark Res. 5:12. doi: 10.1186/s40364-017-0093-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Curran MA, Montalvo W, Yagita H, Allison JP. 2010. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 107:4275–4280. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gajewski TF, Woo SR, Zha YY, Spaapen R, Zheng Y, Corrales L, Spranger S. 2013. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr Opin Immunol. 25:268–276. doi: 10.1016/j.coi.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 54.Dhandapani M, Goldman A. 2017. Preclinical cancer models and biomarkers for drug development: new technologies and emerging tools. J Mol Biomark Diagn. 8:5. doi: 10.4172/2155-9929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jespersen H, Lindberg MF, Donia M, Soderberg EMV, Andersen R, Keller U, Ny L, Svane IM, Nilsson LM, Nilsson JA. 2017. Clinical responses to adoptive T-cell transfer can be modeled in an autologous immune-humanized mouse model. Nat Commun. 8:707. doi: 10.1038/s41467-017-00786-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ye W, Jiang Z, Li GX, Xiao Y, Lin S, Lai Y, Wang S, Li B, Jia B, Li Y, et al. Quantitative evaluation of the immunodeficiency of a mouse strain by tumor engraftments. J Hematol Oncol. 2015;8:59. doi: 10.1186/s13045-015-0156-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cutz JC, Guan J, Bayani J, Yoshimoto M, Xue H, Sutcliffe M, English J, Flint J, LeRiche J, Yee J, et al. Establishment in severe combined immunodeficiency mice of subrenal capsule xenografts and transplantable tumor lines from a variety of primary human lung cancers: potential models for studying tumor progression-related changes. Clinical Cancer Res: Off J Am Assoc Cancer Res. 2006;12:4043–4054. doi: 10.1158/1078-0432.CCR-06-0252. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.