

ABSTRACT
Human antibodies of the IgG2 subclass exhibit complex inter-chain disulfide bonding patterns that result in three structures, namely A, A/B, and B. In therapeutic applications, the distribution of disulfide isoforms is a critical product quality attribute because each configuration affects higher order structure, stability, isoelectric point, and antigen binding. The current standard for quantification of IgG2 disulfide isoform distribution is based on chromatographic or electrophoretic techniques that require additional characterization using mass spectrometry (MS)-based methods to confirm disulfide linkages. Detailed characterization of the IgG2 disulfide linkages often involve MS/MS approaches that include electrospray ionization or electron-transfer dissociation, and method optimization is often cumbersome due to the large size and heterogeneity of the disulfide-bonded peptides. As reported here, we developed a rapid LC-MALDI-TOF/TOF workflow that can both identify the IgG2 disulfide linkages and provide a semi-quantitative assessment of the distribution of the disulfide isoforms. We established signature disulfide-bonded IgG2 hinge peptides that correspond to the A, A/B, and B disulfide isoforms and can be applied to the fast classification of IgG2 isoforms in heterogeneous mixtures.
Keywords: Biosimilars, biotherapeutics, bottom-up, disulfide bond scrambling, disulfide isoforms, disulfide mapping, hinge-region peptides, IgG2, MALDI, mass spectrometry, monoclonal antibodies
1. Introduction
Biotherapeutics based on IgGs are the fastest growing class of human therapeutics. Over 60 IgG monoclonal antibodies (mAbs) have been granted approvals for commercialization in the US or EU as of February 2018, and numerous IgG-based formats, such as bispecifics and antibody-drug conjugates, are currently being developed.1,2 While the IgG1 subclass is the most prevalent, the IgG2 and IgG4 subclasses comprise approximately 20% of all molecules in clinical development.1,3 Structurally, IgGs are tetrameric glycoproteins composed of two heavy chains (HC) and two light chains (LC) bridged by inter-chain disulfide bonds (4 for IgG1 and IgG4; 6 for IgG2).4 The four IgG subclasses (IgG1, IgG2, IgG3, and IgG4) differ structurally with respect to their conserved amino acid sequences, the length of the core hinge region, and the number and configurations of inter-chain disulfide bonds.5,6 The HC and LC are linked by one disulfide bond, and the HCs are linked by two (for IgG1 and IgG4) or four (for IgG2) disulfide bonds located in the hinge region. The inter-chain disulfide bond configuration and non-covalent inter-domain interactions modulate the higher-order structures of each IgG subclass.7–10
IgG2 mAbs produced using standard large-scale bioprocessing conditions are composed of a mixture of three co-purifying inter-chain disulfide isoforms, namely A, A/B, and B (Figure 1).11–13 The inter-chain disulfides of the A isoform are configured in the classical IgG arrangement, where the LC-HC disulfide is found in the same antigen-binding fragment (Fab) region and the four hinge cysteines are bonded in a parallel fashion with the same residue of the opposite HC. The A isoform is furthermore subcategorized as the A1 and A2 forms, which have identical disulfide configurations but the A2 form has one hinge disulfide that is resistant to reduction.14 The disulfide configuration of the B isoform connects the CH1 inter-chain cysteine of one HC to a hinge inter-chain cysteine of the opposite HC and the LC to a hinge cysteine of the proximal HC. The A/B isoform is a hybrid structure that has one Fab arm in the A configuration and the other Fab arm in the B configuration. Although the isoforms only differ in the arrangement of the inter-chain disulfide bonds, each disulfide pattern alters the higher-order structure of the mAb, thereby affecting thermal stability, binding efficiency, surface hydrophobicity, isoelectric point, and possibly influencing developability.15–19 For example, A-isoform enriched panitumumab and denosumab demonstrated higher apparent affinity to the human IgG Fc receptor-like 5 (FCRL5) compared to the B isoform.20 Disulfide isoform distribution may also affect the conjugation profile of cysteine-linked antibody-drug conjugates.21
Figure 1.
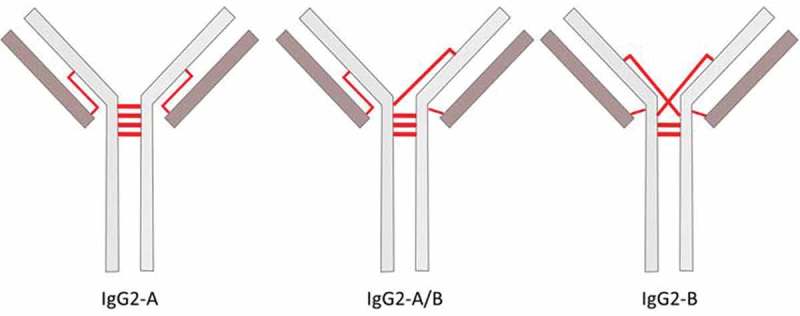
Schematic of IgG2 disulfide isoforms mediated by different configurations of the inter-chain disulfide bonds. The inter-chain disulfide bonds are represented by red lines.
Disulfide mapping and quantitative evaluation of disulfide variants is a core component of product quality attribute assessments, as disulfide variants in IgGs may result in differences in drug substance stability and functional activity.17,20,22–26 The flexible and solvent-accessible hinge region is particularly crucial, as it tends to be labile to reduction and oxidation that can lead to disulfide scrambling or trisulfide formation.15,17,19,27–30 Several mass spectrometry (MS)/MS approaches for characterizing disulfide linkages have been described using collision-induced dissociation (CID), electron-transfer dissociation (ETD), or electron-capture dissociation (ECD).11–13,29,31–35 These approaches often require multiple analysis, comparison of reduced and non-reduced peptide maps, interpretation of complex data sets, or large sample consumption. Electrospray ionization MS/MS characterization of IgG2 hinge disulfides can also be difficult due to the large size of hinge peptides (5 – 10 kDa, depending on disulfide configuration and number of missed cleavages) that generate low-intensity, multiply charged peak clusters that complicate the deconvolution of the MS/MS spectra.11,12,32 Quantification of IgG2 disulfide isoforms requires a separate reverse-phase chromatography or charge distribution analysis of the intact protein.11,12 The advantage of using matrix-assisted laser desorption/ionization (MALDI)-MS/MS for IgG2 disulfide-bonded (DSB) peptide analysis is the predominance of singly-charged ions in the MALDI spectra, which considerably simplifies data analysis and renders it compatible with both semi-quantitative output and automation from a single LC-MS/MS analysis.36–38
MALDI in-source decay (MALDI-ISD) is a fragmentation process that is spontaneously caused by ion-molecule reactions during desorption/ionization in the MALDI plume.39–41 Takayama et al.42 described this mechanism as a hydrogen radical transfer from the matrix to the peptide bond carbonyl group, followed by radical ion rearrangements that predominantly yield c- and z + 2 ions. Based on this concept, MALDI-ISD has been used for protein and peptide sequencing, disulfide bond identification, localization of post-translation modifications (PTMs), and de novo sequencing of a 13.6 kDa protein.43–56
Identification of DSB peptides via MALDI-ISD follows the triplet rule, as described previously and shown in Figure 2.26,37 Briefly, partial reduction in the ion source results in the detection of two reduced peptides (P1-SH and P2-SH) in addition to the original DSB peptide (P1-S-S-P2). The m/z of the DSB peptide is therefore the sum of the m/z values of P1-SH and P2-SH, minus the molecular weight of (H2 + H)+ (Figure 2). Fragmentation of the reduced peptides is performed via CID, and the identity of the DSB peptide is matched to the expected peptides derived from in silico digestion of a provided sequence or by a search engine such as Mascot. Validated DSB peptides detected above a user-defined threshold are tabulated in the Bruker DisulfideDetect software and quantified based on the precursor and ISD-fragment peak intensities. The validation step drastically reduces the number of false-positive triplets, and thus yields a reliable representation of the disulfide structure of a protein. The DSB score represents the abundance of all the 8 peaks expected from the 2 quartet patterns in the DSB-peptide MS/MS spectra, which are fragmentation components of the reduced peptides in the forms of MH+, [MH+]+ S, [MH+]-H2, and [MH+]-SH2.
Figure 2.
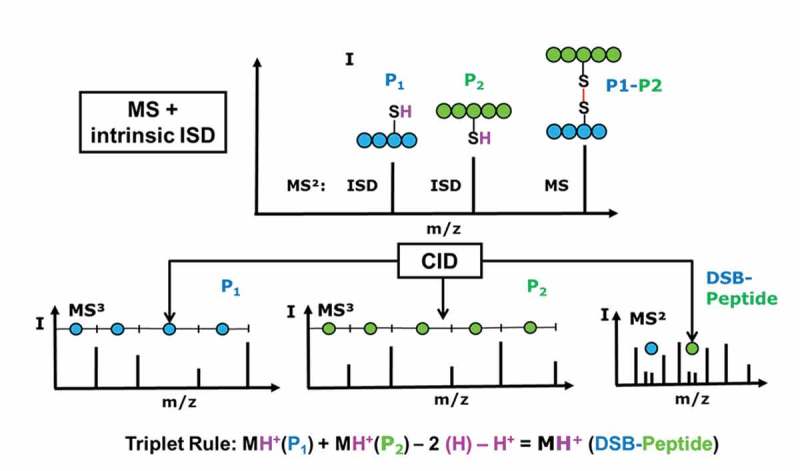
Disulfide-bonded peptide (DSB-peptide) analysis workflow: non-reduced peptides are chromatographically separated and mass analyzed by MALDI-MS.
Here, we explore the application of MALDI-ISD-CID for the identification and semi-quantitation of DSB peptides that pertain to each IgG2 disulfide isoform in heterogeneous samples. We discuss the manual identification and validation of signature hinge peptides that correspond to each disulfide isoform, which can be applied for future automated analysis of IgG2 disulfide isoform distribution. Combining both qualitative and quantitative analyses into a single run with software-led data interpretation can greatly improve the throughput of disulfide characterization at various stages of biotherapeutic development.
2. Results
2.1. Automated assignment of singly disulfide-bonded peptides of adalimumab with native or scrambled disulfides
The automated DSB peptide analysis workflow was initially established using an IgG1 mAb (adalimumab) in both intact and heat-stressed conditions. Disulfide-bonded peptides composed of two asymmetric peptides linked by a single disulfide bond and meeting the triplet rule criteria were automatically identified and quantified by the DisulfideDetect software; the output is displayed in a disulfide connectivity map (Figure 3). Blue lines in the disulfide map denote the canonical disulfide bonds and red lines represent unexpected bonds (either scrambled or non-canonical) based on the Native Disulfide Bond reference table embedded in the DisulfideDetect software. Each DSB is additionally visualized by a blue bubble, where an “N” indicates a native or expected bond. The size of the bubbles reflects the relative abundance of a DSB compared to other DSBs linking to the same cysteine. Because the IgG1 hinge peptide is composed of two identical peptides with two disulfide bonds, validation of the ISD-reduced DSB peptide required manual MS/MS data processing (data not shown). Manual identification of multiply-bonded peptides is based on detection of the MH+, [MH+]+ S, [MH+]-H2, and [MH+]-SH2 quartet in the MS/MS spectrum, as discussed in the following sections addressing the manual validation of the IgG2 hinge peptides.
Figure 3.
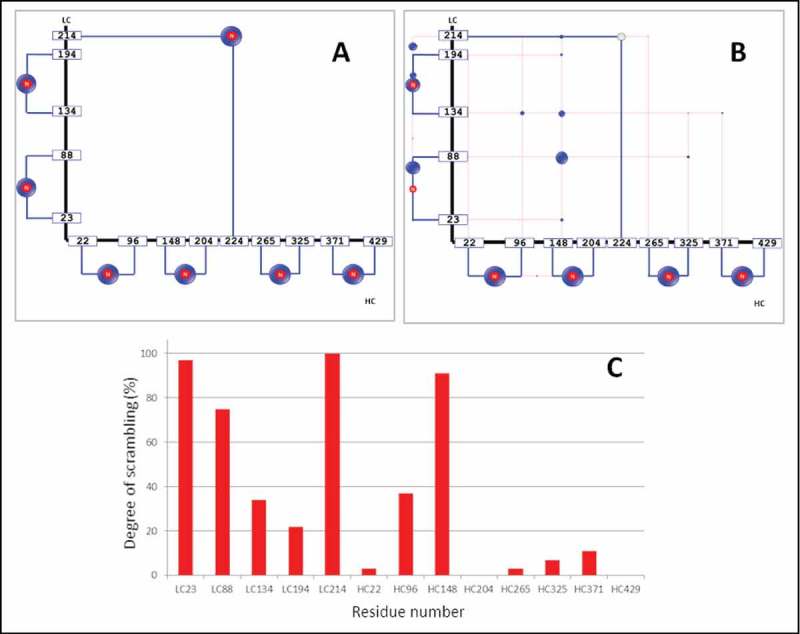
Disulfide connectivity maps from a native IgG1 before (A) and after heat-stress (B), and residue-specific degree of disulfide scrambling observed in the heat-stressed IgG1 mAb (C). Native DSBs are represented by blue lines, and scrambled disulfides by red lines. In the heat-stressed sample (B), the DSB that links the LC and HC was subject to extensive scrambling and the native bond was no longer detectable. The degree of scrambling of cysteine residues in the heat-stressed adalimumab is shown in the histogram (C). Degree of scrambling varied significantly across the DSBs upon heat treatment: from 100% scrambling at the LC214 cysteine to virtually absent scrambling at HC429. The cysteines involved are coded by their residue number in the light or heavy chain, e.g., HC22 refers to the cysteine at residue 22 of the adalimumab HC.
The DSB connectivity maps provide a simple and holistic visualization of the potential disulfide scrambling “hot spots” for every cysteine in the antibody. The detected DSBs of the non-stressed IgG1 were all native, as expected. Heat stress at 70°C induced disulfide bond scrambling at nearly all cysteine residues, with the exception of HC204 and HC429. The connection between LC and HC (LC214-HC224) was no longer detectable after heat stress (no blue bubble present in Figure 3B) and several alternative bonds were instead detected at LC214. Other scrambling hotspots were detected at LC23, LC88, and HC148, which are all located in the Fab region.
The quantitative measure displayed in the histogram in Figure 3C was calculated as the relative percentage of the total peak intensities of all disulfide bonds from each cysteine. The intensity (I) of a DSB-peptide was calculated as the sum of intensities of the peak triplet IP1, IP2, and IDSB-Peptide (Figure 2). For each cysteine residue Cx, the software calculates both the sum of intensities of the all DSB-peptides (Σ Ix), as well as those containing the native bond (Σ INx). The degree of scrambling (Sx) of disulfide bonds involving Cx was calculated as shown in Equation 1. Thus, Sx is a specific determination of the degree of scrambling for Cx, and the two cysteine residues that define a disulfide bond, Cx and Cy, may result in different determinations of Sx and Sy. The accuracy achievable from LC-MALDI datasets was estimated at ± 25–30% for label-free quantitation of peptides.57,58
2.2. Automatic assessment of the DSBs of the IgG2 isoforms
The first step in our analysis of the IgG2 A, B and A/B disulfide isoforms was the automatic software-based assessment of the disulfide organization using the canonical disulfide configuration as the template for the intra-chain and LC-HC inter-chain disulfide bonds. Both the canonical and non-canonical DSB-peptides composed of a single disulfide crosslink were detected via automated analysis (Figure 4). The DSB connectivity map obtained for each of the IgG2 disulfide isoform fractions showed significant differences in the DSB organization of the isoforms, as expected. For example, the LC212-HC129 bond was detected only in the A and A/B forms using this method, which is consistent with previous reports about the LC-HC connectivity in these isoforms.11,12 Most native intra-chain and inter-chain disulfide bonds (12 total for an IgG2) were detected in all isoforms. However, the intra-chain HC22-HC96 could not be detected in neither the A nor the B isoforms, suggesting possible trypsin missed cleavage at this site or low ionization of this particular DSB peptide. The LC-HC DSB peptide in the B configuration could not be detected in the automatic assessment, as this peptide is part of the multiply-crosslinked hinge peptide.
Figure 4.
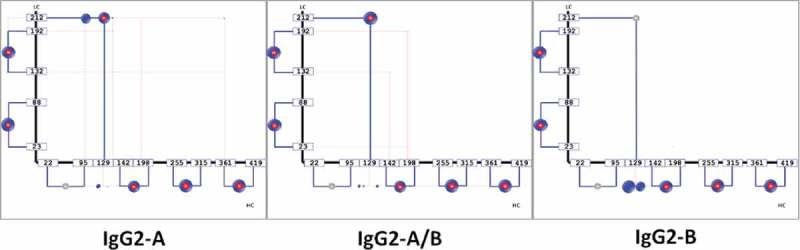
Disulfide connectivity maps of the IgG2 A, A/B, and B disulfide isoforms. Native DSBs (blue, indicated by “N”) as well as scrambled DSBs (red) were observed. The size of the bubble represents the prevalence of each disulfide bond configuration.
In addition to the native disulfide bonds, scrambled disulfides and scrambling hot spots were also detected and quantified automatically by the software (Figure 5). Higher abundance of scrambling was observed in the A isoform, which elutes later than the B and A/B isoforms in the RP-UHPLC method used for separation of disulfide isoforms (Figure 6). The amount of scrambling in the A isoform is consistent with its longer exposure to the 82°C column temperature used during fraction collection. The small size of the blue bubbles in the disulfide map of the A and A/B isoforms indicate that molecules containing scrambled inter-chain disulfides were present in minute amounts relative to the native configurations. The histogram in Figure 5 shows that HC129 and HC142 were involved in scrambling in all 3 isoforms, whereas LC212 scrambling was detected only in the A isoform. The lack of scrambled disulfides involving LC212 in the A/B and B isoforms is likely due to the limitation of the automated software detection of multiply cross-linked peptides. Taken together, the obtained linkage maps alone showed significant differences between the IgG2 isoforms, but they did not allow identification of the isoform-specific hinge peptide structures.
Figure 5.
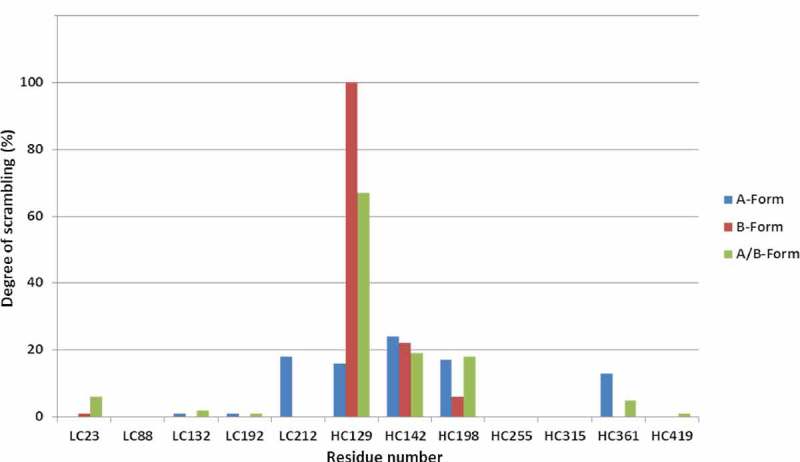
Overview of relative degree of scrambling of DSBs at specific cysteine residues, automatically outputted by DisulfideDetect for the A, A/B, and B isoforms.
Figure 6.
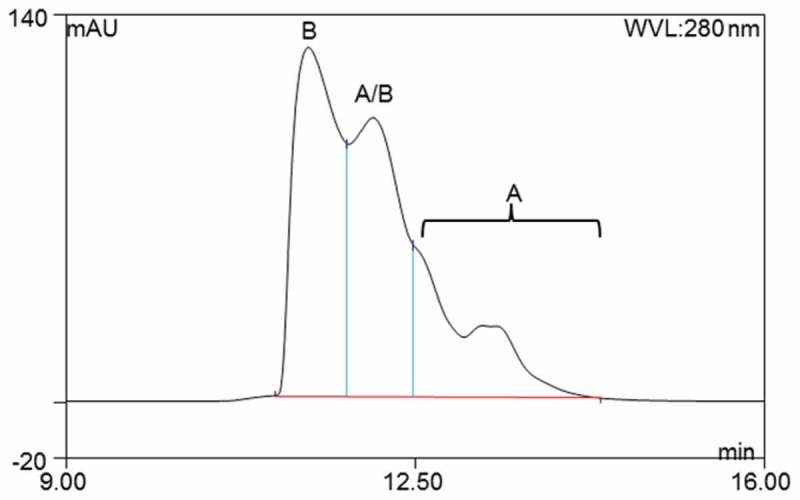
RP-UHPLC separation of the IgG2 A, A/B, and B isoforms. This LC method was used to collect the disulfide isoform fractions for tryptic digestion, and the lack of baseline resolution of the peaks results in some expected cross-contamination from neighboring species. Peaks were assigned based on previous publications.11,12
2.3. Identification of IgG2-A isoform signature DSB peptides
In the IgG2-A isoform, the LC212 residue is connected to HC129 in the CH1 region, in contrast to HC224 in the CH2 region as is the case in IgG1s. The MS/MS spectrum of the respective tryptic dipeptide LC212b-HC129 with m/z 2039.943 is shown in Figure 7. The quartet of fragment ions generated by the CID fragmentation of the HC peptide GPSVFPLAPCSR yielded ions with m/z of 1196.642 (MH+-SH2), 1228.614 (MH+-H2), 1230.630 (MH+), and 1262.602 (MH++ S). The corresponding fragment ion quartet was also detected for the LC peptide with one missed cleavage, SFNRGEC (812.3 Da), albeit at lower intensity than the HC peptide fragments. In addition, sequence-specific b- and y-ions from both peptides were detected. Sequence fragmentation and the presence of the fragment ion quartet were also manually verified for the MS/MS spectra in cases of a lower-scoring DSB triplet.
Figure 7.
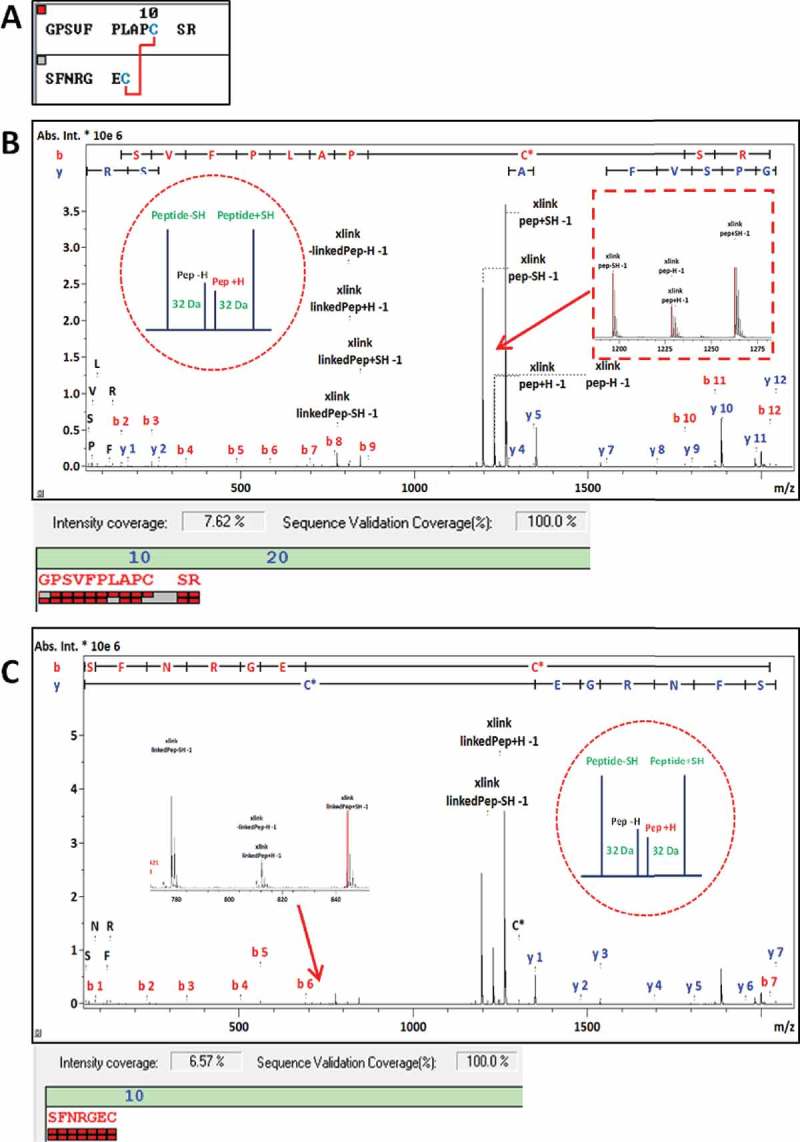
Validation of DSB-peptide LC212b-HC129, with sequence shown in (A). MS/MS spectrum with the matching HC sequence-related fragment ions annotated shown in (B). Same MS/MS spectrum with the matching LC-related fragment ions annotated shown in (C). One missed cleavage was observed in the LC peptide, therefore assigned as LC212b.
The predicted hinge peptide structure for the A isoform is a symmetrical DSB peptide spanning residues HC217 – HC224 of m/z 5351.562. The tryptic peptide sequence of the A isoform hinge is CCVEC PPCPA PPVAG PSVFL FPPKP K with all four Cys residues disulfide-bonded to the opposite HC peptide (Figure 8). The fragment ions in the MS/MS spectrum covered the peptide sequence across residues PA PPVAG PSVFL FPPKP (residues 9 – 26). Since the presence of multiple disulfide bonds prevents efficient fragmentation in MS/MS analyses across the disulfide crosslinked sequences, the full sequence of the hinge peptide is not covered by MS/MS fragments.59,60 However, the presence of MS/MS fragments such as the b8-b25 and y5-y21 ions are consistent with the expected MS/MS fragmentation pattern of the hinge peptide (Figure 8A).
Figure 8.
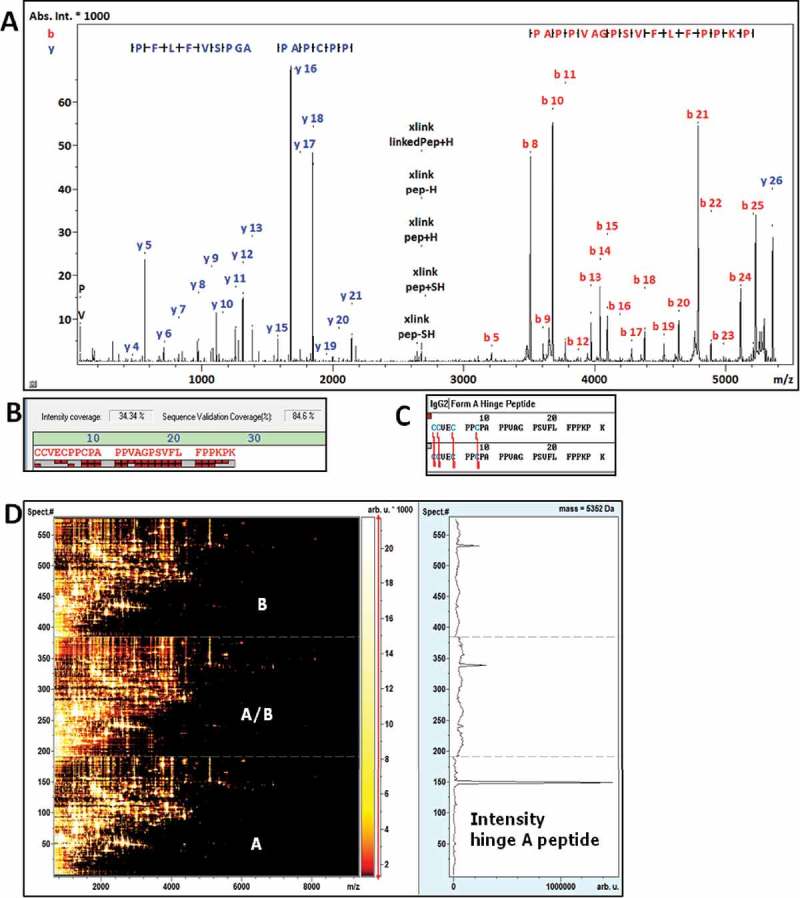
(A) MS/MS spectrum of the IgG2 A hinge peptide with sequence specific fragments annotated that support the identification. (B) Sequence coverage view of the hinge peptide with sequence in (C), the red bricks correspond to N-terminal (upper row of bricks) and C-terminal (lower row) fragment ions. (D) Comparative LC-MALDI heat map of the IgG2 digests of the A, B and A/B isoforms with an Extracted Ion Chromatogram at m/z 5352 across the 3 analyses. The XIC across all three datasets shows a strong prevalence in the A form and was only spuriously detected in case of the B and A/B forms.
In summary, the m/z 5351.5 peptide detected via MALDI-MS can be assigned as a signature for the A isoform (HC217-224)2 DSB hinge peptide. The extracted ion chromatogram at 5352 m/z showed the predominance of this peptide in the A isoform LC-MALDI heat map, and only minor amounts in the A/B and B isoforms. The presence of the A isoform hinge peptides in the A/B and B digests can be attributed to minor cross-contamination during LC fractionation due to the lack of baseline resolution of the isoforms (Figure 6).
2.4. Identification of the IgG2-A/B isoform signature hinge peptide
The asymmetric hinge peptide with cysteines LC212a, HC216, HC129, and (HC217-224)2 with m/z 6886.253 is specific to the A/B isoform. This DSB peptide is composed of the 2 hinge peptides linked via three disulfide bonds at HC217-224, and the HC216 residue is connected to either LC212a or to HC129 (Figure 9). The MH+ ion was observed as m/z 6886.23, which is in agreement with the proposed structure with a −3.4 ppm error. The HC peptide sequences involved were identified in the MS/MS spectrum with high confidence, with 100% sequence coverage for the 12mer peptide containing the CH1 HC129 residue, and 89% coverage for the two hinge HC217-224 peptides. Similar to the results observed for the A hinge sequence, the A/B hinge peptide sequence was confirmed by b5-b25 and y2-y18. The LC tripeptide with no missed cleavages (denoted as LC212a) with sequence GEC was not directly observed with fragment ions in the MS/MS spectrum, but its presence was inferred by detection of the peak quartets at m/z 5658.65 corresponding to [(HC217-224)2,LC212a] and m/z 1230.60 corresponding to HC129a, which add up to the expected mass of the A/B hinge DSB peptide.
Figure 9.
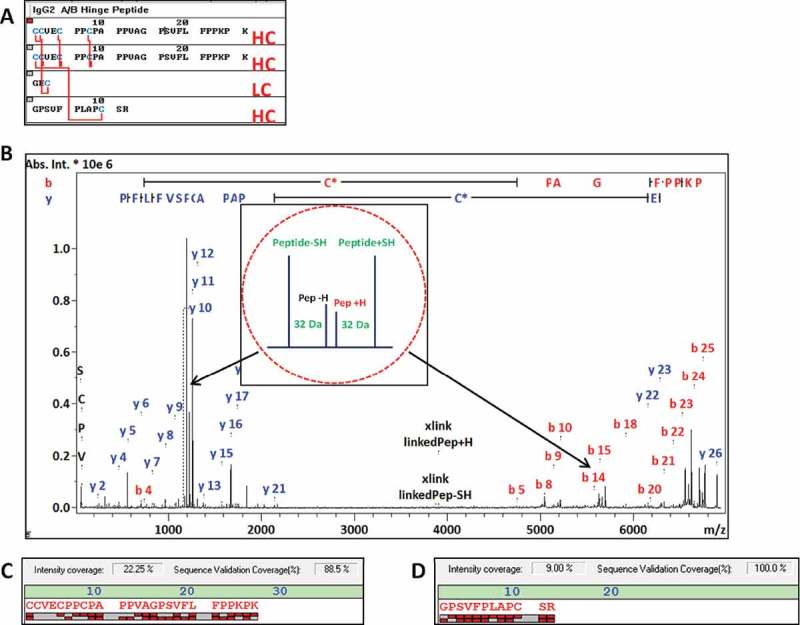
MS/MS spectrum identifying the IgG2 A/B specific hinge peptide (m/z 6886.253) with fragment ions assigned (B). The structure of the asymmetric peptide with 5 disulfide bonds is shown in (A). Bottom: the fragment ion coverage of the hinge peptide (C) and the linked HC 12mer peptide (D) are shown. The peak quartet at m/z 1230.630 corresponds to the 12mer HC peptide and the quartet at m/z 5658.65 to hinge peptide with loss of the LC tripeptide (peak quartets are indicated by red arrows).
Interrogation of the LC-MALDI heat map for the m/z 6886.253 peptide [LC212a,HC129,(HC217-224)2] was detected in preparations from all three isoforms, with the highest abundance detected in the A/B MALDI preparation. This cross-contamination of the A/B hinge peptide was expected as the A/B peak eluted in between the A- and B-form peaks in the LC fractionation (Figure 6). The relative amounts of A/B hinge observed in the A and B isoform MALDI preparations (data not shown) agree with the tailing peak shapes of the RP-UHPLC elution profiles.
2.5. Identification of IgG2-B isoform signature hinge peptides
The IgG2-B hinge peptide has a symmetrical structure involving residues LC212a, HC129a, and HC217-224, and has an expected m/z 8420.944: (LC212a,HC129a,HC217-224)2. The DSB peptide linkage pattern of the B isoform shows the LC212 bonded to HC218, and HC129 bonded to HC217 (Figure 10). Large peptides and proteins prepared in hot matrices such as α-cyano-4-hydroxycinnaminic acid (HCCA) suffer extensive metastable fragmentation in reflector mode MALDI spectra, which complicates further analysis.48,61 This effect was not significant in the 5 – 6 kDa range, but prevented the analysis of 8 – 9 kDa hinge peptides. The B isoform digests were therefore crystallized in super DHB (sDHB) matrix instead of HCCA to reduce the effect of metastable fragmentation on the molecular ion. As sDHB is also compatible with the analysis of the 5 – 6 kDa hinge peptides, future experiments can be directly set up in this matrix.
Figure 10.
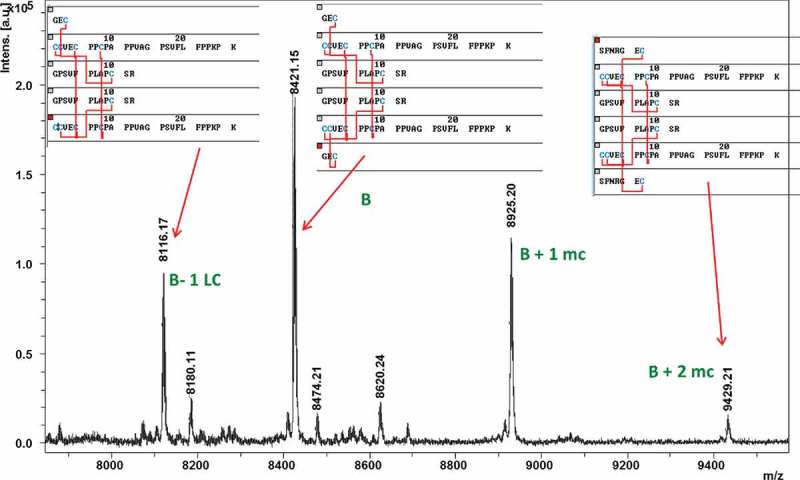
Reflector MALDI-MS spectrum close-up of the hinge peptide fraction from the LC-MALDI sDHB dataset of the IgG2 B isoform. The main peak at m/z 8421.15 corresponds to the symmetric hinge peptide of the B isoform. The peaks at m/z 8925.20 and 9429.21 correspond to the same peptide structure with 1 or 2 LC heptapeptides instead of tripeptides due to incomplete proteolytic digestion (“mc” = missed cleavage). The peak at m/z 8116.17 corresponds to B isoform hinge with loss of one LC tripeptide.
Four DSB peptides that correspond to the B isoform hinge were detected with m/z values 8116.17, 8421.15, 8925.20, and 9429.21 (Figure 10). The symmetric B isoform hinge peptide was found as m/z 8421.15 (theoretical m/z 8420.944). The additional peaks at m/z 8925.20 and m/z 9429.21 correspond to peptides containing one missed cleavage on one or both LCs, respectively, yielding a LC heptapeptides (SFNRGEC) instead of the tripeptide (GEC). The m/z 8116.17 variant of the hinge peptide correlated to the loss of one LC tripeptide, similarly observed in the A/B isoform hinge peptide analysis.
Complex peptide structures consisting of multiple peptides connected by multiple disulfide bonds, as in the case of B-isoform hinge peptides, typically do not generate sufficient peptide backbone fragmentation for their unequivocal identification by MS/MS. Assignment of the observed m/z value to the corresponding structure was accomplished together with the peak quartet at m/z 1196.47 and m/z 7192.59 (Figure 11). The quartet fragment pattern in Figure 11 composed of the peaks at m/z 1196.5, 1230.6, and 1262.6 corresponds to the HC peptide GPSVFPLAPCSR (HC129a). The peak quartet at m/z 7192.59 corresponds to the loss of the HC129a 12mer peptide from the B-form hinge peptide (theoretical m/z 7191.321), which either results from a reductive ISD product or reduced cysteines that were already present prior to enzymatic digestion. Additionally, the peak quartet at m/z 4211.34 matches the peptide (LC212a,HC129a,HC217-224), which is the symmetrically cleaved hinge peptide (theoretical m/z 4211.984). Detection of the m/z 4211.34 peak indicates that one of the two hinge disulfides at the C-terminus may have been present in reduced form prior to enzymatic digestion, allowing ISD reduction of the only other remaining disulfide connecting the two hinge peptides. IgG2 mAbs have been reported to contain higher amounts of free thiol in comparison to IgG1s,62,63 and it is possible that the B isoform contributes greatly to the reported values due to the conformational strain imparted by the crossed-disulfide configuration. The m/z 2957.08 corresponds to the (LC212a,HC129a,HC217-224) peptide with a truncated HC217-224 after Gly-15 prior to the subsequence Pro-16 residue, representing a proline cleavage product of the symmetrically cleaved hinge peptide.
Figure 11.
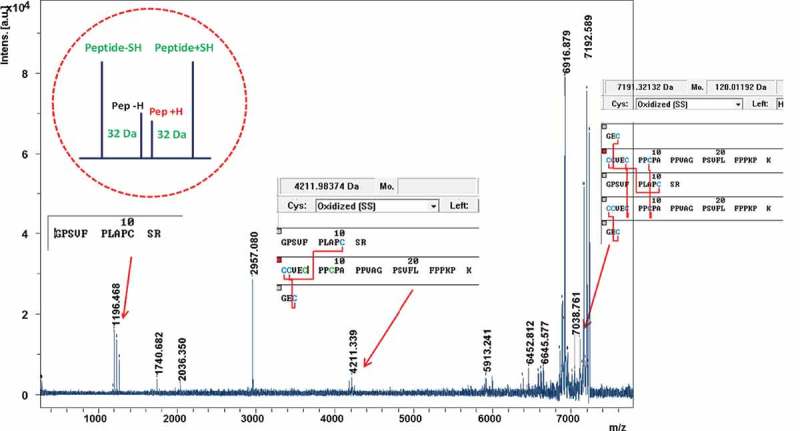
MS/MS spectrum of the IgG2-B hinge peptide (m/z 8420.944) with matching peak quartet identification and corresponding sequences.
Altogether, the hinge peptide fraction of the B isoform in the LC-MALDI dataset contained over 40 peptides, spanning retention times from 55.2 – 56.5 minutes and a mass range of 5 – 10 kDa. Further analysis determined that this fraction contained a heterogeneous mixture of DSB-peptides pertaining to the A, A/B, and B hinge peptide structures (Figure 12). The heterogeneity was expected, and the B and A/B isoforms partially co-elute in the RP-UHPLC fractionation method, several of which were confirmed by MS/MS (not shown). Most of the peaks detected in this LC-MALDI data subset were determined to be peptides containing missed cleavages or losses of singly-disulfide bonded fragments. This structural heterogeneity made the initial determination of disulfide connectivity time consuming, but the established list of signature DSB-peptide structures and molecular weights (MWs) will significantly facilitate further work, as MS/MS data interpretation may be required only sporadically.
Figure 12.
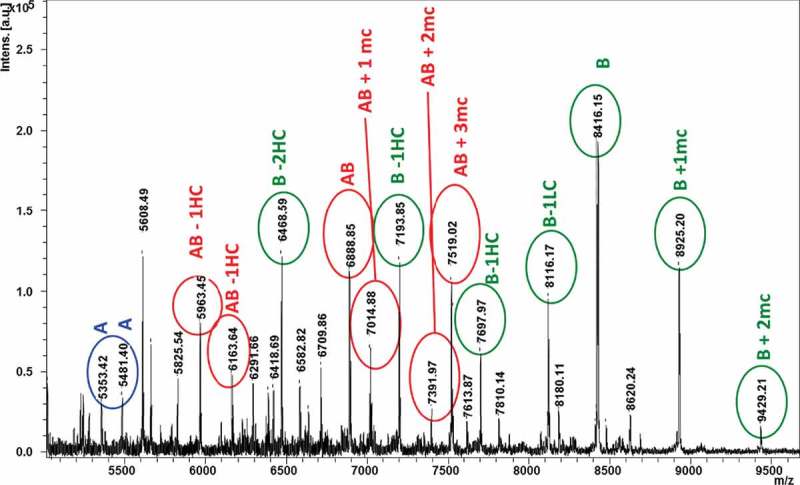
MALDI-MS spectrum (5–10 kDa region) of the hinge peptide fraction from the LC-MALDI dataset of the IgG2 B isoform, obtained from an sDHB matrix preparation. Within this fraction were approximately 40 variants of hinge peptides from all three IgG2 disulfide isoforms. Color-coded IgG2-A (blue), -A/B (red), and -B (green) peaks provide an overview of the isoform-specific hinge peptide fragments in a heterogeneous mixture. “mc” = number of missed cleavage chains in peptides.
2.6. Catalog of IgG2 disulfide isoform signature DSB-peptides
All the peptides verified by their MS and MS/MS spectra are summarized in Table 1. The set of m/z values below can be added to software databases as a template for future automatic detection of each IgG2 disulfide isoform. High sequence conservation near the inter-chain cysteines, regardless of allotype, implies that this collection of signature peptides is widely applicable to other human IgG2s.5,6 The peptides described here do not support discernment between different cross-linking configurations of nearby cysteines (i.e., canonical vs. scrambled hinge disulfides). For example, disulfide bond variants within the lower hinge region of the native B isoform reported elsewhere would not be differentiated using this method.32
Table 1.
IgG2 disulfide isoform-specific hinge peptides directly detected in MALDI-MS spectra in sDHB matrix. Listed peptide configurations were in agreement with MS/MS spectra. Tryptic peptides were named after the cysteine position within light and heavy chain, e.g. HC129 is the cysteine 129 containing tryptic peptide of the heavy chain; a: no miscleavages, b: 1 miscleavage. *MS match only, MS/MS spectrum not interpretable.
| MH+ | A | A/B | B | Peptide configuration |
|---|---|---|---|---|
| 2039.9 | + | + | LC212b-HC129a | |
| 5351.6 | + | (HC217-224)2 | ||
| 5479.7 | + | K(HC217-224)2 | ||
| 6886.3 | + | LC212a, HC129a, (HC217-224)2 | ||
| 8420.9 | + | (LC212a, HC129a, HC217-224)2 | ||
| 8925.2 | + | LC212a, LC212b, (HC129a, HC217-224)2 | ||
| 9429.4* | + | (LC212b, HC129a, HC217-224)2 |
3. Discussion
We developed an approach to characterize IgG2 disulfide bonds using a single trypsin/Lys-C digestion under acidic conditions and in the presence of N-ethylmaleimide (NEM) to prevent artifactual disulfide scrambling, thus eliminating the need for separate reduced and non-reduced peptide maps that are commonly employed for disulfide mapping of proteins.12,24,64,65 The LC-MALDI-TOF/TOF approach described here uses the Bruker DisulfideDetect software to automatically output semi-quantitative maps of the singly disulfide-bonded peptides within a single analysis. This method takes advantage of the partial reductive fragmentation of peptides at the disulfide bond by in-source decay in the MALDI source. Resulting MALDI-MS spectra thus contain both kinds of peaks, the peptide chain’s MH+ reduced by ISD, as well as the disulfide bonded peptide (DSB-peptide) precursor ion, which are all subsequently fragmented by MS/MS in the TOF/TOF ion optics.53 The reduced peptides are directly identified by in-sequence search of the target protein, and the disulfide bond connectivity is assigned based on the triplet rule with the prerequisite that both reduced and DSB-bonded peptides coelute in the LC. False-positives are minimized in the qualified candidates list, as peptide sequences that do not contain cysteine residues are automatically filtered out. Prediction of DSB-peptide m/z values is not necessary in this approach due to the specificity of sequence searches using the MALDI-MS/MS spectra. This method was established using both intact and heat-stressed adalimumab (IgG1), demonstrating both the software-led disulfide bond assignment and that disulfide scrambling did not occur during peptide preparation. Complex scrambling events due to heat stress as low as ~ 1% were detectable using this approach.
Peaks from peptides containing two or more disulfide bonds are contained in the same dataset but cannot be automatically identified by the software. Partial reductive cyanylation was previously described to circumvent this limitation,37 however, we performed manual identification of the multiply cross-linked peptides in order to avoid unwanted chemical modifications. The manual MS/MS data analysis was greatly simplified by the predominance of singly-charged precursor ions in the MALDI spectra, and we identified a list of IgG2 disulfide isoform-specific peptides that will be useful for future IgG2 disulfide isoform assessments. Identification of peptides containing multiple disulfides is based on sequence-specific fragmentation and the typical DSB-related fragment ion quartet composed of [MH+], [MH+]+ S, [MH+]-H2, and [MH+]-SH2 is present in the same MS/MS spectrum. We used this principle to validate DSB-peptide identifications that correspond to the IgG2 isoform hinge peptides. The curated collection of signature peptides corresponding to each IgG2 disulfide isoform can now be used to enable automatic assignments of the DSB peptides. To our knowledge, MALDI MS/MS spectra of the A, A/B, and B isoform-specific hinge peptides and their interpretation have not been previously reported.
LC-MALDI-MS/MS can provide a rapid assessment of the disulfide connectivity in mAbs from a single dataset, without a priori assumptions about the expected disulfide bonds. Relative distributions of the native, scrambled, and partially-reduced isoforms were all demonstrated here using both IgG1 and IgG2 subclasses. Our method consumes relatively low sample amounts (1–2 µg) because ISD fragmentation of disulfide bonds efficiently yields reduced peptides together with the DSB-peptide precursor ion. The approach we presented can also serve as an orthogonal verification to the IgG2 disulfide isoform distribution determined via traditional LC methods. The high level of automation in this workflow also makes it a suitable approach in early development stages. By minimizing the need for tedious manual data analysis, the workflow described here has the potential to become an indispensable tool for both detailed characterization and high-throughput analyses.
4. Materials and methods
4.1. Materials
Fully human IgG2κ mAb was manufactured at Agensys, Inc. using in-house protocols and stored as a frozen, formulated bulk at 25 mg/mL in acetate/sucrose, pH 5.2 buffer. The antibody was produced in Chinese hamster ovary cells and purified using procedures established in-house at Agensys, Inc. The IgG1 adalimumab (Humira®, Abbvie) was purchased in its medication form (local pharmacy) in a concentration of 50 mg/mL, and used in the original injection formulation. The IgG1 with scrambled disulfides were generated by incubating adalimumab in its original buffer at 70°C for 30 min.
Pepsin, guanidine-HCl, HPLC-grade acetonitrile, sodium phosphate dibasic and monobasic were purchased from MilliporeSigma. HPLC-grade NEM and trifluoroacetic acid (TFA) were obtained from Merck. The MALDI matrices HCCA and sDHB were obtained from Bruker Daltonik.
4.2. Chromatographic fractionation of IgG2 disulfide isoforms
Collection of IgG2 disulfide isoforms utilized a Thermo Scientific Ultimate 3000RS UHPLC equipped with an AFC-3000 fraction collector. The starting material containing a mixture of 22% A, 33% A/B, and 45% B isoforms was fractionated using an Agilent Technologies Zorbax RRHD 300SB-C8, 2.1 mm ID x 50 mm L column with a 1.8 µm particle size and 300 Å pore size. The LC conditions were as follows: mobile phase A (MPA) composed of 0.1% TFA, 2% isopropyl alcohol (IPA) in water and mobile phase B (MPB) composed of 0.1% TFA, 25% CH3CN in IPA were flowed through the 82°C column at 0.4 mL/min. The gradient delivered 25.0% – 27.5% MPB over 6 minutes, followed by 27.5% – 28.0% MPB over 1 minute, and finally 28.0% – 30.0% MPB over 1 minute. Fraction collection was programed by time based on the elution time and peak width of each disulfide isoform.
4.3. Trypsin/Lys-C digestion under non-reducing conditions
The adalimumab and IgG2 mAb fractions (~ 50 µg each) were denatured and free thiol groups alkylated via incubation for 3 h at 37°C in 5.8 M guanidine-HCl and 7 mM NEM. The samples were purified and buffer exchanged using Amicon Ultra-0.5 centrifugal filters with 10 kDa MWCO (MilliporeSigma). Denatured and alkylated samples were digested overnight at 37°C with Mass Spec Grade Trypsin/Lys-C mix (Promega), at an enzyme-to-substrate ratio of 1:10 in the presence of 0.175 mM NEM in a 50 mM sodium phosphate buffer, pH 5.8.
4.4. LC separation of digested peptides
The digested mAb (~ 2 µg) was separated by RP-nanoLC (Ultimate 3000 RSLCnano LC system, Thermo Scientific). A Thermo Scientific Acclaim PepMap RSLC C18 column (15 cm x 300 µm, 2 µm particle size, 100 Å pore size) was equilibrated with 98% MPA (water with 0.1% TFA) and 2% MPB (0.1% TFA in CH3CN) flowing at 4 µL/min. A gradient from 2% to 45% B over 30 min was applied to separate the digest.
Fifteen second fractions were collected by the PROTEINEER fc II LC-MALDI fraction collector (Bruker) on MTP AnchorChip sample plates (Bruker) with hydrophilic anchors as follows. In a first screening experiment for the detection of all DSB-peptides containing one SS-bond (therefore, not the hinge peptides), fractions were spotted on 800 µm anchors with a sheath flow of 0.5 µL per spot of a prepared HCCA solution consisting of 36 µL saturated HCCA (in 90% CH3CN/10% water) mixed with 748 µL of 85% acetonitrile/15% water, 8 µL of 1 mM ammonium phosphate, and 8 µL of 10% TFA in water. Calibrant spots were prepared with a mixture of synthetic peptides (peptide calibration standard II, Bruker) plus bovine Insulin, (Sigma) and matrix to cover a calibration range from 700 to 7000 Da, allowing for external calibration with < 10 ppm mass accuracy.
For the detection of hinge peptides greater than 7 kDa, the fractions were spotted on the 2 mm anchors of a MTP BigAnchor 384 target (Bruker) and 0.5 µL of sDHB matrix solution consisting of 50 mg sDHB (Bruker) dissolved in 0.1% TFA in 50% water/50% CH3CN manually on the spots with the dried fractions. Proper crystallization of the matrix was verified visually.
4.5. Mass spectrometry
Data acquisition and processing were generated and processed using Bruker rapifleX-TOF/TOF using compass for flex series 2.0 with standard methods. Spectra were acquired at positive reflector ion mode with an acceleration voltage of 20 kV. Eight thousand laser shots were accumulated per fraction in MS mode at a laser shot rate of 5 kHz, and 5000 shots were accumulated for every MS/MS spectrum with the same laser frequency. LC-MALDI datasets were imported in BioPharma Compass 2.0 (BPC 2.0) and peptides identified using in silico digestion of the sequences of the IgG2 LC and HC. The collection of identified peptides was then further processed in DisulfideDetect 1.2.2 (Bruker) for automatic identification and quantification of the DSB peptides in the LC-MALDI dataset.
Multiply disulfide-bonded peptides (i.e., hinge peptides) required manual matching of the data to the theoretical MWs of hinge peptides. Fractions containing mass spectral peaks matching the theoretical hinge peptide masses of the B form were interrogated again for mis-cleaved proteolytic products or putative reduced forms of the hinge peptide. MS/MS spectra from all possible precursors were matched with their theoretical structures by annotating typical MALDI-MS/MS quartet fragment ion patterns of disulfide-bonded peptides (Figure 9–11). This approach allowed the identification of large DSB-peptides that achieved lower sequence coverage by MS/MS.
Acknowledgments
The authors would like to thank the Agensys Process Sciences and Manufacturing group for providing the IgG2 mAb used in this study and Mike Greig (Bruker) for critical discussion of this manuscript. This work was done in a collaboration between Agensys and Bruker.
Author contributions
The manuscript was written through contributions of AS, LL, DS, and GT. FW, AF, GR, and AM provided technical contributions. FW and AF were employees of Agensys, Inc. at the time of the study. All authors have given approval to the final version of the manuscript.
Disclosure statement
The authors declare they do not have any conflicts of interest.
Abbreviations
- CID
Collision-induced dissociation
- DSB
Disulfide Bond
- DSB peptide
Disulfide-bonded peptide
- HC
Heavy Chain
- IPA
Isopropyl alcohol
- ISD
In-source decay
- LC
Liquid Chromatography, Light Chain
- mAb
Monoclonal antibody
- MALDI
Matrix-Assisted Laser Desorption/Ionization
- MW
Molecular weight
- NEM
N-ethylmaleimide
- sDHB
super DHB: 9:1 (w/w) 2,5-dihydroxybenzoic acid (2,5-DHB) and 2-hydroxy-5-methoxybenzoic acid
- TFA
Trifluoroacetic acid
- XIC
Extracted Ion Chromatogram
References
- 1.The Antibody Society Approved antibodies; [accessed 2018. February 02]. https://www.antibodysociety.org/news/approved-antibodies/
- 2.Sau S, Alsaab HO, Kashaw SK, Tatiparti K, Iyer AK.. Advances in antibody-drug conjugates: a new era of targeted cancer therapy. Drug Discov Today. 2017;22(10):1547–1556. doi: 10.1016/j.drudis.2017.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaplon H, Reichert JM. Antibodies to watch in 2018. mAbs. 2018;10(2):183–203. doi: 10.1080/19420862.2018.1415671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jefferis R. Antibody therapeutics: isotype and glycoform selection. Expert Opin Biol Ther. 2007;7(9):1401–1413. doi: 10.1517/14712598.7.9.1401. [DOI] [PubMed] [Google Scholar]
- 5.Jefferis R, Lefranc M-P. Human immunoglobulin allotypes: possible implications for immunogenicity. mAbs. 2009;1(4):332–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. 2014;5:.520. doi: 10.3389/fimmu.2014.00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saphire EO, Parren PW, Pantophlet R, Zwick MB, Morris GM, Rudd PM, Dwerk RA, Stanfield RL, Burton DR, Wilson IA. Crystal structure of a neutralizing human IgG against HIV-1: a template for vaccine design. Science. 2001;293(5532):1155–1159. doi: 10.1126/science.1061692. [DOI] [PubMed] [Google Scholar]
- 8.Ryazantsev S, Tischenko V, Nguyen C, Abramov V, Zav’yalov V. Three-dimensional structure of the human myeloma IgG2. PLoS ONE. 2013;8:.e64076. doi: 10.1371/journal.pone.0064076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scapin G, Yang X, Prosise WW, McCoy M, Reichert P, Johnston JM, Kashi RS, Strickland C. Structure of full-length human Anti-PD1 therapeutic IgG4 antibody pembrolizumab. Nat Struct Mol Biol. 2015;22(12):953–958. doi: 10.1038/nsmb.3129. [DOI] [PubMed] [Google Scholar]
- 10.Zhang A, Fang J, Chou RYT, Bondarenko PV, Zhang Z. Conformational difference in human IgG2 disulfide isoforms revealed by hydrogen/deuterium exchange mass spectrometry. Biochemistry. 2015;54(10):1956–1962. doi: 10.1021/bi5015216. [DOI] [PubMed] [Google Scholar]
- 11.Dillon TM, Ricci MS, Vezina C, Flynn GC, Liu YD, Rehder DS, Plant M, Henkle B, Li Y, Deechongki S, et al. Structural and functional characterization of disulfide isoforms of the human IgG2 subclass. J Biol Chem. 2008;283(23):16206–16215. doi: 10.1074/jbc.M709988200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wypych J, Li M, Guo A, Zhang Z, Martinez T, Allen MJ, Fodor S, Kelner DN, Flynn GC, Liu YD, et al. Human IgG2 antibodies display disulfide-mediated structural isoforms. J Biol Chem. 2008;283(23):16194–16205. doi: 10.1074/jbc.M709987200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y, Lu Q, Wu S-L, Karger BL, Hancock WS. Characterization and comparison of disulfide linkages and scrambling patterns in therapeutic monoclonal antibodies - using LC-MS with electron transfer dissociation. Anal Chem. 2011;83(8):3133–3140. doi: 10.1021/ac200128d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu YD, Chou RY, Dillon TM, Poppe L, Spahr C, Shi SD, Flynn GC. Protected hinge in the immunoglobulin G2-A2 disulfide isoform. Protein Sci. 2014;23(12):1753–1764. doi: 10.1002/pro.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu YD, Chen X, Enk JZ-V, Plant M, Dillon TM, Flynn GC. Human IgG2 antibody disulfide rearrangement in vivo. J Biol Chem. 2008;283(43):29266–29272. doi: 10.1074/jbc.M804787200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu H, May K. Disulfide bond structures of IgG molecules: structural variations, chemical modifications and possible impacts to stability and biological function. mAbs. 2012;4(1):17–23. doi: 10.4161/mabs.4.1.18347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moritz B, Stracke JO. Assessment of disulfide and hinge modifications in monoclonal antibodies. Electrophoresis. 2017;38(6):769–785. doi: 10.1002/elps.201600425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He Y, Lacher NA, Hou W, Wang Q, Isele C, Starkey J, Ruesch M. Analysis of identity, charge variants, and disulfide isomers of monoclonal antibodies with capillary zone electrophoresis in an uncoated capillary column. Anal Chem. 2010;82(8):3222–3230. doi: 10.1021/ac9028856. [DOI] [PubMed] [Google Scholar]
- 19.Wang X, Kumar S, Singh SK. Disulfide scrambling in IgG2 monoclonal antibodies: insights from molecular dynamics simulations. Pharm Res. 2011;28(12):3128–3144. doi: 10.1007/s11095-011-0503-9. [DOI] [PubMed] [Google Scholar]
- 20.Alabi O, Dement-Brown J, Tolnay M. Human Fc receptor-like 5 distinguishes IgG2 disulfide isoforms and deamidated charge variants. Mol Immunol. 2017;92:.161–168. doi: 10.1016/j.molimm.2017.10.020. [DOI] [PubMed] [Google Scholar]
- 21.Liu-Shin L, Fung A, Malhotra A, Ratnaswamy G. Influence of disulfide bond isoforms on drug conjugation sites in cysteine-linked IgG2 antibody-drug conjugates. mAbs. 2018;10(4):583–595. doi: 10.1080/19420862.2018.1440165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang H, Cui W, Gross ML. Mass spectrometry for the biophysical characterization of therapeutic monoclonal antibodies. FEBS Lett. 2014;588(2):308–317. doi: 10.1016/j.febslet.2013.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grujic O, Stevens J, Chou RY, Weiszmann JV, Sekirov L, Thomson C, Badh A, Grauer S, Chan B, Graham K, et al. Impact of antibody subclass and disulfide isoform differences on the biological activity of CD200R and βklotho agonist antibodies. Biochem Biophys Res Commun. 2017;486(4):985–991. doi: 10.1016/j.bbrc.2017.03.145. [DOI] [PubMed] [Google Scholar]
- 24.Liu H, Lei QP, Washabaugh M. Characterization of IgG2 disulfide bonds with LC/MS/MS and postcolumn online reduction. Anal Chem. 2016;88(10):5080–5087. doi: 10.1021/acs.analchem.5b04368. [DOI] [PubMed] [Google Scholar]
- 25.White AL, Chan HT, French RR, Willoughby J, Mockridge CI, Roghanian A, Penfold CA, Booth SG, Dodhy A, Polak ME, et al. Conformation of the human immunoglobulin G2 hinge imparts superagonistic properties to immunostimulatory anticancer antibodies. Cancer Cell. 2015;27(1):138–148. doi: 10.1016/j.ccell.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wiesner J, Resemann A, Evans C, Suckau D, Jabs W. Advanced mass spectrometry workflows for analyzing disulfide bonds in biologics. Expert Rev Proteomics. 2015;12(2):115–123. doi: 10.1586/14789450.2015.1018896. [DOI] [PubMed] [Google Scholar]
- 27.Wang T, Liu YD, Cai B, Huang G, Flynn GC. 2015. Investigation of antibody disulfide reduction and re-oxidation and impact to biological activities. J Pharm Biomed Anal. 102:519–528. doi: 10.1016/j.jpba.2014.10.023. [DOI] [PubMed] [Google Scholar]
- 28.Bloom JW, Madanat MS, Marriott D, Wong T, Chan SY. Intrachain disulfide bond in the core hinge region of human IgG4. Protein Sci. 1997;6(2):407–415. doi: 10.1002/pro.5560060217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sung W-C, Chang C-W, Huang S-Y, Wei T-Y, Huang Y-L, Lin YH, Chen HM, Chen SF. Evaluation of disulfide scrambling during the enzymatic digestion of bevacizumab at various pH values using mass spectrometry. Biochim Biophys Acta. 2016;1864(9):1188–1194. doi: 10.1016/j.bbapap.2016.05.011. [DOI] [PubMed] [Google Scholar]
- 30.Liu YD, Wang T, Chou R, Chen L, Kannan G, Stevenson R, Goetze AM, Jiang XG, Huang G, Dillon TM, et al. IgG2 disulfide isoform conversion kinetics. Mol Immunol. 2013;54(2):217–226. doi: 10.1016/j.molimm.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 31.Wu S-L, Jiang H, Lu Q, Dai S, Hancock WS, Karger BL. Mass spectrometric determination of disulfide linkages in recombinant therapeutic proteins using online LC-MS with electron-transfer dissociation. Anal Chem. 2009;81(1):112–122. doi: 10.1021/ac801560k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang B, Harder AG, Connelly HM, Maheu LL, Cockrill SL. Determination of fab−hinge disulfide connectivity in structural isoforms of a recombinant human immunoglobulin G2 antibody. Anal Chem. 2010;82(3):1090–1099. doi: 10.1021/ac902466z. [DOI] [PubMed] [Google Scholar]
- 33.Allen MJ, Guo A, Martinez T, Han M, Flynn GC, Wypych J, Liu YD, Shen WD, Dillon TM, Vezina C, et al. Interchain disulfide Bonding in Human IgG2 Antibodies Probed by Site-Directed Mutagenesis. Biochemistry. 2009;48(17):3755–3766. doi: 10.1021/bi8022174. [DOI] [PubMed] [Google Scholar]
- 34.Yen TY, Joshi RK, Yan H, Seto NO, Palcic MM, Macher BA. Characterization of cysteine residues and disulfide bonds in proteins by liquid chromatography/electrospray ionization tandem mass spectrometry. J Mass Spectrom. 2000;35(8):990–1002. doi: 10.1002/1096-9888(200008)35:8<990::AID-JMS27>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 35.Guan X, Zhang L, Wypych J. Direct mass spectrometric characterization of disulfide linkages. mAbs. 2018;10(4):572–582. doi: 10.1080/19420862.2018.1442998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin M, Campbell JM, Mueller DR, Wirth U. Intact protein analysis by matrix-assisted laser desorption/ionization tandem time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 2003;17(16):1809–1814. doi: 10.1002/rcm.1102. [DOI] [PubMed] [Google Scholar]
- 37.Schnaible V, Wefing S, Resemann A, Suckau D, Bucker A, Wolf-Kümmeth S, Hoffman D. Screening for disulfide bonds in proteins by MALDI in-source decay and LIFT-TOF/TOF-MS. Anal Chem. 2002;74(19):4980–4988. [DOI] [PubMed] [Google Scholar]
- 38.Yang H, Liu N, Qiu X, Liu S, New A. Method for analysis of disulfide-containing proteins by matrix-assisted laser desorption ionization (MALDI) mass spectrometry. J Am Soc Mass Spectrom. 2009;20(12):2284–2293. doi: 10.1016/j.jasms.2009.08.020. [DOI] [PubMed] [Google Scholar]
- 39.Brown RS, Lennon JJ. Sequence-specific fragmentation of matrix-assisted laser desorbed protein/peptide ions. Anal Chem. 1995;67(21):3990–3999. [DOI] [PubMed] [Google Scholar]
- 40.Reiber DC, Grover TA, Brown RS. Identifying proteins using matrix-assisted laser desorption/ionization in-source fragmentation data combined with database searching. Anal Chem. 1998;70(4):673–683. [DOI] [PubMed] [Google Scholar]
- 41.Hardouin J. Protein sequence information by matrix-assisted laser desorption/ionization in-source decay mass spectrometry. Mass Spectrom Rev. 2007;26(5):672–682. doi: 10.1002/mas.20142. [DOI] [PubMed] [Google Scholar]
- 42.Takayama M. 2001. N-Cα Bond Cleavage of the Peptide Backbone via Hydrogen Abstraction. J Am Soc Spectrom. 12:1044–1049. doi: 10.1016/S1044-0305(01)00289-6. [DOI] [Google Scholar]
- 43.Demeure K, Quinton L, Gabelica V, De Pauw E. Rational selection of the optimum MALDI matrix for top-down proteomics by in-source decay. Anal Chem. 2007;79(22):8678–8685. doi: 10.1021/ac070849z. [DOI] [PubMed] [Google Scholar]
- 44.Demeure K, Gabelica V, De Pauw EA. New advances in the understanding of the in-source decay fragmentation of peptides in MALDI-TOF-MS. J Am Soc Mass Spectrom. 2010;21(11):1906–1917. doi: 10.1016/j.jasms.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 45.Patterson SD, Katta V. Prompt fragmentation of disulfide-linked peptides during matrix-assisted laser desorption ionization mass spectrometry. Anal Chem. 1994;66(21):3727–3732. [DOI] [PubMed] [Google Scholar]
- 46.Quinton L, Demeure K, Dobson R, Gilles N, Gabelica V, De Pauw E. New method for characterizing highly disulfide-bridged peptides in complex mixtures: application to toxin identification from crude venoms. J Proteome Res. 2007;6(8):3216–3223. doi: 10.1021/pr070142t. [DOI] [PubMed] [Google Scholar]
- 47.Yang H, Liu N, Liu S. 2013. Determination of peptide and protein disulfide linkages by MALDI mass spectrometry. Top Curr Chem. 331:79–116. doi: 10.1007/128_2012_384. [DOI] [PubMed] [Google Scholar]
- 48.Macht M, Asperger A, Deininger SO. Comparison of laser-induced dissociation and high-energy collision-induced dissociation using matrix-assisted laser desorption/ionization tandem time-of-flight (MALDI-TOF/TOF) for peptide and protein identification. Rapid Comm Mass Spectrom. 2004;18(18):2093–2105. doi: 10.1002/rcm.1589. [DOI] [PubMed] [Google Scholar]
- 49.Debois D, Bertrand V, Quinton L, De Pauw-Gillet MC, De Pauw E. MALDI-in source decay applied to mass spectrometry imaging: a new tool for protein identification. Anal Chem. 2010;82(10):4036–4045. doi: 10.1021/ac902875q. [DOI] [PubMed] [Google Scholar]
- 50.Ayoub D, Jabs W, Resemann A, Evers W, Evans C, Main L, Baessmann C, Wagner-Rousset E, Suckau D, Beck A. Correct primary structure assessment and extensive glyco-profiling of cetuximab by a combination of intact, middle-up, middle-down and bottom-up ESI and MALDI mass spectrometry techniques. mAbs. 2013;5(5):699–710. doi: 10.4161/mabs.25423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Resemann A, Wunderlich D, Rothbauer U, Warscheid B, Leonhardt H, Fuchser J, Kuhlmann K, Suckau D. Top-down de novo protein sequencing of a 13.6 kDa camelid single heavy chain antibody by matrix-assisted laser desorption ionization-time-of-flight/time-of-flight mass spectrometry. Anal Chem. 2010;82(8):3283–3292. doi: 10.1021/ac1000515. [DOI] [PubMed] [Google Scholar]
- 52.Resemann A, Jabs W, Wiechmann A, Wagner E, Colas O, Evers W, Belau E, Vorwerg L, Evans C, Beck A, et al. Full validation of therapeutic antibody sequences by middle-up mass measurements and middle-down protein sequencing. mAbs. 2016;8(2):318–330. doi: 10.1080/19420862.2015.1128607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suckau D, Resemann A, Schuerenberg M, Hufnagel P, Franzen J, Holle A, Novel A. MALDI LIFT-TOF/TOF mass spectrometer for proteomics. Anal Bioanal Chem. 2003;376(7):952–965. doi: 10.1007/s00216-003-2057-0. [DOI] [PubMed] [Google Scholar]
- 54.Yoo C, Suckau D, Sauerland V, Ronk M, Ma M. Toward top-down determination of PEGylation site using MALDI in-source decay MS analysis. J Am Soc Mass Spectrom. 2009;20(2):326–333. doi: 10.1016/j.jasms.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 55.Bakalarski CE, Gan Y, Wertz I, Lill JR, Sandoval W. Rapid, semi-automated protein terminal characterization using ISDetect. Nat Biotechnol. 2016;34(8):811–813. doi: 10.1038/nbt.3621. [DOI] [PubMed] [Google Scholar]
- 56.Katta V, Chow DT, Rohde MF. Applications of in-source fragmentation of protein ions for direct sequence analysis by delayed extraction MALDI-TOF mass spectrometry. Anal Chem. 1998;70(20):4410–4416. [DOI] [PubMed] [Google Scholar]
- 57.Maltman DJ, Brand S, Belau E, Paape R, Suckau D, Przyborski SA. Top-down label-free LC-MALDI analysis of the peptidome during neural progenitor cell differentiation reveals complexity in cytoskeletal protein dynamics and identifies progenitor cell markers. Proteomics. 2011;11(20):3992–4006. doi: 10.1002/pmic.201100024. [DOI] [PubMed] [Google Scholar]
- 58.Neubert H, Bonnert TP, Rumpel K, Hunt BT, Henle ES, James IT. Label-free detection of differential protein expression by LC/MALDI mass spectrometry. J Proteome Res. 2008;7(6):2270–2279. doi: 10.1021/pr700705u. [DOI] [PubMed] [Google Scholar]
- 59.Clark DF, Go EP, Toumi ML, Desaire H. Collision induced dissociation products of disulfide-bonded peptides: ions result from the cleavage of more than one bond. J Am Soc Mass Spectrom. 2011;22(3):492–498. doi: 10.1007/s13361-010-0064-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lowe EK, Anema SG, Bienvenue A, Boland MJ, Creamer LK, Jiménez-Flores R. Heat-induced redistribution of disulfide bonds in milk proteins. 2. Disulfide bonding patterns between bovine β-Lactoglobulin and κ-Casein. J Agric Food Chem. 2004;52(25):7669–7680. doi: 10.1021/jf0491254. [DOI] [PubMed] [Google Scholar]
- 61.Padliya ND, Wood TD. A strategy to improve peptide mass fingerprinting matches through the optimization of matrix-assisted laser desorption/ionization matrix selection and formulation. Proteomics. 2004;4(2):466–473. doi: 10.1002/pmic.200300567. [DOI] [PubMed] [Google Scholar]
- 62.Franey H, Brych SR, Kolvenbach CG, Rajan RS. Increased aggregation propensity of IgG2 subclass over IgG1: role of conformational changes and covalent character in isolated aggregates. Protein Sci. 2010;19(9):1601–1615. doi: 10.1002/pro.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang W, Czupryn MJ. Free sulfhydryl in recombinant monoclonal antibodies. Biotechnol Prog. 2002;18(3):509–513. doi: 10.1021/bp025511z. [DOI] [PubMed] [Google Scholar]
- 64.Zhang W, Marzilli LA, Rouse JC, Czupryn MJ. Complete disulfide bond assignment of a recombinant immunoglobulin G4 monoclonal antibody. Anal Biochem. 2002;311(1):1–9. [DOI] [PubMed] [Google Scholar]
- 65.Martinez T, Guo A, Allen MJ, Han M, Pace D, Jones J, Gillespie R, Ketchem RR, Zhang Y, Balland A. Disulfide connectivity of human immunoglobulin G2 Structural isoforms. Biochemistry. 2008;47(28):7496–7508. doi: 10.1021/bi800576c. [DOI] [PubMed] [Google Scholar]