

Abstract
Background/Aims
Atrophic gastritis (AG), intestinal metaplasia (IM), and Helicobacter pylori (HP) are the risk factors for the development of gastric cancer (GC). Chromatin remodeling is one of the epigenetic mechanisms involved in the carcinogenesis of GC. The purpose of this study was to investigate the expression profiles of defined chromatin remodeling genes in gastric mucosal samples and their values as gastric carcinogenesis biomarkers.
Materials and Methods
In total, 95 patients were included in the study. Patients were divided into 3 groups as: GC group (n=34), AG group (n=36), and control group (n=25). AG group was further divided into subgroups based on the presence of HP and IM in gastric mucosa. Chromatin remodeling gene expressions were analyzed using real-time PCR (RT-PCR) array in all groups. Data were evaluated using the RT-qPCR primer assay data analysis software.
Results
EED, CBX3, and MTA1 were more overexpressed, whereas ARID1A, ING5, and CBX7 were more underexpressed in the AG and GC groups compared with the controls. No significant differences were observed between the AG and GC groups concerning the expression of these 6 genes, although the fold change levels of these genes in the GC group were well above than in the AG group. EED, CBX3, and MTA1 were significantly more overexpressed in HP- and IM-positive AG subgroup compared with the HP- or IM-negative AG subgroup.
Conclusion
In conclusion, our results provide an evidence of epigenetic alterations in AG. Expressions of EED, CBX3, MTA1, ARID1A, ING5, and CBX7 may be considered as promising markers to be used in GC screening for patients with AG.
Keywords: Atrophic gastritis, chromatin remodeling, epigenetic, gastric cancer, Helicobacter pylori, intestinal metaplasia
INTRODUCTION
Worldwide, gastric cancer (GC) ranks as the second-most prevalent cause of cancer-related mortality (1). It is known that inflammation is an important factor in GC carcinogenesis. GC carcinogenesis with several genetic and epigenetic changes, triggered by inflammation, follows the sequence of gastritis-atrophy-metaplasia-dysplasia-cancer (2,3). Atrophic gastritis (AG), intestinal metaplasia (IM), and Helicobacter pylori (HP) are well-known carcinogenic pathogens for GC (4). Furthermore, HP infection shows a strong association with chronic AG and IM (5). There were no specific, facilitating, and useful markers for GC screening in precancerous lesions such as AG, IM, and dysplasia. There is a clear need for more predictive and selective markers to detect the development GC in patients with AG.
Gradual genetic and epigenetic alternations occur in several proto-oncogene and tumor suppressor genes that are involved in inflammation-associated carcinogenesis stages (6). Understanding these genetic and epigenetic changes in carcinogenesis stages leads to important information on the development of cancer from precancerous lesions. Screening of precancerous and cancerous cells improves as our limited body of knowledge on cancer molecular biology expands (7). It is known that epigenetic changes have similar effects as genetic changes. They can trigger carcinogenesis by inducing proto-oncogenes or suppressing tumor suppressor genes. Epigenetic alterations, in contrast to genetic ones, are reversible in carcinogenesis (8). Epigenetic treatments may be used for the treatment of cancer or prevention of the formation of precancerous lesions in the future (9).
Epigenetic alternations, such as chromatin remodeling, have been observed and described in the pathways of GC carcinogenesis, which include such as chromosomal instability and microsatellite instability pathway, β-catenin/Wnt, TGFβ/SMAD, and RAF/RAS/MAPK intermediary pathway (10–12). Epigenetic changes, which lead to subsequent aberrant gene expression, play key roles as ultimate predictors in inflammation-induced GC carcinogenesis (11). Chromatin remodeling genes, such as EED, ARID1A, ING5, CTBP1, CBX3, CBX7, MTA1, and NSD1, have key roles in the carcinogenesis of GC (10,11,13–16). There are no studies about chromatin remodeling gene expression profiles in AG and their values as gastric carcinogenesis biomarkers. Further studies are needed to elucidate the value of these defined genes as biomarkers for GC screening. Therefore, the purpose of this study was to investigate the diagnostic value of chromatin remodeling genes in GC progression from AG by comparing the expressions of chromatin remodeling genes in gastric mucosal samples, including normal gastric mucosa, AG, and GC.
MATERIALS AND METHODS
Study location
The study was undertaken by the Departments of Gastroenterology and Medical Biology in Manisa Celal Bayar University between September 2014 and September 2015.
Ethics
This study was conducted by the application of good clinical practice to comply with the Helsinki declaration. Ethical approval (#160) for the study was obtained from the Institutional Ethical Review Board of Manisa Celal Bayar University Medical Center on June 4, 2014. Written informed consent from each subject was obtained prior to any study-related procedure.
Study design and samples
Matched tissue samples were collected from 95 patients who came to the Department of Gastroenterology in Manisa Celal Bayar University between September 2014 and September 2015. Patients were categorized into 3 groups. The first group contained 34 patients with GC, who were confirmed macroscopically and histopathologically. The second group included 36 confirmed patients with AG, whereas the third group was the control group, which was composed of 25 healthy individuals with normal gastric mucosa and without GC or AG. Patients with cancer of any type who received chemotherapy or radiotherapy, non-GC patients, and patients who underwent gastrectomy were excluded from the study. All relevant demographical information was recorded for all participants. In the AG group, the duration of disease, severity of activation, routine examination, relevant pathological assessment results, and proof of HP and/or IM role in gastric mucosa were recorded for comparison. GC staging was made via CT or PET-CT.
Endoscopy
Endoscopy was performed on all patients by experienced gastroenterologists in the Gastroenterology Department of Manisa Celal Bayar University using Olympus Luxera GIF-H260 unit. Complete endoscopic examination, including that of the esophagus, stomach, and duodenum, was performed for the control and AG groups. Endoscopic examination for the GC group was conducted if the tumor growth did not block the access. Distal tumor was evaluated by abdominal CT when it could not be accessed.
Biopsy samples for histopathological and epigenetic (chromatin remodeling gene expression analyses) assessments were taken using Olympus biopsy forceps from tumor tissue, gastric mucosa, and antrum/corpus mucosa in the GC, AG, and control groups, respectively. Pathological examinations of all biopsy samples were accomplished in the Pathology Department of Manisa Celal Bayar University.
RNA isolation from tissue
Tissue samples were quickly frozen on a block of dry ice and stored at −80°C until RNA isolation. Total RNA from the tissue samples was isolated using RNeasy Mini Kit (Qiagen, Germany) following the manufacturer’s protocol with small modifications. Briefly, tissue samples (20–30 mg) were homogenized with 600 μL of RLT buffer for 5 min with the help of a 7-mm-diameter metal ball using a Tissue Lyser II homogenizer (Qiagen, Germany) at 25,000 Hz. Tissue lysate was centrifuged at maximum speed and the supernatant was used in the RNeasy Mini Kit Protocol.
RNA quality
RNA quantification was measured at 260 nm (A260). RNA purity was assessed by the A260/A280 ratio, and the quality of RNA was regarded as satisfactory if A260/A280>2.0 and A260/A230>1.8.
cDNA synthesis
RT First Strand Kit (C-03) (SABioscience, Frederick, MD, USA) was used for cDNA synthesis. Overall, 8 μL of RNA sample was mixed with 2 μL of 5× GE Buffer (gDNA Elimination Buffer) and incubated at 42°C for 5 min. The PCR cocktail composed of 4 μL of BC3 (5× RT Buffer 3), 1 μL of P2 (Primer and External Control mix), 2 μL of RE3 (RT Enzyme Mix 3), and 3 μL of H2O was prepared in another tube and combined with the RNA sample followed by a 15-min incubation at 42°C and 5 min at 95°C.
RT-PCR array
RT-PCR array mix (2300 μL) that is composed of 102 μL of diluted cDNA, 1150 μL of 2X RT2 SYBR Green ROX FAST Master mix, and 1048 μL of H2O was prepared; 20 μL/well of this mix was loaded onto a 96-well Human Epigenetic Chromatin Modification Enzymes RT2 Profiler™ PCR Array (PAHS-085Z) (SABiosciences, Frederick, MD, USA), which allows for the detection of the expression levels of 84 key genes listed in Table 1. Array was run on a Rotor-Gene RG-3000 (Corbett Research, Qiagen, Germany) for an initial period of 10 min at 95°C, followed by a two-step cycle of 15 s at 95°C and 30 s at 60°C for a total of 40 cycles. Resulting data were analyzed, and the most over- and underexpressed 8 genes were chosen to cross-validate by RT-qPCR primer assay.
Table 1.
Human epigenetic chromatin remodeling factors genes to be investigated
| Chromatin remodeling | Genes |
|---|---|
| SWI/SNF Complex Components | ARID1A, INO80 (INOC1), PBRM1, SMARCA2, SMARCA4 |
| Polycomb Group Genes | ASXL1, BMI1 (PCGF4), CTBP1, CTBP2, EED, EZH2, PCGF1, PCGF2, PCGF3, PCGF5, PCGF6, PHC1, PHC2, RING1, RNF2, SUZ12, TRIM27 |
| Chromobox/Heterochromatin Protein | |
| 1 (HP1) Homologs | CBX1, CBX3, CBX4, CBX5, CBX6, CBX7, CBX8 |
| Bromodomain Proteins | BAZ1A, BAZ1B, BAZ2A, BAZ2B, BPTF, BRD1, BRD2, BRD3, BRD4, BRD7, BRD8, BRDT, BRPF1, BRPF3, BRWD1, WDR11, BRWD3, ZMYND8 |
| Chromodomain/Helicase/DNA | |
| -Binding Domain (CHD) Proteins | CDYL, CDYL2, CHD1, CHD2, CHD3, CHD4, CHD5, CHD6, CHD7, CHD8, CHD9 |
| Nucleosome-Remodeling and Histone Deacetylase (NuRD) Complex Components | CHD3, MBD3, MTA1, MTA2, NAB2, SPEN |
| Plant Homeodomain (PHD) Proteins | NSD1, PHF1, PHF2, PHF3, PHF5A, PHF6, PHF7, PHF13, PHF21A, PHF21B |
| Inhibitor of Growth (ING) Family Members | ING1, ING2, ING3, ING4, ING5, RING1 |
| Methyl-CpG DNA-Binding Domain Proteins | MBD1, MBD2, MBD3, MBD4, MECP2, HINFP |
| CCCTC-Binding Factor (Zinc Finger Protein) | CTCF |
HP: Helicobacter pylori
RT-qPCR primer assay
The 8 genes, which were chosen as the most over- or underexpressed, were EED, ARID1A, ING5, CTBP1, CBX3, CBX7, MTA1, and NSD1. Also, a housekeeping gene, HPRT1, was selected for RT-qPCR. PCR reactions were performed by mixing 1 μL of specific primers (10 pmol) with 12.5 μL of SYBR Green Master Mix (SABiosciences, Frederick, MD, USA), 2.2 μL of cDNA sample, and dH2O to bring the volume to 25 μL. The qPCR primer assays were run on Rotor-Gene RG-3000 initially for 5 min at 95°C, followed by a three-step cycle of 1 min at 94°C, 40 s at 61°C, and 1 min at 72°C for 40 cycles; a final step of elongation at 72°C for 2 min was performed. The cycle threshold (Ct) values normalized against the housekeeping HPRT1 gene were analyzed with REST 2009 (Relative Expression Software Tool V.2.0.13) in standard mode.
Data analysis and statistic
RT-PCR and RT-qPCR data were analyzed using Microsoft Excel 2010 and the manufacturer’s online analysis RT2Profiler Plus PCR Array software at http://www.sabiosciences.com/pcrarraydataanalysis.php (Qiagen, Germany). Data analysis is based on the ΔΔCt method with the normalization of the raw data to either housekeeping genes or an external RNA control. The Ct values were used to quantify the expression levels in a PCR array. Any gene with a Ct value over 32 was categorized as not detectable, and a Ct value of 35 was taken during ΔCt calculations for those undetected genes. Genes with a fold change of 2 were considered as upregulated and those as 0.5 were considered as downregulated. Results were expressed as the mean±standard deviation, and the Student t-test was performed between the replicate 2−ΔCt values for each gene in the GC, AG, and control groups. A p-value of ≤0.05 was considered as statistically significant.
RESULTS
In total, 95 patients were included in the study between September 2014 and September 2015. Patients were divided into 3 groups as: GC group (n=34), AG group (n=36), and control group (n=25). The demographic characteristics are described in Table 2 for each of the 3 groups. The mean age of patients was 55.71±10.17 years with a male-to-female sex ratio of 61:35. There were no significant differences related to age and gender between the GC, AG, and control groups (Table 2).
Table 2.
Demographic results of patients
| GC (n=34) | AG (n=36) | Control (n=25) | p | ||
|---|---|---|---|---|---|
| Age | 58,85±7.5 | 55,71±10.17 | 54,84±7.4 | 0.66 | |
| Gender | Male | 68% (n=23) | 67% (n=24) | 56% (n=14) | 0.60 |
| Female | 32% (n=11) | 33% (n=12) | 44% (n=11) |
GC: gastric cancer; AG: atrophic gastritis
The sites of GC were assessed, and the majority of the tumors were found to be located in the proximal stomach. The proportion of patients with GC by the site of involvement was as follows: proximal involvement, 44.1% (n=15); distal involvement, 35.3% (n=12); and diffuse involvement, 20.6% (n=7). Pathological diagnosis showed adenocarcinoma for all patients with GC. Of all the patients with GC, 47% (n=16) had undifferentiated tumors and 53% (n=18) had differentiated tumors. Nearly 20.6% (n=7) of the patients with GC had mucinous and signet ring cell adenocarcinomas. Distant-organ metastases, including peritoneum (26%, n=9), liver (15%, n=5), pancreas (%3, n=1), and pulmonary (3%, n=1), were detected in a total of 47% (n=16) of patients with GC.
The overexpression of EED, CBX3, and MTA1 genes was significantly higher in the GC group than in the control group (fold change>2). The expression of ARID1A, ING5, and CBX7 genes was significantly lower in the GC group than in the control group (fold change<0.5 and fold regulation<−2). There were no significant differences related to the expression of CTBP1 and NSD1 genes between the GC and control groups (fold change<2, p>0.05) (Table 3) (Figure 1, 2).
Table 3.
Chromatin remodeling gene expression analysis
| GC | Intestinal type GC | Non-intestinal type GC | Atrophic Gastritis | |||||
|---|---|---|---|---|---|---|---|---|
| Gene | Fold change | p | Fold change | p | Fold change | p | Fold change p | |
| EED | 12.5222 | 0.000002 | 24.1447 | 0.00001 | 9.018 | 0.000186 | 9.8957 | 0.01312 |
| ING5 | 0.0908 | 0.000005 | 0.1568 | 0.00886 | 0.1781 | 0.000471 | 0.1273 | 0.000016 |
| MTA1 | 10.4114 | 0.000007 | 21.5986 | 0.00001 | 7.2285 | 0.000805 | 8.4081 | 0.009641 |
| CTBP1 | 1.4982 | 0.743963 | 1.1662 | 0.53807 | 1.6961 | 0.500348 | 1.0972 | 0.414009 |
| NSD1 | 1.3354 | 0.684861 | 1.4137 | 0.61029 | 1.3359 | 0.857535 | 0.8017 | 0.467536 |
| CBX3 | 9.4826 | 0.000006 | 20.0195 | 0.00001 | 6.5262 | 0.000571 | 8.7146 | 0.009204 |
| CBX7 | 0.071 | 0.003226 | 0.1255 | 0.09518 | 0.1425 | 0.02124 | 0.1021 | 0.00463 |
| ARID1A | 0.097 | 0.003279 | 0.1636 | 0.09797 | 0.1871 | 0.023799 | 0.1332 | 0.005093 |
| HPRT1 | 1 | - | 1 | - | 1 | - | 1 | - |
Note: Intestinal-type GC group, non-intestinal type GC, and AG group were compared with the control group GC: gastric cancer; AG: atrophic gastritis
Figure 1.

The clustergram creates a heatmap with dendrograms to indicate genes that are co-regulated. The color saturation reflects the magnitude of the change in gene expression. Green squares represent lower gene expression in the experimental samples (ratios<1), black squares represent genes equally expressed (ratios~1), red squares represent higher than control levels of gene expression (ratios>1), and gray squares indicate insufficient or missing data. The x-axis indicates the groups (GC; AG; CTR, control group), and the y-axis indicates the genes
Figure 2.

Overexpressed Genes in GC: Hybridization intensity of each gene in the GC and control groups appears as a log10 base scattered plateau. The x-axis represents the control group, and the y-axis represents the GC group
The overexpression of EED, CBX3, and MTA1 genes was significantly higher in the AG group than in the control group (fold change>2). The expression of ARID1A, ING5, and CBX7 genes was significantly lower in the AG than in the control group (fold change<0.5 and fold regulation<−2). There were no significant differences related to the expression of CTBP1 and NSD1 genes between the AG and control groups (fold change<2, p>0.05) (Table 3) (Figure 1, 3).
Figure 3.

Overexpressed Genes in AG: Hybridization intensity of each gene in the AG and control groups appears as a log10 base scattered plateau. The x-axis represents the control group, and the y-axis represents the AG group
There were no significant differences between the expression levels of EED, CBX3, MTA1, ARID1A, ING5, and CBX7 gene between patients with AG and those with GC, although the expression level (fold change level) of these genes was higher in patients with GC than in those with AG (Table 4, 5) (Figure 4).
Table 4.
Comparison of GC and AG groups for gene expressions
| Gene | Fold change | 95% CI | p |
|---|---|---|---|
| EED | 1.2654 | (0.58, 1.95) | 0.82088 |
| ING5 | 1.3858 | (0.96, 1.81) | 0.453466 |
| MTA1 | 1.2383 | (0.53, 1.94) | 0.376929 |
| CTBP1 | 1.3643 | (0.71, 2.02) | 0.245189 |
| NSD1 | 1.7091 | (0.96, 2.46) | 0.604644 |
| CBX3 | 1.0881 | (0.54, 1.63) | 0.742473 |
| CBX7 | 1.1527 | (0.80, 1.51) | 0.911585 |
| ARID1A | 1.2328 | (0.85, 1.61) | 0.714353 |
| HPRT1 | 1 | (1.00, 1.00) | - |
AG group was compared with the GC group
AG: atrophic gastritis; GC: gastric cancer; CI: confidence interval
Table 5.
Comparison of intestinal-type GC and AG groups for gene expressions
| Gene | Fold change | 95% CI | p |
|---|---|---|---|
| EED | 2.4399 | (1.28, 3.60) | 0.6077 |
| ING5 | 1.2317 | (0.82, 1.64) | 0.88578 |
| MTA1 | 2.5688 | (1.27, 3.87) | 0.91457 |
| CTBP1 | 1.0628 | (0.43, 1.70) | 0.83795 |
| NSD1 | 1.7634 | (0.94, 2.59) | 0.92465 |
| CBX3 | 2.2972 | (1.32, 3.27) | 0.51173 |
| CBX7 | 1.229 | (0.82, 1.64) | 0.88087 |
| ARID1A | 1.2285 | (0.82, 1.64) | 0.88108 |
| HPRT1 | 1 | (1.00, 1.00) | - |
Intestinal-type GC group was compared with the AG group GC: Gastric cancer; AG: atrophic gastritis; CI: confidence interval
Figure 4.
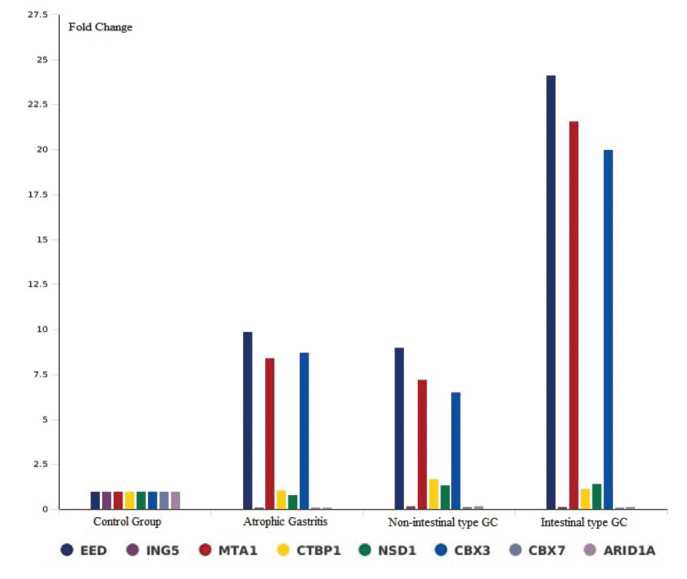
Fold Change Levels of Chromatin Remodeling Genes in All Groups: The multigroup plot provides a bar chart and line graph representation of the expression change for a selected set of genes across all of the tested groups. The x-axis indicates the groups (intestinal-type GC, non-intestinal-type GC, AG, and control group), and the y-axis indicates the fold change levels of chromatin remodeling genes
Patients with AG (n=36) were divided into 2 subgroups by HP invasion in gastric mucosa as follows: patients with positive HP invasion in gastric mucosa (33.3%, n=12) and patients with negative HP invasion in gastric mucosa (66.7%, n=24). The expression level of EED, CBX3, and MTA1 genes was significantly higher in the subgroup of patients with AG with positive HP invasion in gastric mucosa than in the subgroup of patients with AG with negative HP invasion in gastric mucosa. There were no differences related to the expression of ARID1A, ING5, and CBX7 genes between the HP-positive and HP-negative AG subgroups (Table 6).
Table 6.
Comparison of gene expressions in HP-positive and HP-negative AG
| Gene | Fold change | 95% CI | p |
|---|---|---|---|
| EED | 3.9964 | (1.17, 6.82) | 0.009386 |
| ING5 | 1.1947 | (0.49, 1.90) | 0.278388 |
| MTA1 | 8.2199 | (2.42, 14.02) | 0.000072 |
| CTBP1 | 0.7053 | (0.04, 1.37) | 0.901341 |
| NSD1 | 1.3233 | (0.07, 2.57) | 0.054525 |
| CBX3 | 4.9535 | (2.19, 7.72) | 0.000807 |
| CBX7 | 0.9325 | (0.37, 1.49) | 0.562173 |
| ARID1A | 0.9994 | (0.40, 1.60) | 0.467906 |
| HPRT1 | 1 | (1.00, 1.00) | - |
HP-positive AG subgroup was compared with the HP-negative AG subgroup HP: helicobacter pylori; AG: atrophic gastritis; CI: confidence interval
Patients with AG (n=36) were also divided into 2 subgroups by IM positivity in gastric mucosa as follows: patients with positive IM in gastric mucosa (52.8%, n=19) and patients with negative IM in gastric mucosa (47.2%, n=17). The expression level of EED, CBX3, and MTA1 genes was significantly higher in the subgroup of AG patients with positive IM in gastric mucosa than in the subgroup of patients with AG with negative IM in gastric mucosa. There were no differences related to the expression of ARID1A, ING5, and CBX7 genes between the IM-positive and IM-negative AG subgroups (Table 7).
Table 6.
Comparison of gene expressions in IM-positive and IM-negative AsG
| Gene | Fold change | 95% CI | p |
|---|---|---|---|
| EED | 4.8561 | (1.84, 7.88) | 0.021212 |
| ING5 | 1.0662 | (0.57, 1.56) | 0.636688 |
| MTA1 | 3.2664 | (0.55, 5.99) | 0.012231 |
| CTBP1 | 0.9145 | (0.24, 1.59) | 0.812571 |
| NSD1 | 0.9263 | (0.26, 1.59) | 0.19222 |
| CBX3 | 2.9562 | (1.16, 4.76) | 0.028029 |
| CBX7 | 1.0616 | (0.57, 1.56) | 0.644263 |
| ARID1A | 1.0608 | (0.57, 1.56) | 0.643804 |
| HPRT1 | 1 | (1.00, 1.00) | - |
IM-positive AG subgroup was compared with the IM-negative AG subgroup IM: intestinal metaplasia; AG: atrophic gastritis; CI: confidence interval
DISCUSSION
This study compared the expression levels of chromatin remodeling gene candidates for GC biomarkers between the normal gastric mucosa, AG, and gastric adenocarcinoma. In the present study, using real-time PCR array, we demonstrated that particularly EED, CBX3, and MTA1 genes were overexpressed, and ARID1A, ING5, and CBX7 genes were more underexpressed in the mucosa of patients with AG and GC compared with the controls. No significant differences were observed between AG and GC groups concerning the expression of these 6 genes, although the expression level (fold change level) of these genes was higher in patients with GC than in patients with AG. EED, CBX3, and MTA1 were more overexpressed in the HP-positive AG subgroup than in the HP-negative AG subgroup. These 3 genes were also more overexpressed in the IM-positive AG subgroup than in the IM-negative AG subgroup.
Chronic inflammation plays an important role in the mechanism of gastrointestinal cancers including HP-associated GC (6). GC is preceded by a cascade of precancerous lesions (Correa cascade), which are non-atrophic chronic gastritis, chronic AG, IM, and finally dysplasia (3,17). Gradual genetic and epigenetic alternations occur in several proto-oncogene and tumor suppressor genes involved in the inflammation-associated carcinogenesis stages. There are many studies about the chromatin remodeling genes in GC. These studies suggested that EED, CBX3, MTA1, ARID1A, ING5, and CBX7 genes are involved in the epigenetic mechanisms of the gastrointestinal carcinogenesis and also include GC. In these studies, we investigated the effects of chromatin remodeling gene expression on disease progression in patients with GC. The overexpression of EED, CBX3, and MTA1 genes and underexpression of ARID1A, ING5, and CBX7 genes were related with disease progression in GC. It is known that these genes have key roles in the carcinogenesis of GC (6–9, 15,16). There are no studies about the chromatin remodeling gene expression pattern of AG in the literature. In our study, there were no significant differences between the expression levels of EED, CBX3, MTA1, ARID1A, ING5, and CBX7 between patients with AG and patients with GC, although the expression level (fold change level) of these genes was higher in patients with GC than in patients with AG. It can be explained by the inadequate number of subjects or this situation may exist because of the fact that not all of the cancer cases are intestinal-type GC cases. This is the first study demonstrating the overexpression of EED, CBX3, and MTA1 genes or underexpression of ARID1A, ING5, and CBX7 genes in patients with AG, which is a precancerous for GC, although dysplasia was not detected in any of the patients with AG. We think that these genes may play an important role in the early stage of inflammation-related gastric carcinogenesis. Our results suggest that the expression of EED, CBX3, MTA1, ARID1A, ING5, and CBX7 may be considered as diagnostic markers to be used in GC screening for patients with AG. Further studies are needed to predict these genes as novel biomarkers.
Polycomb group (PcG) proteins (Polycomb Repressive Complexes 1 and 2 (PRC1 and PRC2)) have been found to serve important functions during cellular differentiation in the epigenetic regulation of transcription, apoptosis inhibition, and neoplastic progression (14,18–20). PRC1 and PRC2 coordinate epigenetic regulation through histone modification such as the methylation of histone H3K27 and monoubiquitination of histone H2AK119 (14,21–23). The major components of human PRC2 are Enhancer of Zeste Homolog 2 (EZH2), Embryonic Ectoderm Development (EED), and Suppressor of Zeste 12 (SUZ12) (20,23). PRCI and PRC2 were observed to elevate in some tumor types and act as critical factors in neoplastic progression. PRC2-mediated trimethylation of H3K27 involves PRC1 in genomic loci leading to the condensation of chromatin and epigenetic silencing of target genes (24). At the histone modification sites, EED interchanges these epigenetic complexes (14,20,24,25).
ING5 (inhibitor of growth) plays a key role in carcinogenesis and functions like a tumor suppressor gene (15). It interacts with β-catenin/NF-κB pathways, histone acetyl transferase complexes, expression of p21/waf1, cyclin A1 inhibitor, and p53 to suppress cell-cycle progression (15,26–28). It also suppresses the proliferation, migration, and invasion of GC cells. In addition, ING5 induces apoptosis, autophagy, and the differentiation of GC cells (15,29). The expression of ING5 was suggested for employment as a good marker in the determination of gastric carcinogenesis as well as for its subsequent progression by inhibiting proliferation, growth, invasion, and metastasis (15,29,30).
AT-rich interactive domain-containing protein 1A (ARID1A) is a determining part of the multi-protein SWI/SNF chromatin remodeling complex (31). ARID1A, which emerged as a tumor suppressor gene, is involved in the regulation of cellular differentiation and proliferation (13,31). The inactivation of ARID1A has been recently reported in several tumors, including GC. The crosstalk between ARID1A and PI3K/Akt pathways or between ARID1A and p53 has an important role in gastric carcinogenesis. The SWI/SNF complex, which interacts with p53 and PI3K/Akt pathways, regulates the transcription of target genes (31). Some studies presented a significant relationship between the ARIDIA mutations and mismatch repair deficiency in GC (13,31).
Metastasis-associated protein 1 (MTA1) was found to be overexpressed in a variety of gastrointestinal cancers, including GC (16). MTA1 promotes tumor progression by downregulation of E-cadherin (32). The overexpression of the MTA1 showed higher rates of serosal, lymph node, vascular invasion, and metastasis in GC (16).
Chromobox 3 (CBX3) and chromobox 7 (CBX7), which are members of the chromobox/heterochromatin protein 1 homologs, are polycomb family proteins and are involved as key regulators in mitosis (14). Heterochromatin protein 1 (HP1) and non-histone chromosomal proteins were found to play an important role in the chromatin packaging and gene silencing. HP1γ protein, encoded by CBX3, has a key role in cancer-associated processes, such as gene silencing, elongation, splicing, DNA repair, cell growth, and differentiation (33). Several recent publications indicate that CBX7 is a potential tumor suppressor gene (34–36). CBX7 accounts for the maintenance of a wide number of homeostatic processes, such as cell-cycle control, proliferation, apoptosis, differentiation, and DNA damage response (14). The loss of CBX7 expression induces the aggressive behavior of gastrointestinal carcinomas (14,34).
AG is a well-known risk factor for GC (4). In 10% of cases with AG, GC can develop within a duration of 10–20 years. The risk of developing GC in AG cases increases in relation to the disease duration, severity of atrophy, presence of IM, dysplasia, and HP (4,5). Epigenetic changes, such as DNA CpG islet hypermethylation, miRNA expressions, and specific histone modifications, provide cancer cell initiation and promotion during inflammation-associated gastric carcinogenesis with or without HP and IM (6,7,10). In our study, we found that EED, CBX3, and MTA1 genes are significantly elevated in HP-positive human AG tissue. These 3 genes are also significantly elevated in IM-positive human AG tissue. Our study did not identify a link between the underexpression of ARID1A, ING5, and CBX7 genes and HP or IM positivity in AG tissue. It can be explained by the inadequate number of subjects or this situation may depend on the ethnical discrepancy of patients. Otherwise, according to this conclusion, we express that ARID1A, ING5, and CBX7 may play a more independent role than HP or IM positivity on carcinogenesis in AG. According to the recent above-mentioned result found in this study, we believe that these 6 genes have a significant involvement in the progression of GC stages from AG. With reference to these data, we consider that the detection of these genes may confer further benefits in the endoscopic screening of AG. These 6 genes may be used as promising early markers in the development of GC in patients with AG. We think that further and larger prospective studies are needed to predict the value of these genes, which evaluate the change of gene expression patterns after HP eradication treatment in patients with HP-positive AG and long-term follow-up of the patients with AG, who progress to HP positivity, IM, dysplasia, or GC.
In conclusion, we demonstrated the overexpression of EED, CBX3, and MTA1 and underexpression of ARID1A, ING5, and CBX7 genes in patients with AG and GC in the Turkish population. Clarification of the molecular pathophysiology of GC and AG will provide further grounds for the treatment and prognosis of the diseases. Further studies are required to elucidate the roles of these 6 genes in the carcinogenesis associated with AG. We believe that EED, CBX3, MTA1, ARID1A, ING5, and CBX7 genes may be considered as prospective and promising genetic markers.
Footnotes
Ethics Committee Approval: Ethics committee approval was received for this study from the Institutional Ethical Review Board of Manisa Celal Bayar University Medical Center (Decision No: #160; Decision Date: June 4, 2014).
Informed Consent: Written informed consent was obtained all the subjects who participated in this study.
Peer-review: Externally peer-reviewed.
Author Contributions: Concept - F.B., E.G., S.Ö.B., E.K., U.D., H.Y., A.R.B., H. Yüceyar; Design - F.B., E.G., S.Ö.B., E.K., U.D., H.Y., A.R.B., H. Yüceyar; Supervision - F.B., E.G., S.Ö.B., E.K., U.D., H.Y., A.R.B., H. Yüceyar; Resources – H. Yüceyar; Materials - F.B., E.G., S.Ö.B., E.K., U.D., H.Y., A.R.B., H. Yüceyar; Data Collection and/or Processing - F.B., E.G., S.Ö.B., E.K., H. Yüceyar; Analysis and/or Interpretation - F.B., E.G., S.Ö.B., E.K., H. Yüceyar; Literature Search - F.B., E.G., S.Ö.B., E.K., H. Yüceyar; Writing Manuscript - F.B., E.G., S.Ö.B., H. Yüceyar; Critical Review - F.B., E.G., H. Yüceyar.
Conflict of Interest: The authors have no conflict of interest to declare.
Financial Disclosure: This study was supported by the Celal Bayar University Coordinator of the Scientific Research Projects (2014-095) Manisa, Turkey.
REFERENCES
- 1.Siegel R, DeSantis C, Virgo K, et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin. 2012;62:220–41. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Watari J, Chen N, Amenta PS, et al. Helicobacter pylori associated chronic gastritis, clinical syndromes, precancerous lesions, and pathogenesis of gastric cancer development. World J Gastroenterol. 2014;20:5461–73. doi: 10.3748/wjg.v20.i18.5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Correa P. Human gastric carcinogenesis: a multistep and multifactorial process-First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992;52:6735–40. [PubMed] [Google Scholar]
- 4.Schistosomes, liver flukes and Helicobacter pylori. IARC Monogr Eval Carcinog Risks Hum; IARC Working Group on the Evaluation of Carcinogenic Risks to Humans; Lyon. 7–14 June 1994; 1994. pp. 1–241. [PMC free article] [PubMed] [Google Scholar]
- 5.Weck MN, Brenner H. Association of Helicobacter pylori infection with chronic atrophic gastritis: Meta-analyses according to type of disease definition. Int J Cancer. 2008;123:874–81. doi: 10.1002/ijc.23539. [DOI] [PubMed] [Google Scholar]
- 6.Chiba T, Marusawa H, Ushijima T. Inflammation-associated cancer development in digestive organs: mechanisms and roles for genetic and epigenetic modulation. Gastroenterology. 2012;143:550–63. doi: 10.1053/j.gastro.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 7.Guo M, Yan W. Epigenetics of gastric cancer. Methods Mol Biol. 2015;1238:783–99. doi: 10.1007/978-1-4939-1804-1_41. [DOI] [PubMed] [Google Scholar]
- 8.Yang WY, Gu JL, Zhen TM. Recent advances of histone modification in gastric cancer. J Cancer Res Ther. 2014;10( Suppl):240–5. doi: 10.4103/0973-1482.151450. [DOI] [PubMed] [Google Scholar]
- 9.Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov. 2006;5:37–50. doi: 10.1038/nrd1930. [DOI] [PubMed] [Google Scholar]
- 10.Valenzuela MA, Canales J, Corvalan AH, Quest AF. Helicobacter pylori-induced inflammation and epigenetic changes during gastric carcinogenesis. World J Gastroenterol. 2015;21:12742–56. doi: 10.3748/wjg.v21.i45.12742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skierucha M, Milne AN, Offerhaus GJ, Polkowski WP, Maciejewski R, Sitarz R. Molecular alterations in gastric cancer with special reference to the early-onset subtype. World J Gastroenterol. 2016;22:2460–74. doi: 10.3748/wjg.v22.i8.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tomita H, Takaishi S, Menheniott TR, et al. Inhibition of gastric carcinogenesis by the hormone gastrin is mediated by suppression of TFF1 epigenetic silencing. Gastroenterology. 2011;140:879–91. doi: 10.1053/j.gastro.2010.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Inada R, Sekine S, Taniguchi H, et al. ARID1A expression in gastric adenocarcinoma: clinicopathological significance and correlation with DNA mismatch repair status. World J Gastroenterol. 2015;21:2159–68. doi: 10.3748/wjg.v21.i7.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang W, Qin JJ, Voruganti S, Nag S, Zhou J, Zhang R. Polycomb Group (PcG) Proteins and Human Cancers: Multifaceted Functions and Therapeutic Implications. Med Res Rev. 2015;35:1220–67. doi: 10.1002/med.21358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gou WF, Shen DF, Yang XF, et al. ING5 suppresses proliferation, apoptosis, migration and invasion, and induces autophagy and differentiation of gastric cancer cells: a good marker for carcinogenesis and subsequent progression. Oncotarget. 2015;6:19552–79. doi: 10.18632/oncotarget.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yao Y, Feng S, Xiao M, Li Y, Yang L, Gong J. MTA1 promotes proliferation and invasion in human gastric cancer cells. Onco Targets Ther. 2015;8:1785–94. doi: 10.2147/OTT.S85383. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Correa P. A human model of gastric carcinogenesis. Cancer Res. 1988;48:3554–60. [PubMed] [Google Scholar]
- 18.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–9. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simon JA, Kingston RE. Mechanisms of polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol. 2009;10:697–708. doi: 10.1038/nrm2763. [DOI] [PubMed] [Google Scholar]
- 20.Cao Q, Wang X, Zhao M, et al. The central role of EED in the orchestration of polycomb group complexes. Nat Commun. 2014;5:3127. doi: 10.1038/ncomms4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao R, Wang L, Wang H, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–43. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 22.Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002;16:2893–905. doi: 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuzmichev A, Jenuwein T, Tempst P, Reinberg D. Different EZH2-containing complexes target methylation of histone H1 or nucleosomal histone H3. Mol Cell. 2004;14:183–93. doi: 10.1016/S1097-2765(04)00185-6. [DOI] [PubMed] [Google Scholar]
- 24.Wang L, Brown JL, Cao R, Zhang Y, Kassis JA, Jones RS. Hierarchical recruitment of polycomb group silencing complexes. Mol Cell. 2004;14:637–46. doi: 10.1016/j.molcel.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 25.Cao R, Zhang Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol Cell. 2004;15:57–67. doi: 10.1016/j.molcel.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 26.Ullah M, Pelletier N, Xiao L, Zhao SP, Wang K, Degerny C, et al. Molecular architecture of quartet MOZ/MORF histone acetyltransferase complexes. Mol Cell Biol. 2008;28:6828–43. doi: 10.1128/MCB.01297-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doyon Y, Cayrou C, Ullah M, et al. ING tumor suppressor proteins are critical regulators of chromatin acetylation required for genome expression and perpetuation. Mol Cell. 2006;21:51–64. doi: 10.1016/j.molcel.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 28.Shiseki M, Nagashima M, Pedeux RM, et al. p29ING4 and p28ING5 bind to p53 and p300, and enhance p53 activity. Cancer Res. 2003;63:2373–8. [PubMed] [Google Scholar]
- 29.Zhang F, Baumer N, Rode M, et al. The inhibitor of growth protein 5 (ING5) depends on INCA1 as a co-factor for its antiproliferative effects. PLoS One. 2011;6:e21505. doi: 10.1371/journal.pone.0021505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xing YN, Yang X, Xu XY, et al. The altered expression of ING5 protein is involved in gastric carcinogenesis and subsequent progression. Hum Pathol. 2011;42:25–35. doi: 10.1016/j.humpath.2010.05.024. [DOI] [PubMed] [Google Scholar]
- 31.Wu RC, Wang TL, Shih Ie M. The emerging roles of ARID1A in tumor suppression. Cancer Biol Ther. 2014;15:655–64. doi: 10.4161/cbt.28411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weng W, Yin J, Zhang Y, Qiu J, Wang X. Metastasis-associated protein 1 promotes tumor invasion by downregulation of E-cadherin. Int J Oncol. 2014;44:812–8. doi: 10.3892/ijo.2014.2253. [DOI] [PubMed] [Google Scholar]
- 33.Velez G, Lin M, Christensen T, Faubion WA, Lomberk G, Urrutia R. Evidence supporting a critical contribution of intrinsically disordered regions to the biochemical behavior of full-length human HP1gamma. J Mol Model. 2016;22:12. doi: 10.1007/s00894-015-2874-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pallante P, Forzati F, Federico A, Arra C, Fusco A. Polycomb protein family member CBX7 plays a critical role in cancer progression. Am J Cancer Res. 2015;5:1594–601. [PMC free article] [PubMed] [Google Scholar]
- 35.Forzati F, Federico A, Pallante P, et al. CBX7 gene expression plays a negative role in adipocyte cell growth and differentiation. Biol Open. 2014;3:871–9. doi: 10.1242/bio.20147872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pallante P, Sepe R, Federico A, Forzati F, Bianco M, Fusco A. CBX7 modulates the expression of genes critical for cancer progression. PLoS One. 2014;9:e98295. doi: 10.1371/journal.pone.0098295. [DOI] [PMC free article] [PubMed] [Google Scholar]