

Abstract
Lynch syndrome (LS) is an autosomal dominant disorder characterized by an increased risk of extracolonic cancers and early age of onset. It is associated with germline mutations in the DNA mismatch repair (MMR) genes. We report a case of a patient with colorectal cancer referred to our medical genetics department for molecular analysis and genetic counseling. The proband is a 64-year-old woman diagnosed with a tumor of the cecum. Histopathological examination showed a moderately differentiated mucinous adenocarcinoma categorized by pT3 N0. Analysis of her pedigree revealed three siblings who had colon cancer, as well as one relative with brain cancer. Based on these findings, molecular genetic investigation was found to be necessary in order to identify the disease-causing mutation. Immunohistochemistry staining of MMR proteins was performed on the tumor sample of the index proband. Mutational analysis of the MLH1/MSH2 genes was carried out. Analysis was extended to the family members and the general population. This led to the identification of a heterozygous frameshift duplication in the MLH1 gene at position 910 (c.910dupG). Three siblings had inherited the mutation from their mother, two of whom were asymptomatic at the time of diagnosis. To the best of our knowledge, this is a novel pathogenic duplication that has not been reported in the databases and literature. The outcome of the present case suggests that this mutation was the primary cause of LS in the family.
Keywords: Lynch syndrome, MLH1, colorectal cancer, genetic testing
INTRODUCTION
Lynch syndrome (LS) is an autosomal dominant disorder characterized by an increased risk of the early development of colorectal cancers and other extracolonic carcinomas (1,2). Mutations affecting the DNA mismatch repair (MMR) genes (MLH1, MSH2, MSH6, and PMS2) were found to cause this condition (3,4). Indeed, approximately 90% of the alterations are attributable to the MLH1 and MSH2 genes and are spread over all regions without hotspots (5). The remaining 10%–20% are due to mutations in the MSH6 and PMS2 genes (6). The EPCAM gene, a neighboring MSH2 gene, also contributes to the genetic cause of LS. In fact, up to 3% of the LS is due to EPCAM deletions. Loss of the 3′ end of the EPCAM gene results in epigenetic hypermethylation of the MSH2 promoter, producing a loss of MSH2 protein (7).
We report a novel frameshift mutation associated with LS phenotype in a Moroccan family. This is the first attempt of molecular and presymptomatic diagnosis of LS in Morocco.
CASE PRESENTATION
A 64-year-old Moroccan woman was admitted to the gastroenterology unit of CHU Hassan II, Fez with complaints of abdominal pain associated with constipation, melena, and unusual weight loss. Ultrasound revealed a hyperechoic mass of 85/35 mm between the right iliac fossa and the right flank. There was no ascites, and all the abdominopelvic organs were normal. On double-contrast barium enema, a cecal mass of 10 cm was found. The proband underwent a right-sided hemicolectomy with lymph node dissection. Histopathological examination revealed a moderately differentiated mucinous adenocarcinoma infiltrating the subserosa and classified the tumor as pT3 N0. On the family history of the patient, it was found that her son developed colon cancer at the age of 31 years (III-7). In addition, two other relatives, mother and brother, both died of malignant tumors of the colon at ages 56 (I-2) and 50 (II-4) years, respectively. On the other hand, her son died at the age of 18 years due to a brain tumor (III-6) (Figure 1a). However, the disease could not be characterized for the deceased siblings because of limited availability of medical records.
Figure 1. a, b.
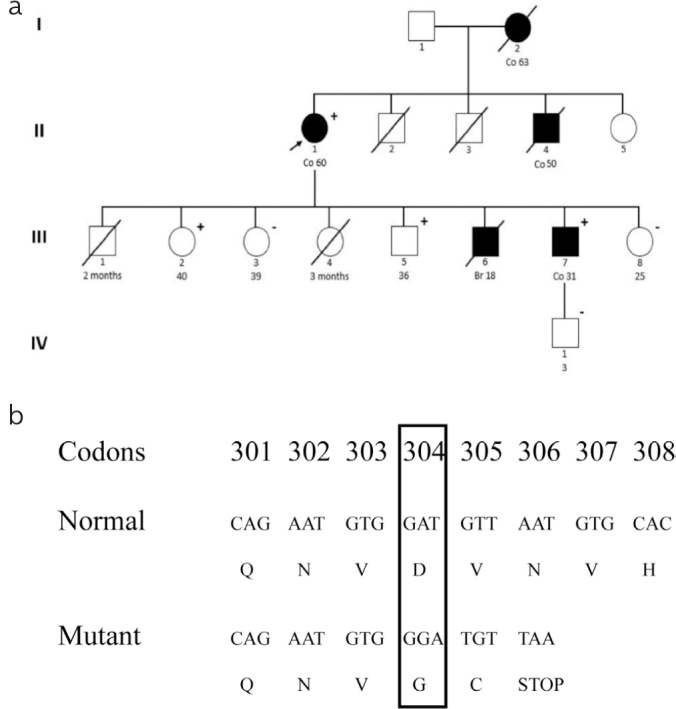
Pedigree of the Moroccan family presenting the segregation of the novel duplication c.911dupG. Square: male, circle: female, open symbol: no tumor, colored symbol: patient with carcinomas, symbol with a diagonal: deceased. Numbers under symbols indicate the age at diagnosis. Plus/Minus signs indicates the presence/Absence of the mutation. The black arrow indicates the index proband (II-1) (a); The duplication results in a frameshift mutation starting from codon 304 and a premature stop three codons downstream (p.D304 fsX306)
In the patient with colonic adenocarcinoma, combined with her family history, it was suspected that she might have hereditary non-polyposis colorectal cancer. These findings propelled us to perform MMR gene mutations screening to the affected family members and other relatives who might be at risk.
Immunohistochemistry analysis of the four MMR proteins (MLH1, MSH2, MSH6, and PMS2) was performed on 4 μm tissue sections from formalin-fixed paraffin-embedded tissue of the index proband (II-1), as previously described (8). The patient exhibited a loss of the MLH1 and PMS2 protein expression. Mutational analysis of the MLH1 and MSH2 genes was first performed in the index patient. Genomic DNA was isolated from the blood sample using a standard salting-out protocol (9). All exonic regions of the MLH1 and MSH2 genes, including flanking intronic boundaries, were amplified using previously reported primers and then sequenced using the 3500 Dx Genetic Analyzer (10). The identified DNA alterations were also validated on other polymerase chain reaction (PCR) products and sequenced in both sense and antisense orientations.
DNA testing identified a heterozygous duplication of (G) nucleotide at position 910 in the MLH1 gene (MLH1: c.910dupG), resulting in a frameshift mutation affecting codon 304 and generating a premature stop three codons downstream (p.D304 fsX306) (Figure 2b). Based on this finding, six family members underwent presymptomatic diagnosis. After providing informed consent, peripheral blood samples were collected from each participant. In addition, 50 healthy individuals were sampled as controls to study the mutation at population level and to demonstrate that the duplication found is not a common polymorphism. Allele-specific PCR of exon 11 was carried out using the forward primer modified at the mutated site (5′-AAATCAGTCCCCAGAATGTGGG-3′) with additional inserted (G) nucleotide in the 3′ end. PCR amplification generated a product at 226 bp (Figure 1b). Results were confirmed by sequencing positive cases. Two asymptomatic relatives (III-5 and III-2) showed a specific product at 226 bp, which was absent in 50 healthy controls (Figure 1b). The two unaffected mutation carriers were referred to the gastroenterology department to start surveillance. Colonoscopic reports of the female patient (III-5) revealed a polyp of 7 mm in the sigmoid colon. Histopathological analysis confirmed a tubular adenoma with high-grade dysplasia. The second patient (III-2) exhibited a 2 mm sessile polyp in the peripapillary region.
Figure 2. a–c.
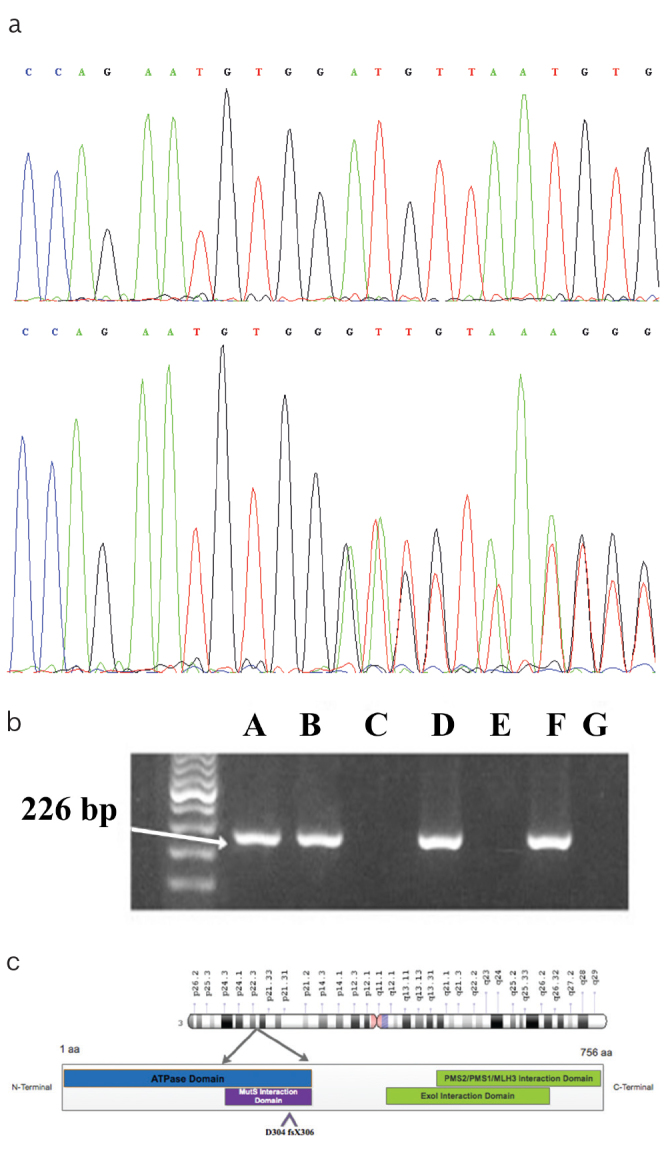
Chromatograms of a normal individual (Wild type), and the index proband (II-1) with the heterozygous duplication of nucleotide G at 910 position (c.910dupG) (a); The electrophoresis profile of amplification product (226 bp) obtained by Allele Specific-PCR. A specific product of 226 bp was found in the index proband and other relatives (A: II-1, B: III-2, D: III-5, F: III-7), and was absent in the remaining siblings (C: III-3, E: III-8, G: IV-1) (b); Protein domain structure of MLH1 comprising N- Terminal ATPase domain (aa 3-338), MutS interaction domain (aa 216-335), ExoI interaction domain (aa 410-650), C-Terminal interaction domain with PMS2/PMS1/MLH3 (c)
DISCUSSION
Lynch syndrome is a hereditary autosomal dominant disorder characterized by considerably increased risks of colonic and extracolonic tumors and earlier age of onset. It was associated with mutations in the MMR genes (3,4). Of the 3000 germline sequence variants reported in the International Society for Gastrointestinal Hereditary Tumors database, the MLH1 and MSH2 genes were the most prevalent with rates at 40% and 34%, respectively, followed by 18% for MSH6 and 8% for PMS2. The majority of the MMR mutations are truncating (nonsense or frameshift), and most of them are specific to a single family (11). However, some founder mutations were also reported (12). In this case report, we have described a novel frameshift mutation (c.910dupG) located in exon 11 of the MLH1 gene among a Moroccan family diagnosed with LS. To our knowledge and according to databases and literature review, this is a previously unreported mutation, leading to truncation of the protein beyond this point. Some previous studies have reported mutations at codon 304 in patients with LS (13,14). In effect, Montera et al. (13) described a deleterious substitution at the same position, leading to a replacement of Asp by Val.
It can be hypothesized that this mutation will develop serious consequences on the formation of mature protein. Since the duplication c.910dupG introduced a premature stop codon early in the translation (at position 306), the dimerization domain with PMS2/MLH3/PMS1, as well as the interaction domain with hExoI, would be lost, leading to a non-functional protein (Figure 2a). This was further supported by the absence of the MLH1 and PMS2 protein expression in the index proband. Moreover, in silico analysis using the MutationTaster prediction tool classified the duplication as disease-causing mutation. Furthermore, the cosegregation of this duplication with the phenotype in the family, in addition to its absence in the normal controls, gives further evidence about the pathogenic nature of the mutation.
It has been proven that the diagnosis of LS has significant implications for surveillance strategy of the patient and extended family. Based on our experience, identification of the underlined mutation in the index proband helped us to provide genetic counseling to her relatives. Indeed, the presymptomatic testing revealed two asymptomatic carriers of the pathogenic mutation, both of them underwent surveillance colonoscopy and were found to carry precancerous lesions.
According to the medical history of the family in this case report, even brain cancer has been reported in a deceased relative (III-6). However, brain cancer does not belong to the spectrum of LS extracolonic tumors. Constitutional MMR deficiency syndrome, which is characterized by a high incidence of childhood cancers (hematologic and brain cancers), may explain the occurrence of this type of malignancy in the proband. Therefore, further genetic exploration in more family members may help understand the manifestation of brain cancer in the family.
The main limitation of the case study was the lack of medical records for the deceased siblings, as well as for illiterate relatives from low socioeconomic areas.
There are a variety of MLH1 mutations reported in different populations. This is the first novel mutation ever reported in a Moroccan family associated with colon cancer susceptibility. Identification of such mutations is crucial and can widely aid in the management of colorectal cancer in predisposed populations through molecular screening, prevention strategies, and follow-up of suspected LS families.
Acknowledgements
The authors would like to thank Pr. Bahia Bennani for revising the manuscript and Pr. El bachir Benjelloun for providing a detailed medical record of the patient.
Footnotes
Peer-review: Externally peer-reviewed.
Author Contributions: Concept - F.Z.M., K.O., M.I.H.; Design - F.Z.M., K.O., M.I.H.; Supervision - L.B. K.O., M.I.H.; Resources - K.O.; Materials - L.B., K.O.; Data Collection and/or Interpretation - F.Z.M., L.B.; Analysis and/or Interpretation - F.Z.M., I.E.B.; Literature Search - F.Z.M., I.E.B., L.B., M.I.H.; Writing Manuscript - F.Z.M.; Critical Review - F.Z.M., L.B., I.E.B., M.I.H.
Conflict of Interest: The authors have no conflict of interest to declare.
Financial Disclosure: The authors declared that this study has received no financial support.
REFERENCES
- 1.Lynch HT, Boland CR, Gong G, et al. Phenotypic and genotypic heterogeneity in the Lynch syndrome: diagnostic, surveillance and management implications. Eur J Hum Genet. 2006;14:390–402. doi: 10.1038/sj.ejhg.5201584. https://doi.org/10.1038/sj.ejhg.5201584 [DOI] [PubMed] [Google Scholar]
- 2.Müller A, Fishel R. Mismatch repair and the hereditary non-polyposis colorectal cancer syndrome (HNPCC) Cancer Invest. 2002;20:102–109. doi: 10.1081/cnv-120000371. https://doi.org/10.1081/CNV-120000371 [DOI] [PubMed] [Google Scholar]
- 3.Lung MS, Trainer AH, Campbell I, Lipton L. Familial colorectal cancer. Intern Med J. 2015;45:482491. doi: 10.1111/imj.12736. https://doi.org/10.1111/imj.12736 [DOI] [PubMed] [Google Scholar]
- 4.Tiwari AK, Roy HK, Lynch HT. Lynch syndrome in the 21st century: clinical perspectives. QJM Mon J Assoc Physicians. 2016;109:151–8. doi: 10.1093/qjmed/hcv137. https://doi.org/10.1093/qjmed/hcv137 [DOI] [PubMed] [Google Scholar]
- 5.Tutlewska K, Lubinski J, Kurzawski G. Germline deletions in the EPCAM gene as a cause of Lynch syndrome-literature review. Hered Cancer Clin Pract. 2013;11:9. doi: 10.1186/1897-4287-11-9. https://doi.org/10.1186/1897-4287-11-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Giardiello FM, Allen JI, Axilbund JE, et al. Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the US Multi-society Task Force on colorectal cancer. Am J Gastroenterol. 2014;109:1159–79. doi: 10.1038/ajg.2014.186. https://doi.org/10.1038/ajg.2014.186 [DOI] [PubMed] [Google Scholar]
- 7.Tomita N, Yamano T, Matsubara N, Tamura K. [A novel genetic disorder of Lynch syndrome - EPCAM gene deletion]. Gan To Kagaku Ryoho. 2013;40:143–7. [PubMed] [Google Scholar]
- 8.Bavi P, Uddin S, Bu R, et al. The biological and clinical impact of inhibition of NF-κB-initiated apoptosis in diffuse large B cell lymphoma (DLBCL) J Pathol. 2011;224:355–66. doi: 10.1002/path.2864. https://doi.org/10.1002/path.2864 [DOI] [PubMed] [Google Scholar]
- 9.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. https://doi.org/10.1093/nar/16.3.1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zahary MN, Kaur G, Abu Hassan MR, Singh H, Naik VR, Ankathil R. Germline mutation analysis of MLH1 and MSH2 in Malaysian Lynch syndrome patients. World J Gastroenterol. 2012;18:814–20. doi: 10.3748/wjg.v18.i8.814. https://doi.org/10.3748/wjg.v18.i8.814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peltomäki P. Update on Lynch syndrome genomics. Fam Cancer. 2016;15:385–93. doi: 10.1007/s10689-016-9882-8. https://doi.org/10.1007/s10689-016-9882-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ponti G, Castellsagué E, Ruini C, Percesepe A, Tomasi A. Mismatch repair genes founder mutations and cancer susceptibility in Lynch syndrome. Clin Genet. 2015;87:507–16. doi: 10.1111/cge.12529. https://doi.org/10.1111/cge.12529 [DOI] [PubMed] [Google Scholar]
- 13.Montera M, Resta N, Simone C, et al. Mutational germline analysis of hMSH2 and hMLH1 genes in early onset colorectal cancer patients. J Med Genet. 2000;37:e7. doi: 10.1136/jmg.37.7.e7. https://doi.org/10.1136/jmg.37.7.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Limburg PJ, Harmsen WS, Chen HH, et al. DNA Mismatch Repair Gene Alterations in a Population-Based Sample of Young-Onset Colorectal Cancer Patients. Clin Gastroenterol Hepatol. 2011;9:497–502. doi: 10.1016/j.cgh.2010.10.021. https://doi.org/10.1016/j.cgh.2010.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]