

Abstract
Background:
Cellular non-coding RNAs are extensively modified post-transcriptionally, with more than 100 chemically distinct nucleotides identified to date. In the past five years, new sequencing based methods have revealed widespread decoration of eukaryotic messenger RNA with diverse RNA modifications whose functions in mRNA metabolism are only beginning to be known.
Results:
Since most of the identified mRNA modifying enzymes are present in the nucleus, these modifications have the potential to function in nuclear pre-mRNA processing including alternative splicing. Here we review recent progress towards illuminating the role of pre-mRNA modifications in splicing and highlight key areas for future investigation in this rapidly growing field.
Conclusions:
Future studies to identify which modifications are added to nascent pre-mRNA and to interrogate the direct effects of individual modifications are likely to reveal new mechanisms by which nuclear pre-mRNA processing is regulated.
Keywords: mRNA modification, pre-mRNA modification, splicing, RNA-modifying enzymes
Author summary:
Modified nucleotides in messenger RNA are abundant, inducible in response to cellular conditions and can affect diverse stages of the mRNA life cycle. In this article we highlight those mRNA modifications that are known or are likely to be deposited in pre-mRNA co-transcriptionally and discuss individual examples of modified nucleotides in pre- mRNA that influence splicing.
INTRODUCTION
Non-coding RNAs, including ribosomal RNAs (rRNAs), transfer RNAs (tRNAs) and spliceosomal small RNAs (snRNAs), are extensively modified with more than 100 chemically distinct nucleotides that promote their activities in translation and splicing. The recent development of transcriptome-wide approaches for the detection of RNA modifications led to the identification of N6-methylade-nosine (m6A) [1,2], pseudouridine (Ψ) [3-6], 5-methyl- cytidine (m5C) [7], 5-hydroxymethylcytidine (hm5C) [8], N1methyladenosine (m1A) [9,10], 2ˊ-O-methylation (Nm) [11] and N6, 2ˊ-O-dimethyladenosine (m6Am) [12,13] as marks on messenger RNAs (mRNA) of varying abundance (Figure 1A). While excitement about their regulatory potential has led to a race to identify new modifications, our understanding of the functions of these modifications in mRNA processing remains limited. As such, future research efforts are likely to be devoted to the functional characterization of individual modifications.
Figure 1. Human nuclear mRNA modifying enzymes.
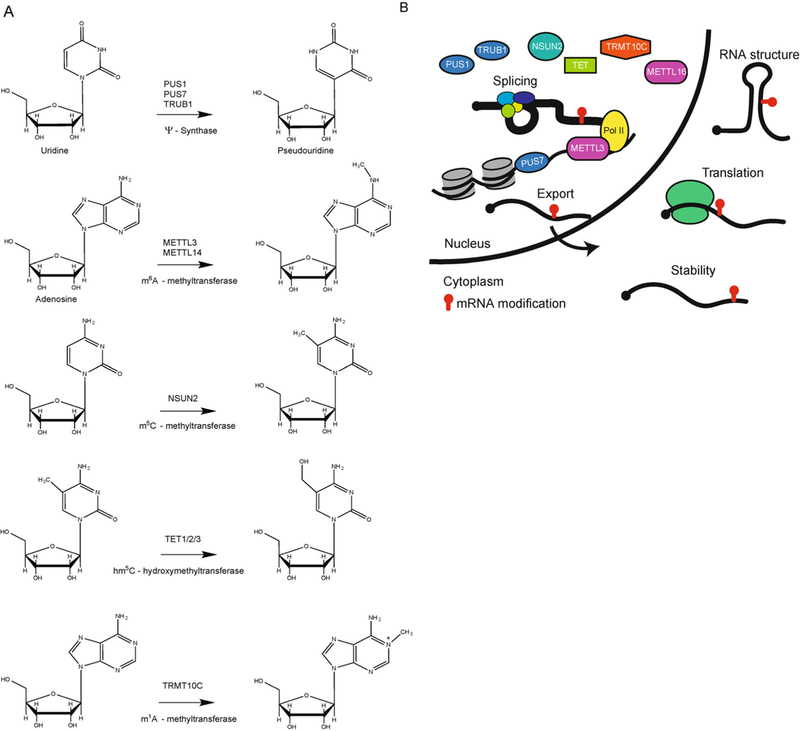
(A) Chemical structures of the modified nucleotides installed by known nuclear human mRNA modifying enzymes. (B) Illustration of the potential function of mRNA modifications in all stages of the mRNA life cycle if they are added co-transcriptionally to nascent pre-mRNA. Depicted are the human mRNA modifying enzymes that are nuclearlocalized PUS1 (Ψ), TRUB1 (Ψ), NSUN2 (m5C), TET (hm5C), TRMT10C (m1A), METTL16 (m6A) and those that are chromatinassociated- PUS7 (Ψ), or interact with Pol II- METTL3 (m6A) suggesting that the modification is added to pre-mRNA.
So far the identified modifications in mRNA have been implicated in regulating diverse steps of the mRNA life cycle including splicing, 3ˊ end processing, export, translation, mRNA stability and decay [14-17]. However, most RNA modifications have been identified in poly(A)+ selected mRNA and their roles in nuclear processing are unknown. Largely indirect evidence suggests that various modifications are added to pre-mRNA in the nucleus where they could function in pre-mRNA processing. In this review, we will discuss what is known and unknown about the prevalence of RNA modifications in pre-mRNA and their roles in splicing. We will also present our view on the key areas of future investigation to determine the functions of RNA modifications in pre-mRNA. m6A was the first modification that was mapped through the transcriptome, and thus there is a richer body of work to contextualize the function of m6A in pre-mRNA processing. We will therefore frame our discussion around m6A as an example to illuminate mechanisms and infer principles of how diverse RNA modifications may affect pre-mRNA processing.
SEQUENCING-BASED METHODS FOR IDENTIFICATION OF RNA MODIFICATIONS
Methods to identify RNA modifications can be grouped into two broad categories: antibody-based enrichment of the RNAs that contain the modification and nucleotide identification from reverse transcriptase stops. The first methods developed for sequencing-based detection of m6A, m1A and hm5C used antibodies that were specific for the respective RNA modifications to immunoprecipitate RNA fragments and sequence the modification- containing RNAs [1,2,8-10]. These methods produced the first transcriptome-wide RNA modification maps but lacked precise single-nucleotide resolution. Newer versions of antibody-based detection methods include the addition of a UV crosslinking step to covalently link antibodies to modified RNA; subsequent analysis of crosslink-induced mutations precisely identifies the modification site [12,18]. Similarly, certain reverse transcriptases that mis-incorporate when they encounter the modified nucleotide have been combined with antibody enrichment to identify modified RNA sites [19,20].
Chemical reactivity that is specific for a given RNA modification has also been exploited to identify modified sites by sequencing. For example, pseudouridine (Ψ) can be selectively modified with the chemical N-cyclohexyl- Nˊ-beta-(4-methylmorpholinium) ethylcarbodiimide p-tosylate (CMCT). The bulky covalent CMC-pseudouri- dine adducts block reverse transcriptase and allow for sequencing-based detection of pseudouridines [3-6]. Bisulfite sequencing has been used to identify m5C sites in RNA based on the fact that this methylation makes cytosine resistant to chemical deamination to uracil in the presence of bisulfite [7]. Other methods for detecting m5C have relied on covalent capture of trapped substrates of the m5C methyltransferase NSUN2. Immunoprecipitation of covalently-bound substrates allows for site-specific detection of m5C-induced reverse transcriptase stops or transversions in the case of substrates captured by covalent trapping with the cytidine analog 5-azacytidine [21,22]. Variations of these techniques using modification-specific antibodies, distinct chemical reactivities and capture of enzymatic substrates are likely to be adapted for genome-wide identification of other mRNA modifications in diverse RNA populations.
EVIDENCE FOR NUCLEAR LOCALIZATION OF RNA-MODIFYING ENZYMES
To aid in the functional characterization of modifications in mRNA, it is important to know when the mark is deposited during the mRNA lifecycle. This information can significantly narrow the investigation to elucidate the functional effects of mRNA modifications. For example, if a modification is added co-transcriptionally, prior to splicing, it has the potential to regulate any step of mRNA processing whether nuclear or cytoplasmic, including pre- mRNA splicing. In contrast, if a modification is added in the nucleoplasm after release from chromatin it will likely have little effect on splicing [23,24], but may participate in nuclear export or nuclear RNA decay. Similarly, if an enzyme that adds or removes RNA modifications (called writers and erasers, respectively) is present in the cytoplasm then it is most likely to regulate cytoplasmic processing such as translation and mRNA stability. Determining the localizations of the modification writers, erasers and readers (proteins that recognize specific modifications) will therefore shed light on the function of a given modification (Figure 1B). Furthermore, this information is likely to reveal whether the fate of a mature mRNA in the cytoplasm can be quickly altered by a modification or if new transcription is required to incorporate the modification and activate a distinct gene expression program.
The m6A methyltransferase complex is nuclear
m6A is present at RRACH motifs throughout coding sequences, but is dramatically enriched in last exons that often contain stop codons [1,2,18]. The mechanism underlying this biased distribution is not understood and may involve restrictions on the activity of either the methyltransferase, a demethylase, or both. The mammalian m6A methyltransferase complex consists of the catalytic subunit methyltransferase like 3 (METTL3), methyltransferase like 14 (METTL14), and a regulatory subunit Wilms’ tumor 1-associating protein (WTAP) [25,26]. WTAP is nuclear and has been previously studied as a splicing factor in human and Drosophila cells [27,28]. The entire methyltransferase complex co-localizes with nuclear speckles, which are nuclear bodies that store splicing factors [25,26]. Analysis of the binding sites of the m6A methyltransferase complex by photoactiva-table ribonucleoside-enhanced crosslinking-immunopre-cipitation (PAR-CLIP) and sequencing revealed that the binding motif for individual components of the methyl-transferase complex matched the consensus RRACH where m6A is found [25]. Altogether, these initial observations suggested that m6A was added to pre- mRNA in the nucleus.
Analysis of transcriptome-wide binding of RNA-modifying enzymes can provide insight into whether a modification is likely to be added to introns or pre- mRNA. For example, METTL3 was shown by overexpression and PAR-CLIP analysis of total RNA to bind primarily to exons, while ~30% of the identified binding sites were in introns [25]. METTL3 binding to introns was at least suggestive of m6A modification of pre-mRNA.
Recent studies found that METTL3 associates with Pol II in human cells, providing a link between transcription and m6A addition (Figure 1B) [29]. Similarly, components of the m6A methyltransferase complex in Drosophila co-localize with Pol II at sites of transcription [30]. In human cells, expression of a slow Pol II mutant increased the levels of total m6A-containing poly(A)+ mRNA that could be immunoprecipitated with an m6A antibody. Analysis of individual mRNAs revealed increased m6A levels when Pol II elongation was slow, providing evidence of coupling between Pol II- mediated transcription and mRNA methylation [29]. One other human m6A methyltransferase, METTL16, was recently identified as the U6 snRNA methyltransferase with MAT2A as its only known direct mRNA methylation substrate. Consistent with its role in regulating MAT2A splicing [31] and in methylating the U6 snRNA [31,32], METTL16 was found to localize to the nucleus (Figure 1B) [33].
Pseudouridine synthases are active in the nucleus
Pseudouridine was found to be relatively uniformly distributed in mRNA with the majority of identified sites within coding regions [3-6]. Pseudouridylation in mRNA has so far been demonstrated to be catalyzed primarily by the standalone pseudouridine synthases (PUS) that also pseudouridylate tRNAs. In yeast, most mRNA pseudouridines have been genetically assigned to two conserved Pus proteins, Pus1 and Pus7, which are nuclear in growing cells. Pus7 was shown to re-localize to the cytoplasm during heat shock coincident with increased mRNA pseudouridylation [4]. Seven out of nine of the known yeast Pus proteins have been proposed to pseudouridylate mRNA targets leading to the presence of this modification in diverse sequence contexts [3-5]. In addition, yeast CBF5 has also been shown to be genetically required for pseudouridylation of a subset of mRNAs [4]. CBF5 encodes the catalytic subunit of the box H/ACA snoRNA-guided pseudouridine synthase that targets nascent pre-rRNA in the nucleolus [34,35].
Human cells from patients with dyskeratosis congenita, which harbor mutations the human ortholog of Cbf5, likewise have decreased pseudouridylation signal at some mRNA pseudouridine sites implying the human enzyme also pseudouridylates mRNAs [4]. Of the human standalone PUS enzymes, only PUS1, PUS7 and TRUB1 have thus far been demonstrated to pseudouridylate mRNA targets [6,36]. All three of these PUS proteins have been shown to at least partially localize to the nucleus or have nuclear isoforms (Figure 1B) [36-38]. PUS7 has been further shown to be chromatin-associated and, more specifically, associated with active Pol II promoters and enhancers [39], suggesting it acts on nascent pre-mRNA. Furthermore, nuclear resident noncoding RNAs such as MALAT1 are pseudouridylated in human cells demonstrating that human pseudouridine synthases are active in the nucleus where they could acton pre-mRNA [3].
m5C and hm5C
As with the pseudouridine synthases, the canonical function of the known mRNA m5C methyltransferase NSUN2 [7,21,22] is in tRNA methylation. The initial m5C profiling by bisulfite sequencing reported thousands of putative m5C sites in mRNA [7]. Low throughput experiments suggested that NSUN2 was an mRNA m5C methyltransferase of a few m5C containing mRNAs. A more recent study, found that depletion of NSUN2 globally reduced the m5C to C ratio in poly(A)-selected and rRNA depleted mRNA based on mass spectrometry analysis [40]. Furthermore, the signal for about half of the m5C sites identified in the same study by bisulfite sequencing was modestly reduced upon depletion of NSUN2. Studies that sought to identify m5C targets of NSUN2 by covalent capture of substrates found far fewer, around three hundred, m5C sites in mRNA [22]. The discrepancy in these results could be explained by increased specificity of the catalytic capture method for direct targets, in which case there should be unidentified m5C methyltransferases whose activity is reduced by depletion of NSUN2. Alternatively, a signal for m5C can represent non-conversion events or other cytidine modifications (e.g., hm5C) that are also resistant to bisulfite- based identification [41]. Although the extent of mRNA m5C remains controversial, the documented m5C mRNA methyltransferase NSUN2 is nuclear and could deposit m5C in nascent RNA (Figure 1B) [38,42].
m5C can be oxidized to form a derivative modification, 5-hydroxymethylcytidine (hm5C), by the ten-eleven translocation (Tet) enzymes. hm5C was mapped in Drosophila by antibody-based enrichment of hm5C- modified RNA. Over 80% of identified sites that were enriched in UC-rich regions were reduced upon knockdown of Drosophila TET (dTET) [8]. All three of the human TET enzymes can convert m5C to hm5C in RNA, as determined from bulk measurements [43,44]. Consistent with their role in DNA hydroxymethylation, the TET proteins reside in the nucleus (Figure 1B) [38,42].
Most of the mRNA modifying enzymes characterized to date reside within the nucleus where they have the potential to interact with nascent pre-mRNA. However, the exact timing and location of mRNA modification has rarely been determined. In principle, analysis of the RNA binding sites of RNA modifying enzymes by methods such as PAR-CLIP in combination with sub-cellular fractionation could address this question. In practice, such analysis of RNA binding targets of enzymes should be interpreted with caution. Except in the case of mutant proteins engineered to trap catalytic intermediates, enzymes are likely to interact transiently with their substrates and no longer interact once the substrate has been modified. This was shown for METTL16, which methylates an adenosine in one of its substrates in SAM- replete conditions. When SAM is depleted there is no methyl donor available and the interaction between METTL16 and interaction with its substrate is stabilized [31]. Therefore, interactions captured by typical CLIP assays may represent weaker or non-substrates. Performing RNA binding analysis on catalytically dead mutants or in the absence of cofactors necessary for modification may better capture the substrate-binding landscape of RNA modification enzymes.
CO-TRANSCRIPTIONAL PRE-mRNA MODIFICATIONS
As noted above, many modifications have been profiled exclusively in poly(A)+ mRNA (pseudouridine, m1A, m5C, m6Am) and thus overlook potential sites in introns (Table 1). Even profiling of total RNA (e.g., by antibody based enrichment) does not give a complete picture of the extent of pre-mRNA modification. Because total RNA is a mixture of mature mRNA, pre-mRNA, and mRNAs containing retained introns, intronic reads may not represent introns that come from pre-mRNA. Moreover, the much greater abundance of mature mRNA likely leads to reduced detection of modifications in introns. Notably, profiling m5C in purified nuclear polyadenylated RNA suggested a much higher prevalence of intronic m5C than similar studies of (predominantly cytoplasmic) poly(A)+ mRNA (~60% vs. ~20%) [48]. Thus, estimates of the fraction of intronic modifications made by total RNA sequencing should be regarded as lower bounds.
Table 1.
Evidence for mRNA modifications in pre-mRNA
| Modification | mRNA | Pre-mRNA | Exons | Introns | Refs. |
|---|---|---|---|---|---|
| m6A | Y | Y | +++ | + | [1,2,12,18,45-47] |
| Ψ | Y | ? | ? | ? | [3-6] |
| m1A | Y | ? | ? | ? | [9,10,19,20] |
| m5C | Y | ? | ++ | ++ | [7,21,22,48] |
| hm5C | Y | ? | +++ | ++ | [8,43,44] |
| Nm | Y | ? | ? | ? | [49] |
| m6Am | Y | ? | ? | ? | [12,13] |
RNA modification: Y- present in;? - unknown if present in mRNA or pre-mRNA. Evidence for distribution of modification in exons relative to introns form poly(A)+ mRNA or total RNA.
While considerable progress has been made in profiling diverse modifications in mRNA, most of the studies have been performed on mature poly(A)+ mRNA, precluding the identification of these modifications in introns and in pre-mRNA. Thus, defining the landscape of pre-mRNA modifications will illuminate whether it is feasible for any modification to have a direct effect on nuclear processing of pre-mRNA. Adapting nucleotide modification profiling techniques to sequence nascent RNA could reveal whether a modification is added co-transcriptionally to pre-mRNA. One promising approach to identify modifications in nascent pre-mRNA is to sequence chromatin- associated RNA. Using non-ionic detergent and 1 mol/L urea, it is possible to separate the soluble, loosely associated RNA and protein components from the insoluble chromatin fraction. Nascent RNA remains tethered to Pol II ternary complexes and thus it remains chromatin bound (based on Ref. [50]). Recently, several groups have coupled this fractionation protocol to high- throughput RNA-sequencing to enrich for unspliced introns containing pre-mRNA and quantify the extent of co-transcriptional splicing [23,24,51-53].
So far only m6A has been definitively shown to be added to pre-mRNA co-transcriptionally (Table 1). A recent study performed single-nucleotide resolution m6A profiling to identify m6A sites from three cellular fractions in HeLa cells: chromatin-associated, nucleoplasmic and cytoplasmic RNA. Surprisingly, although ~75% of chromatin-associated pre-mRNA reads are intronic and most pre-mRNAs in this fraction are incompletely spliced [23,45], ~90% of pre-mRNA m6As reside in exons as compared to introns [45]. This distribution cannot be explained by the occurrence of the RRACH motif recognized by METTL3 in introns compared to exons since the motif occurs more frequently in introns. Nevertheless, although intronic m6As account for only ~10% of total m6A peaks identified in pre-mRNA, over two thousand m6A sites have been reported to reside in introns and may have important functions [45,46]. Analysis of the m6A landscape in each of the three fractions revealed ~90% overlap in detected m6A peaks suggesting limited dynamics of the m6A mark after pre- mRNA release from chromatin [45]. Thus, any regulation of m6A levels by methyltransferases and demethylases is likely almost exclusively co-transcriptional under basal conditions, consistent with their localization. It is still possible that dynamic methylation and demethylation occurs co-transcriptionally in the conditions profiled, but that the steady state levels of total m6A are the same in the cytoplasm. Whether pre-mRNA methylation is more dynamic in other biological conditions remains to be seen.
An outstanding mechanistic question is how cells achieve biased distribution of a modification over genomic features. m6A is largely confined within exon boundaries, which could be achieved if the m6A methyltransferase complex were recruited during exon definition prior to splicing, potentially by cross exon interactions between spliceosomal components. However, METTL3 does not appear to associate with components of the U1 and U2 snRNPs [45]. METTL3 does associate with Pol II [29], which is thought to transcribe exonic and intronic regions at different rates [54]. Therefore, one potential explanation for the exon-restricted distribution of m6A is that METTL3 rides along the gene body with Pol II and slower transcription through exons compared to introns increases the dwell time of METTL3 in the exon long enough to allow catalysis. Alternatively, the specificity of m6A in exons in pre-mRNA may be imparted by demethylases. If demethylase activity were greater in introns, then preferential removal of intronic m6As could lead to the observed exonic bias at steady state. In fact, one m6A demethylase, the fat mass and obesity-associated protein (FTO) was reported to bind primarily to introns and these binding sites correlate with known sites of m6A modification [55]. Therefore, the balance of the expression of the methyltransferase and demethylase could determine the level of net modification in exons versus introns in different cell types and under different conditions.
Two m6A demethylases have been identified to date, FTO and ALKBH5 [47,56]. Recently, FTO was found to preferentially demethylate m6A in the context of m6Am as the first nucleotide of the cap both in vitro and in bulk total mRNA assays [13]. Taken at face value, this result suggests limited potential for regulation of FTO to affect internal intronic methylation levels. However, the bulk assays are likely insensitive to changes in a small set of internal m6As that could be demethylated by FTO, as has been shown for a subset of internal m6A residues in acute myeloid leukemia cells [57]. Going forward, studies on the spatiotemporal relationship between Pol II elongation, spliceosome assembly and modification should clarify the mechanisms by which pre-mRNA modifications are added and removed and how these processes are interconnected.
SPLICING AND SPLICING REGULATION
The spliceosome assembles de novo on the pre-mRNA through interactions with splice site sequences that define the exon/intron boundaries to catalyze intron removal and exon joining. Splicing is achieved by binding of the snRNPs to the splice sites, an interaction which is primarily mediated by base pairing interactions between the snRNA component of snRNPs and the splice site sequences [58]. In mammals these interactions between the snRNPs and the splice site sequences are relatively weak, allowing for regulation. Alternative splicing is the differential inclusion of exons or introns, or a portion of these, into the mature mRNA. Assembly of the spliceo- some at splice sites is regulated by auxiliary factors (splicing factors) recruited to regulatory sequences within an alternative exon or its flanking introns to either promote or repress the snRNP-pre-mRNA interactions. Two major splicing regulator protein families are the serine/arginine rich (SR) proteins and heterogeneous nuclear ribonucleoproteins (hnRNPs) [59,60]. SR proteins have been traditionally thought to bind to exons to enhance splice site usage and exon inclusion, whereas hnRNPs typically induce exon skipping. It is now clear that SR proteins can also repress and hnRNP proteins can also enhance exon inclusion depending on their binding position on the pre-mRNA [61-63]. Many aspects of splicing mechanism and alternative splicing have recently been reviewed in greater detail [62,64]. Here we will focus on specific splicing events that have been demonstrated or are likely to be affected by pre-mRNA modifications.
Modifications in the snRNAs influence splicing
The first evidence that RNA modifications function in splicing came from the study of modifications in the snRNAs. All five of the snRNAs contain modified nucleotides including extensive pseudouridylation and 2ˊ-O-methylation as well as one N6-methyladenosine in U6 snRNA. Pseudouridylation and 2ˊ-O-methylation of the snRNAs are catalyzed primarily by the box H/ACA and box C/D snoRNPs, respectively. snRNA modifications are found in important functional regions and affect splicing activity as exemplified by studies of U2 snRNA. Pseudouridylation of U2 snRNA in the region that base pairs with the pre-mRNA branch point sequence is required for splicing of one tested pre-mRNA substrate in Xenopus oocytes [65]. Consistent with a stimulatory effect on splicing, these pseudouridines were found to stabilize pre-mRNA-snRNA interaction by solution NMR [66]. Endogenous pseudouridylation at two of the positions of the yeast U2 snRNA in the branch site recognition region contributes to pre-mRNA splicing by altering the RNA secondary structure of the branchpoint - interacting stem loop of the U2 snRNA to facilitate the binding of the essential splicing factor Prp5 [67]. U2 snRNA lacking either of three endogenous pseudouridines found in the first 20 nucleotides decreased splicing efficiency and U2 snRNA lacking all three pseudouridines abrogated splicing of a pre-mRNA substrate in human HeLa cell nuclear extracts depleted of U2 snRNA and reconstituted with the modification defective U2 snRNAs [68]. Likewise, four out of five 2ˊ-O-methylated nucleotides in U2 snRNA were shown to be essential for splicing of the same substrate in HeLa nuclear extract [68].
While U2 snRNA is the most heavily modified of the snRNAs, the other snRNAs all contain modified nucleotides. Pseudouridines in the U1 and U5 snRNAs are also clustered around the regions that base pair with the pre- mRNA, which are important for splicing [69]. The pseudouridines and 2ˊ-O-methylated nucleotides in U4 and U6 snRNAs occur in regions where these two snRNAs base pair with each other and could contribute to duplex stabilization. Finally, m6A is present in the U6 snRNA in the region that base pairs with the 5ˊ splice site [70,71]. The m6A nucleotide in the U6 snRNA has been shown to base pair with a pre-mRNA substrate and that adenosine is critical for splicing catalysis. The mechanism by which the m6A in U6 snRNA affects splicing is unknown, however it could influence the strength of base pairing with 5ˊ splice sites or be important for the extensive structural rearrangements in the U6 snRNA during spliceosome assembly and catalysis [72,73].
Some modifications in the snRNAs are induced by changing cellular conditions to influence splicing. The yeast U2 snRNA has stress-inducible pseudouridines: one pseudouridine is added by Pus7 in response to heat shock and nutrient deprivation and the other is added by the box H/ACA snoRNP Cbf5 in response to nutrient deprivation through the mTOR signaling pathway [67,74]. Both pseudouridines are in stem II of U2 snRNA, a region where conformational changes are necessary for splicing catalysis. Recently, it was shown that these pseudouridines alter the conformational dynamics of stem II in a distinct manner [75]. The cellular functions for these inducible modifications remain to be determined, but these results suggest that pseudouridines may be employed to tune snRNA activity and impact splicing. Similarly, an inducible pseudouridine in the U6 snRNA is necessary for efficient splicing of suboptimal introns and is required for filamentous growth in yeast [76]. The evidence presented above supports an important function for snRNA modifications in splicing. The extent to which snRNA modifications influence interactions with individual pre-mRNAs and alternative splicing is still an open question.
Evidence that pre-mRNA modifications function in alternative splicing
Since the first identification of m6A locations transcrip- tome-wide, many studies have reported widespread changes in alternative splicing and isoform expression following depletion of the m6A methyltransferase METTL3 and the demethylase FTO [1,45-47,55,77-79]. However, ~20%−30% of METTL3- and FTO- sensitive alternatively spliced exons have been demonstrated to contain m6A [1,18,46,47,55,78]. This is not surprising given that manipulation of the methyltrans- ferases and demethylases is likely to have a plethora of indirect effects.
The extent to which m6A directly affects splicing regulation is a subject of debate since the reported number of alternative splicing events that are regulated by m6A levels (are affected by METTL3 knockdown/knockout) and also contain m6A varies substantially (from one to several hundred) among published studies [1,18,46,47, 55,78]. These discrepancies may exist for a number of reasons. Different cell lines are likely to have distinct pre- mRNA m6A methylation pattern and thus varying effects on m6A-dependent splicing regulation. Another factor that should be considered when generalizing from splicing analysis of RNA-seq data is that algorithms to detect alternative splicing can differ substantially in the number of splicing events detected, and produce different results with varying user-defined parameters. Some are more sensitive than others to outliers in the data; some are better at detecting complex splicing events and/or rely more/less on annotations of known isoforms [80]. Thus, apparent discrepancies in the prevalence of m6A-sensitive splicing events may reflect data analysis and arbitrary cutoffs rather than biological or experimental differences. Validation of the splicing changes detected by each algorithm by RT-PCR to establish a validation rate with the specified cutoff and datasets could help enhance the confidence in data analysis and conclusions derived from it. Nevertheless, multiple studies point to a limited, but perhaps direct role for m6A in alternative splicing regulation.
Importantly, most of the differential splicing analysis described above has been limited to internal cassette exons and has not included alternative last exons or analysis of the effect of intronic m6As on splicing . Given the enrichment of m6A over last exons, m6A-sensitive alternative last exon usage should be investigated in more detail. Similarly, analysis of the effect of intronic m6As on constitutive and alternative splicing of their neighboring exons may reveal functions for intronic m6As in pre- mRNA splicing regulation as has been shown in a recent study [46].
Broad assertions about the widespread nature of alternative splicing regulation by mRNA modifications have come from analysis of poly(A)+ mRNA-seq following knockdown or knockout of the “writers” and “erasers”. It is difficult to determine the role of pre-mRNA modifications from such experiments because of the high probability of indirect effects. What is needed is further investigation of individual pre-mRNA modifications in mechanistic detail. This will establish paradigms for modification mediated regulation, to then identify coregulated events that share sensitivity to multiple factors (e.g., splicing factor availability and/or binding, position of the modification and sequence context) from high- throughput genomic data sets. In vivo mechanistic studies have proved to be challenging given that knockouts and knockdowns of RNA modification enzymes can lead to a plethora of indirect effects that can confound results. This is especially true for the mRNA modifications that co-opt the tRNA modifying machinery (such as m1A and pseudouridine), because genetically manipulating these enzymes is likely to cause widespread effects downstream of perturbing the biogenesis of functional tRNA and other non-coding RNAs. Orthogonal approaches are urgently needed for site-specifically blocking or adding a modification to determine its ability to induce or rescue the molecular phenotypes identified from knockdown/knockout experiments. Along these lines, Yu and colleagues have engineered box H/ACA snoRNAs to guide site specific pseudouridylation of targets of interest, albeit with limited efficiency [81,82]. Other approaches to direct RNA modification writers or erasers to specific sites or subsets of sites would significantly advance the field.
Minigene reporters are useful tools for dissecting splicing regulatory mechanism and can readily be adapted to test the functional impact of a single modification on pre-mRNA splicing. These reporters, which contain the region of a regulated splicing event including the exon and flanking introns, must be tested to verify that they recapitulate the endogenous gene’s modification pattern and splicing sensitivity to genetic manipulation of RNA modification enzymes. Then, minigene mutagenesis to inhibit modification can be carried out to distinguish direct effects of an RNA modification on splicing from indirect effects of globally perturbing modification writers and erasers. If a local RNA modification directly influences splicing, the pre-mRNA processing effect observed upon modification enzyme knockout/knockdown will be abolished in the context of mutations to the modified nucleotide or mutations in the sequence or structure that is required for modification by the RNA modifying enzyme (e.g., the RRACH motif for m6A or the hairpin loop for TRUBl-dependent pseudouridylation [36]). Mutations of the modified nucleotide that alter splicing could represent the need for modification or the requirement for the nucleotide (modified or unmodified) as part of a regulatory sequence. Thus, similar results from multiple mutant minigene constructs would provide strong evidence that it is indeed loss of modification at the given pre-mRNA site that has a functional consequence for splicing. Furthermore, advances in genome editing with CRISPR/Cas9 now allow testing such mutations in the endogenous context. In vitro splicing of splicing reporters in splicing competent nuclear extracts can serve as a complementary approach to minigene mutagenesis to study the effects of RNA modifications on splicing. Site specific incorporation of a modified nucleotide into an in vitro splicing substrate can easily be achieved in vitro [83], and allows direct testing of the consequence of a modified nucleotide in splicing without knowing the corresponding RNA-modifying enzyme that installs the modification. However, this approach may not be feasible for all pre-mRNA substrates since splicing is inefficient in vitro and does not always recapitulate endogenous regulation. Pre-mRNA modifications that are found to be required for splicing regulation could influence splice site selection by diverse mechanisms.
MOLECULAR MECHANISMS BY WHICH PRE-mRNA MODIFICATIONS REGULATE SPLICING
Pre-mRNA modifications have the potential to impact splicing by three main mechanisms: by altering RNA- RNA interactions, by modulating RNA-protein interactions, and, indirectly, by influencing pre-mRNA-second- ary structure (Figure 2). Pre-mRNA modifications that are deposited within the splice site sequences could influence RNA-RNA interactions between the snRNAs and the pre- mRNA (Figure 2A) and thereby regulate splicing directly, by stabilizing or destabilizing base-pairing between splice sites and spliceosomal snRNAs (as discussed for snRNAs above). m5C has been shown to have structural stabilization properties when present in tRNA [84]. Similarly, pseudouridine [85,86], hm5C [87] and Nm [88,89] have all been shown by thermal denaturation experiments to stabilize RNA duplexes by (1-2 kcal/mol) as compared to their unmodified counterparts, while m6A has been shown to destabilize RNA duplexes to a similar magnitude [90,91]. Regulated modifications in the splice sites could influence alternative splice selection by increasing or decreasing the stability of base pairing interactions between the pre-mRNA the snRNAs. Given the strict RRACH sequence requirement for METTL3-mediated m6A addition, it is unlikely that many m6As are found at the splice site sequences (5ˊ splice site GURAGU, branch site YNYURAC, 3ˊ YAG consensus) under any conditions [45]. However, other modifications such as pseudouridine are added in diverse sequence contexts and as such could be added to the splice site sequences [3]. In one study, 2ˊ-O-methylation of the branch site adenosine inhibited splicing of an intron of the adenovirus pre-mRNA, as expected since the 2ˊ hydroxyl of the adenosine is required for the first step of splicing catalysis [92]. If cryptic sites were available, these were used when the canonical branch point adenosine was methylated. Thus, 2ˊ-O-methylation of an endogenous branch site could inhibit splicing or modulate branch site selection although mammalian branch site identification remains challenging [93-95]. Nm has been identified as a moderately abundant modification in purified human mRNA by mass spec analysis, and a sequencing based method to map the Nm RNA modification found 16% of putative Nm sites in introns [11]. Further validation of intronic Nm sites is needed as the enrichment of a primer sequence motif among these candidate Nm sites suggests a pervasive artefact due to mispriming during reverse transcription [96].
Figure 2. Molecular mechanisms by which pre-mRNA modifications regulate splicing.
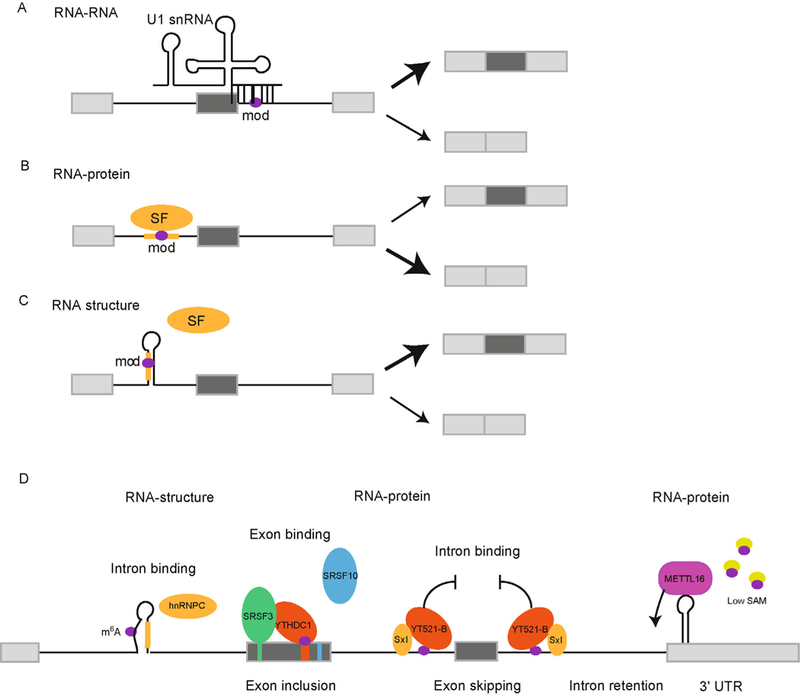
RNA modifications (mod) located in pre-mRNA introns could: (A) alter snRNA-pre-mRNA interactions to change inclusion of an alternative exon (dark grey). U1 snRNA and the 5’ splice site are shown as an example, but modifications could similarily alter pre-mRNA interactions with the U2 or U6 snRNAs; (B) enhance interaction between a splicing factor and its regulatory binding site on the pre-mRNA to change splicing outcome; (C) stabilize or disrupt structures that occlude or expose the binding site of a splicing factor or accessibility of a splice site sequence to the snRNAs. SF-Splicing factor, mod-RNA modification, gold line-SF binding site. (D) Mechanisms by which m6A regulates splicing. Left to right. By modulating structure to increase accessibility of hnRNPC to its binding site in introns. YTHDC1 is a direct m6A reader that binds to m6A in exons, recruits splicing factor SRSF3 to the exon and inhibits SRSF10 binding to enhance exon inclusion. YT521-B (Drosophila YTHDC1) binds the intron of the Sex-lethal pre-mRNA along with the Sex-lethal protein itself to repress exon inclusion and promote female specific splicing. In SAM depleted conditions METTL16 binding to the 3’ UTR is stabalized and METTL16 is sufficient to promote splicing of the upstream intron.
Pre-mRNA modifications could also influence splicing by directly affecting binding of various RNA-binding proteins to their RNA targets (Figure 2B). In the case of m6A, dedicated readers such as the YTH domain family of proteins directly and specifically interact with the methylated adenosine. For example, YTHDC1, the nuclear reader of m6A, preferentially interacts with m6A-containing pre-mRNA (as discussed below). In addition, modifications in pre-mRNA could more subtly alter the affinity of known splicing factors to their binding sites on pre-mRNA. In this manner, controlled deposition of a modification in exonic or intronic splicing enhancer or silencer elements could co-regulate splicing of a subset of targets. Alternatively, modifications that alter the stability of RNA duplexes could indirectly alter splice site accessibility and/or splicing factor binding by changing pre-mRNA secondary structure, as has been demonstrated for certain intronic m6A modification sites (see below) (Figure 2C). Although the exact magnitude of direct splicing regulation by m6A remains to be refined, in individual cases that have been studied in mechanistic detail m6A does function to regulate pre-mRNA splicing for important biological processes. In the following sections, we will review the known mechanisms of m6A-dependent splicing regulation through altered direct and indirect RNA-protein interactions.
METTL16 acts as an m6A reader to promote intron splicing
The primary SAM synthetase in human cells is encoded by the gene MAT2A and serves as the primary methyl donor for most methylation reactions in the cell. In SAM replete conditions, METTL16, the U6 snRNA methyl- transferase, transiently interacts with and specifically methylates an adenosine in a hairpin in the 3ˊ-UTR of the MAT2A gene leading to retention of the upstream intron (Figure 2D) [31]. The intron retained isoform of MAT2A is degraded in the nucleus and consequently MAT2A mRNA and protein levels decrease [97]. In contrast, when SAM levels are low, slowed catalysis caused by lack of the methyl donor increases METTL16 occupancy on the hairpin which promotes splicing of the intron. The positive local effect of METTL16 on splicing was verified by tethering of METTL16 to the 3ˊ-UTR hairpin, which was sufficient to enhance splicing of the upstream intron. Mutational studies provided further support for this regulatory mechanism: mutation of the methylated adenosine or the METTL16 recognition sequence of the hairpin reduced METTL16 binding, abrogated methylation of the hairpin, and decreased METTL16-induced splicing. In addition, METTL16 was shown to directly methylate the MAT2A hairpin in vitro.
METTL16 depletion reduced m6A content at more than two thousand sites in poly(A)+ mRNA, most which were in presumably retained introns. Some of these changes are likely to be an indirect consequence of altered SAM levels in METTL16 knockdown cells, downstream of perturbing MAT2A splicing and expression. Indeed, knockdown or overexpression of MAT2A modulated bulk m6A levels in mRNA similarly, and direct METTL16 binding was not detected at several tested candidate METTL16 m6A sites. Regardless, METTL16 may have additional methylation targets in pre-mRNA and one study reported METTL16 binding primarily to introns by CLIP-seq [32]. Interestingly, METTL16 acts as both a writer and reader of m6A in the case of MAT2A [98-100]. This METTL16- mediated feedback loop that controls SAM synthetase expression in response to SAM levels provides an example of a signal-induced regulation of m6A deposition that in turn directly regulates pre-mRNA splicing and mRNA expression. Most studies to date have focused on basal levels of mRNA modifications and not regulated cases. This example should motivate studies to search for instances in which mRNA modification enzymes are regulated in response to extracellular signals to alter pre- mRNA processing.
YTHDC1 recruits SRSF3 to promote exon inclusion and antagonize SRSF10
YTHDC1 or YT521-B was the inaugural member of the YTH family of proteins and a known splicing factor [98-100]. It was first identified as a splicing factor interacting protein from yeast two-hybrid and co-immunoprecipita- tion experiments [98,101]. It interacts with numerous splicing factors including SRSF2, TRA2A, TRA2B, SRSF3, SRSF10, SAM68 and hnRNPG [78,98,101]. The YTH domain of YTHDC1 was identified and characterized to bind single stranded RNA and found to have homologs across eukaryotes [100]. Later, YTHDC1 was shown to bind methylated RNAs preferentially [102]. Consistent with the view that YTHDC1 is an m6A binding protein in vivo, PAR-CLIP of YTHDC1 showed a significant overlap between its binding sites and known m6A sites identified in HeLa cells, most of which were distributed in exons [102]. YTHDC1 influences alternative splicing both indirectly, through interacting with other splicing factors independent of its YTH domain [101,103], and by direct interaction with pre-mRNA targets dependent on its YTH domain [100]. Specifically, overexpression of YTHDC1 modulates splice site selection of alternative exons of pre-mRNAs and deletion of the YTH domain abrogates this effect.
YTHDC1 interacts with exons in pre-mRNA to recruit SRSF3 to methylated pre-mRNAs and promote exon inclusion. SRSF3 binding to total RNA was reduced following YTHDC1 or METTL3 knockdown suggesting its binding to RNA is sensitive to m6A content. Binding of SRSF3 to a methylated oligo in vitro was enhanced by addition of recombinant YTHDC1. From RNA-seq and PAR-CLIP data it was inferred that SRSF3 and YTDC1 bind and co-regulate alternative splicing events at least in part by antagonizing SRSF10 binding to internal cassette exons [78]. A handful of cassette exons in three pre- mRNAs (SP4, ZNF638 and ALG11) were validated as co-regulated by METTL3, SRSF3 and YTHDC1, and antagonized by SRSF10 (Figure 2D). m6A binding by YTHDC1 is required to promote exon inclusion since a YTH domain mutant does not rescue exon skipping that results from YTHDC1 depletion. Finally, a minigene of ZNF638 recapitulated endogenous co-regulation by YTHDC1, SRSF3 and SRSF10. Importantly, mutations to the predicted binding site of each protein on the alternative exon, including the RRACH motif, had the predicted effect of reducing exon skipping (YTHDC1 and SRSF3) or promoting exon inclusion (SRSF10).
A similar mechanism, involving cooperativity between YTHDC1, SRSF3 and antagonism by SRSF10, was found to influence splicing of pre-mRNAs during lytic replication of Kaposi’s sarcoma-associated herpesvirus (KSHV) [104]. Several stimuli that induce lytic replication in various cell lines increase m6A content. FTO and METTL3 knockdown increased and decreased, respectively, lytic-induced m6A levels of the viral pre-mRNA replication transcription activator (RTA), a key lytic protein. Chemical inhibition of m6A formation with 3- deazaadenosine (DAA) inhibited lytic-induced splicing of the RTA intron, decreased RTA protein expression and attenuated lytic replication. m6A is present in both the regulated intron and the downstream exon of RTA and mutation of three of the methylated RRACH motifs, two in the intron and one in the exon, in a minigene context inhibited intron splicing. UV crosslinking followed by immunoprecipitation and qRT-PCR of wildtype RTA pre- mRNA compared to m6A-deficient mutant RTA demonstrated that lytic-induced m6A addition and interactions with YTHDC1 and SRSF3 were abolished in each of the m6A mutants while SRSF10 interaction was enhanced. Thus, YTHDC1 and SRSF3 have been implicated as positive factors in both host and viral splicing and may represent a common mode of m6A-dependent splicing regulation.
YT521-B, Drosophila YTHDC1, promotes female-specific splicing of Sex-lethal
YT521-B is the Drosophila ortholog of human YTHDC1, and it localizes exclusively to the nucleus [101]. mRNA m6A methylation is widespread in Drosophila and Drosophila has orthologs of all identified members of the human N6-methyladenosine methyltransferase complex: inducer of meiosis (Ime4, homologous to METTL3), karyogamy protein (KAR4, METTL14), female-lethal (fl(2)d, WTAP), Virilizer (Vir, KIAA1429) and Spenito (Nito, RBM15/15B) [30,105,106]. Fl(2)d, Vir, and Spenito were previously characterized splicing factors known to be required for splicing-mediated sex determination in Drosophila [107-113]. Specifically, these proteins promote female-specific splicing of Sex- lethal, the master regulator of sex determination in Drosophila [107,111]. The male-specific isoform of Sex-lethal (Sxl) results from inclusion of a cassette exon that introduces a premature termination codon and thereby decreases Sex-lethal expression. In females, binding of Sex-lethal itself to the introns flanking the male cassette exon represses exon inclusion in a feedback loop.
The sex-specific phenotypes of mutants affecting the methyltransferase complex implicated m6A in splicing regulation of Sxl. Ime4 (METTL3) mutant female flies are flightless, display male specific features and develop ovarian tumors suggesting a link between the putative methyltransferase and sex determination [30,105]. Ime4 and Sxl interact genetically leading to reduced survival of female progeny and YT521-B mutant flies phenocopy these methyltransferase complex mutants [30,105,106]. Ime4 mutants showed reduced overall levels of m6A as expected, and displayed a concomitant shift toward the male-specific, exon-included isoform of Sxl [105,106]. Similarly, Kar4 and YT521-B mutants promoted exon inclusion [105,106].
The effect of the methyltransferase mutants on Sxl splicing is likely to be direct. The introns flanking the Sxl cassette exons were found to contain m6A near the Sxl binding sites [30,105]. The m6A binding protein YT521-B binds to the m6A containing introns of Sex lethal and overexpression of YT521-B is sufficient to repress inclusion of the Sxl male-specific exon (Figure 2D) [30,105]. Furthermore, m6A was found to be widespread in Drosophila based on m6A-seq and miCLIP experiments that revealed a distribution similar to human cells and enrichments of the RRACH motif at m6A peaks [105,106]. Depletion of Ime4 and concomitant loss of m6A led to changes in alternative splicing of additional genes, the majority of which were co-regulated by YT521-B [30,106]. Thus it is likely that YT521-B regulates m6A-dependent alternative splicing of additional genes in Drosophila.
hnRNPC and hnRNPG binding to m6A-sensitive structures influence splicing
Heterogeneous nuclear ribonucleoprotein C (hnRNP C) is an abundant nuclear protein member of the hnRNP family of splicing factors that binds to polyuridine sequences primarily in introns to influence pre-mRNA splicing. One mechanism by which hnRNP C represses exon inclusion is by competing with the core splicing factor U2AF65, which binds to the uridine-rich polypyrimidine tract and promotes 3ˊ splice site recognition [114]. hnRNP C was identified as a protein that preferentially interacts with a hairpin in the abundant nuclear resident ncRNA MALAT1 in an m6A-dependent manner [46]. Structure probing of this MALAT1 hairpin revealed that the methylated adenosine is present in the stem opposite a poly(U) stretch that serves as the binding site for hnRNP C (Figure 2D). RNA duplex destabilization by m6Awas proposed to act as a structural switch: when an adenosine near a poly (U) element is unmethylated, it forms a stable base pair with the hnRNP C binding motif, sequestering the site in a stem and precluding hnRNP C binding. Methylation of the adenosine destabilizes the stem and allows hnRNP C binding. This finding prompted the search for m6A- switches transcriptome wide. Extending the previously reported enrichment of hnRNP C binding in introns [114], PAR-CLIP of hnRNP C followed by m6A-sequencing of bound RNA revealed that hnRNP C binds primarily to intronic m6A sites that resemble the MALAT1 m6A switches [46]. Approximately two thousand hnRNP C- bound m6A sites were no longer bound by hnRNP C following METTL3 and METTL14 knockdown, suggesting this is a widespread mechanism. Finally, a subset of alternatively spliced exons were shown to have neighboring intronic m6A switches that were bound by hnRNP C and co-regulated by hnRNP C, METTL3 and METTL14 (Figure 2D) [46].
Similarly, heterogeneous nuclear ribonucleoprotein G (hnRNP G) was also identified as a protein that preferentially binds m6A-modified MALAT1 [115]. As in the case of hnRNP C, m6A methylation increases the accessibility of the hnRNP G binding site. Interestingly, binding of hnRNP G to RNA in this context appeared to be mediated by a low complexity domain and not its canonical RNA recognition motif (RRM). As with hnRNP C, transcriptome-wide analysis revealed widespread m6A sites that regulate hnRNP G binding, and hnRNP G co-regulated numerous alternative splicing events with METTL3 and METTL14. Overall, these findings suggest that m6A may have widespread indirect effects on splicing factor binding by altering pre-mRNA structure and consequently binding site accessibility. One additional study proposed that another hnRNP, hnRNP A2/B1, binds m6A directly. They identified an RGCA motif similar to the RRACH METTL3 consensus sequence as enriched in hnRNP A2/B1 binding sites. hnRNP A2/B1 associated with m6A containing nuclear RNA with some overlap between specific hnRNP A2/B1 binding events and m6A sites [79]. A more recent study showed by gel shift and isothermal titration calorimetry that hnRNP A2/B1 does not have increased affinity for m6A-containing RNA in vitro [116]. Furthermore, their crystal structure of the hnRNP A2/B1 RRMs in complex with RNA did not reveal the aromatic cage-like surface that is required to directly contact m6A in the m6A “readers” YTHDC1 and YTHDF1 [116]. Therefore, hnRNP A2/B1 binding is likely indirectly affected by m6Aˊs ability to open local structure.
SRSF2 and FTO co-regulate alternative splicing
One study reported that SRSF2 binding at or around m6A sites increased upon FTO knockdown [47]. A panel of target genes that included the RUNX1T1 adipogenesis factor showed an increase in exon inclusion following FTO knockdown, which was correlated with increased m6A content and increased SRSF2 binding. FTO knockdown resulted in a decrease in the short, exon-skipped isoform of RUNX1T1, that was proposed to lead to increased adipogenesis based on isoform-specific overexpression studies. Further analysis is necessary to determine if these alternative splicing events are indeed m6A dependent and not indirectly influenced by FTO depletion.
Potential “readers” of other modifications relevant to splicing
Whether mRNA modifications other than m6A are present in pre-mRNA and regulate pre-mRNA splicing is currently unknown. However, other modifications have already been shown to impact protein binding in endogenous or artificial contexts. For instance, m5C has one nuclear reader, ALYREF, that was shown to facilitate mRNA export from the nucleus [40]. ALYREF has not been implicated in splicing, but establishes precedent for other m5C specific nuclear readers. Although no nuclear reader has been identified for Nm in mRNA, the presence of Nm has likewise been shown to affect protein interactions with mRNA including RIG-I the immune sensor demonstrating the capacity of Nm to influence protein binding [117]. Because this modification is incompatible with Watson-Crick base pairing, m1A has the potential to indirectly affect splicing factor binding via changes in RNA secondary structure.
Pseudouridine has been shown to affect multiple RNA- protein interactions including well-characterized splicing factors. Artificial pseudouridylation of a single uridine at either of two positions in the polypyrimidine tract of an adenoviral pre-mRNA was sufficient to abolish in vitro binding of the core-splicing factor U2AF65 and inhibit splicing of the pseudouridylated intron [83]. This striking effect of pseudouridine was attributed to its rigidifying of the polypyrimidine tract since locked nucleic acid substitutions at the same positions, which constrained the flexibility of the RNA backbone, led to similar inhibition of U2AF65 binding and defective splicing [83]. Likewise, binding of the splicing factor MBNL1 was significantly affected when pseudouridines were artificially added to its recognition motif in pre-mRNA [118]. In another example, pseudouridine indirectly influenced PRP5 binding to the U2 snRNA by stabilizing a structure, the branchpoint-interacting stem loop [67], a mechanism that could also happen in mRNA. Additionally, binding of the cytoplasmic RNA-binding protein Pumilio 2 to its UGUAR binding motif was modestly decreased when the second uridine was pseudouridine [119]. UGYAR is relatively abundant among endogenously pseudouridy- lated mRNAs due to pseudouridylation by PUS7 in the same context [3]. Together, these examples strongly suggest that if pseudouridine, m5C or Nm are identified in the core splicing recognition signals — or in splicing regulatory sequences — they could significantly impact splicing factor binding and splicing outcome.
CONCLUSIONS
Most mRNA modifying enzymes discovered to date localize to the nucleus where they may encounter nascent pre-mRNA. Diverse pre-mRNA modifications are therefore likely to add an additional layer of regulation to pre- mRNA processing as has been demonstrated from studies on m6A. In terms of splicing regulation, pre-mRNA modifications may influence which cis-regulatory elements are bound by a given trans-acting splicing factor and thereby mediate distinct regulation of a subset of targets to allow for fine tuning of splicing regulation against a backdrop of global regulation of splicing factor levels or activity. In this manner pre-mRNA targets of multiple splicing factors could be coordinately regulated and functionally linked in different cell types or under different growth conditions where the activity and/or the balance of RNA modifying enzymes may be different.
Our understanding of the function of pre-mRNA modifications in splicing is currently limited to examples of m6A-mediated splicing regulation. Whether other modifications are added to nascent pre-mRNA and, if so, whether and how they function in splicing regulation is mostly unknown. Moving forward, efforts to determine if and when a particular RNA modification is added to nascent pre-mRNA using approaches such as sequencing of chromatin-associated RNA [23,24,51-53] or metabolic labeling of newly transcribed RNA [49,120-122] will be a critical step towards illuminating the potential functions of additional modifications in pre-mRNA processing. In addition, the development of new methods to site- specifically manipulate RNA modifications including m6A would significantly advance the field by making it possible to distinguish direct versus indirect effects of global perturbations of RNA modifying enzymes on pre- mRNA splicing and nuclear processing. Given the demonstrated ability of modified nucleotides to affect diverse RNA-protein interactions, future studies to identify time and mechanism of addition of RNA modifications to pre-mRNA and to determine the function of individual modifications are likely to reveal new mechanisms by which nuclear pre-mRNA processing is controlled.
ACKNOWLEDGEMENTS
We thank members of the Gilbert lab for helpful discussions. We thank Erin Borchardt and Kristen W Lynch for their reading of the manuscript and suggestions. Funding sources: Jane Coffin Childs Memorial Fund Fellowship to Nicole M. Martinez NIH (GM101316 and CA187236) and the American Cancer Society (RSG-13-396-01-RMC) to Wendy V. Gilbert.
Footnotes
COMPLIANCE WITH ETHICS GUIDELINES
The authors Nicole M. Martinez and Wendy V. Gilbert declare that they have no conflict of interests.
This article is a review article and does not contain any studies with human or animal subjects performed by any of the authors.
REFERENCES
- 1.Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Sal- mon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob Hirsch J, Amariglio N, Kupiec M, et al. (2012) Topology of the human and mouse m6A RNA methylomes revealed by m6A- seq. Nature, 485, 201–206 [DOI] [PubMed] [Google Scholar]
- 2.Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE and Jaffrey SR (2012) Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell, 149, 1635–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carlile TM, Rojas-Duran MF, Zinshteyn B, Shin H, Bartoli KM and Gilbert WV (2014) Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature, 515, 143–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwartz S, Bernstein DA, Mumbach MR, Jovanovic M, Herbst RH, Leon-Ricardo BX, Engreitz JM, Guttman M, Satija R, Lander ES, et al. (2014) Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridyla- tion of ncRNA and mRNA. Cell, 159, 148–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lovejoy AF, Riordan DP and Brown PO (2014) Transcriptome-wide mapping of pseudouridines: pseudouridine synthases modify specific mRNAs in S. cerevisiae. PLoS One, 9, e110799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li X, Zhu P, Ma S, Song J, Bai J, Sun F and Yi C (2015) Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome. Nat. Chem. Biol, 11, 592–597 [DOI] [PubMed] [Google Scholar]
- 7.Squires JE, Patel HR, Nousch M, Sibbritt T, Humphreys DT, Parker BJ, Suter CM and Preiss T (2012) Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res, 40, 5023–5033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delatte B Wang F, Ngoc L, Collignon E, Bonvin E, Deplus R, Calonne E, Hassabi H, Putmans P, Awe S et al. (2016) Transcriptome-wide distribution and function of RNA hydro- xymethylcytosine. Science ,351, 282–285 [DOI] [PubMed] [Google Scholar]
- 9.Dominissini D, Nachtergaele S, Moshitch-Moshkovitz S, Peer E, Kol N, Ben-Haim MS, Dai Q, Di Segni A, Salmon-Divon M, Clark WC, et al. (2016) The dynamic N1- methyladenosine methylome in eukaryotic messenger RNA. Nature, 530, 441–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li X, Xiong X, Wang K, Wang L, Shu X, Ma S and Yi C (2016) Transcriptome-wide mapping reveals reversible and dynamic N1-methyladenosine methylome. Nat. Chem. Biol, 12, 311–316 [DOI] [PubMed] [Google Scholar]
- 11.Dai Q, Moshitch-Moshkovitz S, Han D, Kol N, Amariglio N, Rechavi G, Dominissini D and He C (2017) Nm-seqmaps 2 -O-methylation sites in human mRNA with base precision. Nat. Methods, 14, 695–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE and Jaffrey SR (2015) Single-nucleotide- resolution mapping of m6A and m6Am throughout the transcrip- tome. Nat. Methods, 12, 767–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP, Linder B, Pickering BF, Vasseur J-J, Chen Q, et al. (2017) Reversible methylation of m6Am in the 5’ cap controls mRNA stability. Nature, 541, 371–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gilbert WV, Bell TA & Schaening C (2016) Messenger RNA modifications: form, distribution, and function. Science, 352, 1408–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roundtree IA, Evans ME, Pan T and He C (2017) Dynamic RNA modifications in gene expression regulation. Cell, 169,1187–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patil DP, Pickering BF and Jaffrey SR (2018) Reading m6A in the transcriptome: m6A-binding proteins. Trends Cell Biol, 28, 113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song J and Yi C (2017) Chemical modifications to RNA: anew layer of gene expression regulation. ACS Chem. Biol, 12, 316–325 [DOI] [PubMed] [Google Scholar]
- 18.Ke S, Alemu EA, Mertens C, Gantman EC, Fak JJ, Mele A, Haripal B, Zucker-Scharff I, Moore MJ, Park CY, et al. (2015) A majority of m6A residues are in the last exons, allowing the potential for 3’ UTR regulation. Genes Dev, 29, 2037–2053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Safra M, Sas-Chen A, Nir R, Winkler R, Nachshon A, Bar- Yaacov D, Erlacher M, Rossmanith W, Stern-Ginossar N and Schwartz S (2017) The m1A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nature, 551, 251–255 [DOI] [PubMed] [Google Scholar]
- 20.Li X, Xiong X, Zhang M, Wang K, Chen Y, Zhou J, Mao Y, Lv J, Yi D, Chen X-W, et al. (2017) Base-resolution mapping reveals distinct m1A methylome in nuclear- and mitochondrial-encoded transcripts. Mol. Cell, 68, 993–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khoddami V and Cairns BR (2013) Identification of direct targets and modified bases of RNA cytosine methyltransferases. Nat. Biotechnol, 31, 458–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hussain S, Sajini AA, Blanco S, Dietmann S, Lombard P, Sugimoto Y, Paramor M, Gleeson JG, Odom DT, Ule J, et al. (2013) NSun2-mediated cytosine-5 methylation of vault noncoding RNA determines its processing into regulatory small RNAs. Cell Reports, 4, 255–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhatt DM, Pandya-Jones A, Tong A-J, Barozzi I, Lissner MM, Natoli G, Black DL and Smale ST (2012) Transcript dynamics of proinflammatory genes revealed by sequence analysis of subcellular RNA fractions. Cell, 150, 279–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tilgner H, Knowles DG, Johnson R, Davis CA, Chakrabortty S, Djebali S, Curado J, Snyder M, Gingeras TR and Guigó R (2012) Deep sequencing of subcellular RNA fractions shows splicing to be predominantly co-transcriptional in the human genome but inefficient for lncRNAs. Genome Res, 22, 1616–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, et al. (2014) A METTL3-METTL14 complex mediates mammalian nuclear RNA N-adenosine methylation. Nat. Chem. Biol, 10, 93–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ping XL, Sun B-F, Wang L, Xiao W, Yang X, Wang WJ, Adhikari S, Shi Y, Lv Y, Chen Y-S, et al. (2014) Mammalian WTAP is a regulatory subunit of the RNA N- methyladenosine methyltransferase. Cell Res, 24, 177–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ortega A, Niksic M, Bachi A, Wilm M, Sánchez L, Hastie N and Valcárcel J (2003) Biochemical function of female-lethal (2)D/Wilms’ tumor suppressor-1-associated proteins in alternative pre-mRNA splicing. J. Biol. Chem, 278, 3040–3047 [DOI] [PubMed] [Google Scholar]
- 28.Horiuchi K, Kawamura T, Iwanari H, Ohashi R, Naito M, Kodama T and Hamakubo T (2013) Identification of Wilms’ tumor 1-associating protein complex and its role in alternative splicing and the cell cycle. J. Biol. Chem, 288, 33292–33302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Slobodin B, Han R, Calderone V, Vrielink JAFO, Loayza-Puch F, Elkon R and Agami R (2017) Transcription impacts the efficiency of mRNA translation via co-transcriptional N6-adenosine methylation. Cell, 169, 326–337.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haussmann IU, Bodi Z, Sanchez-Moran E, Mongan NP, Archer N, Fray RG and Soller M (2016) m6A potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature, 540, 301–304 [DOI] [PubMed] [Google Scholar]
- 31.Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP and Conrad NK (2017) The U6 snRNA m6A methyltransferase METTL16 regulates SAM synthetase intron retention. Cell, 169, 824–835.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Warda AS, Kretschmer J, Hackert P, Lenz C, Urlaub H, Höbartner C, Sloan KE and Bohnsack MT (2017) Human METTL16 is a A^-methyladenosine (m6A) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep,18, 2004–2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brown JA, Kinzig CG, DeGregorio SJ and Steitz JA(2016) Methyltransferase-like protein 16 binds the 3’-terminal triple helix of MALAT1 long noncoding RNA. Proc. Natl. Acad. Sci. USA, 113, 14013–14018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lafontaine DLJ, Bousquet-Antonelli C, Henry Y, Caizergues-Ferrer M and Tollervey D (1998) The box H + ACA snoRNAs carry Cbf5p, the putative rRNA pseudouridine synthase. Genes Dev, 12, 527–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zebarjadian Y, King T, Fournier MJ, Clarke L and Carbon J (1999) Point mutations in yeast CBF5 can abolish in vivo pseudouridylation of rRNA. Mol. Cell. Biol, 19, 7461–7472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Safra M, Nir R, Farouq D, Vainberg Slutskin I and Schwartz S (2017) TRUB1 is the predominant pseudouridine synthase acting on mammalian mRNA via a predictable and conserved code. Genome Res, 27, 393–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fernandez-Vizarra E, Berardinelli A, Valente L, Tiranti V and Zeviani M (2007) Nonsense mutation in pseudouridylate synthase 1 (PUS1) in two brothers affected by myopathy, lactic acidosis and sideroblastic anaemia (MLASA). J. Med. Genet, 44, 173–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thul PJ Äkesson L, Wiking M, Mahdessian D, Geladaki A , Blal HA, Alm T, Asplund A, Björk L, Breckels LM, et al. (2017) A subcellular map of the human proteome. Science, 356, eaal3321. [DOI] [PubMed] [Google Scholar]
- 39.Ji X, Dadon DB, Abraham BJ, Lee TI, Jaenisch R, Bradner JE and Young RA (2015) Chromatin proteomic profiling reveals novel proteins associated with histone-marked genomic regions. Proc. Natl. Acad. Sci. USA, 112, 3841–3846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang X, Yang Y, Sun B-F, Chen Y-S, Xu J-W, Lai W-Y, Li A, Wang X, Bhattarai DP, Xiao W, et al. (2017) 5- methylcytosine promotes mRNA export - NSUN2 as the methyltransferase and ALYREF as an m5C reader. Cell Res,27, 606–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Helm M and Motorin Y (2017) Detecting RNA modifications in the epitranscriptome: predict and validate. Nat. Rev. Genet, 18, 275–291 [DOI] [PubMed] [Google Scholar]
- 42.Stadler C, Rexhepaj E, Singan VR, Murphy RF, Pepperkok R, Uhlen M, Simpson JC and Lundberg E (2013) Immunofluorescence and fluorescent-protein tagging show high correlation for protein localization in mammalian cells. Nat. Methods, 10, 315–323 [DOI] [PubMed] [Google Scholar]
- 43.Fu L, Guerrero CR, Zhong N, Amato NJ, Liu Y, Liu S, Cai Q, Ji D, Jin S-G, Niedernhofer LJ, et al. (2014) Tet- mediated formation of 5-hydroxymethylcytosine in RNA. J. Am. Chem. Soc, 136, 11582–11585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huber SM, van Delft P, Mendil L, Bachman M, Smollett K, Werner F, Miska EA and Balasubramanian S (2015) Formation and abundance of 5-hydroxymethylcytosine in RNA. ChemBioChem, 16, 752–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ke S, Pandya-Jones A, Saito Y, Fak JJ, Vagb0 CB, Geula S, Hanna JH, Black DL, Darnell JE Jr and Darnell RB (2017) m A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev, 31, 990–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu N, Dai Q, Zheng G, He C, Parisien M and Pan T (2015) N6-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature, 518, 560–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao X, Yang Y, Sun B-F, Shi Y, Yang X, Xiao W, Hao Y-J, Ping X-L, Chen Y-S, Wang W-J, et al. (2014) FTO- dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res, 24, 1403–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Amort T, Rieder D, Wille A, Khokhlova-Cubberley D, Riml C, Trixl L, Jia X-Y, Micura R and Lusser A (2017) Distinct 5-methylcytosine profiles in poly(A) RNA from mouse embryonic stem cells and brain. Genome Biol, 18, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miller C, Schwalb B, Maier K, Schulz D, Dümcke S, Zacher B, Mayer A, Sydow J, Marcinowski L, Dölken L, et al. (2011) Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol. Syst. Biol, 7, 458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wuarin J and Schibler U (1994) Physical isolation of nascent RNA chains transcribed by RNA polymerase II: evidence for cotranscriptional splicing. Mol. Cell. Biol, 14, 7219–7225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pandya-Jones A, Bhatt DM, Lin C-H, Tong A-J, Smale ST and Black DL (2013) Splicing kinetics and transcript release from the chromatin compartment limit the rate of lipid A-induced gene expression. RNA, 19, 811–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Khodor YL, Rodriguez J, Abruzzi KC, Tang C-HA, Marr II MT and Rosbash M (2011) Nascent-seq indicates widespread cotranscriptional pre-mRNA splicing in Drosophila. Genes Dev, 25, 2502–2512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Khodor YL, Menet JS, Tolan M and Rosbash M (2012) Cotranscriptional splicing efficiency differs dramatically between Drosophila and mouse. RNA, 18, 2174–2186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jonkers I, Kwak H and Lis JT (2014) Genome-wide dynamics of Pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. eLife, 3, e02407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bartosovic M, Molares HC, Gregorova P, Hrossova D, Kudla G and Vanacova S (2017) N’-methyladenosine demethylase FTO targets pre-mRNAs and regulates alternative splicing and 3’-end processing. Nucleic Acids Res, 45, 11356–11370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang C-M, Li CJ, Vagb0 CB, Shi Y, Wang W-L, Song S-H, et al. (2013) ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell, 49, 18–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, Huang H, Nachtergaele S, Dong L, Hu C, et al. (2017) FTO Plays an oncogenic role in acute myeloid leukemia as a N-methyladenosine RNA demethylase. Cancer Cell, 31, 127–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wahl MC, Will CL and Lührmann R (2009) The spliceosome: design principles of a dynamic RNP machine. Cell, 136, 701–718 [DOI] [PubMed] [Google Scholar]
- 59.Long JC and Caceres JF (2009) The SR protein family of splicing factors: master regulators of gene expression. Biochem. J, 417, 15–27 [DOI] [PubMed] [Google Scholar]
- 60.Martinez-Contreras R, Cloutier P, Shkreta L, Fisette J-F, Revil T and Chabot B (2007) hnRNP proteins and splicing control. Adv. Exp. Med. Biol, 623, 123–147 [DOI] [PubMed] [Google Scholar]
- 61.Chen M and Manley JL (2009) Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat. Rev. Mol. Cell Biol, 10, 741–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fu XD and Ares M Jr. (2014) Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet, 15, 689–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martinez NM and Lynch KW (2013) Control of alternative splicing in immune responses: many regulators, many predictions, much still to learn. Immunol. Rev, 253, 216–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nilsen TW and Graveley BR (2010) Expansion of the eukaryotic proteome by alternative splicing. Nature, 463, 457–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhao X and Yu Y-T (2004) Pseudouridines in and near the branch site recognition region of U2 snRNA are required for snRNP biogenesis and pre-mRNA splicing in Xenopus oocytes. RNA, 10, 681–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Newby MI and Greenbaum NL (2001) A conserved pseudouridine modification in eukaryotic U2 snRNA induces a change in branch-site architecture. RNA, 7, 833–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu G, Adachi H, Ge J, Stephenson D, Query CC and Yu Y-T (2016) Pseudouridines in U2 snRNA stimulate the ATPase activity of Prp5 during spliceosome assembly. EMBO J, 35,654–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Donmez G, Hartmuth K and Luhrmann R (2004) Modified nucleotides at the 5’ end of human U2 snRNA are required for spliceosomal E-complex formation. RNA, 10, 1925–1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wu G, Yu AT, Kantartzis A and Yu YT (2011) Functions and mechanisms of spliceosomal small nuclear RNA pseudour- idylation. Wiley Interdiscip. Rev. RNA, 2, 571–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Epstein P, Reddy R, Henning D and Busch H (1980) The nucleotide sequence of nuclear U6 (4.7 S) RNA. J. Biol. Chem, 255, 8901–8906. [PubMed] [Google Scholar]
- 71.Shimba S, Bokar JA, Rottman F and Reddy R (1995) Accurate and efficient N-6-adenosine methylation in spliceoso- mal U6 small nuclear RNA by HeLa cell extract in vitro. Nucleic Acids Res, 23, 2421–2426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Brow DA and Guthrie C (1988) Spliceosomal RNA U6 is remarkably conserved from yeast to mammals. Nature, 334, 213–218 [DOI] [PubMed] [Google Scholar]
- 73.Gu J, Patton JR, Shimba S and Reddy R (1996) Localization of modified nucleotides in Schizosaccharomyces pombe spliceosomal small nuclear RNAs: modified nucleotides are clustered in functionally important regions. RNA, 2, 909–918. [PMC free article] [PubMed] [Google Scholar]
- 74.Wu G, Xiao M, Yang C and Yu YT (2011) U2 snRNA is inducibly pseudouridylated at novel sites by Pus7p and snR81 RNP. EMBO J, 30, 79–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.van der Feltz C, DeHaven AC and Hoskins AA (2018) Stress-induced pseudouridylation alters the structural equilibrium of yeast U2 snRNA stem II. J. Mol. Biol, 430, 524–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Basak A and Query CC (2014) A pseudouridine residue in the spliceosome core is part of the filamentous growth program in yeast. Cell Reports, 8, 966–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V, Peer E, Mor N, Manor YS, et al. (2015) m6A mRNAmethylation facilitates resolution of naive pluripotency toward differentiation. Science, 347, 1002–1006 [DOI] [PubMed] [Google Scholar]
- 78.Xiao W, Adhikari S, Dahal U, Chen Y-S, Hao Y-J, Sun B -F., Sun, H.-Y., Li, A., Ping, X.-L., Lai, W.-Y., et al. (2016) Nuclear m6A reader YTHDC1 regulates mRNA splicing. Mol. Cell, 61, 507–519 [DOI] [PubMed] [Google Scholar]
- 79.Alarcón CR, Goodarzi H, Lee H, Liu X, Tavazoie S and Tavazoie SF (2015) HNRNPA2B1 is a mediator of m6A- dependent nuclear RNA processing events. Cell, 162, 1299–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Norton S, Vaquero-Garcia J and Barash Y (2017) Outlier detection for improved differential splicing quantification from RNA-Seq experiments with replicates. bioRxiv, 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wu G, Huang C and Yu Y-T (2015) Pseudouridine in mRNA: incorporation, detection, and recoding. Methods Enzymol, 560, 187–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fernández IS, Ng CL, Kelley AC, Wu G, Yu Y-T and Ramakrishnan V (2013) Unusual base pairing during the decoding of a stop codon by the ribosome. Nature, 500, 107–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen C, Zhao X, Kierzek R and Yu Y-T (2010) A flexible RNA backbone within the polypyrimidine tract is required for U2AF65 binding and pre-mRNA splicing in vivo. Mol. Cell. Biol, 30, 4108–4119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen Y, Sierzputowska-Gracz H, Guenther R, Everett K and Agris PF (1993) 5-Methylcytidine is required for cooperative binding of Mg2+ and a conformational transition at the anticodon stem-loop of yeast phenylalanine tRNA. Biochemistry, 32, 10249–10253 [DOI] [PubMed] [Google Scholar]
- 85.Kierzek E, Malgowska M, Lisowiec J, Turner DH, Gdaniec Z and Kierzek R (2014) The contribution of pseudouridine to stabilities and structure of RNAs. Nucleic Acids Res, 42, 3492–3501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hudson GA, Bloomingdale RJ and Znosko BM (2013) Thermodynamic contribution and nearest-neighbor parameters of pseudouridine-adenosine base pairs in oligoribonucleotides. RNA, 19, 1474–1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Riml C, Lusser A, Ennifar E & Micura R (2017) Synthesis, thermodynamic properties, and crystal structure of RNA oligonucleotides containing 5-hydroxymethylcytosine. J. Org. Chem. 7b01171. [DOI] [PubMed] [Google Scholar]
- 88.Inoue H, Hayase Y, Imura A, Iwai S, Miura K and Ohtsuka E (1987) Synthesis and hybridization studies on two complementary nona (2’-O-methyl) ribonucleotides. Nucleic Acids Res,15, 6131–6148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Majlessi M, Nelson NC and Becker MM (1998) Advantages of 2’-O-methyl oligoribonucleotide probes for detecting RNA targets. Nucleic Acids Res, 26, 2224–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kierzek E and Kierzek R (2003) The thermodynamic stability of RNA duplexes and hairpins containing N6-alkyladenosines and 2-methylthio- N-alkyladenosines. Nucleic Acids Res, 31, 4472–4480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Roost C, Lynch SR, Batista PJ, Qu K, Chang HY and Kool ET (2015) Structure and thermodynamics of N6- methyladenosine in RNA: a spring-loaded base modification. J. Am. Chem. Soc, 137, 2107–2115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ge J, Liu H and Yu Y-T (2010) Regulation of pre-mRNA splicing in Xenopus oocytes by targeted 2’-O-methylation. RNA, 16, 1078–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mercer TR, Clark MB, Andersen SB, Brunck ME, Haerty W, Crawford J, Taft RJ, Nielsen LK, Dinger ME and Mattick JS (2015) Genome-wide discovery of human splicing branchpoints. Genome Res, 25, 290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gould GM, Paggi JM, Guo Y, Phizicky DV, Zinshteyn B Wang, E. T., Gilbert, W. V., Gifford, D. K. and Burge, C. B. (2016) Identification of new branch points and unconventional introns in Saccharomyces cerevisiae. RNA, 22, 1522–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bitton DA, Rallis C, Jeffares DC, Smith GC, Chen YYC Codlin S, Marguerat S and Bahler J (2014) LaSSO, a strategy for genome-wide mapping of intronic lariats and branch points using RNA-seq. Genome Res, 24, 1169–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gillen AE, Yamamoto TM, Kline E, Hesselberth JR and Kabos P (2016) Improvements to the HITS-CLIP protocol eliminate widespread mispriming artifacts. BMC Genomics, 17, 338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bresson SM, Hunter OV, Hunter AC and Conrad NK (2015) Canonical Poly(A) polymerase activity promotes the decay of a wide variety of mammalian nuclear RNAs. PLoS Genet, 11, e1005610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Imai Y, Matsuo N, Ogawa S, Tohyama M and Takagi T (1998) Cloning of a gene, YT521, for a novel RNA splicing- related protein induced by hypoxia/reoxygenation. Brain Res. Mol. Brain Res, 53, 33–40 [DOI] [PubMed] [Google Scholar]
- 99.Stoilov P, Rafalska I and Stamm S (2002) YTH: a new domain in nuclear proteins. Trends Biochem. Sci, 27, 495–497 [DOI] [PubMed] [Google Scholar]
- 100.Zhang Z, Theler D, Kaminska KH, Hiller M, de la Grange P, Pudimat R, Rafalska I, Heinrich B, Bujnicki JM, Allain FH-T, et al. (2010) The YTH domain is a novel RNA binding domain. J. Biol. Chem, 285, 14701–14710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hartmann AM, Nayler O, Schwaiger FW, Obermeier A and Stamm S (1999) The interaction and colocalization of Sam68 with the splicing-associated factor YT521-B in nuclear dots is regulated by the Src family kinase p59(fyn). Mol. Biol. Cell, 10, 3909–3926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Xu C, Wang X, Liu K, Roundtree IA, Tempel W, Li Y, Lu Z, He C and Min J (2014) Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat. Chem. Biol, 10, 927–929 [DOI] [PubMed] [Google Scholar]
- 103.Rafalska I, Zhang Z, Benderska N, Wolff H, Hartmann AM, Brack-Werner R and Stamm S (2004) The intranuclear localization and function of YT521-B is regulated by tyrosine phosphorylation. Hum. Mol. Genet, 13, 1535–1549 [DOI] [PubMed] [Google Scholar]
- 104.Ye F, Chen ER & Nilsen TW (2017) Kaposi’s sarcoma- associated herpesvirus utilizes and manipulates RNA N6- adenosine methylation to promote lytic replication. J. Virol. JVI.0046617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kan L, Grozhik AV, Vedanayagam J, Patil DP, Pang N, Lim K-S, Huang Y-C, Joseph B, Lin C-J, Despic V, et al. (2017) The m6A pathway facilitates sex determination in Drosophila. Nat. Commun, 8, 15737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lence T, Akhtar J, Bayer M, Schmid K, Spindler L, Ho CH, Kreim N, Andrade-Navarro MA, Poeck B, Helm M, et al. (2016) m6A modulates neuronal functions and sex determination in Drosophila. Nature, 540, 242–247 [DOI] [PubMed] [Google Scholar]
- 107.Granadino B, Campuzano S and Sánchez L (1990) The Drosophila melanogaster fl(2)d gene is needed for the female- specific splicing of Sex-lethal RNA. EMBO J, 9, 2597–2602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Granadino B, Penalva LOF and Sánchez L (1996) The gene fl(2)d is needed for the sex-specific splicing of transformer pre- mRNA but not for double-sex pre-mRNA in Drosophila melanogaster. Mol. Gen. Genet, 253, 26–31 [DOI] [PubMed] [Google Scholar]
- 109.Penalva LOF, Ruiz MF, Ortega A, Granadino B, Vicente L, Segarra C, Valcárcel J and Sánchez L (2000) The Drosophila fl(2)d gene, required for female-specific splicing of Sxl and tra pre-mRNAs, encodes a novel nuclear protein with a HQ-rich domain. Genetics, 155, 129–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Penn JKM, Graham P, Deshpande G, Calhoun G, Chaouki AS, Salz HK and Schedl P (2008) Functioning of the Drosophila Wilms’-tumor-1-associated protein homolog, Fl(2)d, in Sex-lethal-dependent alternative splicing. Genetics, 178, 737–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hilfiker A, Amrein H, Dübendorfer A, Schneiter R and Nöthiger R (1995) The gene virilizer is required for female- specific splicing controlled by Sxl, the master gene for sexual development in Drosophila. Development, 121, 4017–4026 [DOI] [PubMed] [Google Scholar]
- 112.Horabin JI and Schedl P (1996) Splicing of the Drosophila Sex-lethal early transcripts involves exon skipping that is independent of Sex-lethal protein. RNA, 2, 1–10 [PMC free article] [PubMed] [Google Scholar]
- 113.SchüEtt C, Hilfiker A and Nöthiger R (1998) virilizer regulates Sex-lethal in the germline of Drosophila melanogaster. Development, 125, 1501–1507 [DOI] [PubMed] [Google Scholar]
- 114.Zarnack K, König J, Tajnik M, Martincorena I, Eustermann S, Stévant I, Reyes A, Anders S, Luscombe NM and Ule J (2013) Direct competition between hnRNP C and U2AF65 protects the transcriptome from the exonization of Alu elements. Cell, 152, 453–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Liu N, Zhou KI, Parisien M, Dai Q, Diatchenko L and Pan T (2017) N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res, 45, 6051–6063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wu B, Su S, Patil DP, Liu H, Gan J, Jaffrey SR and Ma J (2018) Molecular basis for the specific and multivariant recognitions of RNA substrates by human hnRNP A2/B1. Nat. Commun., 9, 420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Devarkar SC, Wang C, Miller MT, Ramanathan A, Jiang F , Khan AG, Patel SS and Marcotrigiano J (2016) Structural basis for m7G recognition and 2’-O-methyl discrimination in capped RNAs by the innate immune receptor RIG-I. Proc. Natl. Acad. Sci. USA, 113, 596–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.deLorimier E, Hinman MN, Copperman J, Datta K, Guenza M and Berglund JA (2017) Pseudouridine modification inhibits muscleblind-like 1 (MBNL1) binding to CCUG repeats and minimally structured RNA through reduced RNA flexibility. J. Biol. Chem, 292, 4350–4357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Vaidyanathan PP, AlSadhan I, Merriman DK, Al-Hashimi HM and Herschlag D (2017) Pseudouridine and N6- methyladenosine modifications weaken PUF protein/RNA interactions. RNA, 23, 611–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Windhager L, Bonfert T, Burger K, Ruzsics Z, Krebs S, Kaufmann S, Malterer G, L’Hernault A, Schilhabel M, Schreiber S, et al. (2012) Ultrashort and progressive 4sU- tagging reveals key characteristics of RNA processing at nucleotide resolution. Genome Res, 22, 2031–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Duffy EE, Rutenberg-Schoenberg M, Stark CD, Kitchen RR, Gerstein MB and Simon MD (2015) Tracking distinct RNA populations using efficient and reversible covalent chemistry. Mol. Cell, 59, 858–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fuchs G, Voichek Y, Rabani M, Benjamin S, Gilad S, Amit I and Oren M (2015) Simultaneous measurement of genome- wide transcription elongation speeds and rates of RNA polymerase II transition into active elongation with 4sUDRB- seq. Nat. Protoc, 10, 605–618 [DOI] [PubMed] [Google Scholar]