

Abstract
Polyacenes are organic compounds that have multiple, fused, aromatic rings. These highly conjugated molecules often have interesting photonic and/or electronic properties that afford them the potential for application in a host of organoelectronic devices such as sensors, light emitting diodes, photovoltaic devices, and field-effect transistors. Here we show the development and use of the title (domino-HDDA) reaction to synthesize structurally diverse polyacenes from acyclic polyyne precursors. The key event in these transformations is the successive reaction of multiple 1,3-butadiyne units with a series of in situ-generated, diynophilic arynes. The polyyne substrates were designed to allow for rapid engagement of each progressively larger aryne following the initiating (and rate-limiting) production of the first reactive intermediate—the benzyne. We show that aryne trapping reactions are broad in scope and that these cascade or domino processes can be quite efficient.

Highly fused, multicyclic aromatic compounds are important for their potential utility in, for example, organic electronic and photonic applications.1 Benzyne/aryne chemistry has allowed chemists to construct various sub-classes and derivatives of these multi-ring compounds2,3,4. The formation of benzynes—long-standing5 and versatile intermediates in organic chemistry (see ref 6 for a recent comprehensive listing of reviews6)—under purely thermal7,8 (or photochemical9) conditions by the hexadehydro-Diels–Alder (HDDA) reaction10 represents a significant development11. In its simplest form, the HDDA reaction is an intramolecular cycloisomerization that converts a tethered triyne like 1 into the (much more stable!12) fused benzyne 2 (Fig. 1a), which is typically trapped directly by any of a large host of arynophiles. If the trapping agent were to be another 1,3-diyne, then, in principle, a second aryne—a naphthyne (3)—could be formed.
Figure 1. The domino HDDA reaction.
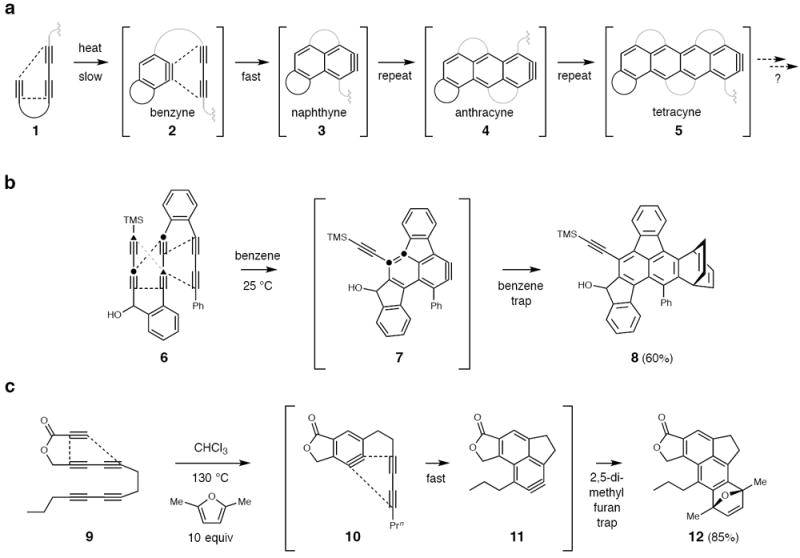
a) Generic scheme of sequential cycloisomerizations in which a new 1,3-diyne unit adds to an aryne to produce the next higher homolog among a series of polyacynes. b) A known example that demonstrates feasibility. c) Our first example, using a substrate designed to initiate reaction by cyclizing in only one sense—that is proceeding from 9 to the initial benzyne 10 and then on to the naphthyne 11 prior to being trapped by an external arynophile. Adduct 12 was the only product observed.
The earliest suggestion of the possibility of a benzyne engaging a 1,3-diyne stems from a study of acetylene pyrolysis13. In fact, Ueda and coworkers14,15,16 demonstrated that by tethering a second diyne unit into a substrate (see 6) it was possible to trap the initial benzyne to produce a naphthyne derivative (i.e., 7), which was then trapped by benzene to give 8 in the example shown (Fig. 1b). They described eight such reactions in which naphthynes were produced and trapped to give products in yields ranging from 10–63%. One potential competing (and efficiency lowering) event in those reactions was the initial cyclization of the “left-hand” tetrayne subunit to a non-productive benzyne—i.e., ring closure to the two atoms labeled as ▲ rather than ●. The reaction of benzyne with 1,3-butadiyne to generate naphthyne has also been studied by density functional theory (DFT) computations.17 Recently, the generation of a naphthyne through trapping of a conventionally generated benzyne (i.e., through reductive elimination of an ortho-iodotriflate by a Grignard reagent) by a tethered 1,3-diyne was described18.
Results and discussion
We envisioned that sequential HDDA reactions within a properly designed polyyne substrate could be used to synthesize higher acenes. We began our studies by designing a substrate—namely, 9—that would be likely to initiate reaction by cyclizing in only one sense (Fig. 1c). That is, we were confident that 9 would efficiently close to 1019 and presumed that it would proceed on to the naphthyne 11 prior to being trapped by an external arynophile. Indeed, when 9 was heated in the presence of 2,5-dimethylfuran, adduct 12 was the only product observed and was isolated in 85% yield following purification. The substrate 9 was prepared in three reactions from commercially available precursors (see Supplementary Section II).
We next explored the hypothesis that extension of the HDDA process to substrates containing more than one additional 1,3-diyne unit would lead to a cascade of cycloisomerizations. If so, this had the potential to greatly expand the versatility of this reaction by allowing rapid construction of complex, polycylic arenes (polyacenes). Moreover, we judged that a key design feature of the requisite polyyne substrates would be that each additional 1,3-diyne moiety be linked to the previous by a two-atom linking unit. Accordingly, we prepared substrate 13 (5 steps, see Supplementary Section II) and heated it in the presence of furan (5 equiv). Adduct 15 obligingly formed, again as the only observed product (Fig. 2a). Presumably (see below), the sequential cyclizations implied by the labels i, ii, and iii in substrate 13 had progressed smoothly to an anthracyne intermediate by way of the naphthyne 14. Similarly, the anthracyne adduct 16, a compound discussed in a later context, was formed when diethyl 3,4-furandicarboxylate20 was used as the trapping agent. To explore whether an aromatic 2-atom linker would be compatible with a similar cascade reaction, we prepared the heptayne 17, now containing a pair of o-substituted benzene rings rather than the two-atom ethano (or dimethylene) linkers along its backbone. The domino reaction of this substrate, being initiated by an essentially identical cyclization to the benzyne intermediate (step i), proceeded at a very similar rate as the other substrates described thus far. Furan trapping led to the isolation of 19 as the major product. We speculate that this reaction was somewhat less efficient because of the potential for further reaction of the more delocalized polyacene present in 19 by its [4+2] cycloaddition with another molecule of the anthracyne 18.
Figure 2. Benzyne to naphthyne to anthracyne.

a) The first example of extension of the domino reaction to a case involving three aryne-forming reactions: i) triyne to benzyne; ii) benzyne to naphthyne; iii) naphthyne to anthracyne; and final trapping event. b) Substrate 17 demonstrates that benzene subunits are suitable 2-atom linkers to conjoin adjacent pairs of 1,3-diynes, a feature that results in the formation of a product having more extended conjugation of the anthracene core in the product. TBS is tert-butyldimethylsilyl; o-DCB is 1,2- or ortho-dichlorobenzene.
To establish the bona fide intermediacy of discreet aryne intermediates in a domino process, we explored the effect of concentration of the external trapping reagent. Furan is a particularly reactive arynophile by virtue of its facile [4+2] cycloaddition, a feature that has been exploited from some of the very earliest investigations of benzyne reactivity21. Furan is also convenient to use as a bulk solvent. Several of our polyyne substrates, when heated in neat furan (ca. 14 M), led to a pair of products—the expected furan adduct of a naphthyne (cf. 12, Fig. 1c) as well as a benzyne/furan adduct, in which the intermediate benzyne was prematurely trapped by a solvent molecule. This bifurcation was most vivid in the case of the octayne 20 (Fig. 3). When heated in neat furan, the benzyne adduct 21 was formed to the near exclusion of the naphthyne-derived product 22 (Fig. 2). In a series of experiments, this reaction was progressively starved of furan; the product ratio favored 22 to an increasing extent, in accord with the expectation that furan trapping is a bimolecular process first-order in the concentration of furan, whereas the competing benzyne to naphthyne cyclization (cf. 10 to 11) is a unimolecular event whose rate is independent of the rate of external trapping. By contrast, we did not observe products of premature trapping by furan for any polyyne substrates containing a benzo-linker; presumably the additional degree of freedom in the ethano-linker slows the rate of each successive aryne homologation event by virtue of population of a larger number of unreactive conformers. As an aside, we note that the three-atom linker in the symmetrical octayne 20 here resides in the center of the molecule and that the domino reaction propagates in one direction only in a process that is, necessarily, desymmetrizing.
Figure 3. Polyyne to benzyne to naphthyne is a stepwise process.

The ratio of products 21:22 is dependent on the concentration of furan, a particularly reactive aryne trapping reagent. This demonstrates that the intermediate benzyne, the precursor to 21, has a sufficiently long lifetime that it can be trapped when the trapping event has a fast enough rate, that rate being dependent upon the inherent rate constant for the trapping event as well as on its concentration. The 21:22 branching ratio changes with [furan] used because, of course, the conversion of the benzyne to naphthyne intermediate is a unimolecular event whereas trapping of either aryne by furan is bimolecular. Ms is mesyl (or methanesulfonyl).
With all external arynophiles that we have examined that are less reactive than furan, intramolecular cyclization of each aryne to its next higher homolog was faster than trapping, and no products of premature interruption of the domino process were observed. We explored most extensively the scope of the reaction by generating the series of naphthynes 24 from substrates 23 (Fig. 4a). Many types of trapping reagents (T1–T2) are effective, providing access to a host of products 25 in good yields. The nature of the ABC tether unit in the polyyne substrates 23 can be varied widely11, and the two-atom linker between adjacent diynes can include substituted arenes (cf. 25h, 25q, 25t–25v) as well as ethano- and 1,2-benzo-linkers. In addition to Diels–Alder products with furan (25a, 25b); [4+2] cycloaddition adducts with a pyrrole (25c), o-dichlorobenzene (o-DCB, 25d), anthracene (25e), cyclopentadienones (25f, 25g), pyrones (25h, 25i), and perylene (25j, 25k) are readily formed. Nucleophilic traps are efficient [carboxylic acid (25l), sulfide (25m), and amine (25n)] as are other types of cycloaddition reactions {[2+2] (25o) and 1,3-dipolar (25p)}. Symmetrical vicinal difunctionalization is also well accommodated [dihalogenation22 (25q and 25r), dihydrogen transfer23 (25s and 25t), and dichalcogenation24,25 (25u and 25v)].
Figure 4. Examples of domino HDDA products via naphthyne intermediates.

a) Generic reaction and reaction conditions for naphthyne generation and trapping. b) Reactions that demonstrate considerable scope in the variety of naphthyne precursors 23 as well as trapping agents (T1–T2) that can be used. Variations of the three-atom (A-B-C) linker allow the formation of products containing the following types of rings newly fused to the initial benzyne intermediate: cyclopentane (cf. 25a), butyrolactone (cf. 25b), indanone (cf. 25c), pyrrolidine (cf. 25e), and indole (cf. 25h). Variations of the classes of trapping reactions include cycloadditions {[4+2] (cf. 25a–25k), [3+2] (cf. 25p), and [2+2] (cf. 25o)}, nucleophilic addition (cf. 25l–25n), oxidative addition (cf. 25q–25r and 25u–25v), and reductive addition (of H2, cf. 25s–25t). Ar is aryl; Ph is phenyl; TMS is trimethylsilyl; Boc is tert-butyloxycarbonyl; TPCPD is tetraphenylcyclopentadienone; Ac is acetyl; THT = tetrahydrothiophene.
aThe reaction solvent was chloroform unless otherwise stated. b25k has the solid bonds (see X-ray diffraction data in Supplementary Section V); 25k’ is the isomeric adduct having, instead, the analogous connectivity implied by the dashed bonds.
A few additional features of these reactions are noteworthy. The furan adduct 25a was also readily formed under photochemical-HDDA conditions9 (at ambient temperature). We computed the internal bond angles at the sp-hybridized carbons26 in some of the naphthyne intermediates [DFT B3LYP/6-31G*] and observed a smaller degree of distortion relative to the benzyne precursors12 (see Supplementary Fig. 1). Trapping with the cyclopentadienone 26 gave an isolable, bis-fused norbornadienone adduct (not shown, see Supplementary Section II and cf. 32, Fig. 6) enroute to 25g. Cheletropic ejection of CO was quite slow (relative to that in the simple o-benzyne adduct27), consistent with the smaller gain in aromatic resonance energy produced upon forming the highly delocalized dibenzotetracene moiety in 25g. The variability in the extent of regioselectivity observed for these reactions suggests that there is a subtle interplay between the inherent reactivity imposed by the degree of distortion of the aryne and the steric bias presented by the two different substituents flanking the aryne carbons as they are approached by trapping agents, themselves of varying size28.
Figure 6. Benzyne to naphthyne to anthracyne to tetracyne.

a) The nonayne 29 cyclizes in four serial (i–iv) events to a tetracyne that is cleanly trapped by anthracene, producing 30. b) Trapping the tetracyne with the dienone 26 provides the adduct 32, which can be more fully aromatized by thermolysis to give the dibenzohexacene 33. c) Absorption spectral data for 33 vis-à-vis 31 and 32. d) Geometry of the heavy atoms from the co-ordinates of the X-ray diffraction analysis of 32-H. d.r. is diastereomeric ratio.
Finally, the two trapping reactions with perylene to give 25j and 25k deserve comment. The former behaves in an expected fashion—namely, benzynes are known to add to the bay region diene portion of perylene29,30 and to eject hydrogen thermally to rearomatize. In contrast, the naphthyne leading to 25k (see Supplementary Section V for X-ray diffraction coordinates) differs from the one that gives 25j by the presence of a larger benzo- rather than ethano-two atom linker, which would encounter a greater steric obstruction were it to add to the bay region diene. DFT calculations (B3LYP/6-31G*, see Supplementary Fig. 2) support this view—the transition structure for concerted addition of that naphthyne to the bay region was computed to be 1.6 kcal•mol-1 higher in energy than that leading to 25k. A lesser amount of an isomeric product in which the naphthyne added in reversed head-to-tail fashion was also formed, and the DFT energetics were consistent with that process as well.
As an aside, we have observed some surprising acid-catalyzed processes for a subset of these polycyclic acenes; these show that unusual reactivity can ensue within certain, structurally complex HDDA products. Two examples are shown in Fig. 5. When exposed to trifluoroacetic acid, the highly fused anthracene derivative 16 gave rise to the doubly oxidized analog 27. When this (mechanistically unusual) transformation was monitored directly by NMR spectroscopy in CDCl3, resonances associated with metastable intermediates were seen, which eventually converged largely to those of the diene 27. This suggests that the oxidation occurrs in stages, likely by way of each of the two possible monoenes. A similar experiment, in which the air in the head space of the NMR sample tube was replaced by nitrogen, resulted in a much slower conversion, implicating dioxygen as the stoichiometric oxidant. Consistent with that and because 27 is more visibly colored than its precursor, 16, the color of the solution turned darker at the top of the NMR sample solution, closer to the air-liquid interface. Finally, in the absence of the TFA, 16 showed no sign of oxidative instability.
Figure 5. Unusual acid-catalyzed transformations of the products 16 and 25s.

a) Brønsted acid-promoted oxidation of the two bis-methylene subunits in 16 (red) to the alkenes in the pair of acenaphthylene subunits (red) in 27. b) Brønsted acid-promoted 1,2-migration within the phenylnaphthalene subunit of 25s, concomitant with formation of the deplanarizing, spirocyclic xanthene moiety. TFA is trifluoroacetic acid.
In a different setting, that of the fluoranthene derivative 25s, methanesulfonic acid was used to remove the trimethylsilyl group by a protodesilylation reaction and to catalyze the introduction of a spirocyclic xanthene (cf. 28) by a Friedel-Crafts addition/dehydration sequence31. Conversion of fluorenones to spirocycles of this sort has been used to confer fluorescence behavior that is rendered dormant by the ketone functional group32; indeed, compound 28 showed emission that was cyan in color (see Supplementary Fig. 10). The one-pot conversion of 25s to 28 was accompanied by a surprising 1,2-migration of the phenyl substituent from its hindered environment in the reactant to the much less encumbered position in 28. The mechanism of Brønsted acid-catalyzed 1,2-phenyl (or –naphthyl) migration on a benzene (or naphthalene) framework has been studied computationally,33,34 which indicates that this process likely proceeds by way of ipso-protonation of the phenyl-bearing carbon followed by phenyl migration to the adjacent carbon atom via a phenonium ion intermediate.
Having established that trapping of domino HDDA products (Fig. 4) is robust and that the benzyne-to-naphthyne-to-anthracyne cascade is viable (Fig. 2), we were motivated to further extend the domino process to see if a tetracyne could be accessed. Toward that end we prepared the nonayne 29. The route of synthesis involved twelve (8 longest linear sequence) reactions (see Supplementary Section II) from commercially sourced chemicals. By contrast, the preparation of nearly all of the other multi-yne substrates in the work reported here required no more than seven synthesis steps.
When 29 was heated in the presence of anthracene, a pleasingly efficient transformation to the adduct 30 transpired (Fig. 6a), establishing that, indeed, a tetracyne can be generated by a domino-HDDA reaction. From 1H NMR spectra, 30 is inferred to exist as a pair of topological isomers [crossed (cf. 31) vs. butterfly] that interconvert slowly on the NMR time scale and whose equilibrium ratio is somewhat solvent dependent. DFT calculations [SMD(CHCl3)/B3LYP-D3BJ/6-31G**//B3LYP/6-31G*] are consistent with these observations, were used to assign structure, and predicted that the desilylated derivative 31 would have a larger preference for one isomer (see Supplementary Figs. 3–5). Indeed, protodesilylation with methanesulfonic acid smoothly gave 31, which was largely a single isomer (d.r. = 47:1).
In a second example (Fig. 6b), the tetracyne from 29 was trapped by the dienone 26, producing the diketone 32. Protodesilylation (MsOH) gave the intermediate 32-H, which, upon thermolytic ejection of CO, smoothly provided the dibenzohexacene product 33. In contrast to relatively unsubstituted polyacenes35, none of these compounds showed signs of sensitivity to air and/or light. The most highly conjugated polyacene 33 has its longest wavelength absorption moved significantly into the infrared (Fig. 6c). Compounds that absorb near-IR light find utility in applications such as organic field-effect transistors, organic photovoltaics, and photodetectors36. Again, topoisomerism was seen37 (NMR) for each of 32–33. Fortunately, evaporative crystallization of a solution of 32-H gave material suitable for single crystal X-ray diffraction analysis (Fig. 6d). The helical twist of the fused tetracene core is clearly evident, a feature mimicked in the lowest energy topoisomer found by DFT calculations (see Supplementary Fig. 6).
In conclusion, we have established that the domino-HDDA reaction is an efficient process having considerable generality. It proceeds through a staged series of cycloisomerization events within substrates containing varying numbers of 1,3-diyne units, each separated by two-atom spacers. The cascade is initiated by a diynophilic alkyne that is connected uniquely by a three-atom tether. This reagent-free thermal reaction is remarkably robust, is broad in scope, and generates considerable structural complexity, as underscored, for example, by the nine newly formed rings present in each of 31–33. A future challenge may be the management of potential product solubility and/or stability issues, but these have not posed significant problems to date.
Methods
General protocol for performing the domino-HDDA reactions
The headspace of a threaded vial containing the polyyne precursor in an organic solvent (initial concentration typically 0.02 M in, often, ethanol-free CHCl3) and the indicated number of equivalents of the trapping reactant was flushed with nitrogen immediately before being sealed with a Teflon-lined cap. The reaction solution was heated at 80-130 °C for ca. 5–16 h. The product(s) was(were) separated/purified by silica gel chromatography.
Data availability
The authors declare that all relevant data supporting the findings of this study are available either within the manuscript itself and/or in the Supplementary Information. Experimental details and full spectroscopic characterization data for all new compounds, a description of the computational methods and results, UV-Vis absorption and emission spectra, a summary of the X-ray diffraction data, and copies of all proton and carbon NMR spectra are provided in the Supplementary Information. Crystallographic data for the structures reported in this Article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 1818244 (25k), 1818243 (28) and 1818245 (32-H). Copies of the data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif.Additionally, the corresponding author can be contacted directly.
Supplementary Material
Acknowledgments
The studies reported here were supported by the U.S. Department of Health and Human Services [National Institute of General Medical Sciences (GM-65597)] and the National Science Foundation (CHE-1665389). The computational work was carried out using hardware and software made available through the University of Minnesota Supercomputing Institute (MSI). Some NMR spectral data were obtained with an instrument purchased with a grant from the NIH Shared Instrumentation Grant program (S10OD011952). We thank Victor G. Young, Jr. (University of Minnesota) for performing the X-ray diffraction analysis and Feng Xu (University of Minnesota) for assistance in collection of absorption and fluorescence spectra.
Footnotes
Author contributions
X.X. and T.H. conceived the experiments, interpreted the data, and co-wrote the manuscript. X.X. executed the experiments and collected the data.
Supplementary information (SI) is available in the online version of the paper.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Ye Q, Chi CY. Recent highlights and perspectives on acene based molecules and materials. Chem Mater. 2014;26:4046–4056. [Google Scholar]
- 2.Perez D, Pena D, Guitian E. Aryne cycloaddition reactions in the synthesis of large polycyclic aromatic compounds. Eur J Org Chem. 2013;2013:5981–6013. [Google Scholar]
- 3.Parker T, Marder S. Synthetic Methods in Organic Electronic and Photonic Materials: A Practical Guide. Royal Society of Chemistry; London: 2015. [Google Scholar]
- 4.Li JB, Zhang QC. Mono- and oligocyclic aromatic ynes and diynes as building blocks to approach larger acenes, heteroacenes, and twistacenes. Synlett. 2013;24:686–696. [Google Scholar]
- 5.Hoffmann RW. Organic Chemistry, A Series of Monographs. Vol. 11 Academic Press; New York: 1967. Dehydrobenzene and Cycloalkynes. [Google Scholar]
- 6.Yoshida S, Hosoya T. The renaissance and bright future of synthetic aryne chemistry. Chem Lett. 2015;44:1450–1460. [Google Scholar]
- 7.Bradley AZ, Johnson RP. Thermolysis of 1,3,8-nonatriyne: Evidence for intramolecular [2+4] cycloaromatization to a benzyne intermediate. J Am Chem Soc. 1997;119:9917–9918. [Google Scholar]
- 8.Miyawaki K, Suzuki R, Kawano T, Ueda I. Cycloaromatization of a non-conjugated polyenyne system: Synthesis of 5h-benzo[d]fluoreno[3,2-b]pyrans via diradicals generated from 1-[2-{4-(2-alkoxymethylphenyl)butan-1,3-diynyl}]phenylpentan-2,4-diyn-1-ols and trapping evidence for the 1,2-didehydrobenzene diradical. Tetrahedron Lett. 1997;38:3943–3946. [Google Scholar]
- 9.Xu F, Xiao X, Hoye TR. Photochemical hexadehydro-Diels–Alder reaction. J Am Chem Soc. 2017;139:8400–8403. doi: 10.1021/jacs.7b03832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoye TR, Baire B, Niu DW, Willoughby PH, Woods BP. The hexadehydro-Diels–Alder reaction. Nature. 2012;490:208–212. doi: 10.1038/nature11518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diamond OJ, Marder TB. Methodology and applications of the hexadehydro-Diels–Alder (HDDA) reaction. Org Chem Front. 2017;4:891–910. [Google Scholar]
- 12.Marell DJ, et al. Mechanism of the intramolecular hexadehydro-Diels–Alder reaction. J Org Chem. 2015;80:11744–11754. doi: 10.1021/acs.joc.5b01356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fields EK, Meyerson S. A new mechanism for acetylene pyrolysis to aromatic hydrocarbons. Tetrahedron Lett. 1967;8:571–575. [Google Scholar]
- 14.Miyawaki K, Kawano T, Ueda I. Multiple cycloaromatization of novel aromatic enediynes bearing a triggering device on the terminal acetylene carbon. Tetrahedron Lett. 1998;39:6923–6926. [Google Scholar]
- 15.Miyawaki K, Kawano T, Ueda I. Domino thermal radical cycloaromatization of non-conjugated aromatic hexa- and heptaynes: Synthesis of fluoranthene and benzo[a]rubicene skeletons. Tetrahedron Lett. 2000;41:1447–1451. [Google Scholar]
- 16.Miyawaki K, Kawano T, Ueda I. Synthesis and properties of functionalized [6] helicenes by the thermal domino radical cycloaromatization of acyclic polyynes. Polycycl Aromat Compd. 2000;19:133–154. [Google Scholar]
- 17.Cahill KJ, Ajaz A, Johnson RP. New thermal routes to ortho-benzyne. Aust J Chem. 2010;63:1007–1012. [Google Scholar]
- 18.Yoshida S, et al. Construction of condensed polycyclic aromatic frameworks through intramolecular cycloaddition reactions involving arynes bearing an internal alkyne moiety. Chem Eur J. 2017;23:15332–15335. doi: 10.1002/chem.201704345. [DOI] [PubMed] [Google Scholar]
- 19.Woods BP, Baire B, Hoye TR. Rates of hexadehydro-Diels–Alder (HDDA) cyclizations: Impact of the linker structure. Org Lett. 2014;16:4578–4581. doi: 10.1021/ol502131r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen JH, Baire B, Hoye TR. Cycloaddition reactions of azide, furan, and pyrrole units with benzynes generated by the hexadehydro-Diels–Alder (HDDA) reaction. Heterocycles. 2014;88:1191–1200. doi: 10.3987/COM-13-S(S)83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wittig G, Pohmer L. Intermediäre Bildung von Dehydrobenzol (Cyclohexadienin) Angew Chem. 1955;67:348–348. [Google Scholar]
- 22.Niu DW, Wang T, Woods BP, Hoye TR. Dichlorination of (hexadehydro-Diels–Alder generated) benzynes and a protocol for interrogating the kinetic order of bimolecular aryne trapping reactions. Org Lett. 2014;16:254–257. doi: 10.1021/ol403258c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Niu DW, Willoughby PH, Woods BP, Baire B, Hoye TR. Alkane desaturation by concerted double hydrogen atom transfer to benzyne. Nature. 2013;501:531–534. doi: 10.1038/nature12492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakayama J, Tajiri T, Hoshino M. Insertion of benzyne and substituted benzynes into the S–S bond of diphenyl and di-p-tolyl disulfides yielding the corresponding o-bis(arylthio)benzenes. Bull Chem Soc Jpn. 1986;59:2907–2908. [Google Scholar]
- 25.Hu YM, et al. Fused multifunctionalized dibenzoselenophenes from tetraynes. Chem Commun. 2017;53:1542–1545. doi: 10.1039/c6cc09732d. [DOI] [PubMed] [Google Scholar]
- 26.Cheong PH-Y, et al. Indolyne and aryne distortions and nucleophilic regioselectivities. J Am Chem Soc. 2010;132:1267–1269. doi: 10.1021/ja9098643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wooi GY, White JM. Structural manifestations of the cheletropic reaction. Org Biomol Chem. 2005;3:972–974. doi: 10.1039/b417538g. [DOI] [PubMed] [Google Scholar]
- 28.Bronner SM, Mackey JL, Houk KN, Garg NK. Steric effects compete with aryne distortion to control regioselectivities of nucleophilic additions to 3-silylarynes. J Am Chem Soc. 2012;134:13966–13969. doi: 10.1021/ja306723r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stork G, Matsuda K. Preparation of benzocoronene and intermediates. 3364275. U S Patent. 1968
- 30.Xu F, Xiao X, Hoye TR. Reactions of HDDA-derived benzynes with perylenes: Rapid construction of polycyclic aromatic compounds. Org Lett. 2016;18:5636–5639. doi: 10.1021/acs.orglett.6b02878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie LH, et al. Unexpected one-pot method to synthesize spiro[fluorene-9,9 ’-xanthene] building blocks for blue-light-emitting materials. Org Lett. 2006;8:2787–2790. doi: 10.1021/ol060871z. [DOI] [PubMed] [Google Scholar]
- 32.Xie LH, Liang J, Song JA, Yin CR, Huang W. Spirocyclic aromatic hydrocarbons (SAHs) and their synthetic methodologies. Curr Org Chem. 2010;14:2169–2195. [Google Scholar]
- 33.Ajaz A, McLaughlin EC, Skraba SL, Thamatam R, Johnson RP. Phenyl shifts in substituted arenes via ipso arenium ions. J Org Chem. 2012;77:9487–9495. doi: 10.1021/jo301848g. [DOI] [PubMed] [Google Scholar]
- 34.Skraba-Joiner SL, McLaughlin EC, Ajaz A, Thamatam R, Johnson RP. Scholl cyclizations of aryl naphthalenes: Rearrangement precedes cyclization. J Org Chem. 2015;80:9578–9583. doi: 10.1021/acs.joc.5b01559. [DOI] [PubMed] [Google Scholar]
- 35.Thorley KJ, Anthony JE. The electronic nature and reactivity of the larger acenes. Isr J Chem. 2014;54:642–649. [Google Scholar]
- 36.Dou LT, Liu YS, Hong ZR, Li G, Yang Y. Low-bandgap near-IR conjugated polymers/molecules for organic electronics. Chem Rev. 2015;115:12633–12665. doi: 10.1021/acs.chemrev.5b00165. [DOI] [PubMed] [Google Scholar]
- 37.Pascal RA., Jr Twisted Acenes. Chem Rev. 2006;106:4809–4819. doi: 10.1021/cr050550l. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all relevant data supporting the findings of this study are available either within the manuscript itself and/or in the Supplementary Information. Experimental details and full spectroscopic characterization data for all new compounds, a description of the computational methods and results, UV-Vis absorption and emission spectra, a summary of the X-ray diffraction data, and copies of all proton and carbon NMR spectra are provided in the Supplementary Information. Crystallographic data for the structures reported in this Article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 1818244 (25k), 1818243 (28) and 1818245 (32-H). Copies of the data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif.Additionally, the corresponding author can be contacted directly.