

Abstract
The xenoestrogen bisphenol A (BPA) is a widespread plasticizer detectable within several ecosystems. BPA is considered a metabolic disruptor, affecting different organs; however, little is known about its mechanism of action in the liver, in which it triggers triglyceride accumulation. Adult zebrafish (Danio rerio) exposed to BPA developed hepatosteatosis, which was associated with an increase in the liver levels of the obesogenic endocannabinoids 2-arachidonoylglycerol and anandamide and a concomitant decrease in palmitoylethanolamide. These changes were associated with variations in the expression of key endocannabinoid catabolic and metabolic enzymes and an increase in the expression of the endocannabinoid receptor cnr1. Acute and chronic in vitro treatments with nano- and micromolar BPA doses showed increased anandamide levels in line with decreased activity of fatty acid amide hydrolase, the main anandamide hydrolytic enzyme, and induced triglyceride accumulation in HHL-5 cells in a CB1-dependent manner. We conclude that BPA is able to produce hepatosteatosis in zebrafish and human hepatocytes by up-regulating the endocannabinoid system.
Industrial progress has resulted in massive environmental contamination with anthropogenic chemicals, which until only recently have been identified as endocrine disruptors (1). This definition encompasses all the exogenous chemicals that interfere with hormonal responses by blocking or activating hormone receptors (2). Among these, bisphenol A (BPA) appears to have a ubiquitous distribution due to its use as plasticizer in numerous products from food and drink packaging to children's toys. Its capability to leak from plastic matrices into water, combined with low volatility and high lipophilicity, results in the massive accumulation of this chemical in the environment, suspended solids, soil and sediments, and subsequent uptake by aquatic wildlife (3). Unconjugated BPA has been frequently detected in human blood with concentrations ranging from 0.3 to 4.4 ng/mL (1–19.4 nM) (4). Similarly, its conjugated form is commonly found in urines samples with average values that fluctuate from 0.4 to 149 μg/L (1.7–653 nM) (5). Estimates of BPA intake from leaching of consumer products ranges from 1 μg/kg·d (6) to almost 5 μg/kg·d (0.325 mg/d per adult) (7). In vivo data in treated rats and human samples confirmed the ability of BPA to bioaccumulate especially in adipose tissue and liver (8, 9).
In the last few years, a different view of endocrine disruptors as metabolic disruptors has been introduced (10). Given the high number of chemical obesogens and their negative consequences on the human population and the environment in general, it has been suggested that high-throughput zebrafish screens be used to readily characterize these compounds.
Also BPA may be viewed as a metabolic disruptor as it acts as an agonist for the proadipogenic nuclear peroxisome proliferator-activate receptor (PPAR)-gamma] (11) but inhibits the release of adiponectin, an important insulin-sensitizing adipokine, from mature adipocytes (12). Moreover, BPA affects glucose-induced calcium signaling in pancreatic α-cells (13) and insulin content and release in β-cells (14).
Despite the alterations that BPA produces in adipose and pancreatic tissues, very little is known about its hepatic effects. However, a recent paper showed the ability of BPA to up-regulate the expression of lipogenic genes and increase de novo fatty acid synthesis in the liver (15).
The autocrine/paracrine lipid-based endocannabinoid signaling system (ECS) is a major player in liver lipid metabolism (16). Peripheral pharmacological blockade of the endocannabinoid receptor CB1 (Cnr1) in mice with diet-induced hepatic steatosis significantly reduced dislypidemia and steatosis (17). Moreover, liver-specific Cnr1 knockout produced similar effects as whole-body Cnr1 deficiency with respect to insulin resistance, indicating a crucial role for the ECS within the liver (18). ECS tone is also influenced by various hepatosteatotic stimuli; a high-fat diet increases hepatic anandamide (AEA) levels, thereby up-regulating the fatty acid synthase pathway and increasing de novo fatty acid production via CB1 activation (19). The ECS also plays a central role in neuronal hypothalamic networks regulating food intake (20). Cnr1−/− mice possess increased CRH and reduced cocaine-amphetamine-related transcript (CART) expression, proving an intimate control of the ECS over peptides involved in the regulation of food intake (21). Furthermore, an inhibitory cross talk between CB1 and melanocortin receptor-4 has been described (22), whereas in the arcuate nucleus, the neuropeptide Y/agouti-related protein system does not seem to be directly influenced by endocannabinoid action (23). Finally, two other acylethanolamides (oleoylethanolamide [OEA] and palmitoylethanolamide [PEA]) were found to be involved in pathways that, independent of the endocannabinoid system, may modulate lipolysis, food intake and inflammation. OEA mediates peripheral regulation of feeding (24) and together with PEA activates PPARα to modulate lipid metabolism (25). Therefore, given the xenoestrogen effects of BPA on hepatic lipid metabolism leading to triglyceride accumulation, we aimed at studying its influence on nonalcoholic fatty liver disease (NAFLD) in the teleost fish, Danio rerio, and human hepatocytes, using the ECS as an early biomarker for disease onset.
Materials and Methods
Maintenance and treatment of adult fish
Adult zebrafish were kept in aquaria at 28°C in oxygenated water and fed twice daily with commercial food (Vipagram) and Artemia salina. Six-month-old female zebrafish were exposed for 48 hours to BPA (Sigma-Aldrich) at a final concentration of 100 μg/L (438.6 nM), ethinyl estradiol (EE2; Sigma-Aldrich) at 200 ng/L (0.887 nM), or vehicle (EtOH). At the end of the treatment, fish were lethally anesthetized by adding into the water 500 mg/L of MS-222 (3-aminobenzoic acid ethyl ester; Sigma-Aldrich) buffered to pH 7.4, and tissues were sampled for further analysis. Procedures were performed in accordance with the Guidelines on the Handling and Training of Laboratory Animals by the Universities Federation for Animal Welfare and with the Italian animal welfare legislation (D.L. 116/92).
Liver morphology
Animals were fixed in neutral 4% paraformaldehyde prepared in PBS (0.1 M, pH 7.4) at 4°C overnight, washed in PBS, dehydrated through a graded series of ethanol, and embedded in paraffin. Consecutive sections were cut at a thickness of 4 μm using a microtome. All sections were stained with Mayer's hematoxylin and eosin, dehydrated, mounted in Eukitt, and examined under an Olympus Vanox photomicroscope. To ascertain the degree of steatosis in liver, we estimated (visually and semiquantitatively) the area of the section occupied by fat vacuoles.
Cell culture and triglyceride analysis
HHL-5 cells are an immortalized human hepatocyte line with a stable primary hepatocyte phenotype and were a gift from Dr A. H. Patel (26). HHL-5 cells were cultured in standard growth media (DMEM [Lonza] supplemented with 10% fetal bovine serum [Lonza], nonessential amino acid [Gibco], and penicillin-streptomycin [Gibco]). Cells were plated in 96-well plates so that they would be 95% confluent at the time of the initiation of the experiments. Cells were washed at least three times with phenol-red free DMEM (Gibco; 11880–028) supplemented with 10% fetal bovine serum, penicillin-streptomycin, and nonessential amino acids and then treated as indicated. Media and drugs were refreshed every day during the chronic BPA treatments. After the indicated exposure times, cells were washed with PBS, stained with AdipoRed (Lonza), and read with a Genios Pro (Tecan) or Envision multilabel (PerkinElmer) plate reader according to the manufacturer's instructions. All the cannabinoid drugs, except for NESS 0327 (Sigma-Aldrich), were purchased from Tocris Bioscience. Drug powders were suspended in 100% dimethylsulfoxide (DMSO) and successively diluted in growth media with a final dilution of 1:1000.
Measurement of endocannabinoids AEA, 2-arachidonoylglycerol (2-AG), and endocannabinoid-like PEA and OEA from adult zebrafish brain and liver
The extraction, purification, and quantification of endocannabinoids has been performed as previously described (23). The amounts of endocannabinoids in zebrafish brain and liver were quantified by isotope dilution with deuterated standards, and data are expressed as picomoles per milligram of tissue weight.
Reverse transcription and quantitative PCR (qPCR) analysis
Total RNA was isolated from zebrafish liver, brain, or HHL-5 cells using Trizol (Invitrogen) and treated with deoxyribonuclease I (Ambion) and reverse transcribed with the SuperScript III reverse transcription reaction kit (Invitrogen) according to the manufacturer's instructions. Ten to 20 ng of starting RNA was then used for qPCR analysis using IQ SybrGreen Supermix (Bio-Rad Laboratories) on a CFX 384 optical thermal cycler (Bio-Rad Laboratories). Data analysis was performed using the CFX Manager software (Bio-Rad Laboratories) using elfa (for Danio rerio) and RNAP (for HHL-5 cells) as a reference genes, and data are expressed as relative mRNA levels with SEs of the mean of triplicate reactions. Statistical significance was determined with the REST 2009 software (QIAGEN GmbH). Primers were designed by AlleleID software (Premier Biosoft) (Table 1).
Table 1.
Summary Table of the Primers Used for the qPCR Analysis With Their Corresponding Gene Target Name and Accession Numbers
| Accession Number | Gene Name | Forward Primer (5′–3′) | Reverse Primer(5′–3′) |
|---|---|---|---|
| AY422992 | elfa | CTTCTCAGGCTGACTGTGC | CCGCTAGCATTACCCTCC |
| XM_692781 | dagla | GAGGGTTTCCGTCGTCAC | TGTTCCTCCAGCAATGATCC |
| NM_200297 | mgll | AAGTGAAGGTGAGAGGAT | AATGTCCAACGATGAAGA |
| NM_001017613 | abdh4 | GCGTCACTCTTATTGAAG | TTAGTCCACCGTATTACA |
| NM_001080613 | nape-pld | CTCAAGGACATGCACTCA | GAGCACAATCTTCAAGACAAT |
| NM_001002700 | faah2a | AACAACGATGCTTGAACA | TCAGAATGCCCTCACTAT |
| NM_212820.1 | cnr1 | TCTGTGGGAAGCCTGTTTC | ACCGAGTTGAGCCGTTTG |
| NM_131074.2 | npy | GGGGACTCTCACAGAAGGGT | TTTCCCATACCTCTGCCTTGTT |
| XM_001923904.3 | agrp | ATCTCATCCACACCTGAGACG | ATTTCAGGCAGTGAGTCTGTGTC |
| NM_173278.1 | mc4r | GGTGGACCGCTACATCACAA | GCGCCAGCATGGTAAAGAAC |
| NM_001082932.1 | cart4 | GGCTGAGGCACTCGATGAA | CCCTACGTCACACCTGGGAAT |
| NM_001128576 | lepa | CATCATCGTCAGAATCAG | GGAATCTCTGGATAATGTC |
| NM_001089466 | srebf2 | ACCATGTCCCAGCAAGTG | TTGGTGGTCAGAAGCAGAG |
| NM_001045425.1 | acrp30 | AGTCCACCTGATGACAGACAGCC | GCCTTTCTCACCTGCTTCACCTTG |
| NM_001020532.1 | cfdl | GCTAAAGCACACTCTCGCCCGT | CACCAGATGTCCTCCCATCCTGAA |
| NM_001161333 | ppara | TCTTCAGGAGAACCATTC | ATCGGCAGTATTGACATT |
| Z27113 | RNAP | AACCAGAAGCGAATCACC | AACGGCGAATGATGATGG |
| XM_011523999 | SREBP | GAAGACTGAGGTGGAGGAC | CAGGACAGGCAGAGGAAG |
| NM_004104 | FASN | ACGATGACCGTCGCTGGAAGG | GGTTGATGCCTCCGTCCACGAT |
| AY237919 | ACACA | TCCAACCTCAACCACTAT | TGGAGTGAATGAGTTGTC |
| NM_016083 | CNR1 | TCTGTTCATCGTGTATGC | CTTGGCTAACCTAATGTCC |
Silencing experiments
Human hepatocytes were plated at 70% confluence and reverse transfected using the Lipofectamine RNA interference transfection system (Thermo Fisher) according to the manufacturer's instructions. Predesigned small interfering RNAs (siRNAs) were used for the negative control (Thermo Fisher; catalog number 4390843) and cnr1 (Thermo Fisher, catalog number 4392420) knockdown group. Once treated with siRNAs, cells were then assayed at the indicated time points for gene expression and triglyceride levels as above.
Fatty acid amide hydrolase (FAAH) enzymatic activity
HHL-5 cells were homogenized at 4°C in 50 mM Tris-HCl buffer (pH 7.0), centrifuged at 800 × g, and then the supernatant was centrifuged at 10 000 × g. Protein concentrations were measured by the Bradford assay (Bio-Rad Laboratories) of 7 μg of membranes from either naïve cells, which were subsequently exposed to BPA (from 11.25 to 90 μM) in the assay buffer, or BPA-treated cells were then incubated with [14C] AEA (10 000 cpm, 1.8 μM) in 50 mM Tris-HCl (pH 9) for 30 minutes at 37°C. [14C]Ethanolamine produced from [14C]AEA hydrolysis was then extracted from the incubation mixture with 2 volumes of CHCl3/CH3OH (1:1 by volume) and the subsequent aqueous phase was measured by scintillation counting to calculate the FAAH activity.
Data analysis
Data are expressed as means ± SEM of the reported number of experiments (n). Statistical significance was calculated using the unpaired Student's t-test or one-way ANOVA, as appropriate.
Results
BPA increases lipid content in zebrafish liver
We first established whether BPA modulates zebrafish liver lipid content using histological analysis. Fish treated with BPA for 48 hours had severe lipid accumulation in livers as compared with controls (Figure 1A). Similarly, the positive control exposed to EE2, a potent synthetic estrogen, produced a comparable, although less dramatic, hepatosteatotic liver. We also noted a slight level of hepatic steatosis in the control group, which may be explained by the high energetic content of the commercial food generally used for breeding zebrafish.
Figure 1.

Histological slices and endocannabinoids quantification revealed intense hepatic steatotic state in adult zebrafish brain and liver exposed to BPA and EE2 for 48 hours. Representative pictures of zebrafish liver slices exposed for 48 hours to BPA and EE2 (A), marked with hematoxylin/eosin stain, unstained structures are composed by lipids, and each picture was taken at ×20 magnification. Scale bar, 20 μm. Liquid chromatography and mass spectrometry quantification of the main endocannabinoids and N-acylethanolamines in zebrafish brain (B) and liver (C) treated for 48 hours with BPA and EE2 is shown. Data are expressed in relative units because every point is the mean of three independent experiments and, each one of it was composed of five animals per group. Values are represented relative to the control group with mean and SEM. *, P < .05 vs EtOH control. Control values were expressed in picomoles per milligram and normalized with total lipid extract from zebrafish tissues. CTR, control.
A. dysregulated endocannabinoid system is present in liver and brain of zebrafish treated with BPA
We went on to determine whether BPA-induced hepatosteatosis is associated with changes in central and peripheral endocannabinoid levels. Brain endocannabinoid levels showed only modest variations, with 2-AG showing a trend toward an increase and AEA displaying a reduction only in the EE2-treated fish (Figure 1B). However, OEA, a potent anorexigenic mediator (24), was decreased in BPA- and EE2-treated animals, whereas PEA did not change (Figure 1B). In contrast, the livers of BPA-treated fish exhibited significant increases of AEA and 2-AG content, similar to those of EE2-exposed fish (Figure 1C), whereas no changes were observed for OEA. Finally, PEA decreased in the livers of BPA- and EE2-treated fish as compared with controls (Figure 1C).
BPA modulates central signals mediating energy homeostasis
In line with the endocannabinoid quantifications described above, we observed moderate variations in the expression of endocannabinoid catabolic and anabolic enzymes in the brain of BPA-treated fish. The 2-AG metabolic enzymes, dagla and mgll, did not show any significant change. However, the synthetic enzymes of AEA, abdh4 and nape-pld, exhibited an opposite trend, with the former being slightly decreased with BPA and significantly down-regulated by EE2 and the latter slightly increased by BPA and EE2 exposure (Figure 2A). In contrast, the AEA catabolic enzyme faah2a was significantly down-regulated in response to BPA and EE2. Cnr1 presented a clear increase in the BPA-treated group, whereas EE2 elicited no change (Figure 2A). Examining the expression of appetite-regulating genes revealed that the orexigenic neuropeptides, neuropeptide Y (npy) and agouti-related peptide (agrp), showed no significant changes in response to BPA, although they were consistently down-regulated in EE2-treated animals (Figure 2B). In line with this, melacortin receptor 4 (mc4r) also displayed a slight but nonsignificant decrease in the EE2 group (Figure 2C). BPA, however, significantly down-regulated the anorexigenic signals leptin (lepa) and cocaine- and amphetamine-regulated transcript 4 (cart4), similar to EE2 (Figure 2C).
Figure 2.

Hypothalamic signals and ECS gene expression in the brain of zebrafish exposed to BPA and EE2. qPCR analysis of genes codifying enzymes for AEA and 2-AG metabolism (A), anabolic effectors (B), and anorexic signals (C) is expressed in the zebrafish CNS after 48 hours to BPA and EE2 estrogen control. Each single point consist of five livers expressed relative to the control (relative expression ± SEM as determined by Bio-Rad Laboratories CFX Manager Software). CTR, control.
BPA induces de novo hepatic fatty acid synthesis by modulation of the ECS and lipogenic and lipolytic gene expression
As seen above, the liver is significantly influenced by BPA with respect to changes in endocannabinoid levels and fat content. There is a discrepancy between the observed increase in 2-AG levels and the expression of regulatory genes. The synthetic enzyme dagla did not show any significant change, whereas the catabolic enzyme mgll increased in expression in zebrafish exposed to BPA (Figure 3A). The AEA synthetic enzymes, abdh4 and nape-pld, showed a massive up-regulation in the livers of BPA-treated animals (Figure 3A). In contrast, EE2 only modestly increased abdh4. No changes were found for the catabolic enzyme faah2a in response to BPA, whereas the EE2 group exhibited a strong down-regulation (Figure 3A). Most interestingly, cnr1, which is a key regulator during conditions of steatosis (18), was also strongly induced by BPA.
Figure 3.

ECS and liver metabolism gene expression of adult zebrafish exposed to BPA and EE2. Expression analysis of genes codifying for AEA and 2-AG catabolic and anabolic enzymes (A), lipogenic markers (B), and lipolytic genes (C) in response to BPA and EE2 exposure for 48 hours is shown. Each single point consist of five livers expressed relative to the control (relative expression ± SEM as determined by Bio-Rad Laboratories CFX Manager Software). CTR, control.
Subsequently, we analyzed the expression level of srebf2, a key lipogenic marker (27), which positively responds to cnr1 activation (19), adiponectin (acrp30), adipsin (cfdl), and leptin (lepa), which are overexpressed in hepatosteatotic livers (28). Both treated groups displayed a higher expression of srebf2, and a similar trend was found for acrp30 but not for cfdl and lepa, which showed significant increases only in the BPA group (Figure 3B). Analysis of ppara mRNA levels revealed an intense down-regulation in the BPA- and EE2-treated groups, indicating a reduced potential for triglyceride hydrolysis (Figure 3C).
BPA increases neutral lipid content in a human hepatocyte cell line in a dose-dependent manner
To confirm the ability of BPA to induce hepatosteatosis through ECS activation, immortalized human hepatocytes (HHL-5) were used as an in vitro model. BPA did not affect HHL-5 cell viability at the tested doses (Supplemental Figure 1A), and oleic acid (OA), a well-known steatogenic compound, at low doses, induced a massive production of lipid droplets after only 24 hours (Supplemental Figure 1B). BPA dose dependently triggered fatty acid accumulation in HHL-5 cells in 24 hours, similar to the estrogenic control EE2 (Figure 4A), and lower doses (≤ of 12 μM) were found ineffective in our acute model (Supplemental Figure 1C).
Figure 4.

Adipored assay and endocannabinoid levels in HHL-5 cells treated with BPA and CB1 antagonist. A, Adipored staining quantification of HHL-5 cells treated with BPA using DMSO as vehicle at the indicated concentrations and EE2 (2 μM) as an estrogen control for 24 hours. B, Combinatory experiment with one effective dose of BPA (45 μM) and two doses of CB1 antagonist AM251 at 1 and 0.3 μM, exposed for 24 hours. C, Liquid chromatography and mass spectrometry quantification of levels of the main endocannabinoids and N-acylethanolamines in the HHL-5 cell line after 72 hours of exposure with BPA and AM251. Individual well fluorescence was measured from nine separate points in a 3 × 3 grid with a Genios Pro plate reader. Data are expressed as the mean and SEM of signal from 96 wells, and liquid chromatography and mass spectrometry quantifications are the mean of five single 100 mm cell plates. *, P < .05 vs DMSO control; **, P < .01).
BPA-mediated steatogenesis is attenuated by CB1 selective antagonism
Considering the ECS regulation of liver lipid metabolism and the changes in the ECS observed in the livers of BPA-treated fish, we investigated whether BPA acted on hepatocytes through this signaling system. We focused our attention on the CB1 receptor by performing a coincubation of an effective dose of BPA with two concentrations of the CB1 antagonist AM251 in HHL-5 cells for 24 hours. The BPA-mediated increase of neutral lipids was dose dependently blocked by AM251, which was inactive per se (Figure 4B).
BPA exposure enhances AEA production through CB1 activation in HHL-5 cells
Consistent with data from zebrafish livers, BPA induced a significant increase of AEA and reduction of PEA in HHL-5 cells (Figure 4C). Notably, AM251 blocked the BPA-mediated overproduction of AEA (Figure 4C). In contrast, AM251 was not able to reverse BPA effects on PEA and OEA reduction and indeed exacerbated them while producing a decrease on its own (Figure 4C). No significant differences in 2-AG were noticed among the groups (Figure 4C).
BPA produces steatosis in hepatocytes via a CB1/AEA positive feedback loop
To rule out the possibility that the effects observed with AM251 were due to its CB1 inverse agonism, we tried to block BPA lipogenic effects with a CB1 neutral antagonist, NESS0327. Both tested doses of this compound significantly inhibited neutral lipid increases produced by BPA (Figure 5A). Similarly, qPCR analysis of key genes involved in de novo hepatic fatty acid synthesis (srebp-1c, fasn, and acaca) confirmed a strong up-regulation in response to BPA exposure (Figure 5B), which was partially or totally blocked by coincubation with NESS0327. Given the above, we hypothesized that exogenous AEA, the endogenous levels of which are elevated by BPA, could induce hepatosteatosis and that CB1 blockade would inhibit this effect. In line with this, we found that several doses of AEA increased neutral lipid content, and this was blocked by NESS0327 (Figure 5C). BPA did not exhibit any noticeable affinity for human recombinant CB1 receptors in a displacement/binding assay up to 100 μM (Supplemental Figure 1D), indicating that it activates CB1 only via its effect on AEA levels.
Figure 5.

BPA triggers an AEA-positive feedback loop in HHL-5 cells by inhibiting FAAH enzymatic activity, involving CB1 activation and its downstream gene mediators. A, Adipored fluorescence signal on HHL-5 cells treated for 24 hours with BPA and two doses of NESS0327 at 1 and 0.3 μM. B, The lowest dose of BPA (22.5 μM) and highest dose of NESS0327 (1 μM) were followed also for the qPCR analysis on srebp-1c, fasn, and acaca, the main cnr1 downstream genes involved in de novo fatty acid synthesis. C, Combinatory experiment with three doses of AEA at 1, 3, and 10 μM and NESS0327 at 0.3 μM on HHL-5 exposed for 24 hours. D, The siRNA experiment was performed in triplicate with the same experimental condition for the control and cnr1 siRNA; after 24 hours of silencing, the cells were treated with BPA at 45 μM and AEA at 3 μM for an additional 24 hours, and then neutral lipid content was evaluated by adipored fluorochrome. Total RNA was collected at the beginning and after the treatment with the aforementioned compounds at 24 hours to confirm the cnr1 knockdown (D). E, FAAH enzymatic activity was measured on HHL-5 membrane fraction after 30 minutes of exposure with BPA at 11.25, 45, and 90 μM, and data are reported as a percentage of inhibition of FAAH activity with respect to the control group, setting as maximal inhibition URB597 at 0.1 μM. An adipored assay was measured as individual well fluorescence from nine separate points in a 3 × 3 grid by Genios pro plate reader, and data are expressed as the average and SEM of a signal from 96 wells, and qPCR data are reported as the relative expression ± SEM by Bio-Rad Laboratories CFX Manager Software. *, P < .05 vs DMSO control.
BPA requires CNR1 to induce steatosis and impairs human FAAH activity
To further investigate the positive feedback loop described above, we carried out knockdown experiments to better evaluate the role of the cnr1 gene in BPA-induced steatosis and assayed BPA effects on FAAH activity to gain an understanding of how it increases AEA levels. In HHL-5 cells, in which CNR1 gene expression was silenced by 80%, BPA was unable to induce lipid accumulation, unlike in mock-transfected cells (Figure 5D). In FAAH activity assays, HHL-5 cell membrane fractions incubated with BPA had 40% reduced FAAH activity with respect to controls (Figure 5E). Moreover, treating HHL-5 cells with BPA for 24 hours prior to membrane isolation also resulted in the reduction of FAAH activity at both concentrations tested, ie, 45 μM (15.16% ± 1.3%, n = 3) and 90 μM (20.93% ± 2.01%, n = 3).
Chronic exposure to low BPA doses produces transient FAAH inhibition and steatosis in HHL-5 cells
In view of the daily BPA human exposure scenario, we wanted to extend our in vitro experiments also to chronic studies with nanomolar doses of BPA. Starting from 1 nM, we tested three log units of BPA concentrations in a 6-day exposure protocol, using EE2 as an estrogen control. Except for the lowest concentration (1 nM), all the BPA-treated groups showed a significant increase in neutral lipid content with respect to the control (Figure 6A). Interestingly, the chronic BPA treatment produced a comparable steatotic induction (an ∼25% increase with respect to the control) as that observed with acute treatments. In parallel, we also assessed FAAH activity in HHL-5 cells treated with 10 and 100 nM BPA. No differences were found in FAAH activity after 6 days of treatment (Figure 6B). Similarly, nanomolar doses of BPA were unable to inhibit FAAH activity after 24 hours of treatment. On the other hand, both tested doses of BPA after 48 hours exposure produced a significant reduction in FAAH activity (Figure 6B), ie, 10 nM (20% ± 4.6%, n = 3) and 100 nM (29% ± 2.4%, n = 3).
Figure 6.
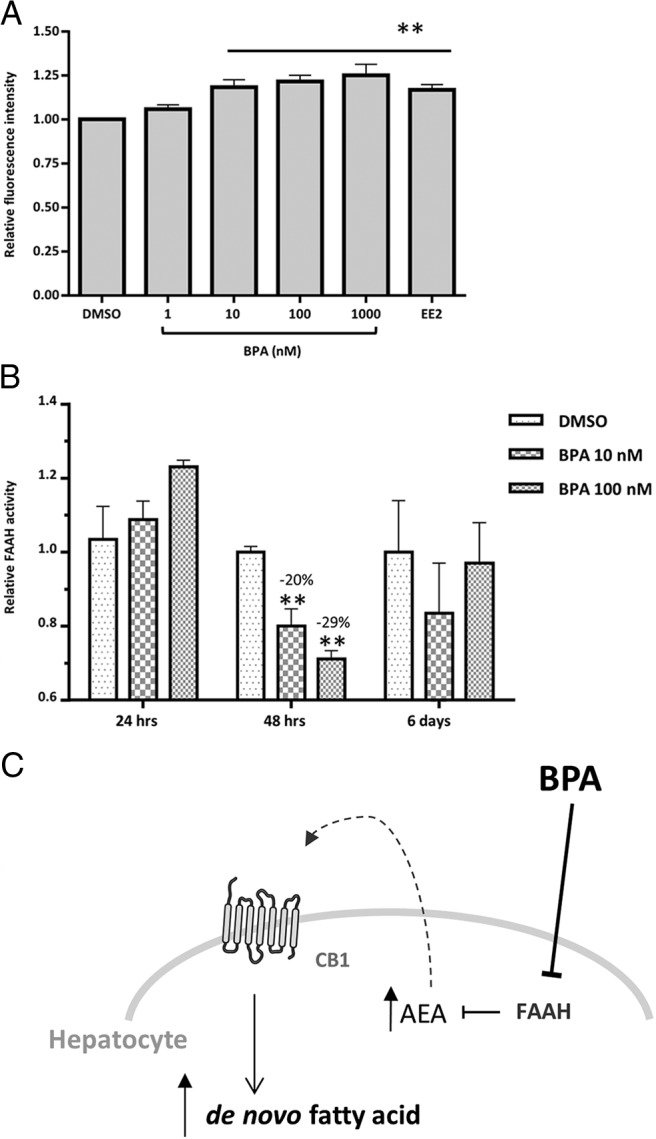
Adipored assay and FAAH activity in HHL-5 cells chronically treated with nanomolar doses of BPA. A, HHL-5 cells were chronically treated with BPA using DMSO as vehicle at the indicated concentrations and EE2 (2 μM) as an estrogen control for 6 days (n = 11). Data are shown as relative values with respect to the corresponding DMSO control. Individual well fluorescence was measured from 12 separate points with an Envision plate reader. B, HHL-5 cells were exposed for 24 hours, 48 hours, or 6 days to 10 and 100 nM of BPA or DMSO vehicle and then harvested and FAAH activity assayed. FAAH activity values were measured by picomoles of hydrolyzed [14C]AEA after 30 minutes of incubation with 7 μg of membranes. Data are expressed as relative value respect to the DMSO control and standard error of the mean. *, P < .05 vs DMSO control; **, P < .01. C, A model of BPA-mediated NAFLD via CB1 activation. BPA exerts its steatogenic effects by inhibiting FAAH activity, resulting in increased AEA levels and subsequent CB1 activation. This initiates a positive feedback loop, which up-regulates CB1 gene expression and its downstream targets involved in de novo fatty acid synthesis.
Discussion
The estrogenic activity of BPA has been known since the 1930s, yet it is still used as an emollient in polycarbonate plastics and epoxy resins, resulting in sustained environmental contamination and daily exposure. BPA has a wide range of biological targets, exerting metabolic interference by antagonizing thyroid receptors (29), modulating insulin and glucagon responses in pancreatic cells (13), and influencing body weight and adipokine release (12, 30). Here we evaluated the effects of BPA environmental contamination on liver energy metabolism using zebrafish as an in vivo model. Furthermore, given the role of ECS dysregulation in metabolic disorders and its conserved function in zebrafish (31), we assessed the effects of BPA on this system in this vertebrate model and in human hepatocytes. We propose the ECS as a target through which BPA acts to induce fatty acid accumulation in the liver and possibly disrupt appetite regulation.
We show for the first time that BPA produces hepatosteatosis in zebrafish, similar to the reported effects on mouse liver (15). These effects were associated with decreased OEA, a potent anorexic signal involved in the peripheral regulation of food intake (24), in the central nervous system (CNS). Also 2-AG, a regulator of feeding behavior via CB1 receptors (32), showed a trend toward an increase in response to BPA. In contrast, AEA brain levels did not change in response to BPA, although they were reduced by EE2. A better understanding of central endocannabinoid tone comes from the gene expression profile of receptors and regulatory enzymes. A consistent increase of cnr1 transcript levels in BPA-treated animals suggests a possible increase of food intake mediated by 2-AG, while no significant changes were found for dagla and mgll genes. It is possible that the elevation of brain 2-AG levels was due to other factors, such as the availability of biosynthetic precursors and substrates for diacylglycerol lipase alpha (DAGLα), or changes in the protein and enzymatic activity levels of this enzyme and monoacylglycerol lipase (MAGL).
Regarding AEA production, we reported a compensation between the two synthesizing enzymes, adbh4 and nape-pld, in treated animals, which might explain why no net difference in AEA levels were found in the brains of treated zebrafish. Faah2a expression, however, did exhibit a significant down-regulation after both BPA and EE2 exposure, underlying a possible estrogen influence on the levels of this gene. Indeed, the presence of an estrogen response element within the faah promoter has been reported, which may lead to a direct (33) or indirect (34) regulation of zebrafish brain faah2a. Among the hypothalamic signals that control food intake, the cart4 gene exhibited a decreased expression correlated with cnr1 up-regulation, in agreement with a relationship already described in zebrafish (35) and mammals (21). Taken together, these findings reveal a general reduction of brain anorexigenic signals linked to the endocannabinoid system after BPA treatment. However, the neuropeptide Y/agouti-related protein system did not change in correlation with the ECS profile, in line with previous studies, which suggested independent stimulation of food intake by CB1 receptors and neuropeptide Y (20). At any rate, because we did not see any significant changes in food intake after zebrafish exposure to BPA (Supplemental Figure 1E), we cannot speculate on the functional consequences of the alterations of the transcripts and of endocannabinoid levels, other than concluding that BPA-induced hepatosteatosis in zebrafish does not appear to be a consequence of increased energy intake.
Despite the role of the ECS in the CNS, peripheral perturbations driven by this signaling system appear to be more important. Given the observed hepatosteatosis, elevated hepatic 2-AG levels in BPA and EE2 groups suggest a possible production of this endocannabinoid from stellate cells working through a CB1-mediated pathway to enhance lipogenic gene expression, similar to alcohol-induced steatosis in mice (36). Increased 2-AG levels were not correlated with changes in the expression of its metabolic enzymes, strengthening the hypothesis that 2-AG is mostly synthesized in stellate cells (which account only for 5%–8% of the total liver cells [37]), and suggests that mgll, the mRNA levels of which were increased, is not involved in 2-AG level control as much as general lipid metabolism (38). Concerning hepatic AEA levels, we reported a concrete increase of these as well, suggesting a potential synergistic action with 2-AG to act as local enhancers of triglyceride levels in hepatocytes through CB1 stimulation (39). In fact, this overproduction of AEA correlated with increased expression of cnr1 transcripts in treated zebrafish livers, similar to results obtained in mouse organotypic liver slices (40), as well as with the up-regulation of the mRNAs of AEA biosynthetic enzymes.
We also observed an up-regulation of srebf2 transcripts in the liver of both treated groups, consistent with data linking cnr1 to srebp-1c expression and elevated de novo fatty acid synthesis in zebrafish livers (41). Additional evidence of intense lipogenesis comes from the elevated expression of leptin, adiponectin (acrp30) and adipsin (cfdl), which are expressed only in fatty livers (28). BPA-mediated NAFLD progression may result not only from enhanced lipogenesis but also thorough reduced lipolysis. Endocannabinoid-like molecules, such as PEA, present noncannabimetic activities, such as the stimulation of PPARα, which mediates peripheral lipolytic activities (25). A significant hepatic reduction of this acylethanolamide was observed in the livers of both treated groups, which correlates with the observed down-regulation of ppara gene expression within the liver, suggesting a possible reduction of lipolysis.
BPA also induced de novo fatty acid production in the human HHL-5 hepatocyte cell line after 24 hours, with a magnitude similar to that of our estrogen control, consistent with observations in mice (15), in which BPA at low doses up-regulates lipogenic genes related to NAFLD. Because CB1 antagonist/inverse agonists have been reported to ameliorate hepatic insulin resistance and steatosis by exerting important effects on lipid metabolism (16), we tried to block the steatogenic effect of BPA with a specific CB1 antagonist (AM251). Importantly, blocking CB1 did reverse the effect of the most effective dose of BPA. Consistent with the findings in zebrafish liver, after exposure to BPA, we observed a reduction of the two acylethanolamides, PEA and OEA, which are involved in lipolysis independent of CB1. These BPA-induced changes were not blocked by AM251, further suggesting CB1-independent effects on lipolytic processes within the liver. However, BPA also caused an increase of AEA levels in HHL-5 cells, which is in line with the in vivo data, and AM251 reversed the BPA-induced increase. CB1 neutral antagonism (NESS0327) also blocked the BPA up-regulation of AEA, supporting an essential role for CB1 activation in BPA lipogenic activity not only by stimulating per se the expression of lipogenic enzymes but also by further enhancing AEA levels and hence causing a potential vicious circle, leading to more triglyceride accumulation in hepatocytes.
Gene expression analysis also confirmed the partial dependence of BPA steatogenic potential on CB1 activity; in fact, the increased expression of key genes involved in hepatic lipogenesis induced by BPA was partially counteracted by NESS0327. Finally, knockdown experiments underlined the essential role of the CB1 receptor in this pathway (19, 42) because BPA and AEA were totally ineffective at triggering de novo fatty acid synthesis when CNR1 was silenced. Once we outlined the involvement of the ECS in acute exposure conditions, we confirmed a similar steatogenic potential of BPA also after chronic treatments, using low doses of this contaminant in line with the human exposure levels (43). After 6 days of treatment with nanomolar doses of BPA, the human hepatocyte showed a significant triglyceride increase comparable with the steatotic insult previously described in the acute experiments. As a biomarker for the chronic BPA obesogenic effect, we monitored FAAH activity over time. Interesting, we noticed a transient inhibition of FAAH activity that culminated after 48 hours of exposure with 10 and 100 nM of BPA, with no differences being found at earlier or later time points. The reduction in FAAH activity (∼25% of inhibition) is comparable with the inhibition of this hydrolytic enzyme after 24 hours of acute BPA exposure, and therefore, we can speculate that this effect is translated to AEA accumulation.
Steatogenic agents increase CB1 activity by stimulating synthesis or reducing the degradation of endocannabinoids (44); therefore, we believe that, in BPA-treated hepatocytes, AEA is feeding a positive feedback loop together with the CB1 receptor, which is necessary for the progression of BPA-induced NAFLD (Figure 6C). Indeed, AEA was able to produce steatosis in HHL-5 cells and this effect was totally abolished by CB1 antagonism. Similarly, BPA was unable to exert its steatotic influence in Cnr1-knockdown hepatocytes. Moreover, similar to a high-fat diet condition, we showed that the pollutant was able to directly inhibit the hydrolytic activity of the human FAAH, thus potentially explaining its selective increase of AEA levels (19). Taken together these data show for the first time that BPA is able to produce NAFLD in adult zebrafish in a manner mediated, at least in part, by endocannabinoid action at CB1 receptors. Furthermore, we showed the steatogenic effect of BPA also in human hepatocytes and confirmed the presence of positive feedback loop between AEA and CB1 resulting from FAAH inhibition, which is essential for the priming of de novo fatty acid production. These results clearly indicate that BPA induces metabolic alterations in humans as well as in Danio rerio hepatocytes. In addition, the present study highlights for the first time the effects of BPA on the ECS, suggesting the use of endocannabinoids and related mediators, as well as their metabolic enzymes, as novel biomarkers for BPA monitoring.
Supplementary Material
Acknowledgments
We are thankful to Dr B. Migliarini for the initial support for the project and Dr Piscitelli for measuring endocannabinoid levels using liquid chromatography-mass spectrometry.
This work was supported by Grant PRIN 2010–2011 prot 2010W87LBJ and Ricerca Finalizzata 2009 Ministero della Salute Grant RF-2009–1536185 (to O.C.).
Disclosure Summary: The authors have nothing disclose.
Abbreviations
- AEA
anandamide
- 2-AG
2-arachidonoylglycerol
- BPA
bisphenol A
- CART
cocaine-amphetamine-related transcript
- CNS
central nervous system
- DMSO
dimethylsulfoxide
- ECS
endocannabinoid signaling system
- EE2
ethinyl estradiol
- FAAH
fatty acid amide hydrolase
- NAFLD
nonalcoholic fatty liver disease
- OEA
oleoylethanolamide
- PEA
palmitoylethanolamide
- PPAR
peroxisome proliferator-activate receptor
- qPCR
quantitative PCR
- siRNA
small interfering RNA.
References
- 1. US Environmental Protection Agency. Memorandum to EDSTAC Members RE: Definition of “Endocrine Disruptor”. Washington DC: 2007. [Google Scholar]
- 2. Hotchkiss AK, Rider CV, Blystone CR, et al. Fifteen years after “Wingspread”—environmental endocrine disrupters and human and widlife health: where we are today and where we need to go. Toxicol Sci. 2008;105:235–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cousins I, Staples C, Kleĉka G, Mackay D. A multimedia assessment of the environmental fate of bisphenol A. Hum Ecol Risk Assess. 2002;8:1107–1135. [Google Scholar]
- 4. Vandenberg LN, Hauser R, Marcus M, Olea N, Welshons WV. Human exposure to bisphenol A (BPA). Reprod Toxicol. 2007;24:139–177. [DOI] [PubMed] [Google Scholar]
- 5. Calafat AM, Ye X, Wong L-Y, Reidy JA, Needham LL. Exposure of the U.S. population to bisphenol A and 4-tertiary-octylphenol: 2003–2004. Environ Health Perspect. 2008;116:39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Scientific Committee on Food. Opinion of the Scientific Committee on Food on Bisphenol A. Brussels: European Commission. Health, Consumer Protection Directorate-General; 2002. [Google Scholar]
- 7. Thomson BM, Cressey PJ, Shaw IC. Dietary exposure to xenoestrogens in New Zealand. J Environ Monit. 2003;5:229–235. [DOI] [PubMed] [Google Scholar]
- 8. Nunez A, Kannan K, Giesy J, Fang J, Clemens L. Effects of bisphenol A on energy balance and accumulation in brown adipose tissue in rats. Chemosphere. 2001;42:917–922. [DOI] [PubMed] [Google Scholar]
- 9. Geens T, Neels H, Covaci A. Distribution of bisphenol-A, triclosan and n-nonylphenol in human adipose tissue, liver and brain. Chemosphere. 2012;87:796–802. [DOI] [PubMed] [Google Scholar]
- 10. Migliarini B, Piccinetti CC, Martella A, Maradonna F, Gioacchini G, Carnevali O. Perspectives on endocrine disruptor effects on metabolic sensors. Gen Comp Endocrinol. 2011;170:416–423. [DOI] [PubMed] [Google Scholar]
- 11. Riu A, Grimaldi M, le Maire A, et al. Peroxisome proliferator-activated receptor γ is a target for halogenated analogs of Bisphenol A. Environ Health Perspect. 2011;119:1227–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hugo ER, Brandebourg TD, Woo JG, Loftus J, Alexander JW, Ben-Jonathan N. Bisphenol A at environmentally relevant doses inhibits adiponectin release from human adipose tissue explants and adipocytes. Environ Health Perspect. 2008;116:1642–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alonso-Magdalena P, Laribi O, Ropero AB, et al. Low doses of bisphenol A and diethylstilbestrol impair Ca2+ signals in pancreatic alpha-cells through a nonclassical membrane estrogen receptor within intact islets of Langerhans. Environ Health Perspect. 2005;113:969–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alonso-Magdalena P, Ropero AB, Carrera MP, et al. Pancreatic insulin content regulation by the estrogen receptor ERα. PLoS One. 2008;3:e2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marmugi A, Ducheix S, Lasserre F, et al. Low doses of bisphenol a induce gene expression related to lipid synthesis and trigger triglyceride accumulation in adult mouse liver. Hepatology. 2012;55:395–407. [DOI] [PubMed] [Google Scholar]
- 16. Silvestri C, Paris D, Martella A, et al. Two non-psychoactive cannabinoids reduce intra-cellular lipid levels and inhibit hepatosteatosis. J Hepatol. 2015;62(6):1382–1390. [DOI] [PubMed] [Google Scholar]
- 17. Tam J, Vemuri VK, Liu J, et al. Peripheral CB1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesity. J Clin Invest. 2010;120:2953–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Osei-Hyiaman D, Liu J, Zhou L, et al. Hepatic CB(1) receptor is required for development of diet-induced steatosis, dyslipidemia, and insulin and leptin resistance in mice. J Clin Invest. 2008;118:3160–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Osei-Hyiaman D, DePetrillo M, Pacher P, et al. Endocannabinoid activation at hepatic CB1 receptors stimulates fatty acid synthesis and contributes to diet-induced obesity. J Clin Invest. 2005;115:1298–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Di Marzo V, Matias I. Endocannabinoid control of food intake and energy balance. Nat Neurosci. 2005;8:585–589. [DOI] [PubMed] [Google Scholar]
- 21. Cota D, Marsicano G, Tschöp M, et al. The endogenous cannabinoid system affects energy balance via central orexigenic drive and peripheral lipogenesis. J Clin Invest. 2003;112:423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Verty ANA, McFarlane JR, McGregor IS, Mallet PE. Evidence for an interaction between CB1 cannabinoid and melanocortin MCR-4 receptors in regulating food intake. Endocrinology. 2004;145:3224–3231. [DOI] [PubMed] [Google Scholar]
- 23. Di Marzo V, Goparaju SK, Wang L, et al. Leptin-regulated endocannabinoids are involved in maintaining food intake. Nature. 2001;410:822–825. [DOI] [PubMed] [Google Scholar]
- 24. De Fonseca FR, Navarro M, Gomez R, et al. An anorexic lipid mediator regulated by feeding. Nature. 2001;414:209–212. [DOI] [PubMed] [Google Scholar]
- 25. O'Sullivan SE. Cannabinoids go nuclear: evidence for activation of peroxisome proliferator-activated receptors. Br J Pharmacol. 2007;152:576–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Clayton RF, Rinaldi A, Kandyba EE, et al. Liver cell lines for the study of hepatocyte functions and immunological response. Liver Int. 2005;25:389–402. [DOI] [PubMed] [Google Scholar]
- 27. Abdelmegeed MA, Yoo S-H, Henderson LE, Gonzalez FJ, Woodcroft KJ, Song B-J. PPARα expression protects male mice from high fat–induced nonalcoholic fatty liver. J Nutr 2011;141:603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yu ST, Matsusue K, Kashireddy P, et al. Adipocyte-specific gene expression and adipogenic steatosis in the mouse liver due to peroxisome proliferator-activated receptor γ1 (PPAR gamma 1) overexpression. J Biol Chem. 2003;278:498–505. [DOI] [PubMed] [Google Scholar]
- 29. Moriyama K, Tagami T, Akamizu T, et al. Thyroid hormone action is disrupted by bisphenol A as an antagonist. J Clin Endocrinol Metab. 2002;87:5185–5190. [DOI] [PubMed] [Google Scholar]
- 30. Rubin BS, Murray MK, Damassa DA, King JC, Soto AM. Perinatal exposure to low doses of bisphenol A affects body weight, patterns of estrous cyclicity, and plasma LH levels. Environ Health Perspect. 2001;109:675–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu LY, Alexa K, Cortes M, et al. Cannabinoid receptor signaling regulates liver development and metabolism. Development. 2016;143:609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mechoulam R, Berry EM, Avraham Y, Di Marzo V, Fride E. Endocannabinoids, feeding and suckling—from our perspective. Int J Obes. 2006;30:S24–S28. [DOI] [PubMed] [Google Scholar]
- 33. Maccarrone M, De Felici M, Bari M, Klinger F, Siracusa G, Finazzi-Agrò A. Down-regulation of anandamide hydrolase in mouse uterus by sex hormones. Eur J Biochem. 2000;267:2991–2997. [DOI] [PubMed] [Google Scholar]
- 34. Waleh NS, Cravatt BF, Apte-Deshpande A, Terao A, Kilduff TS. Transcriptional regulation of the mouse fatty acid amide hydrolase gene. Gene. 2002;291:203–210. [DOI] [PubMed] [Google Scholar]
- 35. Nishio S-I, Gibert Y, Berekelya L, et al. Fasting induces CART down-regulation in the zebrafish nervous system in a cannabinoid receptor 1-dependent manner. Mol Endocrinol. 2012;26:1316–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jeong W-i, Osei-Hyiaman D, Park O, et al. Paracrine activation of hepatic CB1 receptors by stellate cell-derived endocannabinoids mediates alcoholic fatty Liver. Cell Metab. 2008;7:227–235. [DOI] [PubMed] [Google Scholar]
- 37. Geerts A. History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin Liver Dis. 2001;21:311–336. [DOI] [PubMed] [Google Scholar]
- 38. Karlsson M, Contreras JA, Hellman U, Tornqvist H, Holm C. cDNA cloning, tissue distribution, and identification of the catalytic triad of monoglyceride lipase: evolutionary relationship to esterases, lysophospholipases, and haloperoxidases. J Biol Chem. 1997;272:27218–27223. [DOI] [PubMed] [Google Scholar]
- 39. Silvestri C, Ligresti A, Di Marzo V. Peripheral effects of the endocannabinoid system in energy homeostasis: adipose tissue, liver and skeletal muscle. Rev Endocr Metab Disord. 2011;12:153–162. [DOI] [PubMed] [Google Scholar]
- 40. Jourdan T, Demizieux L, Gresti J, et al. Antagonism of peripheral hepatic cannabinoid receptor-1 improves liver lipid metabolism in mice: evidence from cultured explants. Hepatology. 2012;55:790–799. [DOI] [PubMed] [Google Scholar]
- 41. Pai W-Y, Hsu C-C, Lai C-Y, Chang T-Z, Tsai Y-L, Her G. Cannabinoid receptor 1 promotes hepatic lipid accumulation and lipotoxicity through the induction of SREBP-1c expression in zebrafish. Transgenic Res. 2013;22:823–838. [DOI] [PubMed] [Google Scholar]
- 42. Cinar R, Godlewski G, Liu J, et al. Hepatic cannabinoid-1 receptors mediate diet-induced insulin resistance by increasing de novo synthesis of long-chain ceramides. Hepatology. 2014;59:143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goldstone AE, Chen Z, Perry MJ, Kannan K, Buck Louis GM. Urinary bisphenol A and semen quality, the LIFE Study. Reprod Toxicol (Elmsford, NY). 2015;51:7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Purohit V, Rapaka R, Shurtleff D. Role of cannabinoids in the development of fatty liver (Steatosis). AAPS J. 2010;12:233–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.