

Abstract
Extracellular matrix (ECM) components of the brain play complex roles in neurodegenerative diseases. The study of microenvironment of brain tissues with Alzheimer’s disease revealed colocalized expression of different ECM molecules such as heparan sulfate proteoglycans (HSPGs), chondroitin sulfate proteoglycans (CSPGs), matrix metal-loproteinases (MMPs), and hyaluronic acid. In this study, both cortical and hippocampal populations were generated from human-induced pluripotent stem cell-derived neural spheroids. The cultures were then treated with heparin (competes for Aβ affinity with HSPG), heparinase III (digests HSPGs), chondroitinase (digests CSPGs), hyaluronic acid, and an MMP-2/9 inhibitor (SB-3CT) together with amyloid β (Aβ42) oligomers. The results indicate that inhibition of HSPG binding to Aβ42 using either heparinase III or heparin reduces Aβ42 expression and increases the population of β-tubulin III+ neurons, whereas the inhibition of MMP2/9 induces more neurotoxicity. The results should enhance our understanding of the contribution of ECMs to the Aβ-related neural cell death.
Keywords: human-induced pluripotent stem cells, heparan sulfate proteoglycans, chondroitin sulfate proteoglycan, matrix metalloproteinases, hyaluronic acid, neuroprotective
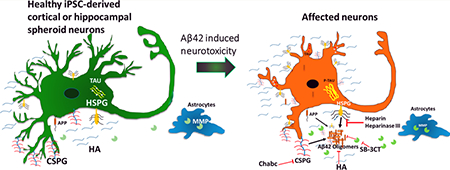
Graphical Abstract
INTRODUCTION
International estimate for the number of Alzheimer’s patients reached ~48 million worldwide in 2015.1 Since there are no disease-modifying therapies available, the improved disease modeling is necessary for evaluating disease pathology and therapeutic interventions. Transgenic animal models do not fully recapitulate microenvironment of endogenous proteins from human; thus, it is essential to create complementary models from human cells. Alzheimer’s disease (AD) is a slowly progressing neurodegenerative disorder characterized by the misfolding, aggregation, and the gain of toxicity due to amyloid-β (Aβ) and tau in the brain.2 Induced pluripotent stem (iPS) cells have emerged as the most promising tools for disease modeling, since they can be derived from the patient’s own body3–6 and have the capacity to self-organize into mini-brain-like organoids.7,8
The pathological mechanism underlying AD is found to be the conformational changes in the Aβ peptides and hyper-phosphorylated tau proteins that assemble to form β-sheeted plates resulting in forming neuritic fibrillatory tangles (NFT). These tangles together with the cytoplasm of neurons form the plaques called senile plaques (SP). The detailed evaluation of the plaques and the surrounding microenvironment of AD brain revealed prominent expression of different ECM molecules such as proteoglycans (PGs), matrix metalloproteinases (MMPs), hyaluronic acid (HA), etc.9 Compared to normal brain, immuno-histochemical analysis revealed colocalized expression of proteoglycans, including heparan sulfate proteoglycans (HSPGs) and chondroitin sulfate proteoglycans (CSPGs), with SPs and NFTs both in the brains of AD patients and in the transgenic animal AD models.10,11
Proteoglycans with high negative charge affect the early stages of fibril formation and enhance the lateral aggregation of fibrils at later stages.12 Distribution of sulfate groups on the proteoglycan is the important feature for the Aβ-PG interaction.12 Perlecan, one type of HSPG and a part of basement membrane, accelerates Aβ fibril formation and maintains Aβ fibril stability by using Perlecan’s HS glycosaminoglycan chains,13 whereas agrin, another kind of HSPG, binds to Aβ, accelerates fibril formation, and protects fibrillar Aβ from proteolysis.14 Different types of sulfated CSPGs are also associated with SPs and NFTs and are responsible for the decreased neurite growth both in in vitro cultures and in response to injury in the central nervous system.15 Studies suggest that CSPGs support the formation of amyloid precursor proteins (APP) that produce Aβ peptide.16 The neuritic density of the AD cells is found to decrease in the CSPG-containing areas compared to other areas, supporting the CSPG-controlled neurite growth.11
Amyloid plaques in the AD patients also showed prominent expression of other extracellular matrix (ECM)-related molecules such as HA17 and matrix metalloproteinase MMP-9.18,19 HA positive neurons in both rat and human brain were found to be resistive to the alteration caused by the AD pathology, indicating the attenuating effect of HA on AD neurons.17,20 MMP-9 was detected near the extracellular amyloid plaques, which when activated, was capable of degrading Aβ peptides.18,21 The enzyme cleaves at three different sites on the Aβ peptide and eliminates the neurotoxic β-sheet-forming capacity of the amyloid peptide.22
Taken together, these studies suggest not only the positive impacts of HA and MMP but also the negative effects of HSPGs and CSPGs on neural degeneration and the needs for evaluating ECMs to identify potential therapeutic targets.9 The objective of this study is to evaluate the impact of these ECM molecules on Aβ42-induced neurotoxicity and thereby on the neural growth and differentiation using human iPS cell-derived neural spheroids.
Forebrain regions of the brain are sensitive to the neural degeneration, suggesting that in vitro forebrain-like models derived from iPS cells could be used for studying AD-related degeneration.3,4,23–26 In this study, two neural differentiation paradigms from human iPS cells through three-dimensional (3D) spheroid formation were investigated to obtain a forebrain-dominant culture. The first type of spheroids used dual Smad inhibition followed by sonic Hedgehog (Shh) pathway inhibition to enrich cortical neurons.27,28 The second type of spheroids used inhibitors of Wnt, Shh, and SMAD signaling followed with Wnt activation to enrich hippocampal dentate gyrus neurons.29,30 Exogenous Aβ42 oligomers were added to different spheroid outgrowth.27,28,31 The impacts of ECM enzymes (to digest proteoglycans), HA, heparin, and the MMP inhibitor on the Aβ-induced neuropathology were examined. The results of this study should enhance our understanding of the contribution of these ECM-related molecules to neuronal degeneration.
MATERIALS AND METHODS
Undifferentiated hiPSC Culture.
Human iPSK3 cells were derived from human foreskin fibroblasts transfected with plasmid DNA encoding reprogramming factors OCT4, NANOG, SOX2, and LIN28 (kindly provided by Dr. S. Duncan, Medical College of Wisconsin).32,33 Human iPSK3 cells were maintained in mTeSR serum-free medium (StemCell Technologies, Inc.) on growth factor reduced Geltrex (Life Technologies).34 The cells were passaged by Accutase every 5−6 d and seeded at 1 × 106 cells per well of six-well plate in the presence of 10 μM Y27632 (Sigma) for the first 24 h.34–36
Cortical and Hippocampal Differentiation of hiPSCs.
Human iPSK3 cells were seeded into Ultra-Low Attachment (ULA) 24-well plates (Corning Inc.) at 3 × 105 cells/well in differentiation medium composed of Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F-12) plus 2% B27 serum-free supplement (Life Technologies). Y27632 (10 μM) was added during the seeding and removed after 24 h. At day 1, the cells formed embryoid bodies (EBs) and were treated with dual SMAD signaling inhibitors 10 μM SB431542 (Sigma) and 100 nM LDN193189 (Sigma).37–39 After 8 d, the cells were treated with fibroblast growth factor (FGF)-2 (10 ng/mL, Life Technologies) and cyclopamine (an Shh inhibitor, 1 μM, Sigma) for cortical differentiation until day 27.27,28 After 27 d, cells were harvested or replated for characterizations or treatments.
For hippocampal differentiation,29 at day 1, the EBs were treated with 10 μM SB431542, 100 nM LDN193189, cyclopamine (1 μM), and IWP4 (a Wnt inhibitor, 2 μM, Stemgent). At day 20, the growth factors were changed to CHIR99021 (a Wnt activator, 5 μM, STEMCELL Technologies Inc.) and brain-derived neurotrophic factor (BDNF; 5 ng/mL, R&D Systems). The cells were treated for another 7 d and then harvested or replated for characterizations or biomolecule treatments.
Aβ42 Oligomer Treatment and Culture Characterizations.
To prepare oligomers of the Aβ42 peptide, biotinylated Aβ42 (Bachem) was fully dissolved at 0.5 mg/mL in hexafluor-2-propanole (HFIP, Sigma).27,28 HFIP Aβ(1−42) solution (10 μL) was dispensed into a siliconized Snap-Cap microtube, put in a desiccator to completely evaporate HFIP, and thereafter stored at −80 °C. Oligomer solutions were prepared freshly for each experiment. The stock was dissolved in 10 μL of dimethyl sulfoxide (DMSO; to 105 μM) and incubated for 3 h at room temperature. Oligomers of Aβ42 were added to the neural cultures derived from human iPSK3 cells at 0 or 1 μM.
Electrophysiology.
Whole-cell patch-clamp recordings were conducted on the cells after 48 d of cortical differentiation. Patch electrodes with resistances of 4−8 MΩ were pulled from borosilicate glass and fire-polished. Current traces were digitized at 20 kHz and filtered at 1 kHz, respectively, with an Axopatch 200B amplifier. Data acquisition and analysis were performed with pCLAMP10 software (Molecular Devices). The bath solution contained 2 mM KCl, 148 mM NaCl, 2 mM MgCl2, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), and 1 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), pH 7.4. The pipet solution contained 130 mM KCl, 10 mM HEPES, and 5 mM EGTA, pH 7.4. Spontaneous postsynaptic currents were recorded in voltage-clamp configuration at a holding potential of −80 mV.
Treatment with Heparin, Heparinase III, Hyaluronic Acid, and Chondroitinase ABC.
The cultures were treated with different molecules in the presence of Aβ42 oligomers: (1) 100 units/mL heparin (Sigma, H3149), which compete with heparan sulfate proteoglycans for Aβ42 binding to reduce Aβ42 uptake; (2) 0.05 units/mL heparinase III (Sigma, H8891), which degrades heparan sulfate proteoglycans, (3) 0.2 mg/mL hyaluronic acid (Sigma), a type of ECM in brain perineuronal net; (4) 0.05 U/mL chondroitinase ABC (Chabc, Sigma), which degrades chondroitin sulfate proteogly-cans, for 72−96 h, respectively. The culture without treatment was used as the control. The treated samples were evaluated by Live/Dead assay, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, immunocytochemistry, etc. The culture supernatants were collected for lactate dehydrogenase (LDH) activity assay.
Biochemical Assays: Live/Dead, MTT, Caspase, and LDH Assay.
The cells were evaluated for viability using Live/Dead staining kit (Molecular Probes). After 72 h, the cells were incubated in DMEM-F12 containing 1 μM calcein-AM (green) and 2 μM ethidium homodimer I (red) for 30 min. The samples were imaged under a fluorescent microscope (Olympus IX70). With ImageJ software, the viability was analyzed and calculated as the percentage of green intensity over total intensity (including both green cells and red cells). For MTT assay, the replated neural cells were incubated with 5 mg/mL MTT (Sigma) solution. The absorbance was measured at 500 nm using a microplate reader (Biorad). Image-iT Live Green Poly Caspase Detection kit (Molecular Probes) was used to detect the expression of caspases. The replated cells were incubated for 1 h with the fluorescent inhibitor of caspases reagent and analyzed by a fluorescence microscope.40,41 The cytotoxicity of cells was assessed using LDH activity assay kit (Sigma, MAK066). Briefly, a total volume of 100 μL of spent medium and LDH reaction mixture was mixed well, and the initial absorbance at 450 nm was measured using a microplate reader (Bio-Rad iMark). The mixture was incubated at 37 °C, and a measurement was taken every 5 min. The LDH activity was calculated through the subtraction of final and initial measurements in comparison to the standard curve.
Immunocytochemistry.
Briefly, the samples were fixed with 4% paraformaldehyde (PFA) and permeabilized with 0.2−0.5% Triton X-100 for intracellular markers.42 The samples were then blocked and incubated with various mouse or rabbit primary antibodies (Supporting Information, Table S1). After they were washed, the cells were incubated with the corresponding secondary antibody: Alexa Fluor 488 goat anti-Mouse IgG or Alexa Fluor 594 goat anti-Rabbit or donkey antigoat IgG (Life Technologies). The samples were stained with Hoechst 33342 and visualized using a fluorescent microscope. The proportion of positive cells was calculated based on the area of a marker of interest normalized to the nuclei using ImageJ analysis, indicating the relative expression among different conditions.
Flow Cytometry.
To quantify the levels of various marker expression, the cells were harvested by trypsinization and analyzed by flow cytometry. Briefly, 1 × 106 cells per sample were fixed with 4% PFA and washed with staining buffer (2% fetal bovine serum in phosphate buffer saline). The cells were permeabilized with 100% cold methanol for intracellular markers, blocked, and then incubated with primary antibodies against Aβ42, β-tubulin III, tau, or MAP2 followed by the corresponding secondary antibody (Supporting Information, Table S1). The cells were acquired with BD FACSCanto II flow cytometer (Becton Dickinson) and analyzed against isotype controls using FlowJo software.
Reverse Transcription-Polymerase Chain Reaction (RT-PCR) Analysis.
Total RNA was isolated from neural cell samples using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol followed by the treatment of DNA-Free RNA Kit (Zymo). Reverse transcription was performed using 2 μg of total RNA, anchored oligodT primers (Operon), and Superscript III (Invitrogen) (according to the protocol of the manufacturer). Primers specific for target genes (Supporting Information, Table S2) were designed using the software Oligo Explorer 1.2 (Genelink). The gene β-actin was used as an endogenous control for normalization of expression levels. Real-time RT-PCR reactions were performed on an ABI7500 instrument (Applied Biosystems), using SYBR1 Green PCR Master Mix (Applied Biosystems). The amplification reactions were performed as follows: 2 min at 50 °C, 10 min at 95 °C, and 40 cycles of 95 °C for 15 s and 55 °C for 30 s, and 68 °C for 30 s. Fold variation in gene expression was quantified by means of the comparative Ct method: which is based on the comparison of expression of the target gene (normalized to the endogenous control β-actin) between the cortical and the hippocampal samples.
Statistical Analysis.
Each experiment was repeated three times, and the representative results were presented. To assess the statistical significance, one-way ANOVA followed by Fisher’s LSD post hoc tests were performed. A p-value <0.05 was considered statistically significant.
RESULTS
Cortical and Hippocampal Differentiation and Characterization.
Two different types of neural populations were derived from human iPS cells to study the effects of Aβ42 treatments. For the first population, iPS cells were induced by dual Smad inhibition followed with cyclopamine (a Shh inhibitor) and FGF-2 treatment in suspension to generate spheroids enriched with cortical telencephalic neurons (referred to as cortical; Figure 1Ai and Supporting Information, Figure S1A). For the second population, iPS cells were treated with IWP4 (an inhibitor of Wnt), cyclopamine, and duel SMAD inhibitors followed with activation of Wnt and BDNF treatment to produce spheroids with identity of hippocampal dentate gyrus neurons (referred to as hippocampal; Figure 1Aii and Supporting Information, Figure S1A).29
Figure 1.

Cortical and hippocampal differentiation from human iPS cells and characterizations. (Ai) Cortical differentiation protocol. (Aii) Hippocampal differentiation protocol. The differentiation was induced through embryoid formation. The day 28 neural spheroids were replated.(B) Fluorescent images of neural patterning marker expression for cells derived from both cortical and hippocampal protocols. Scale bar: 100 μm.(C) Quantitative expression of neural patterning markers. (D) RT-PCR analysis of the gene expression of PROX1 and TBR1. * indicates p < 0.05.
The replated cells were compared for neuron marker MAP2 and different cortical forebrain markers TBR1 (cortical layerVI) and BRN2 (cortical layer II−IV) and hippocampal marker PROX1 (Figure 1B,C). The cells from both protocols expressed mature neuron marker MAP2. The hippocampal protocol resulted in the higher MAP2 expression suggesting the increased postmitotic hippocampal granule neurons. Also, on the one side, the expression of PROX1 was elevated in the cells derived using hippocampal protocol compared to the cortical protocol (Figure 1C). On the other side, the TBR1 expression was elevated in the cortical protocol. No significant difference was observed for BRN2 expression. Consistently, qualitative RT-PCR analysis showed the elevated expression of PROX1 in hippocampal population and the elevated expression of TBR1 in cortical population. (Figure 1D). The vGlut1 (a more mature marker for glutamatergic neurons) expression was comparable for the two groups (Supporting Information, Figure S1B). The expression of MMP-9, CSPG, Perlecan (a HSPG protein), and hyaluronic acid (HA) was detected for cortical and hippocampal groups (Supporting Information, Figure S2).
Effect of Aβ42 Peptide and MMP Inhibitors on Cell Viability.
The telencephalic neurons derived using cortical differentiation protocol were used for this set of experiments. The neural spheroids were replated to geltrex-coated plate at day 20 and treated with Aβ42 oligomers at day 21 to induce cellular death. To evaluate the effects of MMP inhibitor on the exogenous Aβ42 level and thereby on cell viability, the cells were also treated with MMP-2/9 inhibitor SB-3CT and Aβ42 oligomers (0.5 μM). Live/Dead assay and Caspase assay were used to examine cell viability. The neurons treated with the Aβ42 peptide were subjected to more cell death compared to the control cells (Figure 2A,B). Treatment of neurons with SB-3CT together with Aβ42 peptide increased cell death and cytotoxicity. Caspase activity was also elevated after Aβ42 treatment (Figure 2C,D). SB-3CT treatment along with Aβ42 peptide further increased the level of caspase activity. The analysis of Aβ-induced cytotoxicity was quantified using the flow cytometry analysis. Compared to the control, the percentage of live cells was decreased from 73.0% to 51.8% after Aβ42 treatment (Supporting Information, Figure S3A). MAP-2 expression was reduced (from 34.6% to 16.2%) after Aβ42 treatment (Supporting Information, Figure S3B). The amount of glutamatergic neurons decreased after Aβ42 treatment, while the decrease for GABAergic (GABA = γ-aminobutyric acid) neurons was less compared to glutamatergic neurons (Supporting Information, Figure S4). Consistently, electrophysiology analysis showed that treatment of Aβ42 oligomers resulted in a strong reduction in the amplitude and frequency of spontaneous excitatory postsynaptic currents (sEPSCs; Figure 2E) recorded from these cells, indicating the Aβ42 induced loss of excitatory glutamatergic synapses and/or neurons.
Figure 2.

Effect of MMP inhibitor on cell viability of Aβ42-treated neural spheroid outgrowth derived from human iPS cells (cortical). (A) Live/Dead images of neural outgrowth after treatment with Aβ and Aβ+MMP2/9 inhibitor (SB-3CT). (B) Quantification of cell viability. (C) Caspase images of neural outgrowth after treatment with Aβ and Aβ+SB-3CT. Scale bar: 100 μm. (D) Quantification of caspase expression. * indicates p <0.05. (E) Representative traces of patch-clamp recordings of sEPSCs in control and Aβ42 oligomers treated cells (day 48).
The expression of cell-bound Aβ42 peptide, tau protein, and phosphorylated tau (p-tau) was then investigated. The addition of Aβ42 oligomers to the culture increased the expression of cell-bound Aβ42 peptide (Figure 3A,B). To evaluate the hyperphosphorylation of tau, the expression of both tau and p-tau was analyzed. In normal human brain, phosphorylation of tau negatively regulates the attachment of tau to microtubules. In AD patients, tau becomes increasingly phosphorylated, which causes it to detach from micro-tubules.43–45 Currently, it is unclear if the addition of Aβ42 oligomers to the culture induces hyperphosphorylation of tau or not.46 In this study, the results indicate that the tau expression was decreased (as the result of neuron loss) after Aβ42 treatment (Figure 3B,D,E). While the staining of p-tau showed some expression, it was thought not related to the neuritic fibrillary tangles based on staining pattern. MMP-9 was shown to have protective roles in the brain and control the plaque growth,47 and MMP-9 is capable of degrading fibrillary Aβ42 in a mutated APP/PS1 mouse. In this study, the SB-3CT treatment decreased cell viability possibly through increased caspase expression (Figure 2), increased Aβ42 binding, and reduced the expression of tau compared to the cultures with Aβ42 treatment only (Figure 3C–F).
Figure 3.

Effect of MMP inhibitor on cell-bound Aβ42 expression of Aβ-treated neural spheroid outgrowth derived from human iPS cells. Fluorescent images of Aβ42, tau, and p-tau for (A) control (no Aβ42); (B) Aβ42-treated cells; and (C) Aβ42+MMP2/9 inhibitor (SB-3CT) treated cells. Scale bar: 100 μm. Quantification of marker expression for (D) Aβ42 and (E) tau. * indicates p < 0.05.
Effect of ECM Enzymes on the Viability of Aβ-Treated iPS Cell-Derived Neural Spheroids.
The effects of Heparinase III (HepIII, digests the HSPGs) and Chondroitinase ABC (Chabc, digests CSPGs) on the expression of Perlecan (a HSPG protein), CSPG, and HA were first confirmed (Supporting Information Figure S5). HepIII treatment reduced the expression of Perlecan and HA. Chabc treatment reduced the expression of Perlecan and CSPG and altered HA expression pattern (more dotlike). Then, the derived cortical spheroid outgrowth treated with Aβ42 peptide together with different ECM-related biomolecules, including Heparin (Hep, compete with HSPG-Aβ binding),48,49 HepIII, Chabc, and HA, were analyzed for cell viability (Figure 4). The treatment of cells with Aβ42 together with Hep, HepIII, or HA promoted cell survival, indicated by the higher cell viability (Figure 4A,B). The cells were also analyzed for cytotoxicity using LDH assay. The treatment of cells with Hep, HepIII, and HA reduced LDH level and thus the cytotoxic effect of Aβ42 on the cells (Figure 4C), while the treatment with Chabc showed no significant changes. The cell viability was further analyzed using MTT assay. Heparin or HA treatments showed the increased MTT activity compared to the Aβ42-treated cells, but the treatment with HepIII and Chabc had no significant changes (Figure 4D). These results indicate that the addition of heparin to the culture attenuates the effect of Aβ42 on the cell viability, which might be due to the competition of heparin with the HSPGs for binding with Aβ42 peptide.48,49
Figure 4.

Effect of ECM-related molecules on the viability of Aβ42-treated neural spheroid outgrowth derived using cortical protocol (cortical).(A) Live/Dead images of neural spheroid outgrowth treated with Hep, HepIII, Chabc, and HA after Aβ42 treatment. Scale bar: 100 μm. (B) Quantification of the cell viability. (C) Cytotoxicity of LDH activity. (D) MTT activity. * indicates p < 0.05 for the test conditions compared to the Aβ42 control group.
The neural spheroid outgrowth derived using the hippo-campal protocol was also treated with Aβ42 oligomers together with Hep, HepIII, Chabc, or HA. The treatments with Hep, HepIII, or HA promoted cell viability compared to the control cells. Chabc treatment did not increase cell viability (Figure 5A,B). The cytotoxicity analyzed by LDH assay showed the reduced toxicity for the Aβ42-induced cells after treatment with the HepIII and HA, as well as Chabc (Figure 5C). No significant change was observed for the MTT activity with any treatment (Figure 5D).
Figure 5.

Effect of ECM-related molecules on the viability of Aβ42-treated neural spheroid outgrowth derived using hippocampal protocol (hippo).(A) Live/Dead images of neural spheroid outgrowth treated with Hep, HepIII, Chabc, and HA after Aβ42 treatment. Scale bar: 100 μm. (B) Quantification of the cell viability. (C) Cytotoxicity of LDH activity. (D) MTT activity. * indicates p < 0.05 for the test conditions compared to the Aβ42 control group.
Effect of ECM Enzymes on the Expression of Aβ42 and tau.
To evaluate the effect of ECM enzymes on the expression of Aβ42 and tau, the Aβ42-treated cortical spheroid outgrowth was investigated. Immunostaining results indicated that Hep, HepIII, Chabc, and HA treatments reduced the expression of cell-bound Aβ42 (Figure 6A,B). The flow cytometry analysis of heparin-treated cortical neurons showed the decreased Aβ42 expression (from 59.4% to 35.3%), confirming the results from image analysis (Figure 6B). Similarly, heparinase III treatment decreased Aβ42 expression (from 59.4% to 39.3%; Figure 6B). Treatment of Chabc decreased Aβ42 expression to 17.6%, and the addition of HA decreased the Aβ42 bounding to 20.6% (Figure 6B). Also, the cells were costained with late-stage neuronal marker MAP2 and tau. The increased MAP-2 and tau expression were observed for Hep and HepIII treatment. No significant changes were found for Chabc and HA treatment (Figure 6A–C).
Figure 6.

Effect of ECM-related molecules on expression of Aβ42, MAP2, and tau of Aβ42-treated neural spheroid outgrowth derived using cortical protocol (cortical). (A) Fluorescent images of neural spheroid outgrowth treated with Heparin, Heparinase III, Chondroitinase ABC, and hyaluronic acid (Hep, HepIII, Chabc, HA, respectively) after Aβ42 treatment. Aβ42 (red)/p-tau (green)/Hoechst (blue), MAP2 (red)/tau (green)/Hoechst (blue). Scale bar: 100 μm. (B) (i) Quantification of the relative expression for Aβ42. (ii) Flow cytometry analysis of Aβ42 expression with various treatments. Black line: negative control. Red line: marker of interest. (C) Quantification of the relative expression for MAP2 and tau. * indicates p < 0.05 for the test conditions compared to the Aβ42 only group.
The neural spheroids derived from hippocampal protocol were also examined (Figure 7A–C). The treatments with Hep, HepIII, and HA as well as Chabc reduced cell-bound Aβ42 compared to the group of Aβ42-induced control (Figure 7B(i)). Flow cytometry analysis indicated that all four treatments reduced Aβ42 level (from 25.5% to 10−19%; Figure 7B(ii)). These results are consistent with the study showing that neuronal HS deficiency increases Aβ clearance in the hippocampus of amyloid model mice.49 All of the four treatments also increased MAP-2 and tau expression compared to the Aβ42-treated cells (Figure 7C).
Figure 7.

Effect of ECM-related molecules on expression of Aβ42, MAP2, and tau of Aβ42-treated neural spheroid outgrowth derived using hippocampal protocol (hippo). (A) Fluorescent images of neural spheroid outgrowth treated with Heparin, Heparinase III, Chondroitinase ABC, and hyaluronic acid (Hep, HepIII, Chabc, HA, respectively) after Aβ42 treatment. Aβ42 (red)/p-tau (green)/Hoechst (blue), MAP2 (red)/tau (green)/Hoechst (blue). Scale bar: 100 μm. (B) (i) Quantification of the relative expression for Aβ42. (ii) Flow cytometry analysis of Aβ42 expression with various treatments. Black line: negative control. Red line: marker of interest. (C) Quantification of the relative expression for MAP2 and tau. * indicates p < 0.05 for the test conditions compared to the Aβ42 only group.
Effect of ECM Enzymes on the Neuronal Patterning of Aβ-Treated Cells.
Since the addition of various enzymes to the culture had impacts on cell viability and toxicity, their impacts on various regional neuronal marker expression for possible selective cell death were further evaluated. The cortical neurons were examined for β-tubulin III and forebrainhippocampal region markers PROX1 and TBR1. Aβ42 induction reduced the expression of β-tubulin III, indicating that endogenous Aβ42 caused the death of neuronal cells (Figure 8A–C). The treatments with Hep, HepIII, Chabc, and HA promoted β-tubulin III expression. But the expression of PROX1 was similar for all the treatments compared to the Aβ42-induction control. Flow cytometry analysis showed that Hep, HepIII, Chabc, and HA promoted the survival of β-tubulin III-expressing neurons (from 31.4% to 39.6%, 37.5%,49.4%, and 40.9%, respectively) compared to Aβ42 only group (Figure 8B(ii)). The TBR1 expression was reduced by Aβ42 induction and the treatments with Hep, HepIII, Chabc, and HA promoted the survival of TBR1-expressing neurons (Figure 8A,C).
Figure 8.

Effect of ECM-related molecules on expression of neural markers for Aβ42-treated spheroid outgrowth derived using cortical protocol (cortical). (A) Fluorescent images of neural spheroid outgrowth treated with Heparin, Heparinase III, Chondroitinase ABC, and hyaluronic acid (Hep, HepIII, Chabc, HA, respectively) after Aβ42 treatment. PROX1 (red)/β-tubulin III (green)/Hoechst (blue), TBR1 (red)/β-tubulin III (green)/Hoechst (blue). Scale bar: 100 μm. (B) (i) Quantification of the relative expression for PROX1 and β-tubulin III. (ii) Flow cytometry analysis of β-tubulin III expression with various treatments. Black line: negative control. Red line: marker of interest. * indicates p < 0.05 for the test conditions compared to the Aβ42 only group.
For the neural spheroids derived with hippocampal protocol, Aβ42 induction reduced the expression of PROX1 (Figure 9A–C). The treatment with Hep, HepIII, and HA promoted the survival of PROX1-expressing neurons, but the treatment with Chabc did not show the changes compared to the Aβ42 induction control. Flow cytometry analysis showed that Hep, HepIII, and HA promoted the survival of β-tubulin III-expressing neurons (from 25.4% to 36.8%, 32.2%, and 43.2%, respectively) compared to Aβ42 only group (Figure 9B(ii)), but the treatments did not rescue the β-tubulin III expression to the level of no-Aβ42 control. In addition, Chabc treatment did not show the increase of β-tubulin III-expressing neurons by flow cytometry. Slight increase in TBR1 was observed for Hep-treated groups.
Figure 9.

Effect of ECM-related molecules on expression of neural markers for Aβ42-treated spheroid outgrowth derived using hippocampal protocol (hippo). (A) Fluorescent images of neural spheroid outgrowth treated with Heparin, Heparinase III, Chondroitinase ABC, and hyaluronic acid (Hep, HepIII, Chabc, HA, respectively) after Aβ42 treatment. PROX1 (red)/β-tubulin III (green)/Hoechst (blue), TBR1 (red)/β-tubulin III (green)/Hoechst (blue). Scale bar: 100 μm. (B) (i) Quantification of the relative expression for PROX1 and β-tubulin III. (ii) Flow cytometry analysis of β-tubulin III expression with various treatments. Black line: negative control. Red line: marker of interest. (C) Quantification of the relative expression for TBR1 and β-tubulin III. * indicates p < 0.05 for the test conditions compared to the Aβ42 only group.
DISCUSSION
Human iPS cells have been used as an effective tool to generate disease models for neurodegenerative diseases like AD.31,45,50,51 In this study, the response of neural spheroid outgrowth derived from iPS cells (generated from healthy human fibroblasts) to Aβ42 oligomer treatment was investigated. In our separate study, neural degenerative microenvironment was modeled in the cortical organoids using the iPS cells derived from fibroblasts of AD patient with presenilin 1 (PS1) M146 V mutation.52 Since AD progression leads to neurodegeneration in forebrain and the hippocampal area, both cortical forebrain and hippocampal-rich neuronal populations were evaluated. The derived neurons indicated forebrain identity with expression of regional markers PROX1 and TBR1. Since one hallmark of AD is the formation of Aβ plaque and the increased extracellular Aβ42,53 the AD-related neurotoxicity in the iPS cell-derived neurons was induced by exogenous addition of Aβ42 oligomers.23,31 The immunocytochemical analysis showed adequate uptake of Aβ42 peptides by the cells.
To understand the drug screening ability of iPS cell-derived neural models,54 the impacts of various ECM enzymes on Aβ42-treated iPS cell-derived neural spheroid outgrowth were evaluated in the current study. This study mainly focused on the effects on cell viability, Aβ42 expression, and the neural tissue patterning markers. Previous research has shown that pathologically altered tau mediates neurodegeneration in AD, and studies are still going on to elucidate the mechanism underlying tau alteration.43,44,55 One hypothesis is that aberrant tau phosphorylation plays an important role in pathogenesis of AD,56 and another hypothesis is that Aβ oligomers induce tau aggregation and formation of tau oligomers.46,57 The results of this study were not able to delineate the relations between Aβ42 and tau phosphorylation, where genetic mutations might be required.53 The decreased tau expression might be due to the loss of neuronal cells. Tau plays a key role in the regulation of neurite extension. The reduction in tau may result in dysgenesis of axonal tracts and the reduction in β-tubulin III expression.
The development/remodeling of neuronal connections and cell invasion depends on the modification of ECM. MMPs play a key role in both neurogenesis and neurodegeneration in diseases.58,59 MMPs are secreted by the astrocytes and play important roles in the extracellular degradation of both monomeric and fibrillar Aβ during AD progress.21 Astrocytes around the plaques show enhanced expression of MMP-2 and MMP-9 in both post-mortem AD brains and mouse models.60 Also coculturing the human Aβ peptide (both Aβ40 and Aβ42) with astrocytes lowered the peptide concentration, and the MMP inhibition reduced Aβ clearance, suggesting the involvement of MMPs in the Aβ degradation.47,61 MMP-9 cleaves mainly in the C-terminal hydrophobic region of the Aβ peptide. Therefore, by the addition of SB-3CT, a selective gelatinase inhibitor of MMP2/MMP9, to the Aβ42-treated culture, the impact of Aβ42 on the cell cytotoxicity was intensified. Our results showed the decreased cell viability and the increased Aβ42 expression with SB-3CT treatment.
HSPGs are negatively charged and can form insoluble complexes with amyloid filament that protect ligands from proteolysis.13 HSPGs bind to Aβ and accelerate its oligomerization and aggregation.48 Moreover, HSPGs affect cellular uptake of Aβ and mediate neurotoxicity and immune response induced by Aβ peptides. In vivo evidence using a mouse model deficient in neuronal HSPG demonstrates that HSPGs inhibit Aβ clearance and promote Aβ plaque deposition.49 HSPGs are also found to be the target of Aβ-induced oxidative stress production.62 The detailed evaluation reveals that minimal chain length is a prerequisite for efficient fibril polymerization and deposition by HSPGs. Fragmentation of HS using heparinase III was found to inhibit the polymerization and deposition of amyloid peptides.63 It is also proposed that the Aβ-HS interaction is mutually protective, because HS is protected from heparinase degradation after the complex formation. The results from our study showed that the treatment of Aβ42-induced cells with Heparinase III promote cell viability and reduced the expression of cell-bound Aβ42.
The HSPG-targeted treatment provides the possibility for the new therapeutic agent heparin for AD treatment.64 In vitro treatment of neurons with heparin significantly counteracted Aβ-cytotoxicity, revealing the capability of heparin on AD treatment.64,65 Heparin has the capability to bind to the region 12−17 of Aβ (HHQK)66 and can reduce Aβ42 cellular uptake by disrupting HSPG-Aβ42 binding.48 The addition of low molecular weight heparin reduced plaques and Aβ accumulation in a mouse AD model.67 Heparin binds with growth factors (such as FGF-2) with a neurite-promoting activity and induces axonal development, so heparinized surfaces have been used for neuron differentiation of iPS cells.68 The results from this study showed that heparin treatment reduced Aβ42-induced neurotoxicity. Heparin/Heparinase III treatments also supported the survival of neurons derived using cortical and hippocampal protocols.
Perineuronal nets (PNNs) in brain are specialized ECM structures responsible for synaptic stabilization in the adult brain. PNNs are largely negatively charged and are built by CSPGs together with HA.17 While it is expected that Chabc treatment may increase neuron death, our data showed the mixed results. Chabc treatment on the Aβ42-induced cells did not improve cell viability but supported the survival of MAP2 and β-tubulin III positive cells especially for cortical population, indicating that CSPGs may have some neuro-protective effects on neurons in brain. Low molecular weight chondroitin sulfate reduced Aβ-induced neurotoxicity in vitro and in vivo by reducing reactive oxygen species levels and caspase-3 expression.69 Injection of Chabc in young AD models elevated lectican levels, resulting in a reversal of contextual fear memory deficits and restoration to normal long-term potentiation.70 Increased synaptic density surrounding plaques and reduced Aβ peptides in the hippocampus was also observed.71
HA may have neuroprotective effect for brain-like cells.17,72,73 The HA treatment reduced the Aβ42 level of the cells, supported the survival of forebrain neurons, and increased the expression of β-tubulin III in both cortical and hippocampal populations. HA may have attenuation effects on AD-affected neurons, and neurons in HA environment may be resistive to the alteration caused by AD pathology.17,20 Since HA can be fabricated into hydrogels and modified with bioactive molecules such as heparin,74 rational design of biomaterials can be realized with HA.
Taken together, the different ECM-related molecules have differential impacts on the neuron outgrowth from iPS cell-derived neural spheroids and thereby should be explored in future to understand their effects on AD-associated pathology.
CONCLUSION
The study focuses on the impacts of various ECM components, ECM proteolytic enzymes, and related molecules on cell viability of Aβ42-induced iPS cell-derived neural spheroid outgrowth. The exogenous addition of Aβ42 oligomers induces cell death and neurotoxicity in both cortical and hippocampal populations. The matrix metalloproteinase inhibitor increases cell death. The impacts of heparin, heparinase III, and hyaluronic acid on Aβ42-treated cells are more conclusive than the effects of chondroitin sulfate proteoglycans. Heparin/heparinase III and hyaluronic acid treatments reduce Aβ42 oligomer uptake and support neuron cell survival. This study indicates the importance of extracellular matrix in neural degeneration.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank Ms. R. Didier in FSU Dept. of Biomedical Sciences for her help with flow cytometry analysis, Dr. B. K. Washburn and K. Poduch in FSU Dept. of Biological Sciences for their help with RT-PCR analysis, Dr. S. Duncan at Medical College of Wisconsin, Dr. D. Gilbert in FSU Dept. of Biological Sciences for human iPSK3 cell, and Dr. Y. Yan for preparation of cells of electrophysiology study. This work is supported by FSU startup fund and partially from National Science Foundation (CAREER award, Grant No. 1652992 to Y.L.). Research reported in this publication was also supported by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health under Award No. R03NS102640 (to Y.L. and Y.Z.) and National Institute of Mental Health under Award No. R01MH115188 (to Y.Z.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsbiomaterials.8b00021.
Characterization of cortical and hippocampal differentiation from human induced pluripotent stem (iPS) cells. Expression of MMP-9, perlecan (an HSPG protein), CSPG, and HA for cortical and hippocampal groups. Effect of Aβ42 treatment. Glutamate and GABA expression for cortical and hippocampal cells after Aβ42 treatment. Effects of Chabc and heparinase III on the expression of MMP-9, perlecan, CSPG, and HA. A list of antibodies. Primer sequence for target genes (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Prince M; Wilmo A; Guerchet G; Ali G; Wu Y; Prina M World Alzheimer Report 2015 The Global Impact of Dementia. Alzheimer’s Res. Ther 20168 DOI: 10.1186/s13195-016-0188-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Nussbaum JM; Seward ME; Bloom GS Alzheimer disease: a tale of two prions. Prion 2013, 7, 14–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Goldstein LS; Reyna S; Woodruff G Probing the secrets of Alzheimer’s disease using human-induced pluripotent stem cell technology. Neurotherapeutics 2015, 12, 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Raja WK; Mungenast AE; Lin YT; Ko T; Abdurrob F; Seo J; Tsai LH Self-Organizing 3D Human Neural Tissue Derived from Induced Pluripotent Stem Cells Recapitulate Alzheimer’s Disease Phenotypes. PLoS One 2016, 11, e0161969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ooi L; Sidhu K; Poljak A; Sutherland G; O’Connor MD; Sachdev P; Munch G Induced pluripotent stem cells as tools for disease modelling and drug discovery in Alzheimer’s disease. J. Neural Transm 2013, 120, 103–111. [DOI] [PubMed] [Google Scholar]
- (6).Kondo T; Asai M; Tsukita K; Kutoku Y; Ohsawa Y; Sunada Y; Imamura K; Egawa N; Yahata N; Okita K; Takahashi K; Asaka I; Aoi T; Watanabe A; Watanabe K; Kadoya C; Nakano R; Watanabe D; Maruyama K; Hori O; Hibino S; Choshi T; Nakahata T; Hioki H; Kaneko T; Naitoh M; Yoshikawa K; Yamawaki S; Suzuki S; Hata R; Ueno S; Seki T; Kobayashi K; Toda T; Murakami K; Irie K; Klein WL; Mori H; Asada T; Takahashi R; Iwata N; Yamanaka S; Inoue H Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Abeta and differential drug responsiveness. Cell Stem Cell 2013, 12, 487–496. [DOI] [PubMed] [Google Scholar]
- (7).Qian X; Nguyen HN; Song MM; Hadiono C; Ogden SC; Hammack C; Yao B; Hamersky GR; Jacob F; Zhong C; Yoon KJ; Jeang W; Lin L; Li Y; Thakor J; Berg DA; Zhang C; Kang E; Chickering M; Nauen D; Ho CY; Wen Z; Christian KM; Shi PY; Maher BJ; Wu H; Jin P; Tang H; Song H; Ming GL Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell 2016, 165, 1238–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Di Lullo E; Kriegstein AR The use of brain organoids to investigate neural development and disease. Nat. Rev. Neurosci 2017, 18, 573–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ariga T; Miyatake T; Yu RK Role of proteoglycans and glycosaminoglycans in the pathogenesis of Alzheimer’s disease and related disorders: amyloidogenesis and therapeutic strategies−a review. J. Neurosci. Res 2010, 88, 2303–2315. [DOI] [PubMed] [Google Scholar]
- (10).Snow AD; Mar H; Nochlin D; Kimata K; Kato M; Suzuki S; Hassell J; Wight TN The presence of heparan sulfate proteoglycans in the neuritic plaques and congophilic angiopathy in Alzheimer’s disease. Am. J. Pathol 1988, 133, 456–463. PMID: 2974240. [PMC free article] [PubMed] [Google Scholar]
- (11).DeWitt DA; Silver J; Canning DR; Perry G Chondroitin sulfate proteoglycans are associated with the lesions of Alzheimer’s disease. Exp. Neurol 1993, 121, 149–152. PMID: 8339766. [DOI] [PubMed] [Google Scholar]
- (12).McLaurin J; Franklin T; Zhang X; Deng J; Fraser PE Interactions of Alzheimer amyloid-beta peptides with glycosaminoglycans effects on fibril nucleation and growth. Eur. J. Biochem 1999, 266, 1101–1110. PMID: 10583407. [DOI] [PubMed] [Google Scholar]
- (13).Perry G; Siedlak SL; Richey P; Kawai M; Cras P; Kalaria RN; Galloway PG; Scardina JM; Cordell B; Greenberg BD; et al. Association of heparan sulfate proteoglycan with the neurofibrillary tangles of Alzheimer’s disease. J. Neurosci 1991, 11, 3679–3683. PMID: 1941102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Castillo GM; Ngo C; Cummings J; Wight TN; Snow AD Perlecan binds to the beta-amyloid proteins (A beta) of Alzheimer’s disease, accelerates A beta fibril formation, and maintains A beta fibril stability. J. Neurochem 1997, 69, 2452–2465. PMID: 9375678. [DOI] [PubMed] [Google Scholar]
- (15).Properzi F; Asher RA; Fawcett JW Chondroitin sulphate proteoglycans in the central nervous system: changes and synthesis after injury. Biochem. Soc. Trans 2003, 31, 335–336. PMID: 12653631. [DOI] [PubMed] [Google Scholar]
- (16).Shioi J; Refolo LM; Efthimiopoulos S; Robakis NK Chondroitin sulfate proteoglycan form of cellular and cell-surface Alzheimer amyloid precursor. Neurosci. Lett 1993, 154, 121–124. PMID: 8361624. [DOI] [PubMed] [Google Scholar]
- (17).Soleman S; Filippov MA; Dityatev A; Fawcett JW Targeting the neural extracellular matrix in neurological disorders. Neuroscience 2013, 253, 194–213. [DOI] [PubMed] [Google Scholar]
- (18).Backstrom JR; Lim GP; Cullen MJ; Tokes ZA Matrix metalloproteinase-9 (MMP-9) is synthesized in neurons of the human hippocampus and is capable of degrading the amyloid-beta peptide (1−40). J. Neurosci 1996, 16, 7910–7919. PMID: 8987819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ethell IM; Ethell DW Matrix metalloproteinases in brain development and remodeling: synaptic functions and targets. J. Neurosci. Res 2007, 85, 2813–2823. [DOI] [PubMed] [Google Scholar]
- (20).Yasuhara O; Akiyama H; McGeer EG; McGeer PL Immunohistochemical localization of hyaluronic acid in rat and human brain. Brain Res. 1994, 635, 269–282. PMID: 8173963. [DOI] [PubMed] [Google Scholar]
- (21).Yin KJ; Cirrito JR; Yan P; Hu X; Xiao Q; Pan X; Bateman R; Song H; Hsu FF; Turk J; Xu J; Hsu CY; Mills JC; Holtzman DM; Lee JM Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-beta peptide catabolism. J. Neurosci 2006, 26, 10939–10948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Shinagawa R; Masuda S; Sasaki R; Ikura K; Takahata K In vitro neurotoxicity of amyloid beta-peptide cross-linked by transglutaminase. Cytotechnology 1997, 23, 77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Nieweg K; Andreyeva A; van Stegen B; Tanriover G; Gottmann K Alzheimer’s disease-related amyloid-beta induces synaptotoxicity in human iPS cell-derived neurons. Cell Death Dis. 2015, 6, e1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Lee HK; Velazquez Sanchez C; Chen M; Morin PJ; Wells JM; Hanlon EB; Xia W Three Dimensional Human Neuro-Spheroid Model of Alzheimer’s Disease Based on Differentiated Induced Pluripotent Stem Cells. PLoS One 2016, 11, e0163072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Harasta AE; Ittner LM Alzheimer’s Disease: Insights from Genetic Mouse Models and Current Advances in Human IPSC-Derived Neurons. Adv. Neurobiol 2017, 15, 3–29. [DOI] [PubMed] [Google Scholar]
- (26).Birey F; Andersen J; Makinson CD; Islam S; Wei W; Huber N; Fan HC; Metzler KRC; Panagiotakos G; Thom N; O’Rourke NA; Steinmetz LM; Bernstein JA; Hallmayer J; Huguenard JR; Pasca SP Assembly of functionally integrated human forebrain spheroids. Nature 2017, 545, 54–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Yan Y; Bejoy J; Xia J; Guan J; Zhou Y; Li Y Neural patterning of human induced pluripotent stem cells in 3-D cultures for studying biomolecule-directed differential cellular responses. Acta Biomater. 2016, 42, 114–126. [DOI] [PubMed] [Google Scholar]
- (28).Vazin T; Ball KA; Lu H; Park H; Ataeijannati Y; Head-Gordon T; Poo MM; Schaffer DV Efficient derivation of cortical glutamatergic neurons from human pluripotent stem cells: a model system to study neurotoxicity in Alzheimer’s disease. Neurobiol. Dis 2014, 62, 62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Yu DX; Di Giorgio FP; Yao J; Marchetto MC; Brennand K; Wright R; Mei A; McHenry L; Lisuk D; Grasmick JM; Silberman P; Silberman G; Jappelli R; Gage FH Modeling hippocampal neurogenesis using human pluripotent stem cells. Stem Cell Rep. 2014, 2, 295–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Yu DX; Marchetto MC; Gage FH How to make a hippocampal dentate gyrus granule neuron. Development 2014, 141, 2366–2375. [DOI] [PubMed] [Google Scholar]
- (31).Zhang D; Pekkanen-Mattila M; Shahsavani M; Falk A; Teixeira AI; Herland AA 3D Alzheimer’s disease culture model and the induction of P21-activated kinase mediated sensing in iPSC derived neurons. Biomaterials 2014, 35, 1420–1428. [DOI] [PubMed] [Google Scholar]
- (32).Si-Tayeb K; Noto FK; Sepac A; Sedlic F; Bosnjak ZJ; Lough JW; Duncan SA Generation of human induced pluripotent stem cells by simple transient transfection of plasmid DNA encoding reprogramming factors. BMC Dev. Biol 2010, 10, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Si-Tayeb K; Noto FK; Nagaoka M; Li J; Battle MA; Duris C; North PE; Dalton S; Duncan SA Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology 2010, 51, 297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Yan Y; Martin L; Bosco D; Bundy J; Nowakowski R; Sang QX; Li Y Differential effects of acellular embryonic matrices on pluripotent stem cell expansion and neural differentiation. Biomaterials 2015, 73, 231–242. [DOI] [PubMed] [Google Scholar]
- (35).Bejoy J; Song L; Zhou Y; Li Y Wnt-Yes associated protein interactions during neural tissue patterning of human induced pluripotent stem cells. Tissue Eng., Part A 2018, 24, 546–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Song L; Wang K; Li Y; Yang Y Nanotopography promoted neuronal differentiation of human induced pluripotent stem cells. Colloids Surf., B 2016, 148, 49–58. [DOI] [PubMed] [Google Scholar]
- (37).Chambers SM; Fasano CA; Papapetrou EP; Tomishima M; Sadelain M; Studer L Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol 2009, 27, 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Yan Y; Song L; Madinya J; Ma T; Li Y Derivation of cortical spheroids from human induced pluripotent stem cells in a suspension bioreactor. Tissue Eng., Part A 2018, 24, 418–431. [DOI] [PubMed] [Google Scholar]
- (39).Song L; Tsai AC; Yuan X; Bejoy J; Sart S; Ma T; Li Y Neural differentiation of spheroids derived from human induced pluripotent stem cells-mesenchymal stem cells coculture. Tissue Eng., Part A 2018, 24, 915–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Yan Y; Calixto Bejarano F; Sart S; Muroski M; Strouse GF; Grant SC; Li Y Cryopreservation of embryonic stem cell-derived multicellular neural aggregates labeled with micron-sized particles of iron oxide for magnetic resonance imaging. Biotechnol. Prog 2015, 31, 510–521. [DOI] [PubMed] [Google Scholar]
- (41).Sart S; Yan Y; Li Y The microenvironment of embryoid bodies modulated the commitment to neural lineage post-cryopreservation. Tissue Eng., Part C 2015, 21, 356–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Sart S; Yan Y; Li Y; Lochner E; Zeng C; Ma T; Li Y Crosslinking of extracellular matrix scaffolds derived from pluripotent stem cell aggregates modulates neural differentiation. Acta Biomater. 2016, 30, 222–232. [DOI] [PubMed] [Google Scholar]
- (43).LaFerla FM Pathways linking Abeta and tau pathologies. Biochem. Soc. Trans 2010, 38, 993–995. [DOI] [PubMed] [Google Scholar]
- (44).Stancu IC; Vasconcelos B; Terwel D; Dewachter I Models of beta-amyloid induced Tau-pathology: the long and ″folded″ road to understand the mechanism. Mol. Neurodegener 2014, 9, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Ochalek A; Mihalik B; Avci HX; Chandrasekaran A; Teglasi A; Bock I; Giudice ML; Tancos Z; Molnar K; Laszlo L; Nielsen JE; Holst B; Freude K; Hyttel P; Kobolak J; Dinnyes A Neurons derived from sporadic Alzheimer’s disease iPSCs reveal elevated TAU hyperphosphorylation, increased amyloid levels, and GSK3B activation. Alzheimer’s Res. Ther 2017, 9, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Zheng WH; Bastianetto S; Mennicken F; Ma W; Kar S Amyloid beta peptide induces tau phosphorylation and loss of cholinergic neurons in rat primary septal cultures. Neuroscience 2002, 115, 201–211. PMID: 12401334. [DOI] [PubMed] [Google Scholar]
- (47).Yan P; Hu X; Song H; Yin K; Bateman RJ; Cirrito JR; Xiao Q; Hsu FF; Turk JW; Xu J; Hsu CY; Holtzman DM; Lee JM Matrix metalloproteinase-9 degrades amyloid-beta fibrils in vitro and compact plaques in situ. J. Biol. Chem 2006, 281, 24566–24574. [DOI] [PubMed] [Google Scholar]
- (48).Kanekiyo T; Zhang J; Liu Q; Liu CC; Zhang L; Bu G Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal amyloid-beta uptake. J. Neurosci 2011, 31, 1644–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Liu CC; Zhao N; Yamaguchi Y; Cirrito JR; Kanekiyo T; Holtzman DM; Bu G Neuronal heparan sulfates promote amyloid pathology by modulating brain amyloid-beta clearance and aggregation in Alzheimer’s disease. Sci. Transl. Med 2016, 8, 332ra44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Ovchinnikov DA; Wolvetang EJ Opportunities and Limitations of Modelling Alzheimer’s Disease with Induced Pluripotent Stem Cells. J. Clin. Med 2014, 3, 1357–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Yang J; Zhao H; Ma Y; Shi G; Song J; Tang Y; Li S; Li T; Liu N; Tang F; Gu J; Zhang L; Zhang Z; Zhang X; Jin Y; Le W Early pathogenic event of Alzheimer’s disease documented in iPSCs from patients with PSEN1 mutations. Oncotarget 2017, 8, 7900–7913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Yan Y; Song L; Bejoy J; Zhao J; Kanekiyo T; Bu G; Zhou Y; Li Y Modelling neurodegenerative microenvironment using cortical organoids derived from human stem cells. Tissue Eng., Part A 2018. 241125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Choi SH; Kim YH; Hebisch M; Sliwinski C; Lee S; D’Avanzo C; Chen H; Hooli B; Asselin C; Muffat J; Klee JB; Zhang C; Wainger BJ; Peitz M; Kovacs DM; Woolf CJ; Wagner SL; Tanzi RE; Kim DY A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 2014, 515, 274–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Yahata N; Asai M; Kitaoka S; Takahashi K; Asaka I; Hioki H; Kaneko T; Maruyama K; Saido TC; Nakahata T; Asada T; Yamanaka S; Iwata N; Inoue H Anti-Abeta drug screening platform using human iPS cell-derived neurons for the treatment of Alzheimer’s disease. PLoS One 2011, 6, e25788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Bloom GS Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol 2014, 71, 505–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Johnson GV; Stoothoff WH Tau phosphorylation in neuronal cell function and dysfunction. J. Cell Sci. 2004, 117, 5721–5729. [DOI] [PubMed] [Google Scholar]
- (57).Chabrier MA; Blurton-Jones M; Agazaryan AA; Nerhus JL; Martinez-Coria H; LaFerla FM Soluble abeta promotes wild-type tau pathology in vivo. J. Neurosci 2012, 32, 17345–17350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Brkic M; Balusu S; Libert C; Vandenbroucke RE Friends or Foes: Matrix Metalloproteinases and Their Multifaceted Roles in Neurodegenerative Diseases. Mediators Inflammation 2015, 2015, 620581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Szymczak P; Wojcik-Stanaszek L; Sypecka J; Sokolowska A; Zalewska T Effect of matrix metalloproteinases inhibition on the proliferation and differentiation of HUCB-NSCs cultured in the presence of adhesive substrates. Acta Neurobiol Exp (Wars) 2010, 70, 325–336 PMID: 21196941. [DOI] [PubMed] [Google Scholar]
- (60).Yoon SS; AhnJo S-M Mechanisms of Amyloid-beta Peptide Clearance: Potential Therapeutic Targets for Alzheimer’s Disease. Biomol. Ther 2012, 20, 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Ries M; Sastre M Mechanisms of Abeta Clearance and Degradation by Glial Cells. Front. Aging Neurosci. 2016, 8, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Reynolds MR; Singh I; Azad TD; Holmes BB; Verghese PB; Dietrich HH; Diamond M; Bu G; Han BH; Zipfel GJ Heparan sulfate proteoglycans mediate Abeta-induced oxidative stress and hypercontractility in cultured vascular smooth muscle cells. Mol. Neurodegener 2016, 11, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Li JP; Galvis ML; Gong F; Zhang X; Zcharia E; Metzger S; Vlodavsky I; Kisilevsky R; Lindahl U In vivo fragmentation of heparan sulfate by heparanase overexpression renders mice resistant to amyloid protein A amyloidosis. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 6473–6477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Bergamaschini L; Rossi E; Vergani C; De Simoni MG Alzheimer’s disease: another target for heparin therapy. Sci. World J. 2009, 9, 891–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Bergamaschini L; Donarini C; Rossi E; De Luigi A; Vergani C; De Simoni MG Heparin attenuates cytotoxic and inflammatory activity of Alzheimer amyloid-beta in vitro. Neurobiol. Aging 2002, 23, 531–536 PMID: 12009502. [DOI] [PubMed] [Google Scholar]
- (66).Brunden KR; Richter-Cook NJ; Chaturvedi N; Frederickson RC pH-dependent binding of synthetic beta-amyloid peptides to glycosaminoglycans. J. Neurochem 1993, 61, 2147–2154 PMID: 8245966. [DOI] [PubMed] [Google Scholar]
- (67).Bergamaschini L; Rossi E; Storini C; Pizzimenti S; Distaso M; Perego C; De Luigi A; Vergani C; De Simoni MG Peripheral treatment with enoxaparin, a low molecular weight heparin, reduces plaques and beta-amyloid accumulation in a mouse model of Alzheimer’s disease. J. Neurosci 2004, 24, 4181–4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Kuo YC; Wang CT Neuronal differentiation of induced pluripotent stem cells in hybrid polyester scaffolds with heparinized surface. Colloids Surf., B 2012, 100, 9–15. [DOI] [PubMed] [Google Scholar]
- (69).Zhang Q; Li J; Liu C; Song C; Li P; Yin F; Xiao Y; Li J; Jiang W; Zong A; Zhang X; Wang F Protective effects of low molecular weight chondroitin sulfate on amyloid beta (Abeta)-induced damage in vitro and in vivo. Neuroscience 2015, 305, 169–182. [DOI] [PubMed] [Google Scholar]
- (70).Senkov O; Andjus P; Radenovic L; Soriano E; Dityatev A Neural ECM molecules in synaptic plasticity, learning, and memory. Prog. Brain Res 2014, 214, 53–80. [DOI] [PubMed] [Google Scholar]
- (71).Howell MD; Bailey LA; Cozart MA; Gannon BM; Gottschall PE Hippocampal administration of chondroitinase ABC increases plaque-adjacent synaptic marker and diminishes amyloid burden in aged APPswe/PS1dE9 mice. Acta Neuropathol Commun. 2015, 3, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Liang Y; Walczak P; Bulte JW The survival of engrafted neural stem cells within hyaluronic acid hydrogels. Biomaterials 2013, 34, 5521–5529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Nih LR; Moshayedi P; Llorente IL; Berg AR; Cinkornpumin J; Lowry WE; Segura T; Carmichael ST Engineered HA hydrogel for stem cell transplantation in the brain: Biocompatibility data using a design of experiment approach. Data Brief 2017, 10, 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Gwon K; Kim E; Tae G Heparin-hyaluronic acid hydrogel in support of cellular activities of 3D encapsulated adipose derived stem cells. Acta Biomater. 2017, 49, 284–295. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.