

Introduction
Skin, as the outermost layer of the body, is constantly exposed to ultraviolet B (UVB) irradiation and other environmental pollutants. Due to increased exposure to these pollutants, the incidence of skin cancer is steadily increasing worldwide (1–3). In the United States, non-melanoma skin cancer is the most common type of cancer, with more than 1 million new cases per year. Natural phytochemicals have been widely used as potential chemopreventive agents against skin cancers (4–6). Earlier studies suggest that phytochemical supplements, including curcumin (7), green tea (8) and ursolic acid (9), are effective in blocking both chemical-induced and UVB-induced skin carcinogenesis. However, the exact mechanisms by which these chemopreventive agents shift the balance from reactive intermediates to protective mechanisms by promoting detoxification and radical scavenging reactions remain unclear (10, 11).
Epigenomic alterations, including DNA methylation, histone modification, and miRNAs, are associated with skin cancer development. These alterations could be useful biomarkers and novel therapeutic targets for the prevention and treatment of skin cancer. For example, drugs that target histone deacetylases (HDACs) and DNA methyltransferase (DNMT) inhibitors have been approved by the FDA as chemotherapeutics, although their toxicity limits their applications (12, 13). Aberrant epigenetic alterations have been observed in the initiation and progression of skin cancers (14–17). Certain dietary phytochemicals that possess chemopreventive effects have been reported to prevent cancer by targeting epigenetic modifications (18, 19). For example, sulforaphane and phenethyl isothiocyanate, which are derived from cruciferous vegetables and allyl compounds in garlic, have been reported to be potent inhibitors of HDACs and DNMTs (20–24).
In addition, increasing evidence indicates that oxidative stress and chronic inflammatory disorders contribute to the increased risk of many diseases, including skin cancers (26–28). The nuclear factor erythroid-2-related factor-2 (Nrf2) -antioxidant response element (ARE)-mediated anti-oxidative stress and anti-inflammatory pathways appear to play a major role in protection against carcinogenesis (29, 30). Nrf2, which is also termed NFE2L2, is a basic-region leucine zipper (bZIP) transcription factor that regulates the expression of many phase II detoxifying enzymes, including glutathione S-transferases (GSTs). Thus, Nrf2 protects against oxidative stress and electrophilic challenges to maintain cellular chemical homeostasis (31). In the cytosol, Nrf2 binds to Kelch-like ECH-associating protein (Keap1), a substrate adaptor protein for a CUL3-containing E3 ubiquitin ligase (32). In vivo studies have shown that Nrf2-deficient mice exhibit significantly lower levels of cellular defense in various tissues, as summarized in reviews (33, 34), with an increased risk of developing carcinogen-induced colorectal (35) and skin cancers (5, 36–38). Early studies have reported that sulforaphane’s role in phase 2 enzyme inducing activities and disruption of the Nrf2-keap1 interaction (39). However, recent reports have identified effects of Sulforaphane on DNA methylation of the Nrf2 gene promoter, HDACs and DNMTs (25).
Fucoxanthin (FX) and astaxanthin (AST) are two major xanthophyll carotenoids (Figure 1). FX has strong antioxidant properties against obesity and inflammation (40–42). FX reduces the levels of reactive oxygen species (ROS), damage and apoptosis (43). FX increases the level of glutathione (GSH) by inducing glutamate-cysteine ligase catalytic subunit (GCLC) and glutathione synthetase (GSS) expression via the Akt/Nrf2 pathway in human keratinocytes cells (44). Recent reports have shown the protective effects of FX against UVB-induced photoaging and sunburn on the skin in vivo (45, 46). Another important xanthophyll, AST, is well known to have anti-oxidative activities (47–50), antitumor effects (51–53), hepatoprotective effects (54), anti-diabetic effects (55) and anti-inflammatory properties (56). Both in vivo and in vitro studies suggest that AST has health-promoting activities and could potentially be used for the prevention of various diseases, including cancers (57) and Parkinson’s disease (58). AST and its analog AST esters show protective effects against UVB – 7,12-dimethylbenz(a)anthracene (DMBA)-induced skin cancer in rats (59). We have previously reported that AST shows synergistic antioxidant effects with Polyunsaturated fatty acids (PUFAs) at low concentrations via the Nrf2/ARE pathway (60) and that AST decreased the methylation of the glutathione S-transferase P (GSTP1) gene (61). Recently, it has been reported that AST increases chromosomal stability and normalizes epigenetic modifications in oocyte maturation (62). AST inhibited HDAC-9 expression and facilitated the inactivation of hepatic stellate cells, which are critical for liver fibrosis development (63).
Figure 1.

Chemical structures of FX and AST.
However, the effects of FX and AST on the reversal of abnormal epigenetic changes have not yet been clearly described in skin cells. The JB6 P+ is a promotion-sensitive and post-initiated mouse epidermal cell line, an in vitro cell model for tumor promotion induced by tumor promoters such as 12-O-tetradecanoylphorbol-13-acetate (TPA) and epidermal growth factor (64, 65). Human hepatoma HepG2 cells have been used as the model cell line for the study of phase II enzymes induction (66). We hypothesize that two important xanthophylls, FX and AST, inhibit the neoplastic transformation of mouse skin JB6 P+ cells induced by TPA. The molecular mechanism may be mediated by the activation of Nrf2-mediated pathways and epigenetic modification by targeting Nrf2 promoter demethylation by modulating the expression and activity of HDACs or DNMTs. Hence, we aimed to investigate the effects of AST and FX on the Nrf2-ARE signaling pathway and epigenetic modifications to elucidate the chemopreventive effects of AST and FX against skin cancers.
MATERIALS AND METHODS
Chemicals and reagents
FX and AST were purchased from Sigma-Aldrich, Inc. (St. Louis, MO, USA). The MTS [3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt)] test using the CellTiter 96® AQueous One Solution was purchased from Promega (Madison, WI, USA). 5-aza-deoxycytidine (5-aza, a DNMT inhibitor) and trichostatin A (TSA, an HDAC inhibitor) were obtained from Sigma-Aldrich. Fetal bovine serum (FBS), Dulbecco’s Modified Eagle’s Medium (DMEM), Minimum Essential Media (MEM) and trypsin-EDTA solution were purchased from Gibco Laboratories (Waltham, MA, USA). Penicillin G and streptomycin were obtained from Gibco Laboratories (Grand Island, NY, USA).
Cell culture and treatment
The human hepatocellular HepG2-C8 cell line was previously established by stable transfection with the pARE-TI-luciferase construct (provided by Dr. William Fahl, University of Wisconsin). Both the HepG2 immortalized human hepatoma cell line and mouse epidermal JB6 P+ cells were purchased from American Type Culture Collection. The HepG2-C8 cells were maintained in DMEM supplemented with 10% FBS. The JB6 P+ cells were maintained in MEM with 5% FBS. JB6 P+ cells stably transfected with shMock- and shNrf2-knockdown were established using virus-medidated short hairpin RNAs (shRNAs) maintained in MEM supplemented with 5% FBS and 2 ug/mL puromycin as previous described (25). Cells were treated with various concentrations of FX, AST or 5-aza in combination with TSA. The medium was changed every two days. 100 nM TSA was added to the medium with the 5-aza at 500 nM for another 18 hours before the cell harvest. All cells were maintained in the cell culture medium, which was supplemented with 10U/mL penicillin G and 100 μg/mL streptomycin at 37° C in a humidified 5% CO2 atmosphere. FX and AST were dissolved in DMSO. DMSO was used as a vehicle in all experiments at a concentration of 0.1%.
Cell viability test – MTS assay
The JB6 P+ cells were seeded in a 96-well plate at a density of 5 × 103 cells/well for 24 hours and were treated with DMSO 0.1%, FX, or AST at various concentrations for one day, three days or five days. The cell culture medium was changed every other day. The HepG2-C8 cells were seeded in a 96-well plate at a density of 1 × 104 cells/well for 24 hours and then were treated with either DMSO 0.1% or various concentrations of FX for 24 hours in DMEM supplemented with 1% FBS. To determine cell viability, the CellTiter® 96 aqueous non-radioactive cell proliferation assay (Promega, Madison, WI, USA) was used according to the manufacturer’s instructions.
Luciferase reporter activity assay
Stably transfected HepG2-C8 cells were used to study the effects of FX on the Nrf2-ARE pathway. The HepG2-C8 cells were seeded at a density of 1×105 cells/well in 12-well plates for 24 hours. The cells were treated with vehicle or various concentrations of FX in DMEM supplemented with 1% FBS for 24 hours. ARE-luciferase activity was determined using the luciferase activity assay kit (Promega, Madison, WI, USA) according to the manufacturer’s instructions. The cells were lysed by the reporter lysis buffer in each well, and 10 μl of the cell lysate supernatant were analyzed for Nrf2-ARE activity using a Sirius luminometer (Berthold Detection System GmbH, Pforzheim, Germany). The results were normalized against the protein concentration in each sample as determined by a BCA protein assay (Pierce Biotech, Rockford, IL, USA). The results are expressed as an inducible fold change compared with the vehicle control (0.1% DMSO) from three independent replicates.
RNA extraction and quantitative real-time polymerase chain reaction
HepG2-C8 cells were seeded in 6-well plates at a density of 3 × 105 cells/well and then treated with vehicle or FX at various concentrations for another 6 hours. JB6 P+ cells were seeded in 6-well plates at a density of 1 × 104 cells/dish for 3 hours, 6 hours or 24 hours and then treated with the vehicle control, FX or AST at various concentrations for three days or as indicated in the figures. Total RNA was extracted using an RNeasy Micro Kit (Qiagen, Valencia, CA, USA), according to the manufacturer’s protocol. In total, 1 µg of RNA from each sample was used to synthesize the first-strand cDNA by the SuperScript III First-Strand cDNA Synthesis System (Invitrogen, Grand Island, NY, USA). The mRNA expression of Nrf2 and the Nrf2 downstream genes was determined using the Applied Biosystems 7900HT Fast Real-Time PCR System and compared with the vehicle control (0.1% DMSO). The primer pairs used were as previously described (25, 67).
Preparation of protein lysis and western blotting
The protein concentrations of the cleared lysates were determined using the BCA method (Pierce, Rockford, IL); 25 µg of the total protein was resolved by 4% to 15% SDS-PAGE (Bio-Rad, Hercules, CA). After electrophoresis, the proteins were electrotransferred to a polyvinylidene difluoride (PVDF) membrane (Millipore, Bedford, MA). The PVDF membrane was then blocked with 5% BSA in PBS-0.1% Tween 20 and then sequentially incubated with specific primary antibodies and horseradish peroxidase (HRP)-conjugated secondary antibodies. The blots were visualized by the Super Signal enhanced chemiluminescence detection system and documented using the Gel Documentation 2000 system (Bio-Rad, Hercules, CA). The anti-Nrf2, anti-NQO1, anti-HO1, anti-SOD, and anti-β-ACTIN antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Anchorage-independent cell neoplastic transformation assay
JB6 P+ cells were used in the TPA-induced neoplastic transformation assay as previously described (25, 68). Cells were transferred to 1 mL of Basal Medium Eagle medium with 0.33% agar over 3 mL of BME medium with 0.5% agar supplemented with 10% FBS in 6-well plates. These cells were treated with 0.1% DMSO, TPA (20 ng/mL) alone, or TPA in combination with FX or AST. The colonies formed from the transformed cells were photographed using a computerized microscope system with the Nikon ACT-1 program (Version 2.20, LEAD Technologies, Charlotte, NC) and counted using the ImageJ program (Version 1.40g, National Institutes of Health).
DNA extraction and pyrosequencing
Genomic DNA was isolated from the treated JB6 P+ cells using the QIAamp DNA Mini Kit (Qiagen, Valencia, CA). The bisulfite conversion of the genomic DNA was performed using the EZ DNA Methylation Gold Kit (Zymo Research Corp, Irvine, CA) according to the manufacturer’s instructions as previously described (69). The converted DNA was amplified by PCR using the Platinum PCR SuperMix (Invitrogen, Carlsbad, CA) with primers designed for the Nrf2 gene promoter region that spanned the methylated CpG sites from −1226 to −1069, with the translational start site (TSS) referenced as +1, as previously described (70). The sequences of the primers were 5’- AGTAGTAAAAATATTTTTTTAGTTGGAGGT-3’ (sense), 5’ ATATAATCTCATAAAACCCCACCTCT-3’ (antisense), and ATTTTTTTAGTTGGAGGTTATT (sequencing). The following PCR amplification conditions were used: 3 minutes at 94°C; 30 seconds at 94°C, 45 seconds at 70/55°C and 1 minute at 72°C for 15 cycles; 30 seconds at 94°C, 45 seconds at 60°C and 1 minute at 72°C for 25 cycles; and 5 minutes at 72°C for 1 cycle. The pyrosequencing was then carried out on the PCR products with a PyroMark Q 96 ID Sequencer from Qiagen (Valencia, CA, USA) using the pyrosequencing primers, enzymes and substrate (PyroMark Gold®Q96 Reagents kit, Qiagen, CA, USA) per the manufacturer’s protocol.
DNMT and HDAC activity assays
The DNMT activity and HDAC activity assays were conducted using a DNMT Activity/Inhibition Assay Kit (Epigentek, Farmingdale, NY) and an HDAC Activity/Inhibition Direct Assay Kit (Epigentek, Farmingdale, NY), respectively, following the manufacturer’s protocols. The nuclear protein fraction was extracted using the NEPER Nuclear and Cytoplasmic Protein Extraction Kit (Thermo Scientific, Pittsburgh, PA), and the relative DNMT activity was calculated based on the ratio of the treatment group to the control group after the normalization to the amount of protein.
Data presentation and statistical analysis
At least three independent experiments were performed for each analysis. The results are presented as the mean ± standard error of the mean (SEM) of each group unless otherwise specified. The differences between the controls and the treated groups were evaluated using Student’s t-tests. Significance includes *, P < 0.05; **, P < 0.01; #, P < 0.001.
RESULTS
Cytotoxicity of FX and AST in JB6 P+ cells
The cell viability of the JB6 P+ cells was measured by the MTS assay as shown in Figure 2. The JB6 P+ cells were treated with various concentrations of FX or AST for one day, three days or five days to examine their cytotoxicity. Treatment with AST or FX showed time- and dose-dependent effects on the cell viability (Figure 2). The cell viability of JB6 P+ cells that were treated with 50 µM of AST was 91.4% after the 5-day treatment, which was much higher than the 23.5% cell viability after the 5-day treatment with 50 µM of FX. The cell viabilities following the AST (< 50 µM) and the FX (< 12.5 µM) treatments were greater than 50% after the 1-day, three-day and five-day treatments. These doses were used in the subsequent studies.
Figure 2. Cell viability of the JB6 P+ cells after treatment with FX or AST.
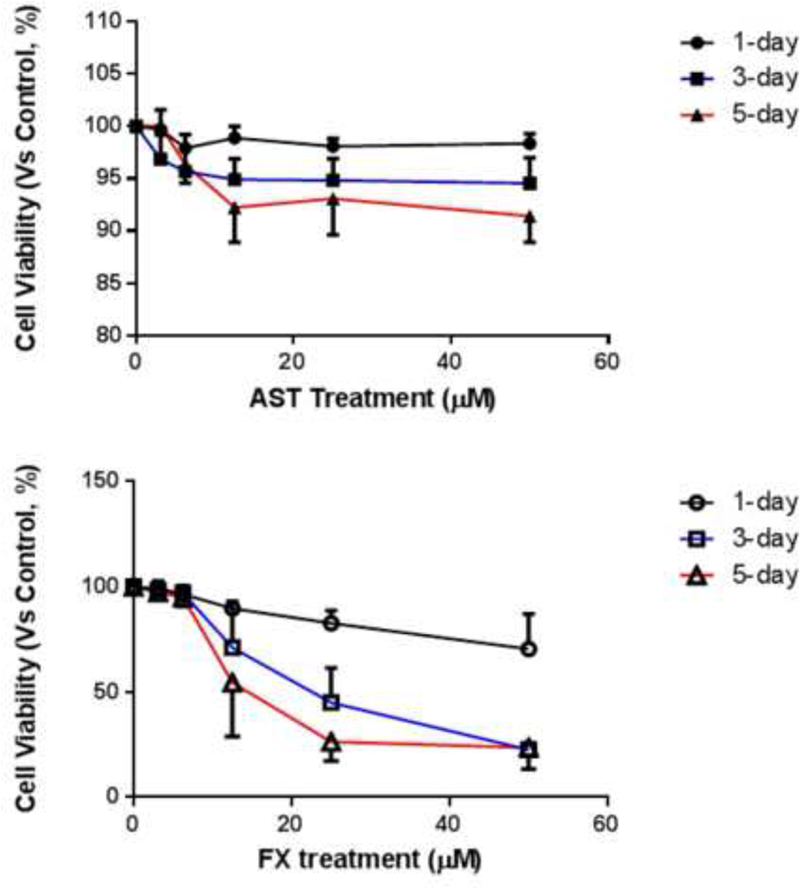
JB6 P+ cells were treated with various concentrations of FX or AST for one-, three- or five-days as described in Materials and Methods. Cell viability was determined by the MTS assay. The data are presented as the means ± SEM.
FX and AST inhibit the TPA-induced cell transformation of JB6 P+ cells
The effects of FX and AST treatment on the TPA-induced anchorage-independent growth of the JB6 P+ cells were examined in soft agar following a 14-day treatment (Figure 3). Compared with the TPA treatment alone, the FX and AST treatment, at various concentrations ranging from 6.25 µM to 25 µM, significantly decreased the number of JB6 P+ transformed cell colonies in Figure 3. Representative figures were selected from three independent replicates and showed that the number and size of the JB6 P+ transformed cell colonies are smaller than those in the group treated with TPA alone in Figure 3A. Both FX and AST, at the selected doses, inhibited the TPA-induced JB6 P+ cell transformation with a relatively low toxicity, suggesting their potential to protect JB6 P+ cells against TPA-induced carcinogenesis.
Figure 3. Inhibitory effects of AST and FX on the anchorage-independent colony formation of JB6 P+ cells.

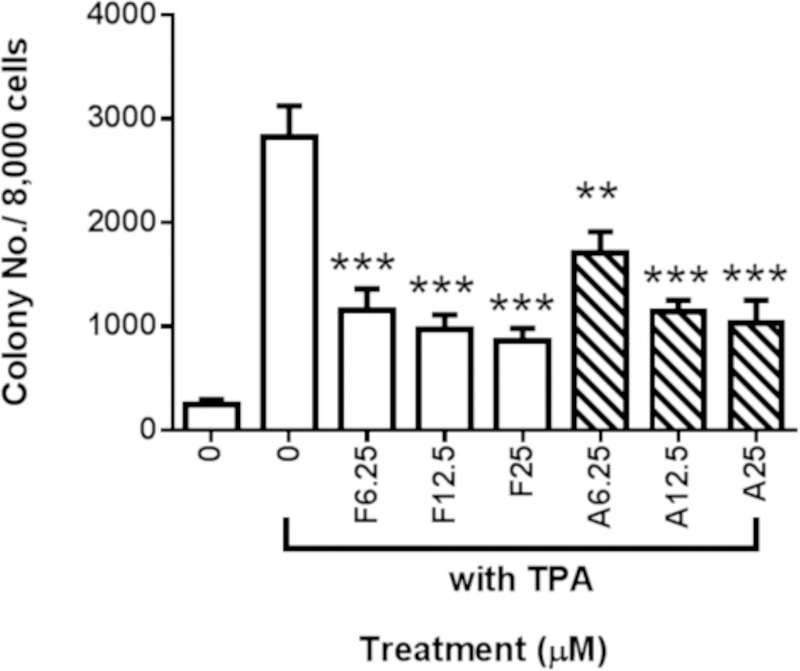
The colonies that exhibited anchorage-independent growth were counted under a microscope using ImageJ software. (A) Representative images under a microscope; (B) The graphical data are presented as the means ± SEM of three independent replicates. **, P < 0.01 and ***, P < 0.001, indicate significant differences between the treatment groups and TPA alone. Student’s t-test was used to calculate the significance of the differences.
FX induces ARE-luciferase reporter activity in HepG2-C8 cells
To investigate the mechanism underlying these effects, we used HepG2-C8 cells that were stably transfected with the ARE-luciferase reporter vector. We have previously reported that AST, in concentrations ranging from 12.5 µM to 100 µM, significantly induced luciferase activity with a relatively low toxicity (60). In this study, HepG2-C8 cells were treated with 100 µM of FX for 24 hours, and the cell viability was approximately 70% compared with that of the vehicle control, which was treated with 0.1% DMSO (Figure 4A). By comparing the cell viability of the FX-treated HepG2 cells with that of the JB6 P+ cells, it seems that the HepG2 cells are more resistant to the treatment toxicity of FX. FX significantly induced luciferase activity in a dose-dependent manner at concentrations ranging from 25 µM to 100 µM (Figure 4B). However, 3.13 µM to 12.5 µM of FX did not significantly induce luciferase activity.
Figure 4. Cell viability and induction of ARE-luciferase activity by FX treatment in HepG2-C8 cells transfected with the ARE-luciferase vector.
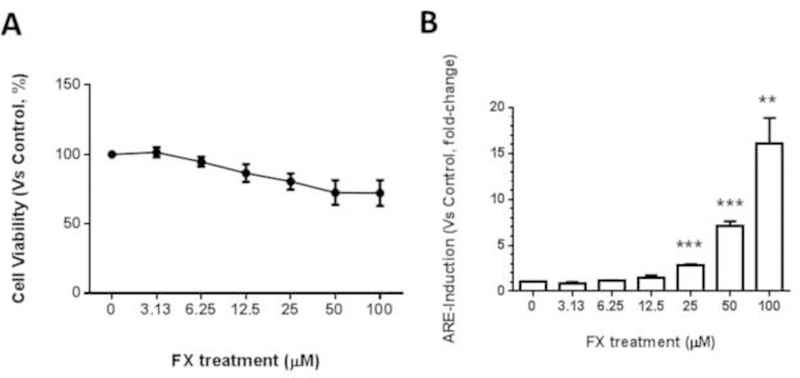
(A) Cell viability of the JB6 P+ cells after treatment with FX. Cells were treated with various concentrations of FX ranging from 3.13 µM to 100 µM for one day. Cell viability was determined by the MTS assay. The data are presented as the means ± SEM. (B) The luciferase activity was normalized based on the protein concentrations in the BCA protein assay. The data are presented as the means ± SEM of three independent experiments. **, P < 0.01 and ***, P < 0.001 indicate significant differences between the treatment groups and the control group (DMSO 0.1% without FX). Student’s t-test was used to calculate the significance of the differences compared with the control.
FX increases the mRNA and protein expression levels of Nrf2 and downstream genes in HepG2-C8 cells
FX treatment increased the mRNA expression of Nrf2 (NFE2L2) at 50 µM significantly and SOD at dose-dependent manner in the HepG2-C8 cells. The mRNA expression levels of HO-1 and NQO1 were increased by FX treatment in a dose-dependent manner, but the increases were not significant (Figure 5A). The protein levels of Nrf2, NQO1, HO-1 and SOD were further evaluated in HepG2-C8 cells with various concentrations of FX by western blotting, as shown in Figure 5B and 5C. FX, in concentrations ranging from 3.13 µM to 50 µM, increased the protein expression levels of Nrf2, NQO1, HO-1 and SOD in a concentration-dependent manner. These results suggest that FX has the potential to activate the Nrf2 pathway and, thus, antioxidant and detoxifying enzymes in HepG2-C8 cells.
Figure 5. Effects of FX on the relative endogenous mRNA and protein expression of Nrf2 and downstream genes in HepG2-C8 cells.
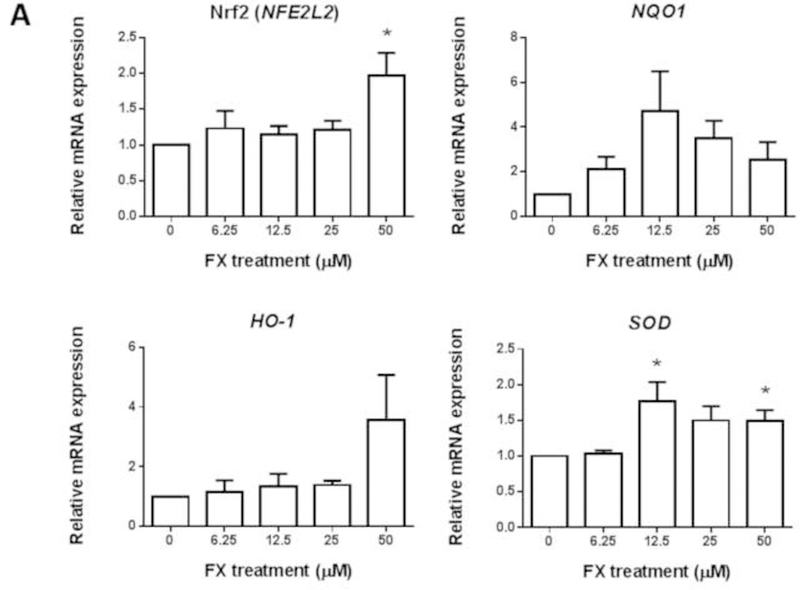
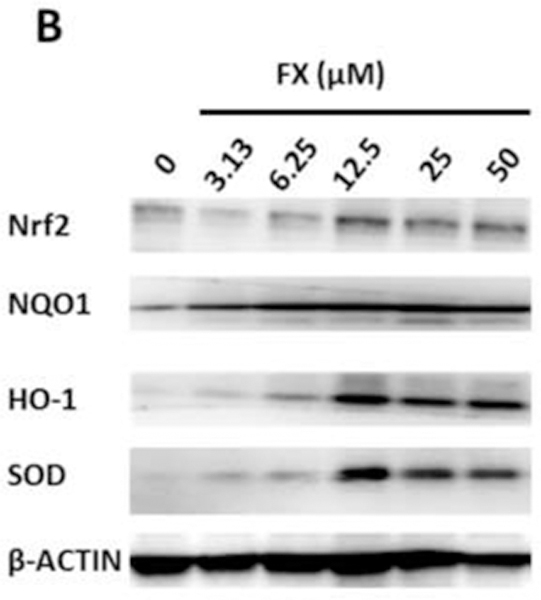
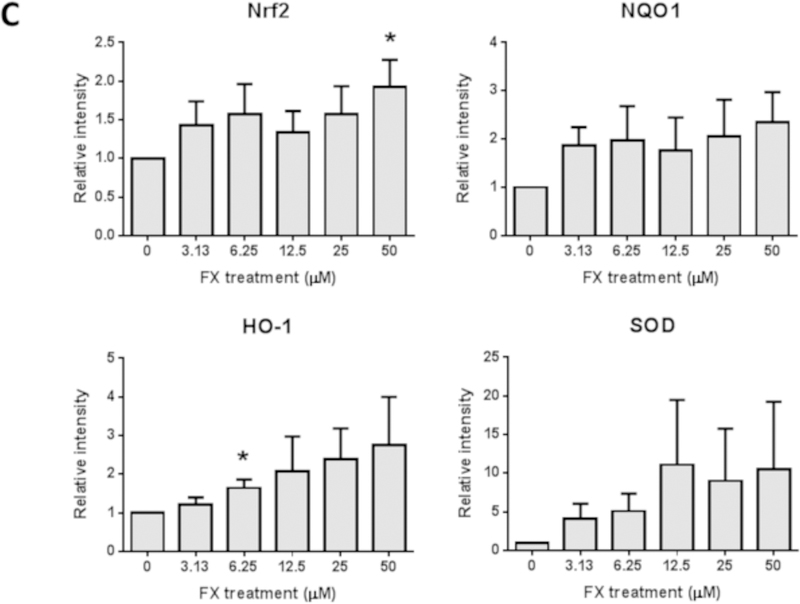
(A) mRNA expression of Nrf2 (NFE2L2), NQO1, HO-1, and SOD. RNA was extracted from HepG2-C8 cells treated with various concentrations of FX for one day. The data are expressed as the means ± SEM of three independent replicates. β-Actin was used as an endogenous housekeeping gene. (B) Protein expression of Nrf2 (NFE2L2), NQO1, HO-1, and SOD. β-ACTIN was used as the housekeeping protein. (C) Quantification by densitometry from at least three independent experiments. The results are presented as the means ± SEM. Student’s t-test was used to calculate the significance of the differences compared with the control*, P < 0.05.
FX increases the mRNA expression levels of Nrf2 and Nqo1
JB6 P+ cells were treated with FX or AST for 3 hours, 6 hours or 24 hours to investigate the dose-response and time course of the mRNA expression of Nrf2 (or Nfe2l2), Nqo1 (Figure 6A) and Dnmts (Figure 6B). As shown in Figure 6A, 24-hour treatment with FX significantly increased the mRNA expression of Nrf2 by 2.8-fold at 12.5 µM (P = 0.012) and 2.0-fold at 25 µM (P = 0.047). Additionally, 25 µM of FX significantly induced the mRNA expression of Nqo1 at all measured time points. Compared with FX, AST significantly increased Nrf2 mRNA expression from 6 hours and only decreased its expression at 25 µM with the 24-hour treatment. Regarding Nqo1, there was no significant change, but AST reduced the mRNA expression of Nqo1 by 0.82-fold at 6.25 µM (P = 0.04). As shown in Figure 6B, FX, at concentrations ranging from 6.25 µM to 25 µM, significantly reduced the mRNA expression of Dnmt1 after 24 hours of treatment. The protein expression of DNMT1 was significantly reduced by the treatment of FX but not AST (Figure 6C). However, 12.5 µM and 25 µM of FX induced the mRNA expression of Dnmt3b after the 24-hour and 3-hour treatments, respectively. AST reduced the mRNA expression of Dnmt3a by 0.81-fold after the 3-hour treatment (P = 0.015) and 0.71-fold after the 24-hour treatment (P = 0.015). However, AST significantly increased the mRNA expression of Dnmt3b after the 6-hour treatment and decreased its expression at 6.25 µM after the 24-hour treatment, but no significant changes occurred with the other concentrations.
Figure 6. Effects of FX and AST on the relative endogenous mRNA expression of Nrf2, Nqo1, and Dnmts and protein expression of Nrf2 and DNMT1 in JB6 P+ cells.
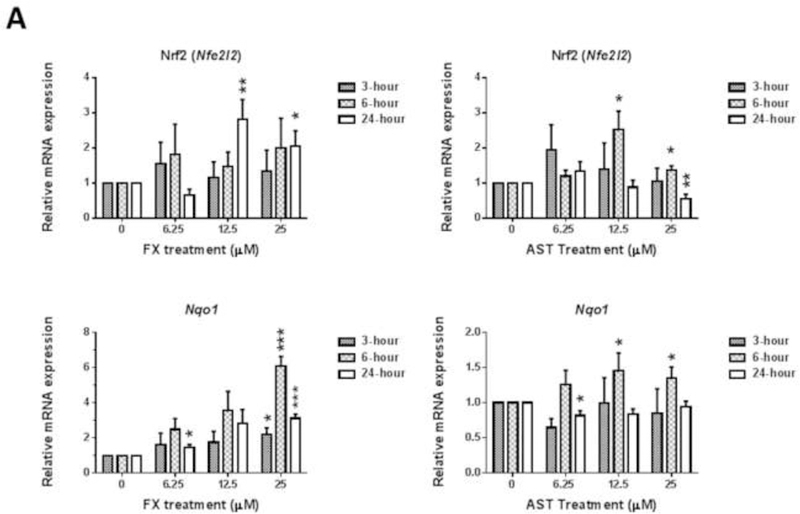
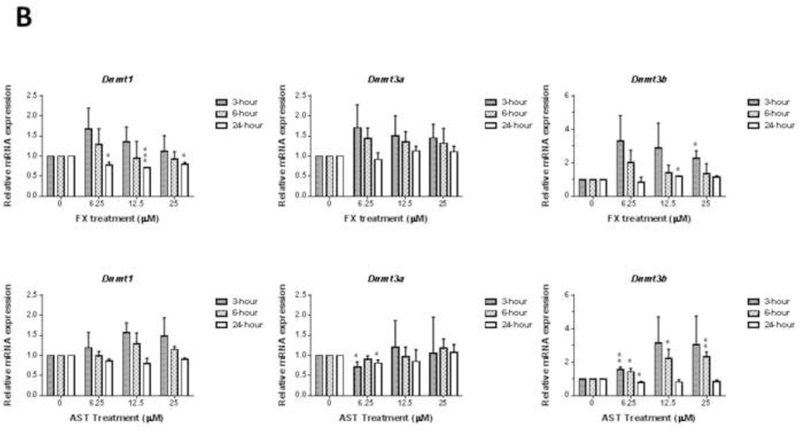
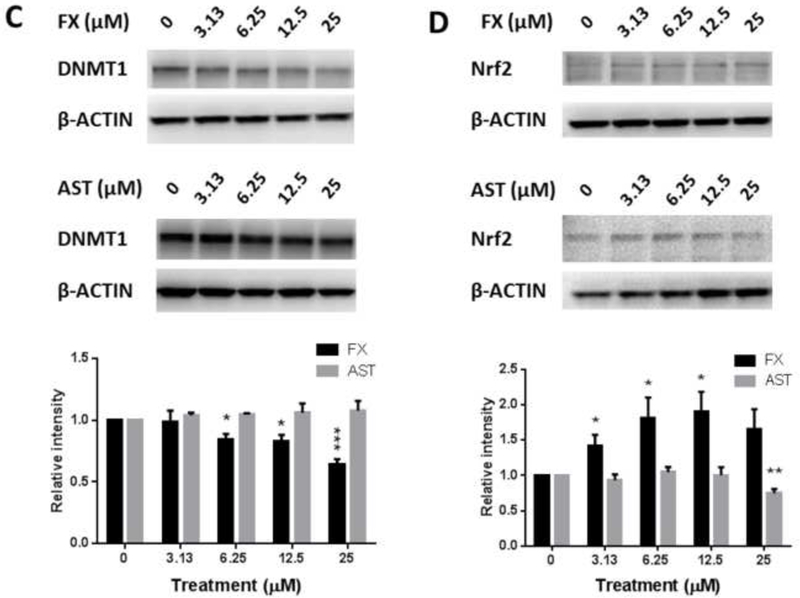
mRNA expression of Nrf2 (Nfe2l2), Nqo1(A), and Dnmts (B). RNA was extracted from JB6 P+ cells treated with various concentrations of FX or AST for 3 hours, 6 hours or 24 hours. β-Actin was used as an endogenous housekeeping gene. Protein expression of Nrf2 (C) and DNMT1 (D) after 24 hours treatment of FX or AST. β-ACTIN was used as the housekeeping protein. The data are expressed as the means ± SEM of at least three independent replicates. Student’s t-test was used to calculate the significance of the differences compared with the control, *, P < 0.05, **, P < 0.01, and ***, P < 0.001.
Knockdown of Nrf2 in TPA-induced JB6 P+ cell transformation in the treatment of FX and AST
The efficiency of shMock and shNrf2 in JB6 P+ cells was examined and the protein expression of Nrf2 was significantly reduced in shNrf2 JB6 P+ cells in Figure 7A (P < 0.01). Treatment of FX from 6.25 µM to 25 µM suppressed the colony number significantly in both shMock and shNrf2 JB6 P+ cells. Compared to FX, the treatment of AST at 25 µM significantly inhibited the TPA-induced anchorage-independent growth of the shMock JB6 P+ cells but not in the shNrf2 JB6 P+ cells. The knockdown of Nrf2 in shNrf2 JB6 P+ cells showed significantly decreased resistance of the cells to TPA-induced cell transformation with the treatment of AST at 25 µM but not FX (Figure 7B&C). The protective role of AST at 25 µM in shNrf2 JB6 P+ cells was significantly reduced compared to shMock JB6 P+ cells. These results suggest preventive effects of AST and FX in TPA-induced JB6 P+ cell transformation are not completely dependent on the Nrf2 on TPA-induced JB6 P+ cell transformation.
Figure 7. Inhibitory effects of AST and FX on the anchorage-independent colony formation of shNrf2 and shMock transfected JB6 P+ cells.
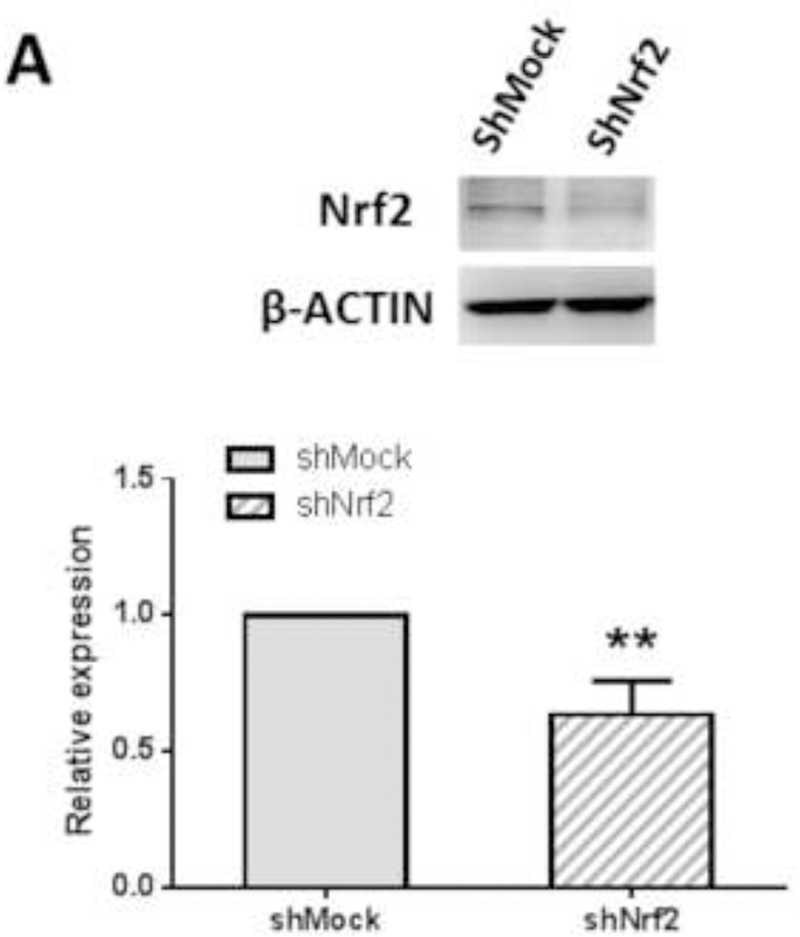

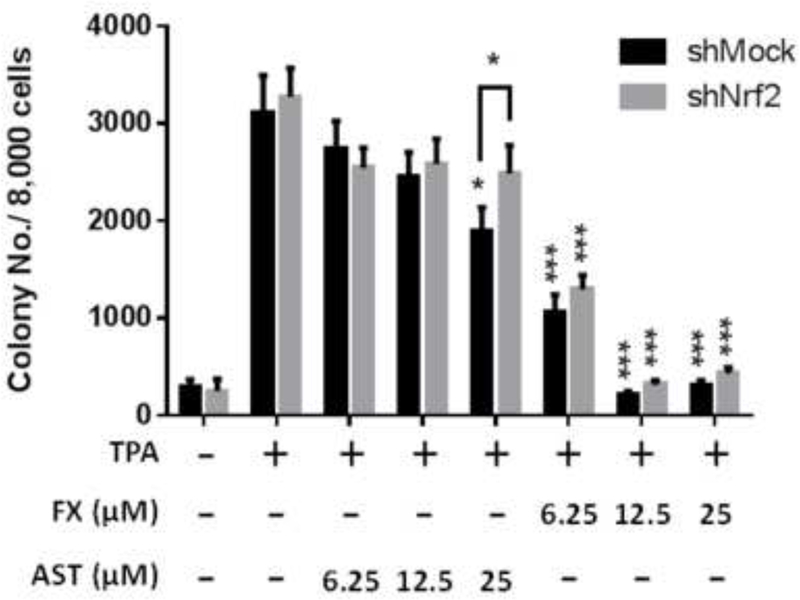
(A) Reduced Nrf2 protein expression in shNrf2 JB6 P+ cells with representative figure and quantification from three independent replicates. The colonies that exhibited anchorage-independent growth were counted under a microscope and further analyzed using ImageJ software. (B) Representative images under a microscope; (C) The graphical data are presented as the means ± SEM of three independent replicates. **, P < 0.01 and ***, P < 0.001, indicate significant differences between the treatment groups and TPA alone. Student’s t-test was used to calculate the significance of the differences.
FX decreased the methylated CpG ratio in the Nrf2 gene promoter region by pyrosequencing
Furthermore, pyrosequencing was performed to determine the methylation status of the Nrf2 promoter region (Figure 8). Hypermethylation in the first five CpG sites was observed, and, on average, 90.1% of these five CpG sites were in the Nrf2 promoter region, which is consistent with previous reports. We previously reported that 5-aza, a DNMT inhibitor, and TSA, an HDAC inhibitor, in combination decreased the CpG methylation levels in the Nrf2 gene promoter region in murine prostate cancer TRAMP C1 cells and human prostate cancer LNCaP cells (69, 70).
Figure 8. Effects of FX and AST on methylation alteration in the Nrf2 promoter regions in JB6 P+ cells by pyrosequencing.
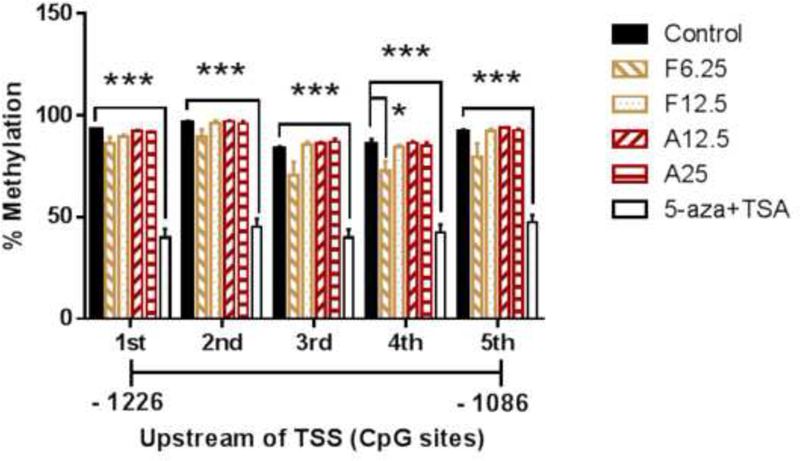
Cells were treated with various concentrations of FX, AST or 5-aza for three days. The medium was changed every two days. 100 nM TSA was added to the medium with the 5-aza at 500 nM for another 18 hours before the cell harvest. The methylation patterns of the first five CpG sites located at positions –1226 to –1086 from the TSS (defined as position 1) in the Nrf2 promoter were determined by pyrosequencing as described in the Materials and Methods section. (A) Methylation of the first five CpG sites from the Nrf2 promoter region. The data are presented as the means ± SEM of at least four independent replicates. *, P < 0.05 and ***, P < 0.001 indicate significant differences between the treatment groups and the control group. Student’s t-test was used to calculate the significance of the differences compared with the control.
HDAC and DNMT activity assays
AST (25 µM) significantly reduced the DNMT activity in the JB6 P+ cells after a three-day treatment (Figure 9A); however, the 3-day AST treatment did not have a significant effect on the HDAC activity (Figure 9B). Compared with AST, FX at a low dose of 6.25 µM significantly reduced the DNMT activity by 67.5% (P = 0.0051), but there was no significant change in the HDAC activity.
Figure 9. Effects of FX and AST on the activities of DNMTs and HDACs.
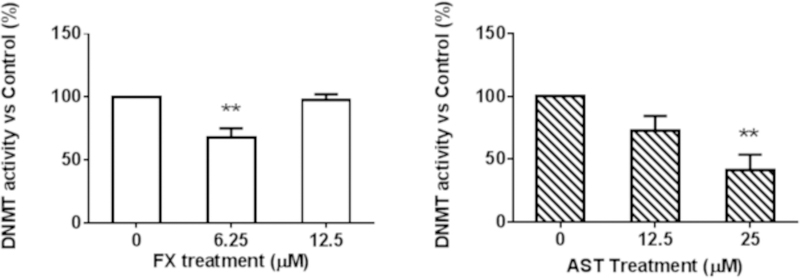
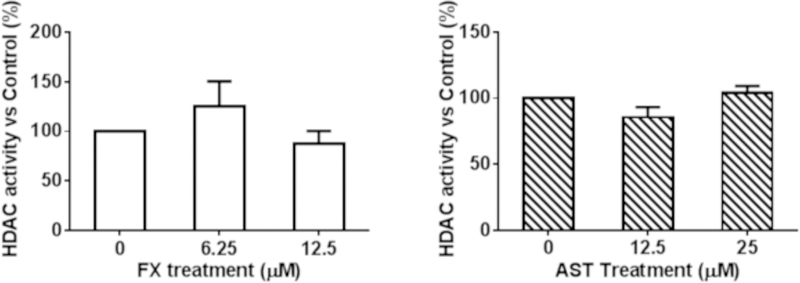
JB6 P+ cells were treated with various concentrations of FX and AST for three days. The relative DNMT and HDAC activities were normalized to the protein amount of each group and were calculated based on the ratio of the FX and AST treatment groups to the control group following the manufacturer’s protocol. The results of (A) DNMT activity and (B) HDAC activity are presented as the means ± SEM. Student’s t-test was used to calculate the significance of the differences compared with the control. **, P < 0.01.
DISCUSSION
Oxidative stress occurs through free radicals that oxidize nucleic acids, proteins, and lipids, causing a gradual deterioration and leading to degenerative diseases, such as cancer and heart disease (71). The main characteristic of an antioxidant is its ability to induce enzymes that trap free radicals (72). Although the biochemical process by which chemoprotective agents protect against cancers that are caused by oxidative stress has not been fully discovered, phase II detoxifying/antioxidant enzymes clearly play a role in fighting oxidative stress and preventing cancers (30). These enzymes detoxify harmful substances by converting them into hydrophilic metabolites that can then be excreted from the human body. Studies have shown that the higher the presence of the phase II detoxifying/antioxidant enzymes, such as NAD(P)H, NQO1, GSTM2, HO-1 and SOD, in the tissue, the less susceptible the tissue is to cancers. Nrf2 and its interaction with AREs increase the transcription of these phase II detoxifying/antioxidant enzymes (30). Our present study shows that FX increased the ARE-luciferase activity in HepG2-C8 cells transfected with the ARE-luciferase vector (Figure 4) and increased the mRNA and protein expression of Nrf2, NQO1, HO-1 and SOD in HepG2-C8 cells. In addition, FX increased the mRNA expression and proteins expression of Nrf2 after 24 hours treatment in JB6 P+ cells (Figure 6). However, the FX-mediate suppression of TPA-induce transformation is not completely dependent on Nrf2, because there was no significant difference between the shMock and shNrf2 JB6 P+ cells. It’s possible that the FX-mediated inhibition in TPA-induced transformation is through other mechanisms including P53-mediated apoptosis and disruption of Nrf2-Keap1 signaling pathway and Nrf2-downstream target genes.
Another possible mechanism by which FX may prevent oxidative stress and, ultimately, cancer is through epigenetic alterations to Nrf2 DNA methylation, which cause changes in gene expression without altering the DNA composition itself. Epigenetic regulation, including DNA methylation and histone modification, is an important process in cancer development. The DNMT enzymes DNMT1, 3a and 3b are directly involved in transferring methyl groups to DNA cytosine. In this study, we observed that both FX and AST inhibited the activity of DNMTs, but there was no significant difference in the activity of HDACs (Figure 8). We found that FX decreased the mRNA and protein expression of DNMT1 significantly, which may be one of the underlying mechanism for the FX-induced demethylation of Nrf2. For the Nrf2 expression, although there was no significant difference on Nrf2 mRNA expression with the treatment of FX at 6.25 µM (Figure 6A), the protein expression of Nrf2 was induced by FX at 6.25 µM significantly (Figure 6D). In addition, DNMT activity can be modulated by other molecular interactions, post-translational modifications and alternative splicing as summarized in review (6). It is possible that FX at higher concentrations may interact with other mechanisms to increase the activity of DNMTs (Figure 9A) to compensate the reduced DNMT1 protein expression (Figure 6C). In our previous study, the epigenetic reactivation of the Nrf2 pathway appeared to be an important mechanism for the inhibition of TPA-induced transformation of JB6 P+ cells (25). We observed that FX significantly reduced the methylation of the first CpG sites in the Nrf2 promoter region, particularly the fourth CpG site (Figure 8), and expression of DNMT1 (Figure 6C). Compared with FX, AST did not change the methylation of the CpG sites in the Nrf2 promoter region, but 5-aza+TSA, the positive control, significantly reduced all five CpG sites (Figure 8).These data suggest that, although AST and FX share similar structures, their underlying mechanisms of Nrf2 activation differ in their inhibition of the TPA-induced transformation of JB6 P+ cells. Further studies will be necessary to elucidate the effects of FX and AST on the expression and methylation other genes involved in the Nrf2-keap1 signaling pathway.
FX and AST are both novel marine carotenoids with antioxidant properties that reduce oxidative stress markers. The protective effects of FX and AST have recently been identified, including the effects of FX against UVB-induced photoaging and sunburn on skin (45, 46) and the effects of AST against UVB-DMBA-induced skin cancer in rats (59). In a recent clinical report, AST helped reduce oxidative stress and free radical production in athletes and healthy subjects during physical exercise [31]. The health-promoting effects of FX and AST make them highly valuable carotenoids and potential dietary/topical treatments for various diseases; however, their effects and mechanisms in skin cancer prevention remain to be further elucidated.
CONCLUSION
In conclusion, our present study shows that FX activates the Nrf2 pathway, thereby contributing to the inhibition of the TPA-induced transformation of JB6 P+ cells. Furthermore, 6.25 µM of FX significantly decreased the DNMT activity, mRNA expression of Dnmt1 after a 24-hour treatment, and methylation of the Nrf2 methylation region. FX and AST activate the Nrf2 pathway, showing a potentially protective role in skin cancer prevention.
ACKNOWLEDGEMENTS
We thank all members of Dr. Ah-Ng Kong’s lab for helpful discussions and preparation of this manuscript. This study was supported in part by R01 CA200129, R01-CA118947, and R01-CA152826 from the National Cancer Institute (NCI), R01 AT009152 from the National Center for Complementary and Integrative Health (NCCIH), and institutional funds awarded to Dr. Ah-Ng Kong.
ABBREVIATIONS
- ARE
anti-oxidant response element
- AST
astaxanthin
- 5-aza
5-aza-deoxycytidine
- DNMT
DNA methyltransferase
- DNMT1
DNA methyltransferase 1
- DNMT3a
DNA methyltransferase 3a
- DNMT3b
DNA methyltransferase 3b
- DMSO
dimethyl sulfoxide
- FBS
fetal bovine serum
- FX
Fucoxanthin
- GCLC
Glutamate-Cysteine Ligase, Catalytic Subunit
- GSH
glutathione
- GSS
Glutathione synthetase
- GST
glutathione-S-transferases
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- HO-1
heme oxygenase-1
- HDAC
histone deacetylases
- NQO1
NAD(P)H: quinone oxidoreductase 1
- Nrf2
nuclear factor (erythroid-derived 2)-like 2, or NFE2L2
- qRT–PCR
quantitative reverse-transcriptase polymerase chain reaction
- ROS
Reactive oxygen species
- SEM
standard error of the mean
Footnotes
CONFLICT OF INTEREST
The authors declare that there are no conflicts of interest
REFERENCES
- 1.Alam M, Ratner D. Cutaneous squamous-cell carcinoma. The New England journal of medicine 2001;344(13):975–83. [DOI] [PubMed] [Google Scholar]
- 2.McGuire S World Cancer Report 2014. Geneva, Switzerland: World Health Organization, International Agency for Research on Cancer, WHO Press, 2015. Advances in nutrition 2016;7(2):418–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Griffin LL, Ali FR, Lear JT. Non-melanoma skin cancer. Clinical medicine 2016;16(1):62–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh RP, Agarwal R. Mechanisms and preclinical efficacy of silibinin in preventing skin cancer. European journal of cancer 2005;41(13):1969–79. [DOI] [PubMed] [Google Scholar]
- 5.Xu C, Huang MT, Shen G, Yuan X, Lin W, Khor TO, et al. Inhibition of 7,12-dimethylbenz(a)anthracene-induced skin tumorigenesis in C57BL/6 mice by sulforaphane is mediated by nuclear factor E2-related factor 2. Cancer Res 2006;66(16):8293–6. [DOI] [PubMed] [Google Scholar]
- 6.Huang MT, Xie JG, Wang ZY, Ho CT, Lou YR, Wang CX, et al. Effects of tea, decaffeinated tea, and caffeine on UVB light-induced complete carcinogenesis in SKH-1 mice: demonstration of caffeine as a biologically important constituent of tea. Cancer research 1997;57(13):2623–9. [PubMed] [Google Scholar]
- 7.Huang MT, Smart RC, Wong CQ, Conney AH. Inhibitory effect of curcumin, chlorogenic acid, caffeic acid, and ferulic acid on tumor promotion in mouse skin by 12-O-tetradecanoylphorbol-13-acetate. Cancer research 1988;48(21):5941–6. [PubMed] [Google Scholar]
- 8.Conney AH, Wang ZY, Huang MT, Ho CT, Yang CS. Inhibitory effect of green tea on tumorigenesis by chemicals and ultraviolet light. Preventive medicine 1992;21(3):361–9. [DOI] [PubMed] [Google Scholar]
- 9.Huang MT, Ho CT, Wang ZY, Ferraro T, Lou YR, Stauber K, et al. Inhibition of skin tumorigenesis by rosemary and its constituents carnosol and ursolic acid. Cancer Res 1994;54(3):701–8. [PubMed] [Google Scholar]
- 10.Talalay P Chemoprotection against cancer by induction of phase 2 enzymes. BioFactors 2000;12(1–4):5–11. [DOI] [PubMed] [Google Scholar]
- 11.Dinkova-Kostova AT, Jenkins SN, Fahey JW, Ye L, Wehage SL, Liby KT, et al. Protection against UV-light-induced skin carcinogenesis in SKH-1 high-risk mice by sulforaphane-containing broccoli sprout extracts. Cancer letters 2006;240(2):243–52. [DOI] [PubMed] [Google Scholar]
- 12.Sigalotti L, Fratta E, Coral S, Cortini E, Covre A, Nicolay HJ, et al. Epigenetic drugs as pleiotropic agents in cancer treatment: biomolecular aspects and clinical applications. Journal of cellular physiology 2007;212(2):330–44. [DOI] [PubMed] [Google Scholar]
- 13.Golabek K, Strzelczyk JK, Wiczkowski A, Michalski M. Potential use of histone deacetylase inhibitors in cancer therapy. Contemp Oncol (Pozn) 2015;19(6):436–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Doorn R, Gruis NA, Willemze R, van der Velden PA, Tensen CP. Aberrant DNA methylation in cutaneous malignancies. Semin Oncol 2005;32(5):479–87. [DOI] [PubMed] [Google Scholar]
- 15.Bachman AN, Curtin GM, Doolittle DJ, Goodman JI. Altered methylation in gene-specific and GC-rich regions of DNA is progressive and nonrandom during promotion of skin tumorigenesis. Toxicol Sci 2006;91(2):406–18. [DOI] [PubMed] [Google Scholar]
- 16.Schinke C, Mo Y, Yu Y, Amiri K, Sosman J, Greally J, et al. Aberrant DNA methylation in malignant melanoma. Melanoma Res 2010;20(4):253–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang AY, Lee JH, Shu L, Zhang C, Su ZY, Lu Y, et al. Genome-wide analysis of DNA methylation in UVB- and DMBA/TPA-induced mouse skin cancer models. Life Sci 2014;113(1–2):45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang CS, Fang M, Lambert JD, Yan P, Huang TH. Reversal of hypermethylation and reactivation of genes by dietary polyphenolic compounds. Nutrition reviews 2008;66 Suppl 1:S18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang AY, Kim H, Li W, Kong AN. Natural compound-derived epigenetic regulators targeting epigenetic readers, writers and erasers. Curr Top Med Chem 2016;16(7):697–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang LG, Beklemisheva A, Liu XM, Ferrari AC, Feng J, Chiao JW. Dual action on promoter demethylation and chromatin by an isothiocyanate restored GSTP1 silenced in prostate cancer. Molecular carcinogenesis 2007;46(1):24–31. [DOI] [PubMed] [Google Scholar]
- 21.Wang LG, Liu XM, Fang Y, Dai W, Chiao FB, Puccio GM, et al. De-repression of the p21 promoter in prostate cancer cells by an isothiocyanate via inhibition of HDACs and c-Myc. International journal of oncology 2008;33(2):375–80. [PubMed] [Google Scholar]
- 22.Nian H, Delage B, Ho E, Dashwood RH. Modulation of histone deacetylase activity by dietary isothiocyanates and allyl sulfides: studies with sulforaphane and garlic organosulfur compounds. Environmental and molecular mutagenesis 2009;50(3):213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Myzak MC, Tong P, Dashwood WM, Dashwood RH, Ho E. Sulforaphane retards the growth of human PC-3 xenografts and inhibits HDAC activity in human subjects. Experimental biology and medicine 2007;232(2):227–34. [PMC free article] [PubMed] [Google Scholar]
- 24.Meeran SM, Patel SN, Tollefsbol TO. Sulforaphane causes epigenetic repression of hTERT expression in human breast cancer cell lines. PloS one 2010;5(7):e11457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Su ZY, Zhang C, Lee JH, Shu L, Wu TY, Khor TO, et al. Requirement and epigenetics reprogramming of Nrf2 in suppression of tumor promoter TPA-induced mouse skin cell transformation by sulforaphane. Cancer Prev Res (Phila) 2014;7(3):319–29. [DOI] [PubMed] [Google Scholar]
- 26.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet 2001;357(9255):539–45. [DOI] [PubMed] [Google Scholar]
- 27.Lund AW, Medler TR, Leachman SA, Coussens LM. Lymphatic Vessels, Inflammation, and Immunity in Skin Cancer. Cancer discovery 2016;6(1):22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh R, Akhtar N, Haqqi TM. Green tea polyphenol epigallocatechin-3-gallate: inflammation and arthritis. [corrected]. Life sciences 2010;86(25–26):907–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh S, Vrishni S, Singh BK, Rahman I, Kakkar P. Nrf2-ARE stress response mechanism: a control point in oxidative stress-mediated dysfunctions and chronic inflammatory diseases. Free Radic Res 2010;44(11):1267–88. [DOI] [PubMed] [Google Scholar]
- 30.Hu R, Saw CL, Yu R, Kong AN. Regulation of NF-E2-related factor 2 signaling for cancer chemoprevention: antioxidant coupled with antiinflammatory. Antioxid Redox Signal 2010;13(11):1679–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kong AN, Yu R, Lei W, Mandlekar S, Tan TH, Ucker DS. Differential activation of MAPK and ICE/Ced-3 protease in chemical-induced apoptosis. The role of oxidative stress in the regulation of mitogen-activated protein kinases (MAPKs) leading to gene expression and survival or activation of caspases leading to apoptosis. Restorative neurology and neuroscience 1998;12(2–3):63–70. [PubMed] [Google Scholar]
- 32.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 1999;13(1):76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee JH, Khor TO, Shu L, Su ZY, Fuentes F, Kong AN. Dietary phytochemicals and cancer prevention: Nrf2 signaling, epigenetics, and cell death mechanisms in blocking cancer initiation and progression. Pharmacol Ther 2013;137(2):153–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suzuki T, Motohashi H, Yamamoto M. Toward clinical application of the Keap1-Nrf2 pathway. Trends in pharmacological sciences 2013;34(6):340–6. [DOI] [PubMed] [Google Scholar]
- 35.Khor TO, Huang MT, Prawan A, Liu Y, Hao X, Yu S, et al. Increased susceptibility of Nrf2 knockout mice to colitis-associated colorectal cancer. Cancer Prev Res (Phila) 2008;1(3):187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saw CL, Yang AY, Huang MT, Liu Y, Lee JH, Khor TO, et al. Nrf2 null enhances UVB-induced skin inflammation and extracellular matrix damages. Cell Biosci 2014;4:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saw CL, Huang MT, Liu Y, Khor TO, Conney AH, Kong AN. Impact of Nrf2 on UVB-induced skin inflammation/photoprotection and photoprotective effect of sulforaphane. Mol Carcinog 2011;50(6):479–86. [DOI] [PubMed] [Google Scholar]
- 38.Gills JJ, Jeffery EH, Matusheski NV, Moon RC, Lantvit DD, Pezzuto JM. Sulforaphane prevents mouse skin tumorigenesis during the stage of promotion. Cancer letters 2006;236(1):72–9. [DOI] [PubMed] [Google Scholar]
- 39.Hu C, Eggler AL, Mesecar AD, van Breemen RB. Modification of keap1 cysteine residues by sulforaphane. Chem Res Toxicol 2011;24(4):515–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quitain AT, Kai T, Sasaki M, Goto M. Supercritical carbon dioxide extraction of fucoxanthin from Undaria pinnatifida. J Agric Food Chem 2013;61(24):5792–7. [DOI] [PubMed] [Google Scholar]
- 41.Gammone MA, D’Orazio N. Anti-obesity activity of the marine carotenoid fucoxanthin. Marine drugs 2015;13(4):2196–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muradian K, Vaiserman A, Min KJ, Fraifeld VE. Fucoxanthin and lipid metabolism: A minireview. Nutr Metab Cardiovasc Dis 2015;25(10):891–7. [DOI] [PubMed] [Google Scholar]
- 43.Zheng J, Piao MJ, Keum YS, Kim HS, Hyun JW. Fucoxanthin Protects Cultured Human Keratinocytes against Oxidative Stress by Blocking Free Radicals and Inhibiting Apoptosis. Biomol Ther (Seoul) 2013;21(4):270–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zheng J, Piao MJ, Kim KC, Yao CW, Cha JW, Hyun JW. Fucoxanthin enhances the level of reduced glutathione via the Nrf2-mediated pathway in human keratinocytes. Mar Drugs 2014;12(7):4214–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matsui M, Tanaka K, Higashiguchi N, Okawa H, Yamada Y, Tanaka K, et al. Protective and therapeutic effects of fucoxanthin against sunburn caused by UV irradiation. J Pharmacol Sci 2016;132(1):55–64. [DOI] [PubMed] [Google Scholar]
- 46.Urikura I, Sugawara T, Hirata T. Protective effect of Fucoxanthin against UVB-induced skin photoaging in hairless mice. Biosci Biotechnol Biochem 2011;75(4):757–60. [DOI] [PubMed] [Google Scholar]
- 47.Aoi W, Naito Y, Sakuma K, Kuchide M, Tokuda H, Maoka T, et al. Astaxanthin limits exercise-induced skeletal and cardiac muscle damage in mice. Antioxidants & redox signaling 2003;5(1):139–44. [DOI] [PubMed] [Google Scholar]
- 48.Iwamoto T, Hosoda K, Hirano R, Kurata H, Matsumoto A, Miki W, et al. Inhibition of low-density lipoprotein oxidation by astaxanthin. Journal of atherosclerosis and thrombosis 2000;7(4):216–22. [DOI] [PubMed] [Google Scholar]
- 49.Kobayashi M In vivo antioxidant role of astaxanthin under oxidative stress in the green alga Haematococcus pluvialis. Applied microbiology and biotechnology 2000;54(4):550–5. [DOI] [PubMed] [Google Scholar]
- 50.Peng J, Yuan JP, Wang JH. Effect of diets supplemented with different sources of astaxanthin on the gonad of the sea urchin Anthocidaris crassispina. Nutrients 2012;4(8):922–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yasui Y, Hosokawa M, Mikami N, Miyashita K, Tanaka T. Dietary astaxanthin inhibits colitis and colitis-associated colon carcinogenesis in mice via modulation of the inflammatory cytokines. Chemico-biological interactions 2011;193(1):79–87. [DOI] [PubMed] [Google Scholar]
- 52.Nagendraprabhu P, Sudhandiran G. Astaxanthin inhibits tumor invasion by decreasing extracellular matrix production and induces apoptosis in experimental rat colon carcinogenesis by modulating the expressions of ERK-2, NFkB and COX-2. Investigational new drugs 2011;29(2):207–24. [DOI] [PubMed] [Google Scholar]
- 53.Jyonouchi H, Sun S, Iijima K, Gross MD. Antitumor activity of astaxanthin and its mode of action. Nutrition and cancer 2000;36(1):59–65. [DOI] [PubMed] [Google Scholar]
- 54.Kang JO, Kim SJ, Kim H. Effect of astaxanthin on the hepatotoxicity, lipid peroxidation and antioxidative enzymes in the liver of CCl4-treated rats. Methods and findings in experimental and clinical pharmacology 2001;23(2):79–84. [DOI] [PubMed] [Google Scholar]
- 55.Uchiyama K, Naito Y, Hasegawa G, Nakamura N, Takahashi J, Yoshikawa T. Astaxanthin protects beta-cells against glucose toxicity in diabetic db/db mice. Redox report : communications in free radical research 2002;7(5):290–3. [DOI] [PubMed] [Google Scholar]
- 56.Ohgami K, Shiratori K, Kotake S, Nishida T, Mizuki N, Yazawa K, et al. Effects of astaxanthin on lipopolysaccharide-induced inflammation in vitro and in vivo. Investigative ophthalmology & visual science 2003;44(6):2694–701. [DOI] [PubMed] [Google Scholar]
- 57.Yuan JP, Peng J, Yin K, Wang JH. Potential health-promoting effects of astaxanthin: a high-value carotenoid mostly from microalgae. Molecular nutrition & food research 2011;55(1):150–65. [DOI] [PubMed] [Google Scholar]
- 58.Lee JJ, Kong M, Ayers GD, Lotan R. Interaction index and different methods for determining drug interaction in combination therapy. Journal of biopharmaceutical statistics 2007;17(3):461–80. [DOI] [PubMed] [Google Scholar]
- 59.Rao AR, Sindhuja HN, Dharmesh SM, Sankar KU, Sarada R, Ravishankar GA. Effective inhibition of skin cancer, tyrosinase, and antioxidative properties by astaxanthin and astaxanthin esters from the green alga Haematococcus pluvialis. J Agric Food Chem 2013;61(16):3842–51. [DOI] [PubMed] [Google Scholar]
- 60.Saw CL, Yang AY, Guo Y, Kong AN. Astaxanthin and omega-3 fatty acids individually and in combination protect against oxidative stress via the Nrf2-ARE pathway. Food Chem Toxicol 2013;62:869–75. [DOI] [PubMed] [Google Scholar]
- 61.Yang Y, Fuentes F, Shu L, Wang C, Pung D, Li W, et al. Epigenetic CpG Methylation of the Promoter and Reactivation of the Expression of GSTP1 by Astaxanthin in Human Prostate LNCaP Cells. The AAPS journal 2016. [DOI] [PubMed] [Google Scholar]
- 62.Li R, Wu H, Zhuo WW, Mao QF, Lan H, Zhang Y, et al. Astaxanthin Normalizes Epigenetic Modifications of Bovine Somatic Cell Cloned Embryos and Decreases the Generation of Lipid Peroxidation. Reprod Domest Anim 2015;50(5):793–9. [DOI] [PubMed] [Google Scholar]
- 63.Yang Y, Bae M, Park YK, Lee Y, Pham TX, Rudraiah S, et al. Histone deacetylase 9 plays a role in the antifibrogenic effect of astaxanthin in hepatic stellate cells. J Nutr Biochem 2016;40:172–7. [DOI] [PubMed] [Google Scholar]
- 64.Takahashi K, Heine UI, Junker JL, Colburn NH, Rice JM. Role of cytoskeleton changes and expression of the H-ras oncogene during promotion of neoplastic transformation in mouse epidermal JB6 cells. Cancer Res 1986;46(11):5923–32. [PubMed] [Google Scholar]
- 65.Suzukawa K, Weber TJ, Colburn NH. AP-1, NF-kappa-B, and ERK activation thresholds for promotion of neoplastic transformation in the mouse epidermal JB6 model. Environ Health Perspect 2002;110(9):865–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yu R, Mandlekar S, Lei W, Fahl WE, Tan TH, Kong AN. p38 mitogen-activated protein kinase negatively regulates the induction of phase II drug-metabolizing enzymes that detoxify carcinogens. J Biol Chem 2000;275(4):2322–7. [DOI] [PubMed] [Google Scholar]
- 67.Su ZY, Khor TO, Shu L, Lee JH, Saw CL, Wu TY, et al. Epigenetic reactivation of Nrf2 in murine prostate cancer TRAMP C1 cells by natural phytochemicals Z-ligustilide and Radix angelica sinensis via promoter CpG demethylation. Chem Res Toxicol 2013;26(3):477–85. [DOI] [PubMed] [Google Scholar]
- 68.Kang NJ, Lee KW, Kwon JY, Hwang MK, Rogozin EA, Heo YS, et al. Delphinidin attenuates neoplastic transformation in JB6 Cl41 mouse epidermal cells by blocking Raf/mitogen-activated protein kinase kinase/extracellular signal-regulated kinase signaling. Cancer Prev Res (Phila) 2008;1(7):522–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu S, Khor TO, Cheung KL, Li W, Wu TY, Huang Y, et al. Nrf2 expression is regulated by epigenetic mechanisms in prostate cancer of TRAMP mice. PloS one 2010;5(1):e8579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Khor TO, Fuentes F, Shu L, Paredes-Gonzalez X, Yang AY, Liu Y, et al. Epigenetic DNA methylation of antioxidative stress regulator NRF2 in human prostate cancer. Cancer Prev Res (Phila) 2014;7(12):1186–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yla-Herttuala S Oxidized LDL and atherogenesis. Annals of the New York Academy of Sciences 1999;874:134–7. [DOI] [PubMed] [Google Scholar]
- 72.Muthuirulappan S, Francis SP. Anti-cancer mechanism and possibility of nano-suspension formulations for a marine algae product fucoxanthin. Asian Pacific journal of cancer prevention : APJCP 2013;14(4):2213–6. [DOI] [PubMed] [Google Scholar]