

Abstract
Purpose
BRAFV600 mutations are frequently found in several glioma subtypes, including pleomorphic xanthoastrocytoma (PXA) and ganglioglioma and much less commonly in glioblastoma. We sought to determine the activity of vemurafenib, a selective inhibitor of BRAFV600, in patients with gliomas that harbor this mutation.
Patients and Methods
The VE-BASKET study was an open-label, nonrandomized, multicohort study for BRAFV600-mutant nonmelanoma cancers. Patients with BRAFV600-mutant glioma received vemurafenib 960 mg twice per day continuously until disease progression, withdrawal, or intolerable adverse effects. Key end points included confirmed objective response rate by RECIST version 1.1, progression-free survival, overall survival, and safety.
Results
Twenty-four patients (median age, 32 years; 18 female and six male patients) with glioma, including malignant diffuse glioma (n = 11; six glioblastoma and five anaplastic astrocytoma), PXA (n = 7), anaplastic ganglioglioma (n = 3), pilocytic astrocytoma (n = 2), and high-grade glioma, not otherwise specified (n = 1), were treated. Confirmed objective response rate was 25% (95% CI, 10% to 47%) and median progression-free survival was 5.5 months (95% CI, 3.7 to 9.6 months). In malignant diffuse glioma, best response included one partial response and five patients with stable disease, two of whom had disease stabilization that lasted more than 1 year. In PXA, best response included one complete response, two partial responses, and three patients with stable disease. Additional partial responses were observed in patients with pilocytic astrocytoma and anaplastic ganglioglioma (one each). The safety profile of vemurafenib was generally consistent with that of previously published studies.
Conclusion
Vemurafenib demonstrated evidence of durable antitumor activity in some patients with BRAFV600-mutant gliomas, although efficacy seemed to vary qualitatively by histologic subtype. Additional study is needed to determine the optimal use of vemurafenib in patients with primary brain tumors and to identify the mechanisms driving differential responses across histologic subsets.
INTRODUCTION
Gliomas represent a heterogeneous group of tumors with a range of behaviors.1 Aggressive malignant diffuse gliomas include WHO grade IV glioblastoma (GBM) and WHO grade III isocitrate dehydrogenase (IDH) 1/2 wild-type anaplastic gliomas.1 For decades, standard of care for GBM, including surgery, chemoradiation with temozolomide, and bevacizumab at recurrence has not significantly improved median overall survival (OS) of 14 to 18 months.2-4 Recurrent GBM is highly resistant, with a historical median progression-free survival (PFS) of 9 weeks and a 6-month PFS of 5% to 15% for nonbevacizumab therapies.5,6 Patients with recurrent grade III malignant diffuse gliomas fare only slightly better, with a median PFS of 13 weeks and 31% 6-month PFS in patients with recurrent anaplastic astrocytoma.5
Patients with IDH1/2-mutant grade II astrocytomas and oligodendrogliomas have better prognoses, although these tumors eventually progress and transform into malignant diffuse gliomas.1 Low-grade gliomas also encompass rarer IDH1/2 wild-type histologies, including pilocytic astrocytoma (PA), pleomorphic xanthoastrocytoma (PXA), and ganglioglioma. These are more indolent, usually occur in younger patients, and can sometimes be cured with surgery and radiation1; however, a subset of tumors exhibit higher-grade histologic features or aggressive biology at initial presentation or relapse. There is no standard effective treatment for these patients.
Irrespective of glioma subtype, radiographic volumetric response to conventional chemotherapies is rare, occurring in 6% of patients with GBM and in 14% with of patients with anaplastic gliomas.5,6 Bevacizumab seems to delay disease progression and ameliorate neurologic symptoms in patients with GBM but provides no survival advantage.4,7,8 Radiographic response rates with bevacizumab may be up to 40%,9,10 but these are often pseudoresponses that result from blood–brain barrier reconstitution and decreased enhancement on magnetic resonance imaging, rather than an indication of true antitumor effects.11
Selective targeting of oncogenic mutations has revolutionized the treatment of genomically defined subtypes of non–small-cell lung cancer (NSCLC), breast, gastric, and ovarian cancers, melanoma, and other solid and hematologic cancers.12 Targeted approaches include selective inhibition of the BRAFV600 oncogene, which is the standard treatment of melanoma, NSCLC, anaplastic thyroid cancer, and Erdheim–Chester disease.13-18 BRAFV600 inhibition has shown promise in BRAFV600-mutant papillary thyroid cancer,19 colorectal cancer,20 and hairy cell leukemia.21 Of importance, BRAFV600 mutations have been identified in several glioma subtypes, specifically in select rare IDH1/2 wild-type gliomas, including PXAs (38% to 100%), gangliogliomas (18% to 57%), anaplastic gangliogliomas (AGGs; 50%), PAs (9%), and less commonly (< 3%) in high-grade gliomas, including GBM.22-26 Despite the BRAFV600 mutation being a recurrent genomic event across multiple glioma subtypes, to our knowledge no prospective therapeutic study has investigated targeted therapy in this setting, although retrospective case series provide some evidence for the activity of RAF inhibitors with or without MEK inhibitors.27-32
Vemurafenib is a selective oral inhibitor of the oncogenic BRAFV600 kinase approved globally for the treatment of patients with BRAFV600-mutant metastatic or unresectable melanoma and in the United States for patients with Erdheim–Chester disease. The VE-BASKET study was a nonrandomized, open-label, histology-agnostic, basket study for patients with nonmelanoma solid tumors and myeloma that harbors BRAFV600 mutations.33 VE-BASKET enrolled 24 patients with glioma. We now report the final efficacy and safety of vemurafenib in this cohort.
PATIENTS AND METHODS
Study Design and Population
The phase II, histology-independent VE-BASKET study was conducted at 23 centers worldwide in patients with a range of BRAFV600 mutation–positive tumor types. Nine centers enrolled one or more patients with glioma. The study design has been described in full elsewhere.33 In brief, the study included six cohorts of patients with prespecified cancers—NSCLC, ovarian, colorectal, and breast cancers, cholangiocarcinoma, and multiple myeloma—as well as a seventh cohort of patients with other BRAFV600 mutation–positive cancers. The other cohort permitted enrollment of patients with cancer types not otherwise specified, including gliomas. As this cohort was anticipated to enroll a heterogeneous patient group, no maximum cohort size was specified. Rather, the cohort remained open until the last disease prespecified cohort closed. Written informed consent was obtained from all participants. The study was performed in accordance with provisions of the Declaration of Helsinki and Good Clinical Practice guidelines. The protocol was approved by institutional review boards or human research ethics committees at each participating center. Eligibility was confirmed by the sponsor on all patients.
Patients with brain tumors were required to have histologically confirmed glioma (any grade) and confirmation of BRAFV600 mutation in tumor material obtained at any point in treatment. Testing for BRAFV600 mutation was performed according to local testing procedures in a Clinical Laboratories Improvement Amendment–accredited laboratory or equivalent for sites outside the United States. Central pathologic confirmation of locally reported glioma subtypes and BRAF mutation was not performed. As the clinical trial database did not capture glioma-specific biomarkers (methylguanine-DNA-methyltransferase [MGMT] promoter methylation, IDH1 mutation, or CDKN2A/B deletion), these data, when available, were extracted directly from pathology reports without source verification by the study sponsor. All patients had recurrent disease after standard therapy; there was no limit on the number of prior therapies, and prior bevacizumab was permitted. Patients had measurable disease (Response Evaluation Criteria in Solid Tumors [RECIST] version 1.134), were age ≥ 16 years, with Eastern Cooperative Oncology Group performance status of 0 to 2 and acceptable laboratory parameters. Patients were excluded if they had prior treatment with a BRAF or MEK inhibitor, were unable to swallow pills, had intractable vomiting, a corrected QT interval of 450 milliseconds or more, or known leptomeningeal metastases.
Treatment
Patients received vemurafenib 960 mg twice per day continuously in 28-day cycles until they experienced disease progression, unacceptable toxicity, or withdrew. The vemurafenib dose could be reduced on the basis of toxicity in decrements of 240 mg at each dose administration to a minimum permitted dose of 480 mg twice per day. Patients who were unable to tolerate this minimum dose were removed from the study. Patients were assessed for response by magnetic resonance imaging and clinical examination every two cycles. As VE-BASKET was not specifically designed for the treatment of primary brain tumors, responses were determined using RECIST.34 Treatment toxicities were evaluated using National Cancer Institute Common Terminology Criteria, version 4.0.35 Patients were required to have dermatologic assessments at baseline, after cycle 1, then every 12 weeks to evaluate for cutaneous squamous cell carcinoma (SCC), keratoacanthoma, basal cell carcinoma, and any other malignancy. Head and neck examinations were performed at baseline and every 12 weeks thereafter to evaluate for noncutaneous SCC. All patients were required to undergo chest computed tomography at baseline and at least every 6 months thereafter to evaluate for noncutaneous SCC.
Statistical Analysis
The primary end point of the study was unconfirmed objective radiographic response rate at week 8 or first assessment, as assessed by individual investigators using RECIST version 1.1. Secondary end points included confirmed objective response rate (ORR), clinical benefit rate (defined as confirmed complete response [CR] or partial response [PR] of any duration or stable disease [SD] lasting ≥ 6 months), PFS, OS, and toxicity. PFS and OS were estimated using the Kaplan–Meier method and 95% CIs (Clopper–Pearson method). The protocol used an adaptive Simon two-stage design36 for all tumor-specific cohorts to minimize the number of patients treated if vemurafenib was deemed to be ineffective for a specific tumor type. A response rate of 15% at week 8 was considered low, a response rate of 45% was considered high, and a response rate of 35% was considered low but still desirable and indicative of efficacy. Assuming response rates as specified in the hypothesis testing, a power of 80% for a high response rate and 70% for the low but still desirable response rate, and a two-sided α level of .1, seven, 13, or 19 patients were required in each cohort, depending on results obtained. However, this analysis only applied to prespecified tumor cohorts 1 to 6. As patients with glioma enrolled in cohort 7 (other solid tumors) were considered an exploratory group, response and survival end points were analyzed and reported descriptively. The study was permanently closed and the final data lock performed on January 12, 2017.
RESULTS
Twenty-four patients with gliomas (median age, 32 years; 18 female patients) were enrolled, including 11 with malignant diffuse glioma (six with GBM and five with anaplastic astrocytoma), seven with PXA, two with PA, three with AGG, and one with a high-grade glioma, not otherwise specified (Table 1). Of the 11 patients with malignant diffuse glioma, four had MGMT testing and all were unmethylated. Across the entire cohort, 18 patients had IDH1 testing (all wild type) and 10 CDKN2A/B testing (nine deleted and one wild type). Of the six patients with GBM, all had received prior temozolomide and two had received bevacizumab. Four of five patients with anaplastic astrocytoma had received prior temozolomide. Among the 13 remaining patients with lower-grade glioma, eight had received prior temozolomide and one had received bevacizumab.
Table 1.
Baseline Characteristics

Aggregate clinical efficacy data are summarized in Table 2. One CR was observed in a patient with PXA, and five patients achieved PR—two with PXA and one each with anaplastic astrocytoma, AGG, and PA—for a confirmed ORR in the overall group of 25% (95% CI, 10% to 47%; Table 2). CR lasted 25.9 months or more (censored at last assessment), and PRs lasted 13.1, 9.9, 7.5, 3.4, and 2.4 months. An additional three patients achieved SD that lasted 6 months or more (12.9, 14.9, and 24.8 [censored at last assessment] months), one each with anaplastic astrocytoma, GBM, and PXA, for an overall confirmed clinical benefit rate of 38% (95% CI, 19% to 59%).
Table 2.
Efficacy Summary

Efficacy data at the individual patient level are shown in Figure 1. In patients with PXA (n = 7), best response included one patient with CR, two with PR, three with SD (one that lasted ≥ 6 months), and one with progressive disease, which yielded a confirmed clinical benefit rate of 57% (95% CI, 18% to 90%). Best response in patients with malignant diffuse glioma (n = 11) included one patient with PR, five with SD (two of whom had SD that lasted ≥ 6 months, thus meeting the definition for clinical benefit), three with progressive disease, and response data unavailable for two as a result of early withdrawal. This yielded a clinical benefit rate of 27% (95% CI, 6% to 61%). In the six patients with GBM, best response was SD in three patients, with two experiencing progression at 3.6 months (censored at the last assessment) and 3.7 months, and one with prolonged SD until 12.9 months. One of five patients with anaplastic astrocytoma achieved PR and two had SD that progressed after 14.9 and 5.6 months. Responses among patients with other tumor types included PR in one patient with PA who was treated for 15.3 months and PR in one patient with AGG who was treated for 13.8 months for a confirmed clinical benefit rate of 33% (95% CI, 4.3% to 77.7%).
Fig 1.

Integrated efficacy and treatment duration by patient. Maximal decrease in sum of the longest diameters (SLD), confirmed best response, treatment duration, and prior regimens in patients with (A) PXA, (B) malignant diffuse glioma, and (C) other tumor types. Numbers that appear above individual waterfall bars indicate the percent maximal increase in SLD from baseline. (*) Unchanged from baseline. (†) Patient had no postbaseline assessments. PD was symptomatic deterioration. (§) Patients with secondary malignant diffuse glioma. CR, complete response; D, deleted; MGMT, methylguanine-DNA-methyltransferase gene promoter methylation; IDH1, isocitrate dehydrogenase 1 gene; N, no; NA, not available; NOS, not otherwise specified; PA, pilocytic astrocytoma; PD, progressive disease; PR, partial response; PXA, pleomorphic xanthoastrocytoma; SD, stable disease; UM, unmethylated; WT, wild type; Y, yes.
Overall median PFS for all patients was 5.5 months (95% CI, 3.7 to 9.6 months; Fig 2). Median PFS durations for the PXA, malignant diffuse gliomas, and other cohorts were 5.7 months (95% CI, 3.0 months to not reached [NR]), 5.3 months (95% CI, 1.8 to 12.9 months), and 3.7 months (95% CI, 2.0 to 13.6 months), respectively. Median OS for all patients was 28.2 months (95% CI, 9.6 to 40.1 months). Median OS durations for PXA, malignant diffuse glioma, and other cohorts were NR (95% CI, 5.0 months to NR), 11.9 months (95% CI, 8.3 to 40.1 months), and 28.2 months (95% CI, 12.8 to 31.6 months), respectively. The longest treatment duration was 39.1 months in a patient with PXA, which was ongoing at study closure (Fig 3) —this was the only patient who had received no radiotherapy or temozolomide before protocol initiation. All patients discontinued the study. Three patients with PXA were enrolled in an extension trial to continue vemurafenib because of ongoing response or SD at the closure of the VE-BASKET study.
Fig 2.

Progression-free survival (PFS) and overall survival (OS) curves in the (A) cohort overall, as well as in patients with (B) pleomorphic xanthoastrocytoma (PXA), (C) malignant diffuse glioma, and (D) other tumor types. NR, not reached.
Fig 3.

Time to events in individual patients. (*) Anaplastic astrocytoma, n = 5; glioblastoma, n = 6. (†) Anaplastic ganglioglioma, n = 3; pilocytic astrocytoma, n = 2; high-grade glioma, not otherwise specified, n = 1. The first patient with pleomorphic xanthoastrocytoma (PXA; top swimmer lane) experienced his or her first progression at 9 months but continued on treatment beyond progression.
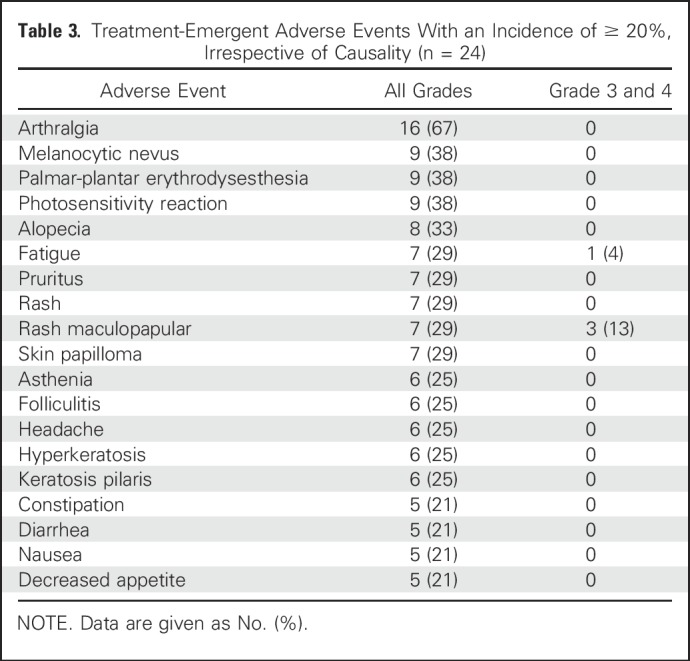
Adverse events, occurring in 20% or more of patients, regardless of cause, are listed in Table 3. Arthralgia (16 of 24 patients; 67%), melanocytic nevus (nine of 24 patients; 38%), palmar-plantar erythrodysesthesia (nine of 24 patients; 38%), and photosensitivity reaction (nine of 24 patients; 38%) were the most common adverse events. Maculopapular rash was the most common grade 3 and 4 event (three of 24 patients; 13%). No grade 5 treatment-related events occurred, and no new vemurafenib safety signals were identified. Ten patients required one or more vemurafenib dose reduction and one discontinued as a result of intolerable adverse effects.
Table 3.
Treatment-Emergent Adverse Events With an Incidence of ≥ 20%, Irrespective of Causality (n = 24)
DISCUSSION
Our data suggest that vemurafenib may have clinically meaningful activity in patients with BRAFV600-mutant gliomas but that this activity varies by histologic subtype. The highest response rate was observed in patients with low-grade tumors, particularly PXA, a histology in which BRAFV600 mutations seem to be a common and early genomic event. In the overall population, including tumors of all grades and histologic subtypes, confirmed ORR was 25% and the clinical benefit rate was 38%—rates numerically higher than those historically observed with other agents used in unselected patients with refractory glioma.37 Although encouraging, these results should be interpreted with caution given the limited number of patients and the descriptive nature of the analysis. These data, however, justify the continued pursuit of this therapeutic strategy through additional dedicated glioma studies.
Although the efficacy reported here is encouraging, the greatest degree of activity was observed in patients with IDH1/2 wild-type low-grade gliomas, specifically PXAs. Historically, PXAs are associated with a better prognosis than GBMs and have been managed with curative intent by surgery, sometimes followed by radiotherapy. For a subset of patients with higher-grade histology or refractory disease, including those enrolled in this study, there is no established standard of care or effective chemotherapy regimen. In our patients, vemurafenib achieved a radiographic response or prolonged stabilization in more than 50% of patients with PXA, which suggests that this strategy may be associated with clinically meaningful benefit. Although one durable response was observed in a patient with PA, only two such patients were enrolled, which precludes the interpretation of efficacy within in this histology.
The 11 patients with high-grade gliomas experienced a more variable response, with PR in one and SD of 6 months or more in two other patients. Although the overall clinical benefit rate was lower than in patients with PXA, AGG, or PA, patients with high-grade glioma were more heavily pretreated, which makes the observed responses even more notable. In addition, patients with PXA, AGG, and PA were younger than those in the high-grade glioma group. The incidence of BRAFV600 mutations is age dependent in patients with gangliogliomas,1 although the etiology that underlies this association is unclear.
The lack of a detailed genomic characterization of the tumors of patients enrolled in this study is a limitation. An important consideration when targeting any oncogene in glioma is whether the previously detected oncogenic alteration is present at the time of treatment and whether, if present, the mutation occurs as the dominant clone. As BRAFV600 mutation status was not confirmed by biopsy immediately before enrollment in the VE-BASKET study, it is unclear whether the mutation was present in the tumors of all patients at the start of vemurafenib treatment. Moreover, GBMs demonstrate substantial temporal and spatial intratumoral heterogeneity,38 and it is possible that in some primary GBMs, BRAFV600 mutations are subclonal or among multiple mutations present and driving tumor growth. These factors, at least in part, may account for the variable efficacy of vemurafenib monotherapy in this subgroup. Of interest, the one patient with GBM who achieved prolonged SD that lasted 12.9 months had a secondary GBM that evolved from a prior low-grade lesion, in keeping with our observation that lower-grade BRAFV600-mutant gliomas seem to be more sensitive to vemurafenib. Another consideration is that the BRAF mutation may not be present in all components of the tumor. This latter mechanism has been potentially implicated in gangliogliomas in which a subset of BRAFV600-mutant gangliogliomas had expression in both neuronal and glial tumor components.39
Of note, as a multihistology basket trial, several characteristics of the VE-BASKET study were suboptimal for the evaluation and treatment of patients with gliomas. The clinical trial was not designed to collect glioma biomarkers, such as MGMT promoter methylation, IDH mutation, or CDKN2A/B deletion status, although we were ultimately able to gather available data on most patients. It is possible that complete biomarker status may have helped provide additional context to the differential activity observed.40-42 MGMT promoter methylation testing is only routine in malignant diffuse gliomas, where it is important for prognostication and in the evaluation of pseudoprogression after chemoradiation. There are no data to suggest that MGMT promoter methylation status would influence radiographic response or PFS with BRAF inhibitors. IDH mutation testing is not currently recommended for PXA, AGG, or PA. Moreover, prior studies have demonstrated mutual exclusivity between IDH and BRAFV600 mutations in gliomas,23,43,44 which indicates that this biomarker might not be relevant in our cohort. Accordingly, IDH1 mutation status was available for 18 (75%) of 24 patients in this study, all of whom all had wild-type IDH1 tumors.
The current study used RECIST, which is designed primarily for the assessment of solid tumors, instead of dedicated brain tumor response criteria, such as the Macdonald or Response Assessment in Neuro-Oncology criteria.11 However, prior studies showed similarity in response assessments between one-dimensional and two-dimensional measurement methods in patients with high-grade gliomas.45,46 Moreover, given the lack of a substantial antiangiogenic effect of vemurafenib, it is unlikely that pseudoresponses might have occurred in our patients. Another limitation is the lack of central review of investigator-reported response assessments. In summary, although the inclusion of patients with primary brain tumors in this study provided the opportunity to evaluate genomically targeted therapy in this relatively large, prospectively accrued group of patients with BRAFV600-mutant gliomas, future histology-agnostic studies should be designed to address brain tumor–specific considerations to optimize the interpretation of the findings.
Despite its shortcomings, the current study serves as an initial proof of concept that BRAFV600 is a targetable oncogene in at least a subset of patients with primary brain tumors. Responses were observed across all glioma subsets, with the strongest signal observed in patients with lower-grade gliomas, particularly the PXA subgroup. Additional evaluation is needed to clarify the precise use of RAF and MEK inhibitors—alone or in combination—in patients with primary brain tumors. Several such studies that permit the enrollment of pediatric or adult patients with glioma are currently ongoing (ClinicalTrials.gov identifiers: NCT01748149, NCT01677741, NCT02124772, NCT02684058, NCT02285439, and NCT03429803). These studies may also help elucidate the underlying mechanisms that drive the differential responses across histologic subsets.
ACKNOWLEDGMENT
The investigators thank the patients who participated in this study and their families. Editorial support was provided by Miller Medical Communications. Funding for editorial support was provided by F Hoffmann-La Roche.
Footnotes
Funded by F Hoffmann-La Roche and supported by Grants No. P30-CA008748 and 1R01-CA201247-01A1 from the National Institutes of Health.
Presented in part at the Annual Meeting of the American Society of Clinical Oncology, Chicago, IL, June 1, 2017.
Clinical trial information: NCT01524978.
AUTHOR CONTRIBUTIONS
Conception and design: Mehdi Touat, Martina Makrutzki, Jose Baselga, David M. Hyman
Administrative support: Martina Makrutzki, Bethany Pitcher, Jose Baselga, David M. Hyman
Provision of study materials or patients: Thomas Kaley, Mehdi Touat, Vivek Subbiah, Franck Bielle, Jean-Yves Blay, David M. Hyman
Collection and assembly of data: Thomas Kaley, Mehdi Touat, Vivek Subbiah, Franck Bielle, Martina Makrutzki, Bethany Pitcher, David M. Hyman
Data analysis and interpretation: Thomas Kaley, Mehdi Touat, Vivek Subbiah, Antoine Hollebecque, Jordi Rodon, A. Craig Lockhart, Vicki Keedy, Ralf-Dieter Hofheinz, Florence Joly, Jean-Yves Blay, Ian Chau, Igor Puzanov, Noopur S. Raje, Jurgen Wolf, Lisa M. DeAngelis, Martina Makrutzki, Todd Riehl, Bethany Pitcher, Jose Baselga, David M. Hyman
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
BRAF Inhibition in BRAFV600-Mutant Gliomas: Results From the VE-BASKET Study
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Thomas Kaley
Research Funding: Eli Lilly, Ludwig Institute for Cancer Research
Mehdi Touat
Consulting or Advisory Role: Agios Pharmaceuticals, Taiho Oncology
Travel, Accommodations, Expenses: Merck Sharp & Dohme
Vivek Subbiah
Consulting or Advisory Role: MedImmune
Research Funding: Novartis (Inst), GlaxoSmithKline (Inst), NanoCarrier (Inst), Northwest Biotherapeutics (Inst), Genentech (Inst), Berg Pharma (Inst), Bayer (Inst), Incyte (Inst), Fujifilm (Inst), PharmaMar (Inst), D3 Oncology Solutions (Inst), Pfizer (Inst), Amgen (Inst), AbbVie (Inst), Multivir (Inst), Blueprint Medicines (Inst), Loxo (Inst), Vegenics (Inst), Takeda (Inst), Alfasigma (Inst), Agensys (Inst), Idera (Inst), Boston Biomedical (Inst)
Travel, Accommodations, Expenses: PharmaMar, Bayer
Antoine Hollebecque
Honoraria: Merck Serono, Amgen
Consulting or Advisory Role: Amgen, Gritstone Oncology
Travel, Accommodations, Expenses: Amgen, Servier
Jordi Rodon
Research Funding: Roche
A. Craig Lockhart
No relationship to disclose
Vicki Keedy
Consulting or Advisory Role: Karyopharm Therapeutics
Research Funding: Pfizer (Inst), CytRx Corporation (Inst), Medpacto (Inst), Plexxikon (Inst), Roche (Inst), Eli Lilly (Inst), BioMed Valley Discoveries (Inst), Immune Design (Inst), GlaxoSmithKline (Inst), Tracon Pharma (Inst), Advenchen Laboratories (Inst), Daiichi Sankyo (Inst)
Franck Bielle
Employment: Celgene (I)
Stock or Other Ownership: Crossject
Research Funding: Sanofi
Ralf-Dieter Hofheinz
Honoraria: Amgen, Boehringer Ingelheim, Bristol-Myers Squibb, Merck, Roche, Medac, Servier, Sanofi
Consulting or Advisory Role: Merck, Roche, Sanofi, Amgen, Bristol-Myers Squibb
Research Funding: Sanofi (Inst), Merck (Inst), Amgen (Inst)
Florence Joly
Consulting or Advisory Role: Roche
Travel, Accommodations, Expenses: Roche
Jean-Yves Blay
Honoraria: Roche
Consulting or Advisory Role: Roche, Novartis, GlaxoSmithKline, Bayer, PharmaMar, Merck
Research Funding: Roche (Inst)
Ian Chau
Honoraria: Eli Lilly
Consulting or Advisory Role: Sanofi, Eli Lilly, Bristol-Myers Squibb, MSD Oncology, Bayer, Genentech, Five Prime Therapeutics, Merck Serono, AstraZeneca, MedImmune
Research Funding: Janssen-Cilag (Inst), Sanofi (Inst), Merck Serono (Inst), Eli Lilly (Inst)
Travel, Accommodations, Expenses: Eli Lilly, Bristol-Myers Squibb
Igor Puzanov
Consulting or Advisory Role: Amgen, Genentech, Bristol-Myers Squibb
Travel, Accommodations, Expenses: Amgen, Merck
Noopur S. Raje
Consulting or Advisory Role: Amgen, Celgene, Takeda, Novartis, Bristol-Myers Squibb, Merck
Research Funding: AstraZeneca (Inst)
Jurgen Wolf
Honoraria: AbbVie, AstraZeneca, Bristol-Myers Squibb, Boehringer Ingelheim, MSD Oncology, Novartis, Roche
Consulting or Advisory Role: AbbVie, AstraZeneca, Bristol-Myers Squibb, Boehringer Ingelheim, Chugai Pharma, Ignyta, Eli Lilly, MSD Oncology, Novartis, Pfizer, Roche
Research Funding: Bristol-Myers Squibb, Novartis, Pfizer
Lisa M. DeAngelis
Consulting or Advisory Role: Genentech, Tocagen, Sapience, BTG
Martina Makrutzki
Employment: F Hoffmann-La Roche
Todd Riehl
Employment: Genentech
Stock or Other Ownership: Genentech
Bethany Pitcher
Employment: F Hoffmann-La Roche
Jose Baselga
Leadership: Infinity Pharmaceuticals, Varian Medical Systems, Bristol-Myers Squibb, Foghorn Therapeutics, Aura Biosciences, Grail
Stock or Other Ownership: PMV Pharma, Juno Therapeutics, Infinity Pharmaceuticals, Grail, Varian Medical Systems, Tango Therapeutics, ApoGen Biosciences, Venthera, Foghorn Therapeutics, Aura Biosciences
Honoraria: Genentech, Novartis, Eli Lilly
Consulting or Advisory Role: Grail, Eli Lilly, Novartis, Genentech, PMV Pharma, ApoGen, Juno Therapeutics
Patents, Royalties, Other Intellectual Property: Patents related to PI3K, PDK1 inhibitors; HER2 receptor, and LIF antibodies, and methods for diagnosis
Travel, Accommodations, Expenses: Genentech, Novartis, Eli Lilly
David M. Hyman
Consulting or Advisory Role: Atara Biotherapeutics, Chugai Pharma, CytomX Therapeutics, Boehringer Ingelheim, AstraZeneca, Pfizer, Bayer, Debiopharm Group, ArQule, Genentech
Research Funding: AstraZeneca, Puma Biotechnology, Loxo
REFERENCES
- 1.Louis DN, Ohgaki H, Wiestler OD, et al. : World Health Organization Histological Classification of Tumours of the Central Nervous System (ed 2). Lyon, France, International Agency for Research on Cancer, 2016 [Google Scholar]
- 2.Stupp R, Hegi ME, Mason WP, et al. : Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 10:459-466, 2009 [DOI] [PubMed] [Google Scholar]
- 3.Stupp R, Taillibert S, Kanner AA, et al. : Maintenance therapy with tumor-treating fields plus temozolomide vs temozolomide alone for glioblastoma: A randomized clinical trial. JAMA 314:2535-2543, 2015 [DOI] [PubMed] [Google Scholar]
- 4.Wick W, Gorlia T, Bendszus M, et al. : Lomustine and bevacizumab in progressive glioblastoma. N Engl J Med 377:1954-1963, 2017 [DOI] [PubMed] [Google Scholar]
- 5.Wong ET, Hess KR, Gleason MJ, et al. : Outcomes and prognostic factors in recurrent glioma patients enrolled onto phase II clinical trials. J Clin Oncol 17:2572-2578, 1999 [DOI] [PubMed] [Google Scholar]
- 6.Lamborn KR, Chang SM, Prados MD: Prognostic factors for survival of patients with glioblastoma: Recursive partitioning analysis. Neuro-oncol 6:227-235, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gilbert MR, Dignam JJ, Armstrong TS, et al. : A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med 370:699-708, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chinot OL, Wick W, Mason W, et al. : Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med 370:709-722, 2014 [DOI] [PubMed] [Google Scholar]
- 9.Kreisl TN, Kim L, Moore K, et al. : Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol 27:740-745, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friedman HS, Prados MD, Wen PY, et al. : Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol 27:4733-4740, 2009 [DOI] [PubMed] [Google Scholar]
- 11.Wen PY, Macdonald DR, Reardon DA, et al. : Updated response assessment criteria for high-grade gliomas: Response assessment in neuro-oncology working group. J Clin Oncol 28:1963-1972, 2010 [DOI] [PubMed] [Google Scholar]
- 12.Hyman DM, Taylor BS, Baselga J: Implementing genome-driven oncology. Cell 168:584-599, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ascierto PA, McArthur GA, Dréno B, et al. : Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol 17:1248-1260, 2016 [DOI] [PubMed] [Google Scholar]
- 14.Robert C, Karaszewska B, Schachter J, et al. : Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 372:30-39, 2015 [DOI] [PubMed] [Google Scholar]
- 15.Planchard D, Besse B, Groen HJM, et al. : Dabrafenib plus trametinib in patients with previously treated BRAFV600E-mutant metastatic non–small-cell lung cancer: An open-label, multicentre phase 2 trial. Lancet Oncol 17:984-993, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Planchard D, Smit EF, Groen HJM, et al. : Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non–small-cell lung cancer: An open-label, phase 2 trial. Lancet Oncol 18:1307-1316, 2017 [DOI] [PubMed] [Google Scholar]
- 17.Diamond EL, Subbiah V, Lockhart AC, et al. : Vemurafenib for BRAFV600-mutant Erdheim–Chester disease and Langerhans cell histiocytosis: Analysis of data from the histology-independent phase 2 open-label VE-BASKET study. JAMA Oncol 4:384-388, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Subbiah V, Kreitman RJ, Wainberg ZA, et al. : Dabrafenib and trametinib treatment in patients with locally advanced or metastatic BRAFV600-mutant anaplastic thyroid cancer. J Clin Oncol 36:7-13, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brose MS, Cabanillas ME, Cohen EE, et al. : Vemurafenib in patients with BRAFV600E-positive metastatic or unresectable papillary thyroid cancer refractory to radioactive iodine: A non-randomised, multicentre, open-label, phase 2 trial. Lancet Oncol 17:1272-1282, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Corcoran RB, Atreya CE, Falchook GS, et al. : Combined BRAF and MEK inhibition with dabrafenib and trametinib in BRAFV600-mutant colorectal cancer. J Clin Oncol 33:4023-4031, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tiacci E, Park JH, De Carolis L, et al. : Targeting mutant BRAF in relapsed or refractory hairy-cell leukemia. N Engl J Med 373:1733-1747, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chi AS, Batchelor TT, Yang D, et al. : BRAFV600E mutation identifies a subset of low-grade diffusely infiltrating gliomas in adults. J Clin Oncol 31:e233-e236, 2013 [DOI] [PubMed] [Google Scholar]
- 23.Brennan CW, Verhaak RG, McKenna A, et al. : The somatic genomic landscape of glioblastoma. Cell 155:462-477, 2013. [Erratum: Cell 157:753, 2014] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dougherty MJ, Santi M, Brose MS, et al. : Activating mutations in BRAF characterize a spectrum of pediatric low-grade gliomas. Neuro-oncol 12:621-630, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schindler G, Capper D, Meyer J, et al. : Analysis of BRAFV600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol 121:397-405, 2011 [DOI] [PubMed] [Google Scholar]
- 26.Dias-Santagata D, Lam Q, Vernovsky K, et al. : BRAFV600E mutations are common in pleomorphic xanthoastrocytoma: Diagnostic and therapeutic implications. PLoS One 6:e17948, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chamberlain MC: Salvage therapy with BRAF inhibitors for recurrent pleomorphic xanthoastrocytoma: A retrospective case series. J Neurooncol 114:237-240, 2013 [DOI] [PubMed] [Google Scholar]
- 28.Bautista F, Paci A, Minard-Colin V, et al. : Vemurafenib in pediatric patients with BRAFV600E mutated high-grade gliomas. Pediatr Blood Cancer 61:1101-1103, 2014 [DOI] [PubMed] [Google Scholar]
- 29.Burger MC, Ronellenfitsch MW, Lorenz NI, et al. : Dabrafenib in patients with recurrent, BRAFV600E mutated malignant glioma and leptomeningeal disease. Oncol Rep 38:3291-3296, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johanns TM, Ferguson CJ, Grierson PM, et al. : Rapid clinical and radiographic response with combined dabrafenib and trametinib in adults with BRAF-mutated high-grade glioma. J Natl Compr Canc Netw 16:4-10, 2018 [DOI] [PubMed] [Google Scholar]
- 31.Brown NF, Carter T, Kitchen N, et al. : Dabrafenib and trametinib in BRAFV600E mutated glioma. CNS Oncol 6:291-296, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Migliorini D, Aguiar D, Vargas MI, et al. : BRAF/MEK double blockade in refractory anaplastic pleomorphic xanthoastrocytoma. Neurology 88:1291-1293, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hyman DM, Puzanov I, Subbiah V, et al. : Vemurafenib in multiple nonmelanoma cancers with BRAFV600 mutations. N Engl J Med 373:726-736, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eisenhauer EA, Therasse P, Bogaerts J, et al. : New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer 45:228-247, 2009 [DOI] [PubMed] [Google Scholar]
- 35.US National Institutes of Health : Common Terminology Criteria for Adverse Events (CTCAE), version 4.0. https://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf
- 36.Lin Y, Shih WJ: Adaptive two-stage designs for single-arm phase IIA cancer clinical trials. Biometrics 60:482-490, 2004 [DOI] [PubMed] [Google Scholar]
- 37.Touat M, Idbaih A, Sanson M, et al. : Glioblastoma targeted therapy: Updated approaches from recent biological insights. Ann Oncol 28:1457-1472, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qazi MA, Vora P, Venugopal C, et al. : Intratumoral heterogeneity: Pathways to treatment resistance and relapse in human glioblastoma. Ann Oncol 28:1448-1456, 2017 [DOI] [PubMed] [Google Scholar]
- 39.Koelsche C, Wöhrer A, Jeibmann A, et al. : Mutant BRAF V600E protein in ganglioglioma is predominantly expressed by neuronal tumor cells. Acta Neuropathol 125:891-900, 2013 [DOI] [PubMed] [Google Scholar]
- 40.Mistry M, Zhukova N, Merico D, et al. : BRAF mutation and CDKN2A deletion define a clinically distinct subgroup of childhood secondary high-grade glioma. J Clin Oncol 33:1015-1022, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huillard E, Hashizume R, Phillips JJ, et al. : Cooperative interactions of BRAFV600E kinase and CDKN2A locus deficiency in pediatric malignant astrocytoma as a basis for rational therapy. Proc Natl Acad Sci USA 109:8710-8715, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lassaletta A, Zapotocky M, Mistry M, et al. : Therapeutic and prognostic implications of BRAFV600E in pediatric low-grade gliomas. J Clin Oncol 35:2934-2941, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cancer Genome Atlas Research Network. Brat DJ, Verhaak RG, et al. : Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med 372:2481-2498, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zehir A, Benayed R, Shah RH, et al. : Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 23:703-713, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Galanis E, Buckner JC, Maurer MJ, et al. : Validation of neuroradiologic response assessment in gliomas: Measurement by RECIST, two-dimensional, computer-assisted tumor area, and computer-assisted tumor volume methods. Neuro Oncol 8:156-165, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shah GD, Kesari S, Xu R, et al. : Comparison of linear and volumetric criteria in assessing tumor response in adult high-grade gliomas. Neuro Oncol 8:38-46, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]