

Abstract
IL-6 is implicated in the development and progression of autoimmune diseases in part by influencing CD4 T cell lineage and regulation. Elevated IL-6 levels drive inflammation in a wide range of autoimmune diseases, some of which are also characterized by enhanced T cell responses to IL-6. Notably, the impact of IL-6 on inflammation is contextual in nature and dependent on the cell type, cytokine milieu and tissue. Targeting the IL-6/IL-6R axis in humans has been shown to successfully ameliorate a subset of autoimmune conditions. In this review, we discuss recent studies investigating how IL-6 regulates the CD4 T cell response in the context of autoimmune disease and highlight how blocking different aspects of the IL-6 pathway is advantageous in the treatment of disease.
Introduction
Interleukin-6 (IL-6) is a pleiotropic cytokine involved in chronic inflammation, autoantibody production, vascular permeability as well as tissue regeneration, metabolism and hematopoiesis. IL-6 is produced by stromal cells, monocytes and lymphocytes, and its expression is increased by IL-1β, TNF-α, as well as stimulation of Toll-like receptors and additional stress response proteins [1]. Elevated IL-6 serum and tissue concentrations are a hallmark of rheumatoid arthritis (RA), systemic lupus erythematosus (SLE) and relapsing-remitting multiple sclerosis (MS), often correlating with disease activity [2–4]. IL-6 signals via three mechanisms: classic, trans- and cluster signaling, each of which lead to distinct immune outcomes. The role of IL-6 in the adaptive immune response is diverse, providing both proinflammatory and immunoregulatory signals based on the cell type, cytokine milieu and the manner through which it is sensed [5]. In this review, we will discuss how the IL-6 signaling pathway influences the adaptive immune response, promotes autoimmunity and how blocking different aspects of this pathway is advantageous in the treatment of disease.
IL-6 promotes Th17 and Tfh cell development while suppressing Treg induction
IL-6 contributes to the development of autoreactive proinflammatory CD4 T cell responses by promoting Th17 cell lineage and function, and by inhibiting the induction of regulatory T cells (Treg) (Figure 1). Th17 cells have been implicated in the pathogenesis of RA, MS, type 1 diabetes (T1D) and SLE [6,7]. IL-6 in combination with TGF-β promotes the development and function of Th17 cells [8], and in mice, IL-6 promotes the expansion of Th17 cells [9]. In addition, a recent study by Zhao et al reports that IL-6 stimulation inhibits expression of RFX1, a transcriptional repressor of IL-17A production in CD4+ T cells [10]. IL-6 also influences Th17 cells via regulation of microRNAs; IL-6 induces miR-183c, which promotes Th17 pathogenicity via upregulation of IL-1R1 [11].
Figure 1. IL-6 is a proinflammatory modulator of T cells.
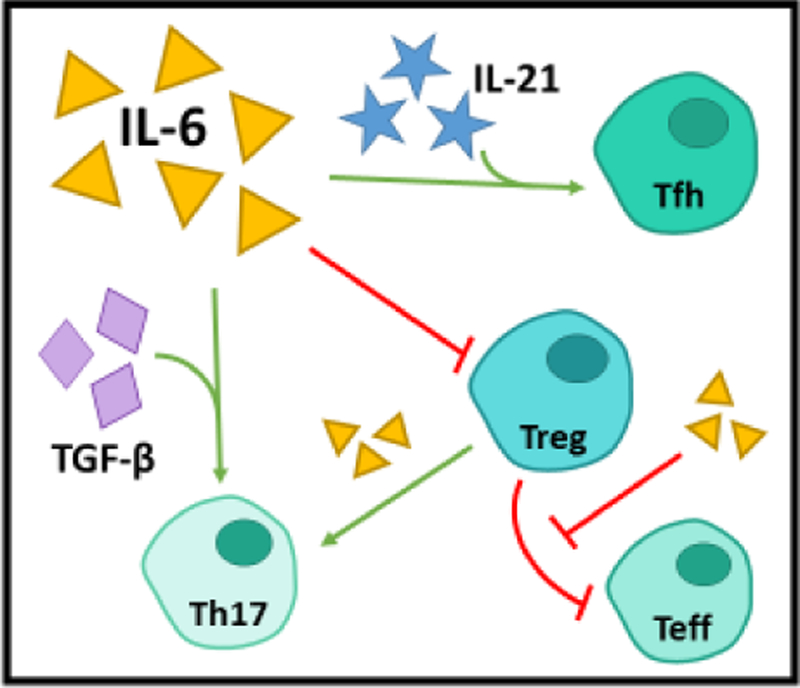
IL-6 contributes to autoimmunity by promoting Tfh, Th17, and Teff lineage and function and by inhibiting the suppressive capacity and induction of Tregs. In the presence of IL-21, IL-6 promotes commitment to the Tfh lineage, which is capable of stimulating B cell proliferation and class switching. In addition to bolstering Teff resistance to suppression by Tregs, IL-6 also promotes the conversion of Tregs to Th17 and may reduce Treg suppressive capacity. Lastly, in the presence of TGF-β, IL-6 enhances commitment and function of Th17 cells, a well-established pathogenic cell type in autoimmunity.
IL-6 is implicated in the regulation of T cell responses both by inhibiting the generation of Foxp3+ Tregs and promoting effector CD4 T cells (Teff) resistant to suppression [8,12–14]. IL-6R is highly expressed on Tregs; it has been proposed that the suppressive capacity of a Foxp3+ TIGIT- IL-6Rhi Treg population could be ‘disarmed’ in the presence of IL-6-associated inflammation, allowing for the activation of effector functions and tissue damage [15]. Foxp3+ Treg can also convert to Th17 upon exposure to IL-6 [16]. This is regulated in part by miR-125a, which reduces IL6R making Treg less sensitive to IL-6 and able to retain regulatory features [17]. Exposure of Teff cells to IL-6 is known to bolster their resistance to suppression by Tregs; Teff resistance has been previously established in T1D, MS, juvenile idiopathic arthritis (JIA), SLE and psoriasis [14,18–21]. STAT3 appears to play a central role in the resistance of Teff to Treg. Studies in MS demonstrated the ability to revert Teff resistance in vitro through the use of a STAT3 inhibitor [14]; more recently Ihantola et al. found Teff resistance was STAT3-dependent in T1D [20].
A recent study shows an influence of IL-6 on naïve T cell activation via TCR, which may have broad implications with respect to the activation and development of autoreactive T cells. Tu et al. demonstrate that TβR1 expression regulates T cell responsiveness in naïve T cells [22]. TGF-β blunts the response to TCR stimulation, enforcing a quiescent state. The response to TGF-β in naïve T cells is regulated through the expression of cell surface TβR1. Strong stimuli overcome the suppression of T cell activation by reducing TβR1 and allowing cells to become activated and mature. IL-6 blocks TβR1 upregulation, allowing for increased T cell activation, a diminished naïve T cell population and the potential to promote the activation of low affinity T cells; this may be a mechanism through which IL-6 could promote Teff resistance [22].
In the context of humoral immunity, IL-6 functions as a central link between T cell and B cell responses. IL-6 promotes the survival, expansion, and maturation of B cells and plasmablasts, in part by promoting the development of T follicular helper (Tfh) cells [5,23]. The activation of STAT3 by IL-6 and IL-21 enhances expression of the transcriptional repressor, Bcl-6, and promotes commitment to the Tfh lineage. Tfh cells localize to B cell follicles where they promote B cell proliferation and immunoglobulin class switching [24]. B cells are also an important source of IL-6. B cell-derived IL-6 drives a murine model of MS by supporting the development of Th17 cells in the central nervous system [25]. B cell-derived IL-6 is required for spontaneous germinal center formation and development of murine lupus [26]. IL-6 produced by B cells promotes the development of Tfh cells [27].
The IL-6 pathway: Unique roles for classic, trans- and cluster signaling
As noted above IL-6 signals via three distinct mechanisms (Figure 2). Classic signaling is mediated by the membrane bound IL-6 receptor (mbIL-6R). IL-6 binds the mbIL-6R leading to recruitment of gp130, which then activates JAK1 and JAK2, leading to phosphorylation of the transcription factors, STAT1 and STAT3 [5]. Trans-signaling is mediated by the soluble IL-6 receptor (sIL-6R), instead of mbIL-6R. Here, IL-6 binds to sIL-6R then forms a complex with cell surface gp130, this allows IL-6 to act on cells that express gp130 but which lack mbIL-6R [28]. Notably gp130 is broadly expressed and soluble gp130 (sgp130) can act to block trans-signaling [29]. The expression of mbIL-6R and sIL-6R are central to the regulation of these signaling pathways. Two distinct mechanisms have been identified for the generation of sIL-6R: alternative splicing and shedding. In humans, a splice variant lacking the transmembrane domain leads to expression of sIL-6R [30]. Ectodomain shedding refers to proteolytic cleavage of the extracellular domain of IL-6R, which is tightly regulated and mediated by ADAMs (a disintegrin and metalloprotease). ADAM17 has been identified as the major protease involved in IL-6R shedding following TCR activation in human CD4 T cells [31].
Figure 2. Signaling modes, JAK/STAT cascade and therapeutic targets.
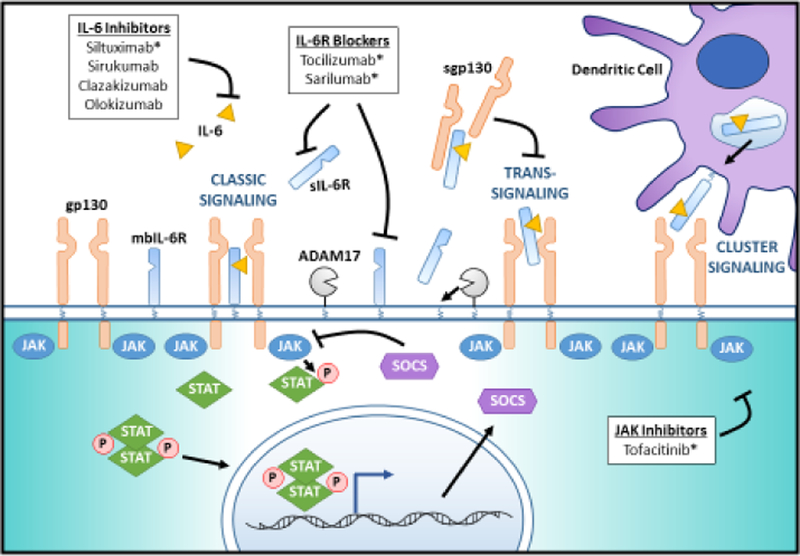
IL-6 signals via three mechanisms: classic signaling mediated by the membrane bound IL-6 receptor (mbIL-6R), trans-signaling mediated by the soluble IL-6 receptor (sIL-6R) and cluster signaling in dendritic cells (DCs). In classic signaling, IL-6 binds the mbIL-6R leading to recruitment of gp130, which then activates JAK1 and JAK2, leading to phosphorylation of the transcription factors, STAT1 and STAT3. Activated STAT3 induces a negative-feedback molecule, suppressor of cytokine signaling (SOCS), SOCS1 and SOCS3. In trans-signaling, IL-6 binds extracellular sIL-6R before complexing with gp130 and initiating the JAK/STAT cascade. Trans-signaling is inhibited by extracellular sgp130, which can complex with sIL-6R and prevent it from binding to the membrane-bound gp130. Classic signaling and trans-signaling may be augmented by ADAM17, which cleaves mbIL-6R to generate sIL-6R. In cluster signaling, IL-6 is complexed with IL-6R within intracellular compartments in DCs before being transported to the membrane to activate gp130 in target cells. Current IL-6 targeting therapies inhibit either IL-6, the IL-6R, or JAK. IL-6 inhibitors include siltuximab, sirukumab, clazakizumab, and olokizumab. Siltuximab is approved for the treatment of Castleman’s disease. Il-6R blocking drugs tocilizumab and sarilumab are both approved for the treatment of RA. Tofacitinib, a JAK inhibitor, is approved for the treatment of RA and has a demonstrated therapeutic effect in ulcerative colitis, alopecia areata, and psoriasis. *indicates FDA approval for an autoimmune disease
Cluster signaling, a third form of IL-6 signaling, was described in 2017 by Heink et al. [16]. This IL-6 cluster signaling occurs in dendritic cells where IL-6 is complexed with the IL-6R in intracellular compartments before being transported to the membrane to activate gp130 in target cells. While sgp130 can interfere with IL-6 trans-signaling, it does not impact cluster signaling; this mode of IL-6 signaling contributes to the generation of Th17 cells via the induction of STAT3 and the upregulation of the IL-23R in the presence of TGF-β1 [8,32]. Importantly, cluster signaling induces faster and more robust activation of STAT3 compared to classic IL-6 signaling [16].
Both IL-6 trans-signaling and cluster signaling play more detrimental roles in adaptive immunity by regulating the differentiation of Th17 cells, suppressing Tregs and contributing to chronic inflammation [16,33,34]. This suggests that Th17 cell differentiation requires multiple IL-6 sources and signaling modes that function as a safeguard to minimize unwanted Th17 cell-dependent immunopathology [35]. IL-6 classic signaling suppresses the differentiation of Foxp3+ Tregs and plays a central role in the development of Tfh cells and germinal centers [5,34]. Blockade of IL-6 classic signaling, but not trans-signaling, alleviated multiorgan autoimmunity in a murine model of enhanced IL-6 expression in follicular B cells dependent on IL-6-driven Tfh [27].
The IL-6/IL-6R axis is dysregulated in autoimmunity
Significant differences in the expression of mbIL6R and sIL6R are found among individuals. Twin studies have demonstrated that up to 72% of these differences can be attributed to genetics with 51% linked to a single genetic variant the Asp358Ala (rs2228145) [36,37]. This genetic variant alters the amino acid in the ADAM17 cleavage site resulting in increased shedding, increased sIL-6R in serum and decreased mbIL-6R levels on CD4 T cells [38,39]. Alterations in mbIL6R and sIL6R are associated with autoimmunity. The IL6R, Asp358Ala (rs2228145) genetic variant is associated with T1D and RA, where increased mbIL-6R expression could lead to higher IL-6 signaling capacity in CD4 effector T cells [39]. There is also a significant reduction in transcript and protein levels of ADAM17 in T cells from subjects with T1D, which correlate with expression of mbIL-6R and suggests a role for ADAM17 in the mechanism of altered IL-6 signaling in T1D [40]. How cluster signaling may be implicated in autoimmunity is still under investigation.
Targeting the IL-6 pathway has proven to be therapeutic in autoimmunity
Anti-IL-6R antibodies
The first approved drug to block the IL-6 signaling pathway in autoimmunity was a humanized anti-IL-6R monoclonal antibody, tocilizumab (TCZ) [41]. The FDA has approved the use of TCZ in the treatment of RA and JIA. In Japan, TCZ is approved for the treatment of Castleman’s disease [1]. Tocilizumab blocks both the classic and trans-signaling pathways by preventing IL-6 from binding to mbIL-6R and sIL-6R [42]. The efficacy of TCZ in treating other autoimmune disease is being explored in ongoing clinical trials including those in systemic sclerosis [43], neuromyelitis optica [44] and new-onset T1D (clinicaltrials.gov NCT02293837). Most recently, a second anti-IL-6R antibody, sarilumab, which binds to the IL-6R with up to a 40-fold higher affinity than TCZ, was approved for the treatment of RA [45].
Anti-IL-6 antibodies
There is also a class of anti-IL-6 antibodies being developed for the treatment of autoimmune diseases. One of the first anti-IL-6 antibodies, siltuximab, functions by inhibiting IL-6 binding to the IL-6R and is approved for the treatment of Castleman’s disease [46]. Treatment with sirukumab has improved symptoms in RA patients that previously failed anti-TNF therapy [47]. A third anti-IL-6 antibody, clazakizumab, improved musculoskeletal manifestations in patients with psoriatic arthritis [48]. Additionally, some of these therapies are also being tested in the setting of malignancy and transplantation [49,50].
JAK inhibitors
Janus kinase (JAK) family enzymes participate in signal transduction of multiple cytokines. Inhibitors of JAKs, Jakinibs, have been developed to for the treatment of inflammatory diseases and cancer. Tofacitinib is a JAK3 specific inhibitor that also inhibits JAK1 and JAK2 and thus has the potential to block IL-6 signaling. Tofacitinib is approved for RA and ulcerative colitis and is effective in a broad range of dermatological conditions including alopecia areata [51] and psoriasis [52]. There is ongoing development of additional Jakinibs including the JAK3-specific inhibitor, peficitinib, which has shown promise in RA [53].
In addition to improving treatment of autoimmunity, blockade of the IL-6 pathway has allowed us to develop a broader understanding of how IL-6 influences the lymphocyte population and its function in vivo. Treg and Th17 cells are altered with TCZ treatment in RA patients. An increase in the ratio of Treg to Th17 cells with therapy is a common finding in these studies; this is predominantly due to an increase in Treg [54,55] though there is limited evidence of decreased Th17 cells [34]. Moreover, Treg function is improved with TCZ therapy in RA and correlates with clinical response, in part due to an increase in the frequency of functionally suppressive CD39+ Tregs [54,56]. Further, natural killer cells are also found to inversely correlate with DAS28 disease score after three months of TCZ treatment [57]. Anti-IL-6 therapies have not been available long enough for their impact to be known; limited studies of tofacitinib demonstrate increases in B cell counts but impaired plasmablast development [58,59]. These findings support the multiple roles played by IL-6 in defining the character of the CD4 T cell and B cell response in human health and disease.
Concluding Comments
Due to its pleiotropic nature, IL-6 has multiple roles in host defense and in human disease, from autoimmunity to cancer. Our understanding of how IL-6 influences the immune response continues to grow, exemplified by the recent discovery of IL-6 cluster signaling and its unique impact on T cell responses [35] alongside the many studies describing the immunologic consequences of IL-6R blockade in humans [33,56]. As the IL-6/IL-6R axis continues to be implicated in more autoimmune diseases, the need to better understand the dysregulation of the signaling pathways driving these diseases also grows. Ultimately, therapeutics targeting the IL-6/IL-6R axis may need to be further tailored to specific cell types or tissue compartments. An example of this is the development of a bispecific antibody against Il-6R and IL-17A, a novel therapeutic approach that could be used to target the Th17 lineage and its pathogenicity in autoimmunity [60]. Future investigation into the cell types, tissues and dysregulated signaling pathways involved in the IL-6/IL-6R axis will enable a more streamlined process of drug development and therapeutic selection.
HIGHLIGHTS.
IL-6 promotes development of Th17 and Tfh cells and suppresses induction of Treg.
IL-6 signals via three distinct mechanisms: classical, trans- and cluster signaling.
IL-6/IL-6R axis is dysregulated in autoimmunity.
Targeting the IL-6 pathway has proven to be therapeutic in autoimmunity.
Acknowledgements
This work was supported by the National Institutes of Health (Grant numbers: 1 DP3 DK104466 and 5 R01 AI132774). The authors wish to thank Anne Hocking for editing and proofing the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
Papers of particular interest, published with the period of review, have been highlighted as:
● Of special interest
●● Of outstanding interest
- 1.Tanaka T, Narazaki M, Kishimoto T: IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol 2014, 6:a016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hirano T, Matsuda T, Turner M, Miyasaka N, Buchan G, Tang B, Sato K, Shimizu M, Maini R, Feldmann M, et al. : Excessive production of interleukin 6/B cell stimulatory factor-2 in rheumatoid arthritis. Eur J Immunol 1988, 18:1797–1801. [DOI] [PubMed] [Google Scholar]
- 3.Linker-Israeli M, Deans RJ, Wallace DJ, Prehn J, Ozeri-Chen T, Klinenberg JR: Elevated levels of endogenous IL-6 in systemic lupus erythematosus. A putative role in pathogenesis. J Immunol 1991, 147:117–123. [PubMed] [Google Scholar]
- 4.Maimone D, Guazzi GC, Annunziata P: IL-6 detection in multiple sclerosis brain. J Neurol Sci 1997, 146:59–65. [DOI] [PubMed] [Google Scholar]
- 5.Hunter CA, Jones SA: IL-6 as a keystone cytokine in health and disease. Nat Immunol 2015, 16:448–457. [DOI] [PubMed] [Google Scholar]
- 6.Kimura A, Kishimoto T: IL-6: regulator of Treg/Th17 balance. Eur J Immunol 2010, 40:1830–1835. [DOI] [PubMed] [Google Scholar]
- 7.Shao S, He F, Yang Y, Yuan G, Zhang M, Yu X: Th17 cells in type 1 diabetes. Cell Immunol 2012, 280:16–21. [DOI] [PubMed] [Google Scholar]
- 8.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK: Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441:235–238. [DOI] [PubMed] [Google Scholar]
- 9.Choi S-C, Xu Z, Li W, Yang H, Roopenian DC, Morse HC, Morel L: Relative Contributions of B Cells and Dendritic Cells from Lupus-Prone Mice to CD4+ T Cell Polarization. J Immunol 2018:ji1701179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.● Zhao M, Tan Y, Peng Q, Huang C, Guo Y, Liang G, Zhu B, Huang Y, Liu A, Wang Z, et al. : IL-6/STAT3 pathway induced deficiency of RFX1 contributes to Th17-dependent autoimmune diseases via epigenetic regulation. Nat Commun 2018, 9:583.Novel mechanism by which IL-6 drives Th17 function via the transcription factor RFX1.
- 11.Ichiyama K, Gonzalez-Martin A, Kim B-S, Jin HY, Jin W, Xu W, Sabouri-Ghomi M, Xu S, Zheng P, Xiao C: The microRNA-183–96-182 cluster promotes T helper 17 cell pathogenicity by negatively regulating transcription factor Foxo1 expression. Immunity 2016, 44:1284–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pasare C, Medzhitov R: Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science 2003, 299:1033–1036. [DOI] [PubMed] [Google Scholar]
- 13.Goodman WA, Levine AD, Massari JV, Sugiyama H, McCormick TS, Cooper KD: IL-6 signaling in psoriasis prevents immune suppression by regulatory T cells. J Immunol 2009, 183:3170–3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schneider A, Long SA, Cerosaletti K, Ni CT, Samuels P, Kita M, Buckner JH: In active relapsing-remitting multiple sclerosis, effector T cell resistance to adaptive T(regs) involves IL-6-mediated signaling. Sci Transl Med 2013, 5:170ra115. [DOI] [PubMed] [Google Scholar]
- 15.Ferreira RC, Rainbow DB, Rubio Garcia A, Pekalski ML, Porter L, Oliveira JJ, Waldron-Lynch F, Wicker LS, Todd JA: Human IL-6R(hi)TIGIT(−) CD4(+)CD127(low)CD25(+) T cells display potent in vitro suppressive capacity and a distinct Th17 profile. Clin Immunol 2017, 179:25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.●● Heink S, Yogev N, Garbers C, Herwerth M, Aly L, Gasperi C, Husterer V, Croxford AL, Moller-Hackbarth K, Bartsch HS, et al. : Trans-presentation of IL-6 by dendritic cells is required for the priming of pathogenic TH17 cells. Nat Immunol 2017, 18:74–85.Initial characterization of cluster signaling in dendritic cells.
- 17.Li D, Kong C, Tsun A, Chen C, Song H, Shi G, Pan W, Dai D, Shen N, Li B: MiR-125a-5p Decreases the Sensitivity of Treg cells Toward IL-6-Mediated Conversion by Inhibiting IL-6R and STAT3 Expression. Sci Rep 2015, 5:14615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Venigalla RK, Tretter T, Krienke S, Max R, Eckstein V, Blank N, Fiehn C, Ho AD, Lorenz HM: Reduced CD4+,CD25- T cell sensitivity to the suppressive function of CD4+,CD25high,CD127 -/low regulatory T cells in patients with active systemic lupus erythematosus. Arthritis Rheum 2008, 58:2120–2130. [DOI] [PubMed] [Google Scholar]
- 19.Schneider A, Rieck M, Sanda S, Pihoker C, Greenbaum C, Buckner JH: The effector T cells of diabetic subjects are resistant to regulation via CD4+ FOXP3+ regulatory T cells. J Immunol 2008, 181:7350–7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.●● Ihantola E-L, Viisanen T, Gazali AM, Näntö-Salonen K, Juutilainen A, Moilanen L, Rintamäki R, Pihlajamäki J, Veijola R, Toppari J: Effector T Cell Resistance to Suppression and STAT3 Signaling during the Development of Human Type 1 Diabetes. J Immunol 2018:ji1701199.Teff resistance in T1D patients is STAT3-dependent but not linked to the capacity of Teffs to produce or respond to IL-6.
- 21.Haufe S, Schepp C, Kuemmerle-Deschner J, Hansmann S, Rieber N, Tzaribachev N, Hospach T, Maier J, Dannecker GE, Holzer U: Impaired suppression of synovial fluid CD4(+) CD25(−) T cells from patients with juvenile idiopathic arthritis by CD4(+) CD25(+) regulatory T cells. Arthritis Rheum 2011. October;63(10):3153–62. [DOI] [PubMed] [Google Scholar]
- 22.●● Tu E, Chia CP, Chen W, Zhang D, Park SA, Jin W, Wang D, Alegre M-L, Zhang YE, Sun L: T Cell Receptor-Regulated TGF-β Type I Receptor Expression Determines T Cell Quiescence and Activation. Immunity 2018, 48:745–759. e746.Novel mechanism involving IL-6 in the enforcement of quiescence in naïve T cells.
- 23.Rosser EC, Oleinika K, Tonon S, Doyle R, Bosma A, Carter NA, Harris KA, Jones SA, Klein N, Mauri C: Regulatory B cells are induced by gut microbiota–driven interleukin-1β and interleukin-6 production. Nat Medicine 2014, 20:1334. [DOI] [PubMed] [Google Scholar]
- 24.Ma J, Zhu C, Ma B, Tian J, Baidoo SE, Mao C, Wu W, Chen J, Tong J, Yang M, et al. : Increased frequency of circulating follicular helper T cells in patients with rheumatoid arthritis. Clin Dev Immunol 2012, 2012:827480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barr TA, Shen P, Brown S, Lampropoulou V, Roch T, Lawrie S, Fan B, O’Connor RA, Anderton SM, Bar-Or A, et al. : B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6-producing B cells. J Exp Med 2012, 209:1001–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.● Arkatkar T, Du SW, Jacobs HM, Dam EM, Hou B, Buckner JH, Rawlings DJ, Jackson SW: B cell-derived IL-6 initiates spontaneous germinal center formation during systemic autoimmunity. J Exp Med 2017, 214:3207–3217.IL-6 produced by B cells drives formation of germinal centers and Tfh differentiation.
- 27.de Valle E, Grigoriadis G, O’Reilly LA, Willis SN, Maxwell MJ, Corcoran LM, Tsantikos E, Cornish JK, Fairfax KA, Vasanthakumar A: NFκB1 is essential to prevent the development of multiorgan autoimmunity by limiting IL-6 production in follicular B cells. J Exp Med 2016, 213:621–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Narazaki M, Yasukawa K, Saito T, Ohsugi Y, Fukui H, Koishihara Y, Yancopoulos GD, Taga T, Kishimoto T: Soluble forms of the interleukin-6 signal-transducing receptor component gp130 in human serum possessing a potential to inhibit signals through membrane-anchored gp130. Blood 1993, 82:1120–1126. [PubMed] [Google Scholar]
- 29.Jostock T, Mullberg J, Ozbek S, Atreya R, Blinn G, Voltz N, Fischer M, Neurath MF, Rose-John S: Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur J Biochem 2001, 268:160–167. [DOI] [PubMed] [Google Scholar]
- 30.Lust JA, Donovan KA, Kline MP, Greipp PR, Kyle RA, Maihle NJ: Isolation of an mRNA encoding a soluble form of the human interleukin-6 receptor. Cytokine 1992, 4:96–100. [DOI] [PubMed] [Google Scholar]
- 31.Briso EM, Dienz O, Rincon M: Cutting edge: soluble IL-6R is produced by IL-6R ectodomain shedding in activated CD4 T cells. J Immunol 2008, 180:7102–7106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B: TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 2006, 24:179–189. [DOI] [PubMed] [Google Scholar]
- 33.Rabe B, Chalaris A, May U, Waetzig GH, Seegert D, Williams AS, Jones SA, Rose-John S, Scheller J: Transgenic blockade of interleukin 6 transsignaling abrogates inflammation. Blood 2008, 111:1021–1028. [DOI] [PubMed] [Google Scholar]
- 34.Samson M, Audia S, Janikashvili N, Ciudad M, Trad M, Fraszczak J, Ornetti P, Maillefert JF, Miossec P, Bonnotte B: Brief report: inhibition of interleukin-6 function corrects Th17/Treg cell imbalance in patients with rheumatoid arthritis. Arthritis Rheum 2012, 64:2499–2503. [DOI] [PubMed] [Google Scholar]
- 35.Quintana FJ: Old dog, new tricks: IL-6 cluster signaling promotes pathogenic TH17 cell differentiation. Nat Immunol 2016, 18:8–10. [DOI] [PubMed] [Google Scholar]
- 36.van Dongen J, Jansen R, Smit D, Hottenga JJ, Mbarek H, Willemsen G, Kluft C, Collaborators A, Penninx BW, Ferreira MA, et al. : The contribution of the functional IL6R polymorphism rs2228145, eQTLs and other genome-wide SNPs to the heritability of plasma sIL-6R levels. Behav Genet 2014, 44:368–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Amaral WZ, Krueger RF, Ryff CD, Coe CL: Genetic and environmental determinants of population variation in interleukin-6, its soluble receptor and C-reactive protein: insights from identical and fraternal twins. Brain Behav Immun 2015, 49:171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garbers C, Monhasery N, Aparicio-Siegmund S, Lokau J, Baran P, Nowell MA, Jones SA, Rose-John S, Scheller J: The interleukin-6 receptor Asp358Ala single nucleotide polymorphism rs2228145 confers increased proteolytic conversion rates by ADAM proteases. Biochim Biophys Acta 2014, 1842:1485–1494. [DOI] [PubMed] [Google Scholar]
- 39.Ferreira RC, Freitag DF, Cutler AJ, Howson JM, Rainbow DB, Smyth DJ, Kaptoge S, Clarke P, Boreham C, Coulson RM, et al. : Functional IL6R 358Ala allele impairs classical IL-6 receptor signaling and influences risk of diverse inflammatory diseases. PLoS Genet 2013, 9:e1003444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.● Hundhausen C, Roth A, Whalen E, Chen J, Schneider A, Long SA, Wei S, Rawlings R, Kinsman M, Evanko SP, et al. : Enhanced T cell responses to IL-6 in type 1 diabetes are associated with early clinical disease and increased IL-6 receptor expression. Sci Transl Med 2016, 8:356ra119.A role for dysregulated IL-6 responsiveness in the pathogenesis of type 1 diabetes.
- 41.Nishimoto N, Yoshizaki K, Miyasaka N, Yamamoto K, Kawai S, Takeuchi T, Hashimoto J, Azuma J, Kishimoto T: Treatment of rheumatoid arthritis with humanized anti–interleukin‐ 6 receptor antibody: a multicenter, double‐ blind, placebo‐ controlled trial. Arthritis Rheum 2004, 50:1761–1769. [DOI] [PubMed] [Google Scholar]
- 42.Calabrese LH, Rose-John S: IL-6 biology: implications for clinical targeting in rheumatic disease. Nat Rev Rheumatol 2014, 10:720–727. [DOI] [PubMed] [Google Scholar]
- 43.Khanna D, Denton CP, Jahreis A, van Laar JM, Frech TM, Anderson ME, Baron M, Chung L, Fierlbeck G, Lakshminarayanan S, et al. : Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): a phase 2, randomised, controlled trial. Lancet 2016, 387:2630–2640. [DOI] [PubMed] [Google Scholar]
- 44.Araki M, Matsuoka T, Miyamoto K, Kusunoki S, Okamoto T, Murata M, Miyake S, Aranami T, Yamamura T: Efficacy of the anti-IL-6 receptor antibody tocilizumab in neuromyelitis optica: a pilot study. Neurology 2014, 82:1302–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Genovese MC, Fleischmann R, Kivitz AJ, Rell‐ Bakalarska M, Martincova R, Fiore S, Rohane P, van Hoogstraten H, Garg A, Fan C: Sarilumab plus methotrexate in patients with active rheumatoid arthritis and inadequate response to methotrexate: results of a phase III study. Arthritis Rheum 2015, 67:1424–1437. [DOI] [PubMed] [Google Scholar]
- 46.van Rhee F, Wong RS, Munshi N, Rossi JF, Ke XY, Fossa A, Simpson D, Capra M, Liu T, Hsieh RK, et al. : Siltuximab for multicentric Castleman’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol 2014, 15:966–974. [DOI] [PubMed] [Google Scholar]
- 47.Torices S, Julia A, Munoz P, Varela I, Balsa A, Marsal S, Fernandez-Nebro A, Blanco F, Lopez-Hoyos M, Martinez-Taboada V, et al. : A functional variant of TLR10 modifies the activity of NFkB and may help predict a worse prognosis in patients with rheumatoid arthritis. Arthritis Res Ther 2016, 18:221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mease PJ, Gottlieb AB, Berman A, Drescher E, Xing J, Wong R, Banerjee S: The Efficacy and Safety of Clazakizumab, an Anti–Interleukin‐ 6 Monoclonal Antibody, in a Phase IIb Study of Adults With Active Psoriatic Arthritis. Arthritis Rheum 2016, 68:2163–2173. [DOI] [PubMed] [Google Scholar]
- 49.Orlowski RZ, Gercheva L, Williams C, Sutherland H, Robak T, Masszi T, Goranova-Marinova V, Dimopoulos MA, Cavenagh JD, Spicka I, et al. : A phase 2, randomized, double-blind, placebo-controlled study of siltuximab (anti-IL-6 mAb) and bortezomib versus bortezomib alone in patients with relapsed or refractory multiple myeloma. Am J Hematol 2015, 90:42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shah JJ, Feng L, Thomas SK, Berkova Z, Weber DM, Wang M, Qazilbash MH, Champlin RE, Mendoza TR, Cleeland C, et al. : Siltuximab (CNTO 328) with lenalidomide, bortezomib and dexamethasone in newly-diagnosed, previously untreated multiple myeloma: an open-label phase I trial. Blood Cancer J 2016, 6:e396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kennedy Crispin M, Ko JM, Craiglow BG, Li S, Shankar G, Urban JR, Chen JC, Cerise JE, Jabbari A, Winge MC, et al. : Safety and efficacy of the JAK inhibitor tofacitinib citrate in patients with alopecia areata. JCI Insight 2016, 1:e89776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bachelez H, van de Kerkhof PC, Strohal R, Kubanov A, Valenzuela F, Lee JH, Yakusevich V, Chimenti S, Papacharalambous J, Proulx J, et al. : Tofacitinib versus etanercept or placebo in moderate-to-severe chronic plaque psoriasis: a phase 3 randomised non-inferiority trial. Lancet 2015, 386:552–561. [DOI] [PubMed] [Google Scholar]
- 53.Kivitz A, Gutierrez‐ Ureña S, Poiley J, Genovese M, Kristy R, Shay K, Wang X, Garg J, Zubrzycka‐ Sienkiewicz A: Peficitinib, a JAK Inhibitor, in the Treatment of Moderate‐to‐Severe Rheumatoid Arthritis in Patients With an Inadequate Response to Methotrexate. Arthritis Rheum 2017, 69:709–719. [DOI] [PubMed] [Google Scholar]
- 54.Kikuchi J, Hashizume M, Kaneko Y, Yoshimoto K, Nishina N, Takeuchi T: Peripheral blood CD4(+)CD25(+)CD127(low) regulatory T cells are significantly increased by tocilizumab treatment in patients with rheumatoid arthritis: increase in regulatory T cells correlates with clinical response. Arthritis Res Ther 2015, 17:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.● Tada Y, Ono N, Suematsu R, Tashiro S, Sadanaga Y, Tokuda Y, Ono Y, Nakao Y, Maruyama A, Ohta A, et al. : The balance between Foxp3 and Ror-gammat expression in peripheral blood is altered by tocilizumab and abatacept in patients with rheumatoid arthritis. BMC Musculoskelet Disord 2016, 17:290.The ratio of Foxp3/Ror-ɣt decreased after abatacept treatment and increased after treatment with tocilizumab.
- 56.Thiolat A, Semerano L, Pers Y, Biton J, Lemeiter D, Portales P, Quentin J, Jorgensen C, Decker P, Boissier MC: Interleukin‐ 6 receptor blockade enhances CD39+ regulatory T cell development in rheumatoid arthritis and in experimental arthritis. Arthritis Rheum 2014, 66:273–283. [DOI] [PubMed] [Google Scholar]
- 57.Daïen CI, Gailhac S, Audo R, Mura T, Hahne M, Combe B, Morel J: High levels of natural killer cells are associated with response to tocilizumab in patients with severe rheumatoid arthritis. Rheumatology 2014, 54:601–608. [DOI] [PubMed] [Google Scholar]
- 58.Weinhold KJ, Bukowski JF, Brennan TV, Noveck RJ, Staats JS, Lin L, Stempora L, Hammond C, Wouters A, Mojcik CF, et al. : Reversibility of peripheral blood leukocyte phenotypic and functional changes after exposure to and withdrawal from tofacitinib, a Janus kinase inhibitor, in healthy volunteers. Clin Immunol 2018, 191:10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rizzi M, Lorenzetti R, Fischer K, Staniek J, Janowska I, Troilo A, Strohmeier V, Erlacher M, Kunze M, Bannert B, et al. : Impact of tofacitinib treatment on human B-cells in vitro and in vivo. J Autoimmun 2017, 77:55–66. [DOI] [PubMed] [Google Scholar]
- 60.● Lyman M, Lieuw V, Richardson R, Timmer A, Stewart C, Granger S, Woods R, Silacci M, Grabulovski D, Newman R: A bispecific antibody that targets IL-6 receptor and IL-17A for the potential therapy of patients with autoimmune and inflammatory diseases. J Biol Chem 2018, 293:9326–9334.Novel therapeutic targeting of IL-17A and IL-6R with potential for enhanced efficiency.