

Abstract
Positive modulators of NMDA receptors are important candidates for therapeutic development to treat psychiatric disorders including autism and schizophrenia. Sulfated neurosteroids have been studied as positive allosteric modulators of NMDA receptors for years, but we understand little about the cellular fate of these compounds, an important consideration for drug development. Here we focus on a visualizable sulfated neurosteroid analogue, KK-169. As expected of a pregnenolone sulfate analogue, the compound strongly potentiates NMDA receptor function, is an antagonist of GABAA receptors, exhibits occlusion with pregnenolone sulfate potentiation, and requires receptor domains important for pregnenolone sulfate potentiation. KK-169 exhibits somewhat higher potency than the natural parent, pregnenolone sulfate. The analogue contains a side-chain alkyne group, which we exploited for retrospective click labeling of neurons. Although the anionic sulfate group is expected to hinder cell entry, we detected significant accumulation of KK-169 in neurons with even brief incubations. Adding a photolabile diazirine group revealed that the expected plasma membrane localization of KK-169 is likely lost during fixation. Overall, our studies reveal new facets of the structure-activity relationship of neurosteroids at NMDA receptors, and their intracellular distribution suggests that sulfated neurosteroids could have unappreciated targets in addition to plasma membrane receptors.
Keywords: Pregnenolone sulfate, glutamate, NMDA, GABA, click chemistry, photolabeling
1. Introduction
Positive modulators of NMDA receptors (NMDARs) are important candidates for therapeutic development to treat psychiatric disorders (Hansen et al., 2017; Reddy, 2010; Smith et al., 2014; Strong et al., 2014; Sun et al., 2015). Sulfated neurosteroids are of particular interest because they modulate both inhibition and excitation through effects on GABAA receptors (GABARs) and NMDARs respectively. Effects on NMDARs depend on both subunit composition and on steroid structure. Pregnane sulfates produce mainly NMDAR inhibition, and pregnenolone sulfate (PREGS) induces potentiation (Korinek et al., 2011; Malayev et al., 2002; Park-Chung et al., 1994; Park-Chung et al., 1997). Although modulators of NMDARs may have clinical utility (Abdallah et al., 2015; Collingridge et al., 2013; Lai et al., 2011; Sun et al., 2015; Vyklicky et al., 2016), our understanding of the effects of PREGS at these receptors remains limited, including basic structure-activity information (Burnell et al., 2018). How PREGS-like compounds are trafficked or compartmentalized in neurons is also unclear, even though compartmentalization may affect receptor access (Jiang et al., 2016) and may hint at additional targets. For instance, in some cases neuroactive compounds may sequester in intracellular organelles, followed by release into the extracellular space (Tischbirek et al., 2012). An important barrier to understanding is the lack of biologically active, visualizable analogues to address cellular distribution. Such compounds may also yield new insights into structure-activity relationships. We have begun to address these fundamental issues with novel, biologically active probes.
Previous work has been equivocal regarding the ability of NMDAR-active sulfated neurosteroids to enter cells and sequester therein. The electronegativity of the sulfate group would seem to prohibit cell entry (Eisenman et al., 2007), compared with un-sulfated GABAA receptor potentiators, which sequester in specific intracellular organelles (Jiang et al., 2016). However, unknown carrier molecules, active transport mechanisms, or other routes of cell entry may exist for sulfated neurosteroids.
Previous efforts to evaluate subcellular distribution have utilized fluorescently labeled neurosteroid analogues (Borovska et al., 2012; Eisenman et al., 2007). Because fluorescent moieties themselves may alter the physical properties, cell permeability and trafficking of steroid analogues, here we took the approach of a more modest chemical alteration that permitted retrospective click chemistry for visualization. This alteration retained modulatory potency and efficacy at NMDARs compared with PREGS and the oxysterol class of NMDAR modulators. One lead compound, KK-169, exhibited readily reversible NMDAR potentiation. Other analogues with longer carbon side-chain lengths exhibited slowly reversible potentiation. KK-169 showed expected features of PREGS neuroactivity with no discernible presynaptic activity at concentrations employed. Retrospective click chemistry revealed that KK-169 accumulates inside neurons, a feature associated with its hydrophobicity based on comparison with other analogues. Our results begin to reveal advantages and limits of chemical biology tools for probing the pharmacology of NMDAR positive allosteric modulators.
2. Material and Methods
2.1. Xenopus oocytes
Stage V–VI oocytes were obtained from sexually mature female Xenopus laevis (Xenopus One) anesthetized with 0.1% tricaine (3-aminobenzoic acid ethyl ester). To achieve defolliculation, oocytes were incubated for 20 min at 37°C in a calcium-free collagenase (2 mg/ml) solution containing (in mM): NaCl (96), KCl (2), MgCl2 (1), and HEPES (5) at pH 7.4. cRNA for receptor subunits was prepared using the mMESSAGE mMachine in vitro transcription kit (Ambion, Austin, TX, USA). Capped mRNA, encoding rat NMDAR GluN1a and GluN2A subunits or rat GABAA receptor α1, β2, γ2L subunits were injected in equal parts (3–13 ng RNA for each receptor subunit). Oocytes were incubated at 18°C in ND96 medium containing (in mM): NaCl (96), KCl (1), MgCl2 (1), CaCl2 (2) and HEPES (10) at pH 7.4, supplemented with pyruvate (5 mM), penicillin (100 U ml−1), streptomycin (100 µg ml−1) and gentamycin (50 µg ml−1) for up to 5 days.
2.2. Cell cultures
All animal care and experimental procedures were consistent with National Institutes of Health guidelines and were approved by the Washington University Animal Studies Committee. Studies involving animals are reported in accordance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath et al., 2010). Sprague-Dawley rat pups (postnatal day 1 to 3), were anesthetized with isoflurane. Hippocampal slices (500 µm thickness) were digested with 1 mg/ml papain in oxygenated Leibovitz L-15 medium (Life Technologies, Gaithersburg, MD), followed by mechanical trituration in modified Eagle’s medium (MEM; Life Technologies) containing 5% horse serum, 5% FCS, 17 mM Dglucose, 400 µM glutamine, 50 U/ml penicillin, and 50 µg/ml streptomycin. Combined astrocytes and neurons were seeded at a density of 600 cells/mm2 onto 35-mm dishes or 25-mm cover glasses coated with 0.1 mg/ml poly-D-lysine and 1 mg/ml laminin. Cultures subsequently developed at 37°C in a chamber with 5% CO2 and controlled humidity. Glial proliferation was inhibited by adding cytosine arabinoside (6.7 µM) 3–4 days after plating. The following day, culture medium was switched to Neurobasal medium (Life Technologies) plus B27 supplement (Life Technologies).
2.3. Click labeling
Cell cultures were incubated with 10 µM KK-169, KK-181 or MQ-189 for 15 min in saline containing (in mM): NaCl (138 ), KCl (4), CaCl2 (2), MgCl2 (1), glucose (10), HEPES (10), pH 7.25 with NaOH, 1 µM 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione (NBQX) and 50 µM D-(−)-2-Amino-5phosphonopentanoic acid (D-APV). For some experiments cells were exposed to 365 nm light using a 0.15 A Blak-Ray lamp for 15 min at a distance of 2 cm. Afterward, cells were immediately fixed for click-chemistry labeling with 4% paraformaldehyde plus 0–0.05% glutaraldehyde in phosphate buffered saline at room temperature for 10 minutes. After a phosphate-buffer saline (PBS) wash, fixed cells were incubated in 1 µM AlexaFluor 488-azide, a click reagent for fluorescence visualization (ThermoFisher Scientific), in 100 µM Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine, 2 mM ascorbic acid,1 mM CuSO4 for 1 h in the dark, followed by three washes in PBS. Cells were coverslipped for subsequent imaging. Epifluorescence photomicrographs were acquired using a Nikon TE2000 inverted microscope with a PlanFluor 40x microscope objective (0.6 N.A.) and a Coolsnap ES2 camera (Photometrics). Images were analyzed using the Fiji distribution of ImageJ imaging software (http://fiji.sc/Fiji).
2.4. Electrophysiology recordings in Xenopus oocytes
Evaluation of receptor modulation in Xenopus oocytes was performed with an OC725 amplifier (Warner Instruments) 2–5 d after RNA injection, in a two-electrode voltage-clamp configuration at −70 mV. Oocytes were transferred to a chamber containing unsupplemented ND96 solution. For NMDA recordings, Ba2+ was substituted for Ca2+ to prevent contributions of endogenous Ca2+ activated current, MgCl2 was omitted, and 10 µM D-serine was added to the bath. Recording pipettes were filled with 3 M KCl and had open tip resistances of ~1 MΩ. Drug application was achieved using a gravitydriven multibarrel delivery system with a common tip. Each drug was applied for 30 s and current elicited by each compound was evaluated at the peak current or the current at the end the application (as indicated).
2.5. Electrophysiology recordings in neuronal cultures and N2a cells
Current recordings were made with an Axopatch 200B amplifier (Molecular Devices) in whole-cell configuration, from neurons 6–13 days in vitro. 6–8 day-old hippocampal cells were used for recordings of exogenous agonist responses because of improved spatial voltage clamp in younger neurons with limited processes; 10–13 day-old cultures were used for EPSC recordings because of synaptic maturity. N2a cells were used 24 h following transfection. For recordings, cells were transferred to an extracellular solution containing (in mM): NaCl (138), KCl (4), CaCl2 (2), glucose (10), HEPES (10), Dserine (0.02), NBQX (0.001), and D-APV (0.01), bicuculline (0.01) at pH 7.25. NMDAR autaptic current was recorded in a D-APV-free solution, whereas AMPA current was recorded in NBQX free solution and GABAA autaptic current in bicuculline free solution. No antagonists were used for N2a cell recordings. The open-tip resistance of patch-pipette for recordings was 2.5–3.5 MΩ when filled with an internal solution containing (in mM): KCl (140), NaCl (4), EGTA (0.1), and HEPES (10) at pH 7.25, adjusted with KOH. Access resistance (8–10 MΩ) was compensated 80–100% for synaptic recordings. For autaptic responses, cells were stimulated with 1.5-ms pulses to 0 mV from −70 mV to evoke transmitter release. Drugs were applied using a multibarrel, gravity-driven local perfusion system with a common tip placed 0.5 mm from the center of the microscope field. For recordings using exogenous drug application, patch-pipettes were filled with a solution containing in (mM): cesium methanesulfonate (120), HEPES (20), EGTA (10) at pH 7.25 adjusted with CsOH. For these experiments, cells were clamped at −70 mV. All electrophysiology recordings were performed at room temperature.
2.6. Chimeric receptors and cell transfection
Recombinant chimeric receptors were as described (Wilding et al., 2014, 2016). HEK cells were propagated in 25 cm2 flasks with MEM plus 10% fetal bovine serum and passaged once each week with protease XXIII (Sigma-Aldrich). Cells used for transfection were seeded onto 12-well plates and transfected the following day. cDNAs were expressed by transient transfection in HEK 293 cells using Lipofectamine 2000 (Invitrogen). Coexpression of GFP from a second vector was used to identify transfected cells. The day after transfection, cells were plated at low density on 35 mm plates coated with nitrocellulose; recordings were obtained on the following 2 d.
Neuro2A (N2A) cells were propagated in 25 cm2 flasks with DMEM plus 10% fetal bovine serum and passaged with 0.05% trypsin-EDTA (Life Technologies). Cells were seeded onto 35 mm dishes and, the following day cDNAs for NMDAR subunits (GluNR1/GluNR2A-D) were expressed by transient transfection along with GFP plasmid using Lipofectamine 2000 (Invitrogen).
2.7. Chemistry
Novel steroid analogue synthesis was through a multi-step process described in the supplemental material. LogP values were calculated using the freely available ALOGPS 2.1 software available from the Virtual Computational Chemistry Lab: http://www.vcclab.org/. Detailed chemical synthesis is supplied as Supplemental material.
2.8. Data analysis
Data were acquired and initially analyzed with pCLAMP 10 software (Molecular Devices) for electrophysiology and ImageJ for images. Data were subsequently processed with Microsoft Excel and are presented as means ± S.E. Statistical significance was determined using a Student’s twotailed t-test, ANOVA, or Mann-Whitney test as indicated in figure legends. Statistical analysis and fitting were executed with GraphPad Prism (GraphPad Software, La Jolla, CA) or Sigma Plot (San Jose, CA). For fitting of memantine onset and offset kinetics a weighted time constant was calculated from bi-exponential fits using the calculated as ΣAi*τi, where Ai is the fractional amplitude and τi is the time constant of the component. p values are indicated in Figures as * p < 0.05, ** p < 0.01, *** p < 0.005.
2.9. Drugs.
All commercial drugs and salts were obtained from Sigma (St. Louis, MO), except for D-APV and NBQX, which were obtained from Tocris.
3. Results
3.1. Bioactivity of KK-169 and related compounds at NMDARs
Based on the ability of sulfated steroids to increase NMDAR function allosterically during agonist activation, we studied the effect of newly synthetized compounds with different side chain lengths at carbon 17. These modifications supported an alkyne group for retrospective click chemistry (Figure 1A). We tested the NMDAR activity of these new compounds in Xenopus oocytes co-expressing GluN1a and GluN2A subunits. Oocytes were challenged with NMDA (5 µM) and with increasing concentrations of test compounds (0.3 µM up to 30 µM) (Figure 1B). Each compound increased NMDA current in a concentration-dependent manner. Among the active compounds, KK-169 exhibited reversible potentiation of NMDA current. Other compounds such as KK-171 and KK-181 increased NMDA current more effectively than KK-169 (1 to 3 fold), but such potentiation was not efficiently removed upon wash (Figure 1B), perhaps because longer side chains increased lipophilicity and promoted retention. We prepared MQ-154 as a potential photolabeling compound (Figure 1A). However, the changes made to permit attachment of the photolabeling group on the side chain abolished NMDAR activity (Figure 1B). Because of its strong biological activity and favorable reversibility, we selected KK-169 as a click-accessible lead compound to study NMDA potentiation by sulfated steroids.
Figure 1.

Biological activity at NMDARs is retained with varied side chain lengths in PREGS analogues. A. The structures of first-generation alkyne-conjugated PREGS analogues. MQ-154 differed from others most notably in the presence of an amide linkage to a side-chain diazirine group. B. Effect of varied concentrations of respective compounds on NMDA-induced current in Xenopus oocytes expressing recombinant GluN1 and GluN2A subunits. NMDA (5 µM) was applied for 50 s in the absence of compound to establish baseline responsiveness, followed by 50 s co-application. Each application/co-application was followed by a 25 s wash period before the ensuing application. Responses are displayed normalized to the original NMDA current (dotted line). N = 4–7 oocytes per compound.
Representative traces for KK-169 potentiation of glutamate-gated current are shown in Figure 2A. In the absence of glutamate, KK-169 did not detectably gate a current (Figure 2B). Analysis of potentiation at varied KK-169 concentrations suggested an EC50 concentration of 6.2 µM (Figure 2C). We next tested KK-169 while varying the concentration of the endogenous agonist glutamate (Figure 3). Responses to low glutamate concentration (e.g., 1 µM) were strongly potentiated, producing a desensitizing response at a concentration of glutamate that did not desensitize in the absence of KK169 (Figure 3A, B). The primary effect of the modulator was to shift the concentration-response for peak glutamate responses to lower agonist concentrations (from ~10 µM to ~3 µM), and also shifted the Hill coefficient (limiting slope) to a lower value (Figure 3C). We acknowledge that strong desensitization at the highest concentrations of glutamate and KK-169 may affect accuracy of measurements, given that drug delivery to oocytes may be slow relative to desensitization rate.
Figure 2.

Potency and efficacy of KK-169. A. Responses of a GluN1 and GluN2A-expressing oocyte to 5 µM NMDA and increasing concentrations of KK-169, co-applied with NMDA. The traces in panel A are color coded according to concentrations in panel C. B. 30 µM KK-169 alone (blue trace) did not gate a current in oocytes with a confirmed response to 5 µM NMDA (black trace). C. A fit of the Hill equation to the responses to increasing KK-169 concentrations yielded an EC50 value of 6.2 µM ± 0.8 and a Hill coefficient of 0.6 ± 0.1. N = 4 cells at 100 µM and 8–11 cells for other concentrations.
Figure 3.

Effect of KK-169 on glutamate (Glu) concentration-response parameters. A. Baseline NMDA responses in the absence of KK-169 from an oocyte expressing GluN1 and GluN2A subunits. B. Response to the lowest concentration of glutamate co-applied with 30 µM KK-169. C. Full glutamate concentration response for peak current recorded in the same cells in the absence and presence of 30 µM KK-169. Responses were normalized to peak current evoked by 100 µM glutamate in the absence of KK-169. Fits to the Hill equation yielded EC50 values of 4.8 vs. 0.22 µM for glutamate alone and plus KK-169 respectively, and Hill coefficients of 1.2 and 0.85 respectively. N = 4 oocytes for each condition.
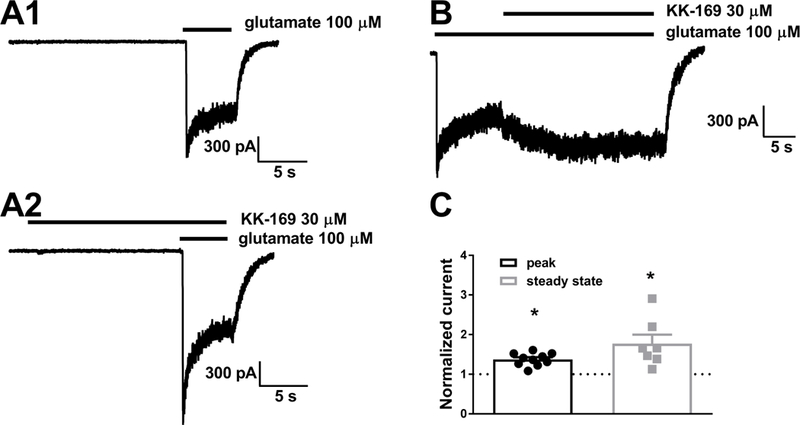
To evaluate whether, in addition to increasing the potency of glutamate, KK-169 also increases agonist efficacy, we challenged native receptors in young cultured hippocampal neurons, where more rapid applications of drug could be achieved, thereby allowing more accurate measurement of peak current. Figure 4 shows that 30 µM KK-169 had modest but statistically significant effects on both peak and steady-state responses to 100 µM glutamate. We conclude that KK-169 increases agonist efficacy, although this effect alone is unlikely to account for the shift in agonist EC50.
Figure 4.

Effect of KK-169 on saturating agonist responses. A. A cultured hippocampal neuron was challenged with indicated concentrations and exposure times of glutamate and KK-169. B. To explicitly test the effect of KK-169 on steady-state responses to saturating glutamate, KK-169 was applied during the NMDA application in another cell. C. Summary of results from the protocols in A and B on peak and steady state NMDA responses respectively. Both peak and steady state were significantly potentiated by KK-169. N = 7 cells. Asterisks indicate paired t-tests, p < 0.05.
Previous work has shown that prolonged incubation in PREGS at low nM concentrations potentiates NMDA currents in Xenopus oocytes, suggesting high-affinity interaction (Kostakis et al., 2013). Consistent with these observations, we found that hippocampal neurons incubated for 3 min in 10 nM (n = 9 cells) or 100 nM (n = 11 cells) KK-169 exhibited modest potentiation over baseline NMDA responses (1.2 ± 0.09; p = 0.04 and 1.6 ± 0.16, p = 0.007 respectively, paired one-sample t tests).
Based on the presence of the sulfate group at carbon 3, we expected that KK-169 may inhibit GABAA receptor activity since the structural demands for GABAA receptor inhibition by sulfated steroids are low (Mennerick et al., 2001; Nilsson et al., 1998) and are evolutionarily conserved (Twede et al., 2007). In hippocampal neurons, GABA (10 µM) co-applied with KK-169 elicited a rapidly decaying current with hallmarks expected of sulfated steroid inhibition (Eisenman et al., 2003; Shen et al., 2000) (Figure 5A). Testing a concentration series for KK-169 produced an IC50 at 10 µM GABA of 2.2 µM for KK-169 (Figure 5B). These data suggest that KK-169 retains features of other sulfated neuroactive steroids, including actions at both NMDA and inhibition of GABAA receptors.
Figure 5.

Effect of KK-169 on GABAA receptor function. A. Effect of co-application of KK-169 at the indicated concentrations with GABA (10 µM) in a hippocampal neuron. The peak GABA current showed little inhibition, consistent with a PREGS-like mechanism (Eisenman et al., 2003; Shen et al., 2000). B. Summary of effects on ending GABA current during the co-application protocol shown in A. N = 9 neurons. Fit to the data with the Hill equation (solid line) predicts an IC50 of 2.2 µM.
3.2. Effects on synaptic transmission
To explore the impact of KK-169 on neuronal transmission, including potential off-target effects, we evaluated the effects of KK-169 on EPSCs in hippocampal neurons (Figures 6, 7). KK-169 significantly increased the amplitude and decay of recurrent (autaptic) evoked NMDA EPSCs (Figure 6). These effects were readily reversible (Figure 6A, B). The concentration response relationship for KK-169 on evoked NMDAR EPSCs was similar to that expected from effects observed on agonist-evoked currents in oocytes (Figure 6D).
Figure 6.

Effect of KK-169 and PREGS on NMDAR EPSCs. A-C. Influence of 30 µM KK-169 on the indicated parameters of pharmacologically isolated, evoked NMDAR EPSCs. Decay time was measured from 10–90% of the peak response. (N = 6 autaptic neurons). Asterisks denote results of paired t tests. D. Summary of concentration-response effects on normalized peak NMDAR EPSCs (N = 7–9 cells per condition, except 2 cells at the 5 µM concentration, EC50 = 3.7 µM with a Hill slope of 3.7. E-G. Effect of equimolar concentrations (30 µM) of KK-169 and PREGS on evoked NMDAR EPSCs (N = 6 comparisons by paired t-test).
Figure 7.
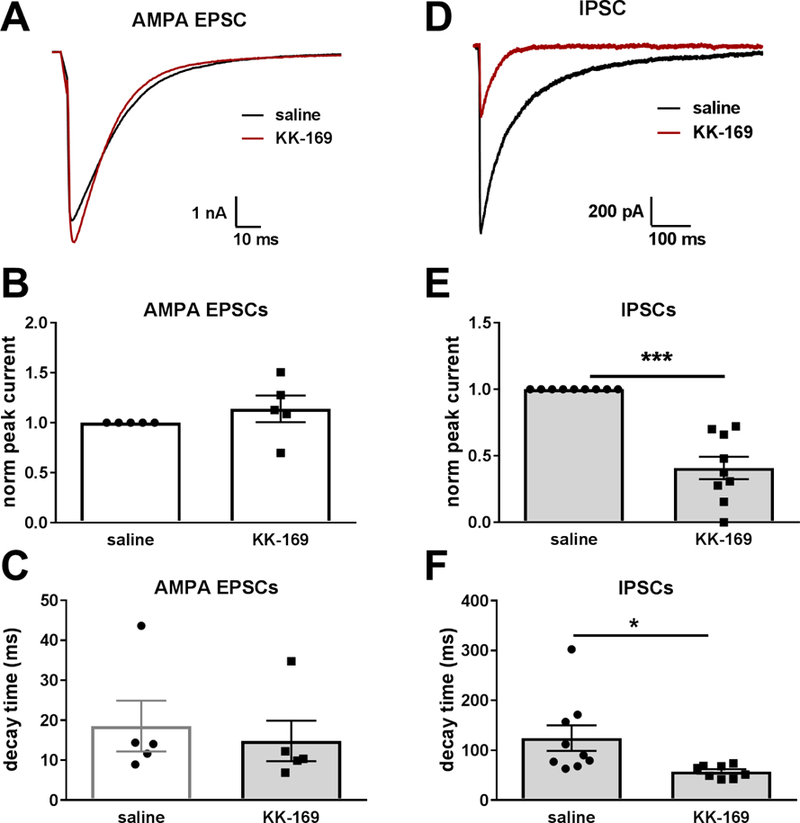
Effect of KK-169 on evoked AMPAR and GABAAR PSCs. A-C. Effect of 30 µM KK-169 on parameters of pharmacologically isolated, evoked autaptic AMPAR EPSCs (N = 5 cells tested in the absence and presence of KK-169). D-F. Effect of 30 µM KK-169 on parameters of pharmacologically isolated autaptic evoked GABAAR IPSCs. Pipette solution included KCl (N = 6 cells tested in the absence and presence of KK-169).
To compare KK-169 with the effect of a canonical positive allosteric modulator, we compared effects of equimolar PREGS (30 µM) on individual cells’ evoked NMDAR EPSCs. Effects on peak current EPSCs observed in the presence of KK-169 were compared to PREGS (Figure 6E-G). KK-169 increased peak current ~ 1.5 fold more than PREGS (Figure 6F). EPSC decay time in the presence of both drugs was approximately doubled compared to control conditions, and no statistical difference between effects of the two drugs on this parameter was found (Figure 6G).
To test selectivity, we tested KK-169 on pharmacologically isolated AMPAR EPSCs (in saline lacking NBQX but with D-APV added). Neither peak current nor decay time was altered by 30 µM KK-169 (Figure 7A-C). These results effectively exclude an important effect of KK-169 on presynaptic glutamate release or on AMPAR function. A presynaptic effect would affect NMDAR and AMPAR EPSCs similarly. The result also suggests a difference from PREGS, which inhibits AMPARs and kainate receptors (Wu and Chen, 1997; Yaghoubi et al., 1998). As expected, KK-169 (30 µM) also inhibited and speeded pharmacologically isolated inhibitory postsynaptic current (IPSCs) mediated by GABAA receptors (Figure 7D).
3.3. Mechanistic features of KK-169
A characteristic of many NMDAR positive allosteric modulators is that they alter the kinetics of openchannel trapping blockers such as ketamine and memantine (Emnett et al., 2015), suggesting a mechanism by which the positive modulator increases channel open probability and thus increases access of the channel to blocker. Consistent with this idea, we found that the presence of KK-169 decreased the time constant for memantine onset and offset during receptor activation by exogenous NMDA (Figure 8). These results support a mechanism by which KK-169 increases NMDAR channel open probability, although we cannot exclude a role for an increase in surface NMDAR number, as observed for recombinant NMDARs expressed in oocytes exposed to prolonged incubation of PREGS (Kostakis et al., 2013).
Figure 8.

KK-169 speeds kinetics of open-channel trapping block. A, B. Sample traces showing the effect of the trapping blocker memantine (10 µM) applied during the steady-state response to 10 µM NMDA of a hippocampal neuron. Panel A shows current in the absence of KK-169, and panel B shows current in the presence of 10 µM KK-169. Artifacts of electronic valve activation have been removed for clarity C. Summary of the effect of KK-169 on the amplitude of steady-state current in the absence of memantine and the alteration of block onset and offset kinetics (N = 10 neurons). The first bar shows the effect of KK-169 normalized to baseline NMDA current amplitude (dotted line), and the other bars show the memantine-induced onset and offset weighted time constants in the presence of KK-169 normalized to values in the absence (dotted line).
To test the assumption that KK-169 is a PREGS-like allosteric modulator, we performed a pharmacological occlusion test (Figure 9). In the presence of a near-saturating concentration of KK169, PREGS lost its ability to potentiate NMDAR currents (Figure 9A, B, E). As a negative comparator, we employed another class of NMDAR positive modulator, SGE-201, an analogue of the oxysterol cholesterol metabolite 24(S)-hydroxycholesterol (Paul et al., 2013). In the presence of KK169, SGE-201 retained potentiation. (Figure 9C, D, F). These results are consistent with a shared mechanism between PREGS and KK-169.
Figure 9.

KK-169 occlusion of PREGS potentiation and KK-169 subunit selectivity suggest a shared mechanism with PREGS. A-D. Traces show the effect of modulators at the indicated concentrations in the presence or absence of a high KK-169 concentration (30 µM). E-F. Summary of effects of modulators during baseline (E, N = 9 cells) and KK-169 incubations (F, N = 9 cells). G-J. Representative responses to 10 µM NMDA alone (black traces) or NMDA + 30 µM KK-169. Responses were acquired from the indicated GluN2 subunits expressed with GluN1a in N2a cells. K. Summary of potentiation for each combination. A two-way ANOVA followed by Dunnett’s multiple comparison test revealed that GluN2A and GluN2B containing NMDARs were increased above baseline (n = 12 and 8 respectively; p < 0.0001 for both conditions) but GluN2C and GluN2D receptors were not (n = 7 and 8 respectively; p = 0.54 and 0.26).
Because PREGS differently potentiates NMDAR based on subunit combination (Kostakis et al, 2013), we expressed NMDAR subunits in murine neuronal N2A cells in order to assess KK-169 effect (Figure 9G-K). Cells were challenged with 10 µM NMDA and 30 µM KK-169 and a significant potentiation was observed when GluN2A and GluN2B subunits were expressed, but not for GluN2C and GluN2D subunits. This pattern of results is consistent with PREGS-like potentiation but not with oxysterol-like potentiation (Paul et al., 2013)
As an independent test of KK-169’s mechanism, we turned to chimeric receptors (Wilding et al., 2016) that distinguish oxysterol from sulfated steroid potentiation. We used constructs that contained GluN1 and GluN2B domains, representing receptors sensitive to oxysterol and PREGS potentiation, and kainate receptor (GluK2) subunit domains, representing receptors insensitive to oxysterol and PREGS potentiation. Previous work has shown that the transmembrane domain alone from GluN subunits, with the balance of domains from the kainate receptor, can sustain oxysterol potentiation, but GluN transmembrane and ligand-binding domains are required for PREGS potentiation (Wilding et al., 2016). Other receptors tested included WT homomeric kainate-gated GluK2 unedited Q and edited R subunits, as well as chimeric subunits with GluN extracellular amino terminal and ligand binding domains linked to the transmembrane and cytoplasmic domains from GluK2 Q (Wilding et al., 2016). As expected, we observed that potentiation by KK-169 (Figure 10A) closely matched the pattern previously elucidated for PREGS-like potentiation (Wilding et al., 2014). NMDAR inhibition, which apparently requires the GluN ligand-binding domain, was evident at high concentrations of KK-169 (Figure 10B).
Figure 10.
For LBD + TMD (teal): EC50 = 22 ± 7 µM, n = 1.6 ± 0.2. IC50= 92 ± 26 µM, b = 1.4 ± 0.2, M = 364 ± 84.
For ATD + LBD (green): EC50 = 3 ± 1 µM, n = 1.5± 0.3. IC50 = 11 ± 9 µM, b =0.87 ± 0.07 M = 100 ± 62.
3.4. KK-169 accumulation and retention
Extracellularly applied sulfated steroids might be expected to lack intracellular access, as a result of the charged sulfate group (Eisenman et al., 2007). To test this, we incubated cells for different time periods in the presence of KK-169. We then fixed cells and performed retrospective in situ click chemistry using an azide conjugated fluorophore (Emnett et al., 2016; Jiang et al., 2016). The method employs copper-catalyzed cycloaddition of an azide-containing fluorophore to the alkyne group contained on KK-169. To our surprise, all incubation times yielded intracellular staining above background levels (Figure 11A-C). These results suggest that KK-169 in fact enters cells at detectable levels despite the anionic sulfate group. To avoid loss of steroid, we did not wash cells prior to incubation in fixative. We cannot exclude the possibility that dilute KK-169 enters cells due to fixativeinduced permeabilization of the membrane. However, the increase in fluorescence with increased incubation time of live cells in KK-169 suggests that a good fraction of KK-169 fluorescence is due to entry of live neurons.
Figure 11.

Intracellular accumulation of KK-169 as revealed by retrospective click chemistry. A, B. Photomicrographs of cells that were incubated in the absence or presence of unlabeled KK-169 for 15 min, then fixed with paraformaldehyde/glutaraldehyde for click labeling with an azide-conjugated fluorophore (see Methods). Bottom panels show phase-contrast images of the fields. Scale bar = 25 µm. C. Summary of normalized cytoplasmic fluorescence intensity as a function of different incubation times in the unlabeled compound. Each symbol color represents an independent experiment. A repeated-measures one-way ANOVA revealed a significant effect of incubation time on labeling intensity (P < 0.01). D-H. Strong accumulation and retention of lipophilic analogue KK-181. D-G. Each panel represents a different field of cells incubated for 15 min in the indicated unlabeled compound, fixed, then click labeled. Top rows were immediately fixed. Bottom rows were washed for 2 min with saline in the absence of compound before fixation. Underlying fluorescence in G represents astrocyte accumulation, which was variable in the wash and non-wash conditions. H. Summary of cytosolic fluorescence from 4 experiments, normalized to the t=0, KK-169 condition. Each color represents an independent experiment of 5 fields of cells per condition. Open and gray bars represent immediate fixation and fixation following a 2 min saline rinse respectively. ANOVA analysis revealed a significant difference main effect of compound (p < 0.05), and a significant effect of wash (p < 0.05). A post-hoc Bonferroni corrected t test revealed a significant difference between the control and wash conditions of only KK-169 (p<0.01).
It is unclear how KK-169 is retained upon fixation. One possibility is that aldehyde cross-linking prevents KK-169 efflux. We found that cold methanol fixation also fostered retention of KK-169, visualized by retrospective click labeling. Intracellular fluorescence in methanol-fixed neurons (n = 24) was 0.93 ± 0.08 that of cells fixed in parallel with the standard protocol (n = 22 cells). To address whether a permeabilized membrane fosters efflux of KK-169 following labeling, we incubated cells in 0.1% Triton-X 100 detergent for 10 min following fixation. Permeabilized cells (n = 26) showed 1.23 ± 0.06 of the fluorescence of cells treated in parallel with the standard aldehyde fixation protocol. We conclude that fixation permanently retains the steroid analogue and renders it mostly non-diffusible, but not necessarily through aldehyde-mediated protein cross-linking.
To examine whether lipophilicity is a factor in the intracellular accumulation, we compared KK-169 accumulation and retention with one of the more lipophilic sulfated compounds, KK-181 (Figure 1), using retrospective click labeling (Figure 11D-H). Cells were either immediately fixed following compound incubation, to assay initial accumulation (Figure 11D, E, H), or were washed with saline for 2 min prior to fixation to evaluate retention (Figure 11F, G, H). We found that saline wash significantly reduced KK-169 retention, suggesting significant efflux of KK-169 in the absence of continued incubation. However, washing following KK-181 incubation had almost no effect on retention of this compound (Figure 11).
It is surprising that KK-169 and KK-181 both exhibited intracellular accumulation. There appeared to be little evidence for preferential plasma membrane accumulation/retention (Figure 11), the location of target GABAA receptors and NMDARs. We hypothesized that the plasma-membrane pool of KK-169 is not fixed in place and that we therefore failed to observe plasma-membrane associated steroid. To test this hypothesis, we prepared another analogue, MQ-189, which has a similar structure to KK-169 except for a diazirine photolabeling group at carbon 6 to allow covalent linkage to proteins and/or lipids prior to fixation (Figure 12A, B). We have previously used this strategy to explore localization of other classes of neuroactive steroids (Jiang et al., 2016). We avoided diazirine placement in the side chain, given the loss of activity in MQ-154 (Figure 1). Unlike MQ-154, MQ-189 (10 µM) potentiated NMDA (10 µM) currents in hippocampal neurons by 38 ± 10% (N = 8, p = 0.007, paired t test), demonstrating that biological activity was retained. Cells incubated in MQ-189 or in KK-169 were irradiated with 365 nm light for 15 min during compound incubation. Subsequently, cells were fixed and click labeled as in previous experiments. We found that photolinkage increased plasma-membrane associated fluorescence compared with KK-169 labeling, which remained localized primarily to intracellular compartments (Figure 12). Although the mean intracellular fluorescence intensity for KK-169 trended toward higher values than for MQ-189, the difference did not reach statistical significance (Figure 12F). Thus, the major difference between compounds was the presence of a plasma membraneassociated component for MQ-189. We conclude that although considerable intracellular KK-169 is retained intracellularly following fixation, the plasma-membrane pool is only readily observable with photo-attachment.
Figure 12.

Enhanced membrane labeling by a photoaffinity click analogue. A, B. Structures of a photoaffinity analogue (MQ-189) and KK-169. C, D. Representative results from incubation in unlabeled analogue in the presence of ultraviolet irradiation (see Methods), followed by fixation and retrospective click fluorescence. D1 and D2 represent fluorescence intensity of the images through the straight lines drawn in C1 and C2. E. Summary of the peak fluorescence in membrane regions expressed as a ratio to cytoplasmic fluorescence from 11–12 fields in 4 experiments. MQ-189 showed a significantly higher ratio of membrane to cytoplasmic labeling (N = 11–12 fields in 4 experiments, Mann-Whitney test, p = 0.001). F. In these same fields the absolute level of intracellular fluorescence trended higher for KK-169 compared to MQ-189 but this difference did not reach statistical significance (Mann-Whitney test, p = 0.13).
4. Discussion
Positive NMDAR modulation is of interest to basic and clinical neuroscience for several reasons. Modest increases in NMDAR activation enhance memory and learning and may benefit cognitive symptoms in schizophrenia (Collingridge et al., 2013; Sun et al., 2015). Exogenous PREGS or its precursor pregnenolone enhances learning and memory (Akwa et al., 2001; Petit et al., 2011; Vallee et al., 1997) and may reduce symptoms in schizophrenia patients (Marx et al., 2009). For development of therapeutics that capitalize on these effects, the field requires more information about cellular actions of the compounds. Novel chemical biology approaches, such as ours, represent one way forward. Our work represents an initial exploration of the pharmacological activity and cellular level distribution of a new class of tools for exploring sulfated neurosteroids. Our results provide new information about structure-activity relationships while revealing some of the structural properties of compounds important for cellular retention and accumulation. As with other recent results (Krausova et al., 2018), our data show that NMDAR potentiation can be retained with alterations to the PREGS structure. Specifically, our results demonstrate that bioactivity tolerates side chain alterations to add a click label (KK-169, KK-181) or click label plus photolabel (MQ-189). Core features of PREGS activity are retained without detectable off target effects.
PREGS has complex actions at NMDARs (Chopra et al., 2015; Korinek et al., 2011), and the binding site remains elusive. The site has been assumed to be extracellular (Park-Chung et al., 1997), and the M3-M4 extracellular loop of GluN2 subunits was initially implicated in sulfated steroid action (Horak et al., 2006). More recent work has demonstrated that both the ligand-binding and transmembrane domains of GluN subunits are required (Wilding et al., 2016). The concentration of intracellular Ca2+ may determine the direction of PREGS modulation of GluN1/2A receptors (Chopra et al., 2015), but this does not imply an intracellular PREGS site. In general, it is unclear whether any previously identified domain is essential for binding or rather for translating the binding of modulator elsewhere into a change in channel open probability. The balance of evidence suggests an external site of interaction in the plasma membrane, so although we found evidence for intracellular accumulation/retention, the basis for currently known neuroactive actions is likely through external, membrane-associated sites.
The primary goal of this study was to develop chemical biology tools for the exploration of NMDAR modulators of the PREGS class. An unexpected outcome of the work was the identification of potent and efficacious analogues (Figure 1), which suggest new aspects of the structure-activity relationship between steroids and NMDARs. Previous work has explored mostly steroid inhibitors of NMDAR function (Adla et al., 2017). Based on the strong activity of multiple analogues with a hydrophobic side chain, we conclude that side chain hydrophobicity is likely to increase potentiation. The hydrophobic chain may foster a steroid orientation that promotes interaction with a hydrophobic (e.g., transmembrane) PREGS site on the receptor. It remains unclear whether introducing lipophilicity in other parts of the steroid structure would exhibit similar benefit. It is possible that strong activity requires hydrophobicity at the opposite end of the molecule from the sulfate charge.
Off-target effects at GABAA receptors, presynaptic sites, and at AMPA/kainate receptors represent a potential disadvantage of sulfated steroids as therapeutics (Lee et al., 2010; Seljeset et al., 2015; Yaghoubi et al., 1998; Zamudio-Bulcock and Valenzuela, 2011). At concentrations that have maximum NMDAR effect, we found little evidence that KK-169 has presynaptic or AMPAR effects. Unfortunately, GABAergic actions are quite ubiquitous among anionic lipophilic compounds (Chisari et al., 2010; Mennerick et al., 2001), so it seems unlikely that this off target effect will be eliminated by additional chemical modifications.
Our visualization approach included in situ variants of click chemistry and photo-labeling, chemical biology techniques that have gained increasing traction in recent years (Baskin et al., 2007; Hofmann et al., 2014; Hou et al., 2012). Our results include the new observation that the negative charge of sulfated steroids does not prevent their rapid intracellular accumulation. Possible explanations include active transport mechanisms or a small amount of passive influx across the membrane through lipophilic interactions. Based on calculated logP values KK-169 is marginally more lipophilic than PREGS, in either its neutral or anionic form (clogP~2.6–2.9). Thus, KK-169 should give a reasonable reflection of the ability of the natural compound PREGS to penetrate membranes through passive mechanisms. Intracellular targets may be involved in some of the reported actions of PREGS, including presynaptic effects. On the other hand, KK-181 is predicted to be considerably more lipophilic (clogP~4), explaining its slow reversibility.
As with our work with KK-169, other cellular studies have localized intracellular compounds by virtue of cellular retention in the absence of photolabeling (Jao et al., 2015; Peyrot et al., 2014; Viertler et al., 2012). In those cases, high-affinity protein interactions may drive retention. Our results do not completely exclude a role for protein targets in accumulation and retention. Presumed membrane localization of compound was rapidly lost before and/or after fixation, and plasma-membrane labeling was revealed only with photo-labeling (Figure 12). This suggests that the plasma membrane pool of KK-169 is more labile than the intracellular pool. Thus, although the precise targets and mechanisms remain elusive, our series of structural analogues provide initial clues of relevant properties responsible for KK-169 accumulation and retention.
5. Conclusions
In summary our work characterizes optically and biochemically accessible pharmacological tools to study the behavior of sulfated steroids as modulators of NMDAR function. Given the importance of NMDARs to neuronal function, and given the interest in NMDAR ligands to therapeutic applications, these tools may help advance understanding of pharmacological actions and aid the rational design of therapeutics.
Supplementary Material
KK-169 is a potent and efficacious positive allosteric modulator of NMDA receptors.
KK-169 is a clickable analogue of pregnenolone sulfate.
KK-169 reveals intracellular accumulation of sulfated steroids.
Acknowledgements:
The authors thank Amanda Taylor for technical help and George Elias for early oocyte experiments. The authors thank members of the Taylor Family Institute for Innovative Psychiatric Research for discussion. CFZ is a member of the scientific advisory board for Sage Therapeutics, and DFC and CFZ hold stock in Sage Therapeutics. Sage Therapeutics was not involved in the design or analysis of experiments herein.
Funding: NIH grants MH101874 (SM, CFZ), MH110550 (DFC), and NS30888 (JEH).
Abbreviations:
- D-APV
D-(-)-2-Amino-5-phosphonopentanoic acid
- GABAR
γ-amino-butyric acid receptor
- NBQX
2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione
- NMDAR
N-methyl-D-aspartate receptor
- PREGS
pregnenolone sulfate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6 References
- Abdallah CG, Sanacora G, Duman RS, Krystal JH, 2015. Ketamine and rapid-acting antidepressants: A window into a new neurobiology for mood disorder therapeutics. Annual Review of Medicine 66, 509–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adla SK, Slavikova B, Smidkova M, Tloustova E, Svoboda M, Vyklicky V, Krausova B, Hubalkova P, Nekardova M, Holubova K, Vales K, Budesinsky M, Vyklicky L, Chodounska H, Kudova E, 2017. Physicochemical and biological properties of novel amide-based steroidal inhibitors of NMDA receptors. Steroids 117, 52–61. [DOI] [PubMed] [Google Scholar]
- Akwa Y, Ladurelle N, Covey DF, Baulieu EE, 2001. The synthetic enantiomer of pregnenolone sulfate is very active on memory in rats and mice, even more so than its physiological neurosteroid counterpart: distinct mechanisms? Proceedings of the National Academy of Sciences of the United States of America 98, 14033–14037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR, 2007. Copper-free click chemistry for dynamic in vivo imaging. Proceedings of the National Academy of Sciences of the United States of America 104, 16793–16797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borovska J, Vyklicky V, Stastna E, Kapras V, Slavikova B, Horak M, Chodounska H, Vyklicky L Jr., 2012. Access of inhibitory neurosteroids to the NMDA receptor. Br J Pharmacol 166, 10691083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnell ES, Irvine M, Fang G, Sapkota K, Jane DE, Monaghan DT, 2018. Positive and Negative Allosteric Modulators of N-Methyl-d-aspartate (NMDA) Receptors: Structure-Activity Relationships and Mechanisms of Action. J Med Chem [DOI] [PMC free article] [PubMed]
- Chisari M, Shu HJ, Taylor A, Steinbach JH, Zorumski CF, Mennerick S, 2010. Structurally diverse amphiphiles exhibit biphasic modulation of GABAA receptors: similarities and differences with neurosteroid actions. Br J Pharmacol 160, 130–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra DA, Monaghan DT, Dravid SM, 2015. Bidirectional Effect of Pregnenolone Sulfate on GluN1/GluN2A N-Methyl-D-Aspartate Receptor Gating Depending on Extracellular Calcium and Intracellular Milieu. Mol Pharmacol 88, 650–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL, Volianskis A, Bannister N, France G, Hanna L, Mercier M, Tidball P, Fang G, Irvine MW, Costa BM, Monaghan DT, Bortolotto ZA, Molnar E, Lodge D, Jane DE, 2013. The NMDA receptor as a target for cognitive enhancement. Neuropharmacology 64, 13–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenman LN, He Y, Fields C, Zorumski CF, Mennerick S, 2003. Activation-dependent properties of pregnenolone sulfate inhibition of GABAA receptor-mediated current. J Physiol 550, 679691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenman LN, Shu HJ, Akk G, Wang C, Manion BD, Kress GJ, Evers AS, Steinbach JH, Covey DF, Zorumski CF, Mennerick S, 2007. Anticonvulsant and anesthetic effects of a fluorescent neurosteroid analog activated by visible light. Nat Neurosci 10, 523–530. [DOI] [PubMed] [Google Scholar]
- Emnett C, Li H, Jiang X, Benz A, Boggiano J, Conyers S, Wozniak DF, Zorumski CF, Reichert DE, Mennerick S, 2016. A clickable analogue of ketamine retains NMDA receptor activity, psychoactivity, and accumulates in neurons. Sci Rep 6, 38808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emnett CM, Eisenman LN, Mohan J, Taylor AA, Doherty JJ, Paul SM, Zorumski CF, Mennerick S, 2015. Interaction between positive allosteric modulators and trapping blockers of the NMDA receptor channel. Br J Pharmacol 172, 1333–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Yi F, Perszyk RE, Menniti FS, Traynelis SF, 2017. NMDA receptors in the central nervous system. Methods Mol Biol 1677, 1–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann K, Thiele C, Schött H-F, Gaebler A, Schoene M, Kiver Y, Friedrichs S, Lütjohann D, Kuerschner L, 2014. A novel alkyne cholesterol to trace cellular cholesterol metabolism and localization. J Lipid Res 55, 583–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horak M, Vlcek K, Chodounska H, Vyklicky L Jr., 2006. Subtype-dependence of N-methyl-Daspartate receptor modulation by pregnenolone sulfate. Neuroscience 137, 93–102. [DOI] [PubMed] [Google Scholar]
- Hou J, Liu X, Shen J, Zhao G, Wang PG, 2012. The impact of click chemistry in medicinal chemistry. Expert Opin Drug Discov 7, 489–501. [DOI] [PubMed] [Google Scholar]
- Jao CY, Nedelcu D, Lopez LV, Samarakoon TN, Welti R, Salic A, 2015. Bioorthogonal probes for imaging sterols in cells. Chembiochem 16, 611–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Shu HJ, Krishnan K, Qian M, Taylor AA, Covey DF, Zorumski CF, Mennerick S, 2016. A clickable neurosteroid photolabel reveals selective Golgi compartmentalization with preferential impact on proximal inhibition. Neuropharmacology 108, 193–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG, 2010. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol 8, e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korinek M, Kapras V, Vyklicky V, Adamusova E, Borovska J, Vales K, Stuchlik A, Horak M, Chodounska H, Vyklicky L Jr., 2011. Neurosteroid modulation of N-methyl-D-aspartate receptors: molecular mechanism and behavioral effects. Steroids 76, 1409–1418. [DOI] [PubMed] [Google Scholar]
- Kostakis E, Smith C, Jang MK, Martin SC, Richards KG, Russek SJ, Gibbs TT, Farb DH, 2013. The neuroactive steroid pregnenolone sulfate stimulates trafficking of functional NMDA receptors to the cell surface via a non-canonical G-protein and Ca++ dependent mechanism. Mol Pharmacol 84, 261–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krausova B, Slavikova B, Nekardova M, Hubalkova P, Vyklicky V, Chodounska H, Vyklicky L, Kudova E, 2018. Positive Modulators of the N-Methyl-d-aspartate Receptor: Structure-Activity Relationship Study of Steroidal 3-Hemiesters. J Med Chem 61, 4505–4516. [DOI] [PubMed] [Google Scholar]
- Lai TW, Shyu WC, Wang YT, 2011. Stroke intervention pathways: NMDA receptors and beyond. Trends Mol Med 17, 266–275. [DOI] [PubMed] [Google Scholar]
- Lee KH, Cho JH, Choi IS, Park HM, Lee MG, Choi BJ, Jang IS, 2010. Pregnenolone sulfate enhances spontaneous glutamate release by inducing presynaptic Ca2+-induced Ca2+ release. Neuroscience 171, 106–116. [DOI] [PubMed] [Google Scholar]
- Malayev A, Gibbs TT, Farb DH, 2002. Inhibition of the NMDA response by pregnenolone sulphate reveals subtype selective modulation of NMDA receptors by sulphated steroids. Br J Pharmacol 135, 901–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx CE, Keefe RS, Buchanan RW, Hamer RM, Kilts JD, Bradford DW, Strauss JL, Naylor JC, Payne VM, Lieberman JA, Savitz AJ, Leimone LA, Dunn L, Porcu P, Morrow AL, Shampine LJ, 2009. Proof-of-concept trial with the neurosteroid pregnenolone targeting cognitive and negative symptoms in schizophrenia. Neuropsychopharmacology 34, 18851903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL, 2010. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol 160, 1573–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennerick S, Zeng CM, Benz A, Shen W, Izumi Y, Evers AS, Covey DF, Zorumski CF, 2001. Effects on γ-aminobutyric acid (GABA)A receptors of a neuroactive steroid that negatively modulates glutamate neurotransmission and augments GABA neurotransmission. Mol Pharmacol 60, 732–741. [PubMed] [Google Scholar]
- Nilsson KR, Zorumski CF, Covey DF, 1998. Neurosteroid analogues. 6. The synthesis and GABAA receptor pharmacology of enantiomers of dehydroepiandrosterone sulfate, pregnenolone sulfate, and (3α,5β)-3-hydroxypregnan-20-one sulfate. J Med Chem 41, 2604–2613. [DOI] [PubMed] [Google Scholar]
- Park-Chung M, Wu FS, Farb DH, 1994. 3α-Hydroxy-5β-pregnan-20-one sulfate: a negative modulator of the NMDA-induced current in cultured neurons. Mol Pharmacol 46, 146–150. [PubMed] [Google Scholar]
- Park-Chung M, Wu FS, Purdy RH, Malayev AA, Gibbs TT, Farb DH, 1997. Distinct sites for inverse modulation of N-methyl-D-aspartate receptors by sulfated steroids. Mol Pharmacol 52, 1113–1123. [DOI] [PubMed] [Google Scholar]
- Paul SM, Doherty JJ, Robichaud AJ, Belfort GM, Chow BY, Hammond RS, Crawford DC, Linsenbardt AJ, Shu HJ, Izumi Y, Mennerick SJ, Zorumski CF, 2013. The major brain cholesterol metabolite 24(S)-hydroxycholesterol is a potent allosteric modulator of N-methyl-Daspartate receptors. Journal of Neuroscience 33, 17290–17300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit GH, Tobin C, Krishnan K, Moricard Y, Covey DF, Rondi-Reig L, Akwa Y, 2011. Pregnenolone sulfate and its enantiomer: differential modulation of memory in a spatial discrimination task using forebrain NMDA receptor deficient mice. Eur Neuropsychopharmacol 21, 211–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyrot SM, Nachtergaele S, Luchetti G, Mydock-McGrane LK, Fujiwara H, Scherrer D, Jallouk A, Schlesinger PH, Ory DS, Covey DF, Rohatgi R, 2014. Tracking the subcellular fate of 20(s)-hydroxycholesterol with click chemistry reveals a transport pathway to the Golgi. J Biol Chem 289, 11095–11110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy DS, 2010. Neurosteroids: endogenous role in the human brain and therapeutic potentials. Progress in Brain Research 186, 113–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seljeset S, Laverty D, Smart TG, 2015. Inhibitory neurosteroids and the GABAA receptor. Adv Pharmacol 72, 165–187. [DOI] [PubMed] [Google Scholar]
- Shen W, Mennerick S, Covey DF, Zorumski CF, 2000. Pregnenolone sulfate modulates inhibitory synaptic transmission by enhancing GABAA receptor desensitization. Journal of Neuroscience 20, 3571–3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CC, Gibbs TT, Farb DH, 2014. Pregnenolone sulfate as a modulator of synaptic plasticity. Psychopharmacology (Berl) 231, 3537–3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strong KL, Jing Y, Prosser AR, Traynelis SF, Liotta DC, 2014. NMDA receptor modulators: an updated patent review (2013–2014). Expert Opin Ther Pat 24, 1349–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun MY, Linsenbardt AJ, Emnett CM, Eisenman LN, Izumi Y, Zorumski CF, Mennerick S, 2015. 24(S)-Hydroxycholesterol as a modulator of neuronal signaling and survival. Neuroscientist [DOI] [PMC free article] [PubMed]
- Tischbirek CH, Wenzel EM, Zheng F, Huth T, Amato D, Trapp S, Denker A, Welzel O, Lueke K, Svetlitchny A, Rauh M, Deusser J, Schwab A, Rizzoli SO, Henkel AW, Muller CP, Alzheimer C, Kornhuber J, Groemer TW, 2012. Use-dependent inhibition of synaptic transmission by the secretion of intravesicularly accumulated antipsychotic drugs. Neuron 74, 830844. [DOI] [PubMed] [Google Scholar]
- Twede VD, Covey DF, Tartaglia AL, Bamber BA, 2007. The neurosteroids dehydroepiandrosterone sulfate and pregnenolone sulfate inhibit the UNC-49 GABA receptor through a common set of residues. Mol Pharmacol 72, 1322–1329. [DOI] [PubMed] [Google Scholar]
- Vallee M, Mayo W, Darnaudery M, Corpechot C, Young J, Koehl M, Le Moal M, Baulieu EE, Robel P, Simon H, 1997. Neurosteroids: deficient cognitive performance in aged rats depends on low pregnenolone sulfate levels in the hippocampus. Proceedings of the National Academy of Sciences of the United States of America 94, 14865–14870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viertler M, Schittmayer M, Birner-Gruenberger R, 2012. Activity based subcellular resolution imaging of lipases. Bioorg Med Chem 20, 628–632. [DOI] [PubMed] [Google Scholar]
- Vyklicky V, Smejkalova T, Krausova B, Balik A, Korinek M, Borovska J, Horak M, Chvojkova M, Kleteckova L, Vales K, Cerny J, Nekardova M, Chodounska H, Kudova E, Vyklicky L, 2016. Preferential inhibition of tonically over phasically activated NMDA receptors by pregnane derivatives. Journal of Neuroscience 36, 2161–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilding TJ, Lopez MN, Huettner JE, 2014. Radial symmetry in a chimeric glutamate receptor pore. Nat Commun 5, 3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilding TJ, Lopez MN, Huettner JE, 2016. Chimeric glutamate receptor subunits reveal the transmembrane domain is sufficient for NMDA receptor pore properties but some positive allosteric modulators require additional domains. Journal of Neuroscience 36, 8815–8825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu FS, Chen SC, 1997. Mechanism underlying the effect of pregnenolone sulfate on the kainateinduced current in cultured chick spinal cord neurons. Neuroscience Letters 222, 79–82. [DOI] [PubMed] [Google Scholar]
- Yaghoubi N, Malayev A, Russek SJ, Gibbs TT, Farb DH, 1998. Neurosteroid modulation of recombinant ionotropic glutamate receptors. Brain Res 803, 153–160. [DOI] [PubMed] [Google Scholar]
- Zamudio-Bulcock PA, Valenzuela CF, 2011. Pregnenolone sulfate increases glutamate release at neonatal climbing fiber-to-Purkinje cell synapses. Neuroscience 175, 24–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.