

Abstract
There is a growing appreciation that the extracellular matrix (ECM) contributes to both the maintenance of immune tolerance in healthy tissues and to its loss at sites of autoimmunity. Here, we review recent literature on the role of ECM and particularly the glycosaminoglycans hyaluronan and heparan sulfate in the development of autoimmune, type 1 diabetes (T1D). Data from transplant models suggest that healthy islets are embedded within an intact ECM that supports beta-cell homeostasis and provides physical and immunoregulatory barriers against immune infiltration. However, studies of human insulitis as well as the non-obese diabetic (NOD) and DORmO mouse models of T1D indicate that autoimmune insulitis is associated with the degradation of basement membrane structures, the catabolism of the islet interstitium, and the accumulation of a hyaluronan-rich, pro-inflammatory ECM. Moreover, in these models of autoimmune diabetes, either the pharmacologic inhibition of heparan sulfate catabolism, the reduction of hyaluronan synthesis, or the targeting of the pathways that sense these ECM changes can all prevent beta-cell destruction. Together these data support an emerging paradigm that in healthy islets the local ECM contributes to both immune tolerance and beta-cell homeostasis while in chronic inflammation the islet ECM is permissive to immune infiltration and beta cell destruction. Therapies that support ECM-mediated “barrier tolerance” may have potential as adjunctive agents in combination regimens designed to prevent or treat autoimmunity.
Keywords: hyaluronan, heparan sulfate, diabetes, T1D, extracellular matrix
Introduction
The extracellular matrix (ECM) surrounds cells and tissues throughout the human body and it has long been known that these structures contribute to local homeostasis and function. However, there is a growing appreciation the ECM also provides barriers against immune infiltration in healthy tissues.
The contributions of the ECM to tissue homeostasis and peripheral tolerance are perhaps best understood for type 1 diabetes (T1D), an autoimmune disease characterized by lymphocyte-mediated destruction of insulin producing β-cells within the pancreatic islets (1). Most current models of the pathogenesis of T1D invoke the progressive, sequential loss of B-cell and T-cell tolerance to islet auto-antigens as well as a failure of immunoregulatory mechanisms that normally control potentially auto-reactive lymphocytes (1–3). Alongside mechanisms of central and peripheral tolerance that are presumably interrupted in this progression, there are suggestions that tissues may themselves provide barriers against immune-mediated destruction and that these are likewise lost in T1D. This can be inferred from histologic data from human T1D (4) as well experimental models of the disease, including the Non-Obese Diabetic (NOD) (5) and DORmO mouse models (Fig.1).
Figure 1: Autoimmune insulitis in the DORmO mouse model of T1D is associated with the progressive, sequential loss of local tissue barriers against autoimmunity.

H&E staining of islets from DORmO mice at various ages and stages of progressive insulitis: (A) no infiltration (3–4 weeks of age), (B) peri-insulitis (4–5 weeks), (C) insulitis (8 weeks), and (D) islet destruction (12 weeks).
Here, we review recent literature on the role of ECM and particularly the glycosaminoglycans hyaluronan (HA) and heparan sulfate (HS) in the development of T1D. We first briefly review what is known about the ECM in healthy islets as well as data suggesting that it supports beta-cell homeostasis as well as providing physical and immunoregulatory barriers against immune infiltration. Next we review studies indicating that autoimmune insulitis is associated with the degradation of basement membrane structures, the catabolism of the islet interstitium, and the accumulation of a hyaluronan-rich, pro-inflammatory matrix and that these changes are permissive to immune infiltration and beta cell destruction. Finally we propose that agents that support ECM integrity may have therapeutic potential for prevention or possibly treatment of T1D.
The ECM in healthy pancreatic islets
In pancreatic islets, as in all tissues, cells exist within an ECM - a complex network of proteins, polysaccharides, and proteoglycans that constitute basement membrane (BM) and interstitial matrix structures. The detailed biochemistry and histologic distribution of islet ECM components are the subjects of several excellent recent studies and reviews (6–8).
In brief, the BM that surrounds capillaries and encases each islet are mostly comprised of tightly interconnected networks of type IV collagen and laminin. These are interwoven with lesser amounts of heparan sulfate proteoglycans (HSPGs) such as perlecan, glycoproteins such as nidogens, and glycosaminoglycans such as hyaluronan (7–10). The islet BM differs somewhat between species, being continuous in mice and discontinuous in humans (11). In contrast to the BM matrix, the islet interstitial matrix present within the islet stroma is more diffuse and mostly contains collagens I, II, and IV, and fibrillin-2 (10,12). HSPGs are present as well, including HS polymers attached to the core proteins collagen type XVIII, versican, and syndecan-1 (8,13).
Together these ECM components provide critical structure and support to islet-resident cells. In particular, transplant studies indicate that the islet ECM communicates chemical and mechanical signals that mediate key aspects of islet physiology including survival (14–18), differentiation (19–22), proliferation (23–25),and insulin secretion (14,15,23,26,27). The ECM signals that support these cells are communicated in part via interactions between cell membrane receptors, such as integrins, and ECM polysaccharides and proteins, such as laminin, that contain the peptide signaling sequence arginine-glycine-aspartic acid (RGD)(28,29). In addition to providing structural support, the interstitial ECM modulates cellular behavior via the binding of growth factors to sulfated glycosaminoglycans such as HS (30,31) and it has been suggested that this may contribute to β-cell homeostasis (32). Capitalizing on these insights, ECM platforms are increasingly utilized to support islet transplantation efforts (33) as well as the development of stem-cell derived β-cells (34).
ECM catabolism and the loss of tissue integrity in autoimmune insulitis
In comparison to healthy islets, the ECM at sites of insulitis is altered in multiple ways. These include the loss of BM integrity, the catabolism of interstitial matrix, and the deposition of a pro-inflammatory matrix dominated by HA.
The transition between non-destructive peri-insulitis (Fig. 1B) and destructive insulitis (Fig. 1C) is accompanied by a breakdown in the islet BM, as demonstrated conclusively by Dr. Lydia Sorokin and her colleagues. In particular, there is a loss of laminin and perlecan staining within the BM in both humans with T1D (10) as well as in the NOD (9,10) and DORmO mouse models of the disease (Fig.2). This catabolism occurs in association with increased expression of cathepsins S, W, and C, and heparanase (9,10,35). In transplant studies, loss of islet BM integrity has major, adverse effects on islet function and viability (36) while inhibition of BM degradation can preserve islet function (37). Modeling studies suggest that the balance between degradation and repair of the BM may be a critical determinant in the progression to clinical diabetes (38). Perhaps consistent with the pathophysiology observed in T1D, disruption of the BM structures that maintain the blood-brain barrier is likewise seen in Multiple Sclerosis (MS) and in the experimental autoimmune encephalitis (EAE) model of that disease (39,40). Together, these data suggest that the islet BM functions as a physical barrier against leucocyte migration into islets and that degradation of this barrier is a critical step in progression to diabetes (Fig.3).
Figure 2: Autoimmune insulitis is associated with the degradation of islet basement membranes, the catabolism of the interstitial matrix and the deposition of a pro-inflammatory matrix dominated by hyaluronan.

Histologic staining of the islet ECM molecules in healthy BalbC mice and in the DORmO mouse model of T1D. In DORmO mice, basement membrane integrity is lost, as evidenced by the break-down in perlecan staining (left). Interstitial matrix likewise undergoes catabolism, as evidenced by the loss of heparan sulfate structures (center). Finally, there occurs the deposition of a pro-inflammatory matrix dominated by hyaluronan (right).
Figure 3: A schematic of the sequential loss of ECM barriers against in autoimmune insulitis.
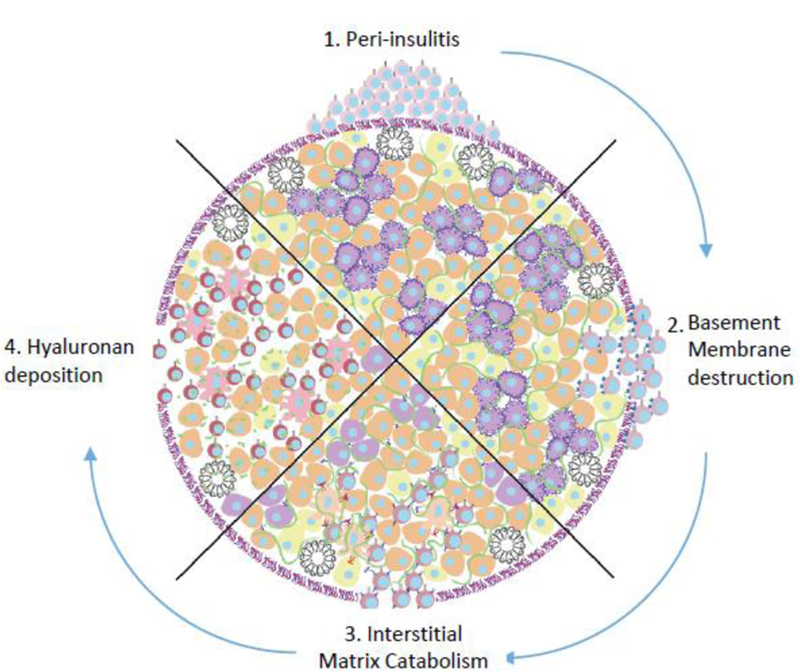
In healthy islets, the ECM provides physical and immunoregulatory barriers against immune infiltration. However, in autoimmune diabetes, effector T cells evade central and peripheral tolerance and accumulate around islets (top). These auto-reactive cells require a break in the laminin rich basement membrane for entry (right). Degradation of islet HS leads to beta cell death (bottom). Deposition of HA allows for further infiltration, effector cell activation, and inhibition of Treg expansion (left). Along with effects on infiltrating leukocytes, this remodeling of the islet ECM also adversely impacts β-cell health.
The subsequent infiltration of leukocytes into the islet stroma (Fig. 1C) is also associated with the catabolism of the interstitial matrix, in particular the loss of HS content, as demonstrated in beautiful work by Dr. Charmaine Simeonovic and her colleagues. They demonstrated that intra-islet HS is lost in both NOD mice(35) as well as in humans with T1D (13) in association with expression of heparanase by leukocytes (41). We observe similar findings in DORmO mice (Fig.2). This catabolism of HS has been reported to contribute to local destruction of β-cells via an increase in local reactive oxygen species that would otherwise be absorbed by HS, a conclusion based on the finding that HS protects islets from oxidative stress in vitro and the observation that much of the missing HS was intracellular (35). However, HS catabolism may contribute to β-cell destruction in additional ways, given HS’s overlapping roles in innate immunity and as a depot for β-cell growth factors (32,42,43). Moreover, glycosaminoglycan synthesis is complex and typically involves extracellular production before intracellular uptake such that the site of HS catabolism and function is uncertain.
The strongest support for an important, functional role for HS in local immune regulation is the finding that treatment with either exogenous HS or a small molecule inhibitor of heparanase, PI-88, prevented progression of established insulitis in NOD mice and prevented β-cell death (35). These treatments of course would also be expected to preserve BM integrity as well, given the abundance of HSPGs there, such that the in vivo site of action (BM versus interstitial matrix) is unclear. However, in vitro, HS protected β-cells themselves from oxidative damage. Consistent with a role for heparanase produced by infiltrating leukocytes in autoimmunity, we find that T-cells isolated from mice lacking heparanase (HPSE−/− mice) are substantially delayed in causing autoimmunity in EAE mice (unpublished results).
In addition to the loss of HS, other changes within the islet interstitial matrix have been reported as well including the loss of inter-alpha-inhibitor (IαI)(44) and tumor necrosis factor-stimulated gene-6 (TSG-6)(44,45), a pair of molecules with complex but generally anti-inflammatory properties (46,47).
In sum, these data suggest that, as with the loss of BM integrity, degradation of the islet interstitial matrix is another critical step in the progression to autoimmune diabetes (Fig.3).
Deposition of pro-inflammatory matrix at sites of autoimmune insulitis
In addition to degradation of the normal islet EMC, robust autoimmune insulitis (Fig. 1C, 1D) is also characterized by the deposition of a pro-inflammatory matrix dominated by HA, as originally shown by Dr. Tom Wight (48). In healthy tissues, HA provides structural support to skin, joints, and other tissues (49). At these sites, HA is typically bound to a diverse group of binding proteins, called hyaladherins, including TSG-6 and IαI (8), and these complexes are typically structurally stable and pro-tolerogenic (50,51). However, at sites of inflammation, both HA production and catabolism are greatly upregulated (49) leading to the accumulation of HA (Fig.2). These promote leukocyte migration and activation within inflamed tissues (49,52). HA deposits are present at sites of autoimmune insulitis in both human T1D (48) as well as the NOD (53) and DORmO (44) mouse models of the disease and most of this HA consists of small fragments (54), consistent with the previously noted depletion of local hyaladherins that protect HA from catabolism. Similar HA deposits are present at sites of autoimmune attack in RA (55), MS (56), and other autoimmune diseases (57,58).
The strongest support for an important, functional role for HS in local immune regulation is the finding that treatment with 4-methylumbelliferone (4-MU), an inhibitor of HA synthesis (59), prevented progression of established insulitis in both DORmO and NOD mice (44,60). This effect was associated with the suspension of cytolytic killing of β-cells and the expansion of CD4+Foxp3+ regulatory T-cells. However, when 4-MU treatment was stopped and HA synthesis resumed, diabetes rapidly ensued (44). Consistent with these results, treatment with recombinant hyaluronidase prevents diabetes in NOD mice (61). Treatment with an antibody clone (IM7) that causes cell surface shedding of CD44, the primary HA receptor, likewise prevents diabetes in the NOD model (61). Moreover, this same suite of treatments (4-MU, hyaluronidase, and anti-CD44 antibodies) are effective treatments in mouse models of Rheumatoid Arthritis (62,63) and MS (56,64,65) as well.
The ECM influences Treg number and function. HA triggers CD44-mediated AKT and ERK1/2 signaling that suppresses Treg expansion (44,64). Consistent with this, immunized CD44−/− mice have increased FoxP3+ Treg (64,66,67). However, 4-MU has only modest effects on Treg numbers in naïve mice and uninflamed tissues (unpublished data) while un-immunized CD44−/− mice have normal Treg numbers (68), suggesting that HA may only suppress Treg expansion in the setting of inflammation. Indeed, culture plates coated with HA actually promote Treg survival (69,70). These seemingly contradictory findings (that HA could both inhibit Treg expansion as well as promote Treg survival) are consistent with established roles for AKT and ERK1/2 in both the inhibition of Treg expansion as well as promotion of Treg survival (71,72). Together these data suggest that HA limits Treg expansion at sites of active inflammation but preserves the capacity for immune tolerance at a later time when HA is cleared.
Along with effects on Treg, HA-CD44 interactions also influence other lymphocyte populations in ways that may impact autoimmunity. This includes well-established effects on costimulation, polarization towards a Th1 phenotype, and trafficking (49). This biology may occur in draining lymph nodes as well as sites of insulitis given that HA accumulates in pancreatic lymph nodes in T1D as well as in islets themselves (48).
HA/CD44 interactions may also directly impact β-cells adversely. In a fascinating study, it was reported that NOD mice deficient for CD44 were protected from autoimmune diabetes. In transfer experiments this protection was associated with the absence of CD44 in recipient mice rather than on donor lymphocytes. Moreover, islets cultured in vitro with HA demonstrated enhanced apoptosis (73). The mechanisms behind these observations are unclear. Nonetheless, they raise the intriguing possibility that the HA-rich matrix present at sites of autoimmune insulitis may contribute directly to β-cell demise in T1D.
Together these data suggest that deposition of a HA-rich matrix contributes to the loss of immune tolerance in pancreatic islets by creating a pro-inflammatory milieu that drives immune dysregulation (Fig.3).
ECM barrier effects in peripheral immune tolerance
The data presented here suggest that intact ECM may promote β-cell homeostasis as well as provide barriers against immune infiltration. These mechanisms of barrier tolerance may complement previously characterized mechanisms of central and peripheral tolerance (Fig. 4).
Figure 4: Peripheral and barrier tolerance mechanisms.
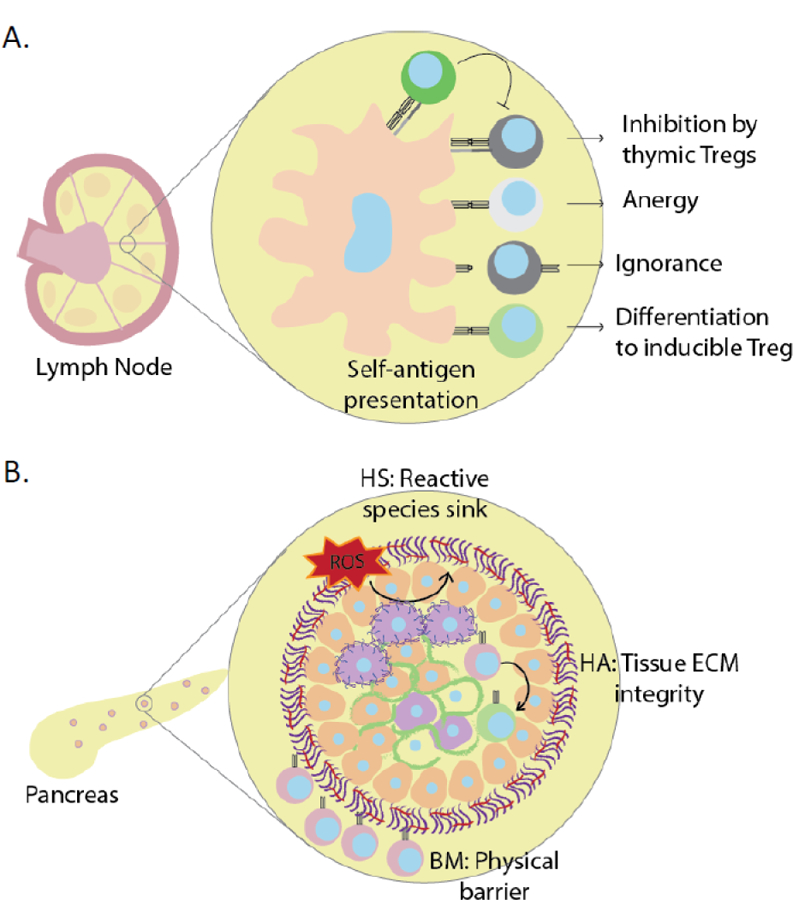
(A) Peripheral tolerance of autoreactive effector T cells is maintained via inhibition by thymic Tregs, induction of anergy, antigenic ignorance, or differentiation to inducible Tregs. (B) The intact islet ECM provides physical and immunologic barriers (“barrier tolerance”) that contribute to immune tolerance. These include a basement membrane that prevents immune infiltration, protective barriers against reactive oxygen species as with HS, and immunomodulatory barriers like the stabilization and sequestration of HA that limit immune activation.
While this is perhaps an appealing model, numerous questions remain. The contributions of lymphocytes versus other leukocyte subsets to ECM catabolism and deposition are unclear, as are the temporal aspects of these processes and their relationship to other local inflammatory events. In NOD mice, autoimmune insulitis begins not long after the islet remodeling that occurs after weaning (74) while in humans viral infections precede T1D in some cases (75). It may be that the local inflammation associated with such events causes critical lapses in barrier tolerance that help make the local ECM permissive to autoimmunity. Alternatively, activated T-cells themselves can catabolize the local ECM as well as priming other cell populations to secrete a pro-inflammatory matrix (76,77). Sorting out the “chicken and egg” question regarding the timing of these events and the responsible cells and molecules is likely to be important to understanding barrier tolerance and why it fails.
Other questions remain as well. How important are different ECM components to barrier tolerance? Are the stages of insulitis progression functionally distinct or are they interdependent? How does the ECM in draining lymph nodes contribute to immune activation? There are some evidence that similar ECM structures provide barrier tolerance in CNS tissue and that these likewise are disrupted in MS (78,79) but it would be important to know whether the same phenomenon are present in other tissues as well. Clearly there remains much to learn about the ECM at sites of autoimmunity and its contributions to barrier tolerance.
Targeting the ECM to Promote Immune Tolerance in T1D – The Potential Path Forward
The observations discussed here may be relevant to autoimmunity prevention. There is great interest in identifying mechanisms of peripheral tolerance that can be targeted therapeutically in T1D. Since screening for auto-antibodies can identify individuals at risk of T1D after the initial loss of tolerance but before clinical diabetes, the hope is that by re-establishing or reinforcing immunoregulatory checkpoints, further disease progression can be forestalled (80). Despite great progress, however, effective and benign prophylactic regimens have not been identified (81,82). An emerging strategy is to develop immunomodulatory combination therapies with complimentary synergistic mechanisms (83). The data reviewed here suggest that it may be possible to target the ECM to promote local immune tolerance in ways that are fundamentally different from existing T- or B-cell directed therapies. Moreover, several of the agents in question, particularly PI-88 and 4-MU are either in clinical trials currently or have an extensive clinical track record in humans (84,85).
However, here again multiple questions remain. Is targeting the ECM a safe strategy in adolescents and children who are still growing and extensively remodeling their tissues? What are the consequences of inhibiting ECM synthesis or breakdown in humans? Would protection from T1D require indefinite targeting of these pathways or would more targeted therapy be sufficient?
While further work clearly remains to be done and numerous questions remain, the ECM is clearly an exciting frontier in our understanding of immune tolerance and T1D.
Highlights:
Islet extracellular matrix provides homeostatic support to pancreatic islets as well as physical and immunologic barriers against immune infiltration.
Degradation of islet basement membrane and interstitial heparan sulfate are required for islet immune infiltration and insulitis
Increased hyaluronan deposition and fragmentation promotes pro-inflammatory lymphocyte responses and islet destruction
Treatment preserving or restoring ECM halts disease progression and promotes regulatory T cells
Acknowledgements
This work was supported in part by the Deutsche Forschungsgemeinschaft (DFG) NA 965/2–1 to NN; and National Institutes of Health grants R01 DK096087–01, R01 HL113294–01A1, and U01 AI101984 to PLB. This work was also supported by grants from the JDRF 3-PDF-2014–224-A-N to NN and 1-SRA-2018–518-S-B Innovation Award to PLB and by grants from the Harrington Institute, and Stanford SPARK to PLB, and by a grant from the Stanford Diabetes Research Center to NN.
Abbreviations:
- HA
Hyaluronan
- HS
Heparan Sulfate
- ECM
Extracellular Matrix
- T1D
Type 1 Diabetes
- BM
Basement Membrane
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interest
PLB and NN are co-founders of Hyalos Therapeutics, a company developing novel small molecules to inhibit HA synthesis.
Bibliography
- 1.Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature (2010) 464:1293–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ziegler A-G, Nepom GT. Prediction and pathogenesis in type 1 diabetes. Immunity (2010) 32:468–478. doi:10.1016/j.immuni.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenblum MD, Remedios KA, Abbas AK. Mechanisms of human autoimmunity. J Clin Invest (2015) 125:2228–2233. 10.1172/JCI78088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Atkinson MA, Herrath von M, Powers AC, Clare-Salzler M. Current concepts on the pathogenesis of type 1 diabetes--considerations for attempts to prevent and reverse the disease. Diabetes Care (2015) 38:979–988. 10.2337/dc15-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Atkinson MA, Leiter EH. The NOD mouse model of type 1 diabetes: as good as it gets? Nat Med (1999) 5:601–604. 10.1038/9442. [DOI] [PubMed] [Google Scholar]
- 6.Bogdani M, Korpos E, Simeonovic CJ, Parish CR, Sorokin L, Wight TN. Extracellular matrix components in the pathogenesis of type 1 diabetes. Curr Diab Rep (2014) 14:552–11. 10.1007/s11892-014-0552-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. *.Naba A, Clauser KR, Mani DR, Carr SA, Hynes RO. Quantitative proteomic profiling of the extracellular matrix of pancreatic islets during the angiogenic switch and insulinoma progression. Sci Rep (2017) 7:40495 10.1038/srep40495.Utilizing peptide labeling, fractionation, and LC-MS/MS, the authors created a pipeline through which full quantitative proteomic profiling can be done on islet samples at various stages of disease progression.
- 8.Hull RL, Johnson PY, Braun KR, Day AJ, Wight TN. Hyaluronan and hyaluronan binding proteins are normal components of mouse pancreatic islets and are differentially expressed by islet endocrine cell types. J Histochem Cytochem (2012) 60:749–760. 10.1369/0022155412457048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Irving-Rodgers HF, Ziolkowski AF, Parish CR, Sado Y, Ninomiya Y, Simeonovic CJ, Rodgers RJ. Molecular composition of the peri-islet basement membrane in NOD mice: a barrier against destructive insulitis. Diabetologia (2008) 51:1680–1688. 10.1007/s00125-008-1085-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korpos E, Kadri N, Kappelhoff R, Wegner J, Overall CM, Weber E, Holmberg D, Cardell S, Sorokin L. The peri-islet basement membrane, a barrier to infiltrating leukocytes in type 1 diabetes in mouse and human. Diabetes (2013) 62:531–542. 10.2337/db12-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Deijnen JH, Hulstaert CE, Wolters GH, Van Schilfgaarde R. Significance of the peri-insular extracellular matrix for islet isolation from the pancreas of rat, dog, pig, and man. Cell Tissue Res (1992) 267:139–146. [DOI] [PubMed] [Google Scholar]
- 12.Van Deijnen JH, Van Suylichem PT, Wolters GH, Van Schilfgaarde R. Distribution of collagens type I, type III and type V in the pancreas of rat, dog, pig and man. Cell Tissue Res (1994) 277:115–121. [DOI] [PubMed] [Google Scholar]
- * 13.Simeonovic CJ, Popp SK, Starrs LM, Brown DJ, Ziolkowski AF, Ludwig B, Bornstein SR, Wilson JD, Pugliese A, Kay TWH, et al. Loss of intra-islet heparan sulfate is a highly sensitive marker of type 1 diabetes progression in humans. PLoS ONE (2018) 13:e0191360 10.1371/journal.pone.0191360This manuscript demonstrates that HS depletion occurs in human islets in pre-diabetic subjects, complementing the seminal work by this group of investigators on HS in murine models of T1D.
- 14.Lucas-Clerc C, Massart C, Campion JP, Launois B, Nicol M. Long-term culture of human pancreatic islets in an extracellular matrix: morphological and metabolic effects. Mol Cell Endocrinol (1993) 94:9– 20. [DOI] [PubMed] [Google Scholar]
- 15.Navarro-Alvarez N, Rivas-Carrillo JD, Soto-Gutierrez A, Yuasa T, Okitsu T, Noguchi H, Matsumoto S, Takei J, Tanaka N, Kobayashi N. Reestablishment of microenvironment is necessary to maintain in vitro and in vivo human islet function. Cell Transplant (2008) 17:111–119. [DOI] [PubMed] [Google Scholar]
- 16.Pinkse GGM, Bouwman WP, Jiawan-Lalai R, Terpstra OT, Bruijn JA, de Heer E. Integrin signaling via RGD peptides and anti-beta1 antibodies confers resistance to apoptosis in islets of Langerhans. Diabetes (2006) 55:312–317. [DOI] [PubMed] [Google Scholar]
- 17.Wang RN, Rosenberg L. Maintenance of beta-cell function and survival following islet isolation requires re-establishment of the islet-matrix relationship. J Endocrinol (1999) 163:181–190. [DOI] [PubMed] [Google Scholar]
- 18.Vernon RB, Preisinger A, Gooden MD, D’Amico LA, Yue BB, Bollyky PL, Kuhr CS, Hefty TR, Nepom GT, Gebe JA. Reversal of diabetes in mice with a bioengineered islet implant incorporating a type I collagen hydrogel and sustained release of vascular endothelial growth factor (2012) 21:2099–2110. 10.3727/096368912X636786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang F-X, Harrison LC. Extracellular signals and pancreatic beta-cell development: a brief review. Mol Med (2002) 8:763–770. [PMC free article] [PubMed] [Google Scholar]
- 20.Oberg-Welsh C Long-term culture in matrigel enhances the insulin secretion of fetal porcine islet-like cell clusters in vitro. Pancreas (2001) 22:157–163. [DOI] [PubMed] [Google Scholar]
- 21.Crisera CA, Kadison AS, Breslow GD, Maldonado TS, Longaker MT, Gittes GK. Expression and role of laminin-1 in mouse pancreatic organogenesis. Diabetes (2000) 49:936–944. [DOI] [PubMed] [Google Scholar]
- 22.Beattie GM, Rubin JS, Mally MI, Otonkoski T, Hayek A. Regulation of proliferation and differentiation of human fetal pancreatic islet cells by extracellular matrix, hepatocyte growth factor, and cell-cell contact. Diabetes (1996) 45:1223–1228. [DOI] [PubMed] [Google Scholar]
- 23.Beattie GM, Montgomery AMP, Lopez AD, Hao E, Perez B, Just ML, Lakey JRT, Hart ME, Hayek A. A novel approach to increase human islet cell mass while preserving beta-cell function. Diabetes (2002) 51:3435–3439. [DOI] [PubMed] [Google Scholar]
- 24.Parnaud G, Bosco D, Berney T, Pattou F, Kerr-Conte J, Donath MY, Bruun C, Mandrup-Poulsen T, Billestrup N, Halban PA. Proliferation of sorted human and rat beta cells. Diabetologia (2008) 51:91– 100. 10.1007/s00125-007-0855-1. [DOI] [PubMed] [Google Scholar]
- 25.Rutti S, Sauter NS, Bouzakri K, Prazak R, Halban PA, Donath MY. In vitro proliferation of adult human beta-cells. PLoS ONE (2012) 7:e35801 10.1371/journal.pone.0035801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bosco D, Meda P, Halban PA, Rouiller DG. Importance of cell-matrix interactions in rat islet beta-cell secretion in vitro: role of alpha6beta1 integrin. Diabetes (2000) 49:233–243. [DOI] [PubMed] [Google Scholar]
- 27.Kaido T, Yebra M, Cirulli V, Rhodes C, Diaferia G, Montgomery AM. Impact of defined matrix interactions on insulin production by cultured human beta-cells: effect on insulin content, secretion, and gene transcription. Diabetes (2006) 55:2723–2729. 10.2337/db06-0120. [DOI] [PubMed] [Google Scholar]
- 28.Llacua A, de Haan BJ, Smink SA, de Vos P. Extracellular matrix components supporting human islet function in alginate-based immunoprotective microcapsules for treatment of diabetes. J Biomed Mater Res A (2016) 104:1788–1796. 10.1002/jbm.a.35706. [DOI] [PubMed] [Google Scholar]
- 29.Tashiro K, Sephel GC, Greatorex D, Sasaki M, Shirashi N, Martin GR, Kleinman HK, Yamada Y. The RGD containing site of the mouse laminin A chain is active for cell attachment, spreading, migration andneurite outgrowth. J Cell Physiol (1991) 146:451–459. 10.1002/jcp.1041460316. [DOI] [PubMed] [Google Scholar]
- 30.Simon Davis DA, Parish CR. Heparan sulfate: a ubiquitous glycosaminoglycan with multiple roles in immunity. Front Immunol (2013) 4:470 10.3389/fimmu.2013.00470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kilkenny DM, Rocheleau JV. Fibroblast growth factor receptor-1 signaling in pancreatic islet beta-cells is modulated by the extracellular matrix. Mol Endocrinol (2008) 22:196–205. 10.1210/me.2007-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Theodoraki A, Hu Y, Poopalasundaram S, Oosterhof A, Guimond SE, Disterer P, Khoo B, Hauge-Evans AC, Jones PM, Turnbull JE, et al. Distinct patterns of heparan sulphate in pancreatic islets suggest novel roles in paracrine islet regulation. Mol Cell Endocrinol (2015) 399:296–310. 10.1016/j.mce.2014.09.011. [DOI] [PubMed] [Google Scholar]
- 33.*.Llacua LA, Faas MM, de Vos P. Extracellular matrix molecules and their potential contribution to the function of transplanted pancreatic islets. Diabetologia (2018) 61:1261–1272. 10.1007/s00125-017-4524-8.The authors evaluate the potential of specific ECM components in supporting islet function and survival in an alginate-encapsulation matrix. This is the first study demonstrating beneficial effects of specific ECM proteins in this biomaterials-based capsule system.
- 34.*.Ribeiro D, Kvist AJ, Wittung-Stafshede P, Hicks R, Forslöw A. 3D-Models of Insulin-Producing β-Cells: from Primary Islet Cells to Stem Cell-Derived Islets. Stem Cell Rev (2018) 14:177–188. 10.1007/s12015-017-9783-8.The authors evaluate the benefits and drawbacks of current 2D and 3D in vitro systems for the study of islet cells. Due to the complex role of ECM on islet health, a complex culture system is required to recapitulate in vivo models.
- 35.Ziolkowski AF, Popp SK, Freeman C, Parish CR, Simeonovic CJ. Heparan sulfate and heparanase play key roles in mouse β cell survival and autoimmune diabetes. J Clin Invest (2012) 122:132–141. 10.1172/JCI46177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.*.Cross SE, Vaughan RH, Willcox AJ, McBride AJ, Abraham AA, Han B, Johnson JD, Maillard E, Bateman PA, Ramracheya RD, et al. Key Matrix Proteins Within the Pancreatic Islet Basement Membrane Are Differentially Digested During Human Islet Isolation. Am J Transplant (2017) 17:451– 461. 10.1111/ajt.13975.The authors critique current human islet isolation techniques, arguing that the process of isolation itself drastically alters basement membrane composition, with major implications for both in vitro and transplant studies using isolated islets.
- 37.Lingwal N, Padmasekar M, Samikannu B, Bretzel RG, Preissner KT, Linn T. Inhibition of gelatinase B (matrix metalloprotease-9) activity reduces cellular inflammation and restores function of transplanted pancreatic islets. Diabetes (2012) 61:2045–2053. 10.2337/db11-1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wedgwood KCA, Richardson SJ, Morgan NG, Tsaneva-Atanasova K. Spatiotemporal Dynamics of Insulitis in Human Type 1 Diabetes. Front Physiol (2016) 7:633 10.3389/fphys.2016.00633.The authors create a novel and intriguing proof-of-concept mathematical model of beta cell, CD8 T cell and CD20 B cell interactions in human type 1 diabetes based on data taken from current literature. This model enables researchers to view the effects that varying a single or multiple factors (e.g. number of B cells, degradation rate, membrane repair rate) is predicted to have on disease.
- 39.Sixt M, Engelhardt B, Pausch F, Hallmann R, Wendler O, Sorokin LM. Endothelial cell laminin isoforms, laminins 8 and 10, play decisive roles in T cell recruitment across the blood-brain barrier in experimental autoimmune encephalomyelitis. J Cell Biol (2001) 153:933–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gerwien H, Hermann S, Zhang X, Korpos E, Song J, Kopka K, Faust A, Wenning C, Gross CC, Honold L, et al. Imaging matrix metalloproteinase activity in multiple sclerosis as a specific marker of leukocyte penetration of the blood-brain barrier. Sci Transl Med (2016) 8:364ra152–364ra152. 10.1126/scitranslmed.aaf8020. [DOI] [PubMed] [Google Scholar]
- 41.Parish CR, Freeman C, Ziolkowski AF, He YQ, Sutcliffe EL, Zafar A, Rao S, Simeonovic CJ. Unexpected new roles for heparanase in Type 1 diabetes and immune gene regulation. Matrix Biol (2013) 32:228–233. 10.1016/j.matbio.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 42.Brennan TV, Lin L, Huang X, Cardona DM, Li Z, Dredge K, Chao NJ, Yang Y. Heparan sulfate, an endogenous TLR4 agonist, promotes acute GVHD after allogeneic stem cell transplantation. Blood (2012) 120:2899–2908. 10.1182/blood-2011-07-368720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu D, Young JH, Krahn JM, Song D, Corbett KD, Chazin WJ, Pedersen LC, Esko JD. Stable RAGE-heparan sulfate complexes are essential for signal transduction. ACS Chem Biol (2013) 8:1611–1620. 10.1021/cb4001553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagy N, Kaber G, Johnson PY, Gebe JA, Preisinger A, Falk BA, Sunkari VG, Gooden MD, Vernon RB, Bogdani M, et al. Inhibition of hyaluronan synthesis restores immune tolerance during autoimmune insulitis. J Clin Invest (2015) 125:10.1172/JCI79271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kvezereli M, Michie SA, Yu T, Creusot RJ, Fontaine MJ. TSG-6 protein expression in the pancreatic islets of NOD mice. J Mol Histol (2008) 39:585–593. 10.1007/s10735-008-9199-5. [DOI] [PubMed] [Google Scholar]
- 46.Petrey AC, la Motte de CA. Thrombin Cleavage of Inter-α-inhibitor Heavy Chain 1 Regulates Leukocyte Binding to an Inflammatory Hyaluronan Matrix. J Biol Chem (2016) 291:24324–24334. 10.1074/jbc.M116.755660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.*.Day AJ, Milner CM. TSG-6: A multifunctional protein with anti-inflammatory and tissue-protective properties. Matrix Biol (2018) 10.1016/j.matbio.2018.01.011.Extensive review of TSG-6 expression, function, and therapeutic potential in health and across a wide range of diseases. TSG-6 binds to and modifies a number of ECM molecules, including heparan sulfate proteoglycans and hyaluronan.
- 48.Jiang D, Liang J, Noble PW. Hyaluronan as an immune regulator in human diseases. Physiol Rev (2011) 91:221–264. 10.1152/physrev.00052.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Day AJ, Prestwich GD. Hyaluronan-binding proteins: tying up the giant. J Biol Chem (2002) 277:4585– 4588. 10.1074/jbc.R100036200. [DOI] [PubMed] [Google Scholar]
- 50.Day AJ, la Motte de CA. Hyaluronan cross-linking: a protective mechanism in inflammation? Trends Immunol (2005) 26:637–643. 10.1016/j.it.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 51.Cyphert JM, Trempus CS, Garantziotis S. Size Matters: Molecular Weight Specificity of Hyaluronan Effects in Cell Biology. Int J Cell Biol (2015) 2015:563818–8. 10.1155/2015/563818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bogdani M, Johnson PY, Potter-Perigo S, Nagy N, Day AJ, Bollyky PL, Wight TN. Hyaluronan and hyaluronan-binding proteins accumulate in both human type 1 diabetic islets and lymphoid tissues and associate with inflammatory cells in insulitis (2014) 63:2727–2743. 10.2337/db13-1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bollyky PL, Bogdani M, Bollyky JB, Hull RL, Wight TN. The Role of Hyaluronan and the Extracellular Matrix in Islet Inflammation and Immune Regulation. Curr Diab Rep (2012) 12:471–480. 10.1007/s11892-012-0297-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.*.Nagy N, la Zerda de A, Kaber G, Johnson PY, Hu KH, Kratochvil MJ, Yadava K, Zhao W, Cui Y, Navarro G, et al. Hyaluronan content governs tissue stiffness in pancreatic islet inflammation. J Biol Chem (2018) 293:567–578. 10.1074/jbc.RA117.000148.Using atomic force microscopy, the authors show that islet HA content governs islet stiffness in a mouse model of type 1 diabetes. This is the first study to implicate a role for mechanosensing in islet inflammation.
- 55.Dahl LB, Dahl IM, Engström-Laurent A, Granath K. Concentration and molecular weight of sodium hyaluronate in synovial fluid from patients with rheumatoid arthritis and other arthropathies. Ann Rheum Dis (1985) 44:817–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Back SA, Tuohy TMF, Chen H, Wallingford N, Craig A, Struve J, Luo NL, Banine F, Liu Y, Chang A, et al. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nat Med (2005) 11:966–972. 10.1038/nm1279. [DOI] [PubMed] [Google Scholar]
- 57.Shan SJC, Douglas RS. The pathophysiology of thyroid eye disease. J Neuroophthalmol (2014) 34:177–185. 10.1097/WNO.0000000000000132. [DOI] [PubMed] [Google Scholar]
- 58.The Role of Hyaluronan and CD44 in the Pathogenesis of Lupus Nephritis. The Role of Hyaluronan and CD44 in the Pathogenesis of Lupus Nephritis (2012) 2012:207190–9. 10.1155/2012/207190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nagy N, Kuipers HF, Frymoyer AR, Ishak HD, Bollyky JB, Wight TN, Bollyky PL. 4-methylumbelliferone treatment and hyaluronan inhibition as a therapeutic strategy in inflammation, autoimmunity, and cancer. Front Immunol (2015) 6:123 10.3389/fimmu.2015.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kuipers HF, Nagy N, Ruppert SM, Sunkari VG, Marshall PL, Gebe JA, Ishak HD, Keswani SG, Bollyky J, Frymoyer AR, et al. The Pharmacokinetics and Dosing of Oral 4-Methylumbelliferone for Inhibition of Hyaluronan Synthesis in Mice. Clin Exp Immunol (2016) 185:372–381. 10.1111/cei.12815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weiss L, Slavin S, Reich S, Cohen P, Shuster S, Stern R, Kaganovsky E, Okon E, Rubinstein AM, Naor D. Induction of resistance to diabetes in non-obese diabetic mice by targeting CD44 with a specific monoclonal antibody. Proc Natl Acad Sci USA (2000) 97:285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yoshioka Y, Kozawa E, Urakawa H, Arai E, Futamura N, Zhuo L, Kimata K, Ishiguro N, Nishida Y. Suppression of hyaluronan synthesis alleviates inflammatory responses in murine arthritis and in human rheumatoid synovial fibroblasts. Arthritis Rheum (2013) 65:1160–1170. 10.1002/art.37861. [DOI] [PubMed] [Google Scholar]
- 63.Hutás G, Bajnok E, Gál I, Finnegan A, Glant TT, Mikecz K. CD44-specific antibody treatment and CD44 deficiency exert distinct effects on leukocyte recruitment in experimental arthritis. Blood (2008) 112:4999–5006. 10.1182/blood-2008-04-150383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.*.Kuipers HF, Rieck M, Gurevich I, Nagy N, Butte MJ, Negrin RS, Wight TN, Steinman L, Bollyky PL. Hyaluronan synthesis is necessary for autoreactive T-cell trafficking, activation, and Th1 polarization. Proc Natl Acad Sci USA (2016) 113:1339–1344. 10.1073/pnas.1525086113.This manuscript demonstrates that HA polarizes T-cells towards a Th1 phenotype while the inhibition of HA synthesis polarizes T-cells toward Th2 and Foxp3+ Treg, indicating that the ECM can be targeted in ways that impact t-cell cell development.
- 65.Brocke S, Piercy C, Steinman L, Weissman IL, Veromaa T. Antibodies to CD44 and integrin alpha4, but not L-selectin, prevent central nervous system inflammation and experimental encephalomyelitis byblocking secondary leukocyte recruitment. Proc Natl Acad Sci USA (1999) 96:6896–6901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guan H, Nagarkatti PS, Nagarkatti M. Role of CD44 in the differentiation of Th1 and Th2 cells: CD44-deficiency enhances the development of Th2 effectors in response to sheep RBC and chicken ovalbumin. J Immunol (2009) 183:172–180. 10.4049/jimmunol.0802325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guan H, Nagarkatti PS, Nagarkatti M. CD44 Reciprocally regulates the differentiation of encephalitogenic Th1/Th17 and Th2/regulatory T cells through epigenetic modulation involving DNA methylation of cytokine gene promoters, thereby controlling the development of experimental autoimmune encephalomyelitis. J Immunol (2011) 186:6955–6964. 10.4049/jimmunol.1004043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bollyky PL, Falk BA, Long SA, Preisinger A, Braun KR, Wu RP, Evanko SP, Buckner JH, Wight TN, Nepom GT. CD44 costimulation promotes FoxP3+ regulatory T cell persistence and function via production of IL-2, IL-10, and TGF-beta. J Immunol (2009) 183:2232–2241. 10.4049/jimmunol.0900191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bollyky PL, Lord JD, Masewicz SA, Evanko SP, Buckner JH, Wight TN, Nepom GT. Cutting edge: high molecular weight hyaluronan promotes the suppressive effects of CD4+CD25+ regulatory T cells. J Immunol (2007) 179:744–747. [DOI] [PubMed] [Google Scholar]
- 70.Bollyky PL, Falk BA, Wu RP, Buckner JH, Wight TN, Nepom GT. Intact extracellular matrix and the maintenance of immune tolerance: high molecular weight hyaluronan promotes persistence of induced CD4+CD25+ regulatory T cells. J Leukoc Biol (2009) 86:567–572. 10.1189/jlb.0109001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Haxhinasto S, Mathis D, Benoist C. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J Exp Med (2008) 205:565–574. 10.1084/jem.20071477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu H, Yao S, Dann SM, Qin H, Elson CO, Cong Y. ERK differentially regulates Th17- and Treg-cell development and contributes to the pathogenesis of colitis. Eur J Immunol (2013) 43:1716–1726. 10.1002/eji.201242889. [DOI] [PubMed] [Google Scholar]
- 73.Assayag-Asherie N, Sever D, Bogdani M, Johnson P, Weiss T, Ginzberg A, Perles S, Weiss L, Sebban LE, Turley EA, et al. Can CD44 Be a Mediator of Cell Destruction? The Challenge of Type 1 Diabetes. PLoS ONE (2015) 10:e0143589 10.1371/journal.pone.0143589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol (2005) 23:447–485. 10.1146/annurev.immunol.23.021704.115643 [DOI] [PubMed] [Google Scholar]
- 75.Coppieters KT, Boettler T, Herrath von M. Virus infections in type 1 diabetes. Cold Spring Harb Perspect Med (2012) 2:a007682–a007682. 10.1101/cshperspect.a007682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bollyky PL, Evanko SP, Wu RP, Potter-Perigo S, Long SA, Kinsella B, Reijonen H, Guebtner K, Teng B, Chan CK, et al. Th1 cytokines promote T-cell binding to antigen-presenting cells via enhanced hyaluronan production and accumulation at the immune synapse. Cell Mol Immunol (2010) 7:211–220. 10.1038/cmi.2010.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gaucherand L, Falk BA, Evanko SP, Workman G, Chan CK, Wight TN. Crosstalk Between T Lymphocytes and Lung Fibroblasts: Generation of a Hyaluronan-Enriched Extracellular Matrix Adhesive for Monocytes. J Cell Biochem (2017) 118:2118–2130. 10.1002/jcb.25842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nagy N, Kuipers HF, Marshall PL, Wang E, Kaber G, Bollyky PL. Hyaluronan in immune dysregulation and autoimmune diseases. Matrix Biol (2018) 10.1016/j.matbio.2018.03.022 [DOI] [PMC free article] [PubMed]
- 79.van Horssen J, Dijkstra CD, de Vries HE. The extracellular matrix in multiple sclerosis pathology. J Neurochem (2007) 103:1293–1301. 10.1111/j.1471-4159.2007.04897.x [DOI] [PubMed] [Google Scholar]
- 80.Ehlers MR, Nepom GT. Immune-directed therapy for type 1 diabetes at the clinical level: the Immune Tolerance Network (ITN) experience. Rev Diabet Stud (2012) 9:359–371. 10.1900/RDS.2012.9.359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Herold KC, Bluestone JA. Type 1 diabetes immunotherapy: is the glass half empty or half full? Sci Transl Med (2011) 3:95fs1–95fs1. 10.1126/scitranslmed.3002981 [DOI] [PubMed] [Google Scholar]
- 82.Lernmark A, Larsson HE. Immune therapy in type 1 diabetes mellitus. Nat Rev Endocrinol (2013) 9:92– 103. 10.1038/nrendo.2012.237 [DOI] [PubMed] [Google Scholar]
- 83.Skyler JS. Immune therapy for treating type 1 diabetes: challenging existing paradigms. J Clin Invest (2015) 125:94–96. 10.1172/JCI79190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liu C-J, Lee P-H, Lin D-Y, Wu C-C, Jeng L-B, Lin P-W, Mok K-T, Lee W-C, Yeh H-Z, Ho M-C, et al. Heparanase inhibitor PI-88 as adjuvant therapy for hepatocellular carcinoma after curative resection: a randomized phase II trial for safety and optimal dosage. J Hepatol (2009) 50:958–968. 10.1016/j.jhep.2008.12.023. [DOI] [PubMed] [Google Scholar]
- 85.Abate A, Dimartino V, Spina P, Costa PL, Lombardo C, Santini A, Del Piano M, Alimonti P. Hymecromone in the treatment of motor disorders of the bile ducts: a multicenter, double-blind, placebo-controlled clinical study. Drugs Exp Clin Res (2001) 27:223–231. [PubMed] [Google Scholar]