

Abstract
Alcohol excitation of the ventral tegmental area (VTA) is important in neurobiological processes related to the development of alcoholism. The ionotropic receptors on VTA neurons that mediate ethanol-induced excitation have not been identified. Quinidine blocks ethanol excitation of VTA neurons, and blockade of two-pore potassium channels is among the actions of quinidine. Therefore two-pore potassium channels in the VTA may be potential targets for the action of ethanol. Here, we explored whether ethanol activation of VTA neurons is mediated by the twopore potassium channel KCNK13. Extracellular recordings of the response of VTA neurons to ethanol were performed in combination with knockdown of Kcnk13 using a short hairpin RNA (shRNA) in C57BL/6J mice. Real-time PCR and immunohistochemistry were used to examine expression of this channel in the VTA. Finally, the role of KCNK13 in binge-like drinking was examined in the drinking in the dark test after knockdown of the channel. Kcnk13 expression in the VTA was increased by acute ethanol exposure. Ethanol-induced excitation of VTA neurons was selectively reduced by shRNA targeting Kcnk13. Importantly, knockdown of Kcnk13 in the VTA resulted in increased alcohol drinking. These results are consistent with the idea that ethanol stimulates VTA neurons at least in part by inhibiting KCNK13, a specific two-pore potassium channel, and that KCNK13 can control both VTA neuronal activity and binge drinking. KCNK13 is a novel alcohol-sensitive molecular target and may be amenable to the development of pharmacotherapies for alcoholism treatment.
Graphical Abstract
Introduction
Activation of dopaminergic (DA) neurons of the ventral tegmental area (VTA) is a property of alcohol that is important for its rewarding and reinforcing actions (Koob and Volkow, 2010). VTA neuronal activity is subject to regulation by several different ionic currents (Canavier et al., 2007; Korotkova et al., 2004) and by synaptic activity, especially activity related to γaminobutyric acid (GABA) (Guan et al., 2012; Theile et al., 2011) or glutamate (Deng et al., 2009; Xiao et al., 2009) neurotransmission. However, acutely dissociated neurons devoid of synaptic connections are still activated by ethanol, indicating that ethanol can excite VTA neurons independent of extrinsic afferents (Brodie et al., 1999). Furthermore, our recent comparison of toluene and ethanol excitation of VTA neurons demonstrated that the effect of ethanol is not diminished by combinations of antagonists of GABAA, GABAB, NMDA, AMPA, metabotropic glutamate, muscarinic and nicotinic cholinergic receptors (Nimitvilai et al., 2016), indicating that these receptors, although important for modulating ethanol excitation, are not primary mediators of the excitatory action of ethanol on VTA neurons in vitro. Several ion channels present on VTA neurons, e.g. h-channels (HCN) (Okamoto et al., 2006; Rivera-Meza et al., 2014) and G protein-coupled potassium channels (GIRK) (Herman et al., 2015), have been suggested as targets of ethanol in the VTA, but pharmacological antagonists of these channels do not reduce ethanol excitation in the VTA (Appel et al., 2003; Koyama et al., 2007; McDaid et al., 2008), so their role in ethanol excitation is likely to be subordinate to a different mechanism that is primarily responsible for the excitation.
One class of ion channels that has been neglected with respect to studies of alcohol is the large family of two-pore channels often called leak potassium channels. These channels are constitutively open and help maintain a negative resting membrane potential (Goldstein et al., 2001). Modulation of leak channel conductance could effectively regulate neuronal activity; specifically, inhibition of leak channels would produce excitation. Depolarization and increased spontaneous firing of VTA neurons, therefore, might occur if ethanol inhibited a leak potassium channel. Different two-pore channels have diverse pharmacological profiles; for example, some are activated by volatile anesthetics and others are inhibited by those same anesthetics (Enyedi and Czirjak, 2010). The tandem-pore halothane-inhibited potassium (THIK) channels, KCNK12 and KCNK13, are inhibited by isoflurane, halothane, quinine, quinidine, and by high extracellular calcium concentration (Enyedi and Czirjak, 2010). These channels are expressed in many brain areas (Rajan et al., 2001), but there are few reports characterizing the function of THIK channels in the central nervous system. As ethanol excitation is blocked by quinidine (Appel et al., 2003) and quinine (Nimitvilai et al., 2016), we hypothesized that THIK leak potassium channels play an important role in mediation of ethanol excitation of VTA neurons. If ethanol action on leak potassium channels in the VTA is behaviorally important, then interfering with this target should alter alcohol drinking. Here, we provide evidence that one THIK channel, KCNK13, affects ethanol excitation and regulates binge-like ethanol consumption. KNCNK13 may be a novel and significant target of ethanol action on VTA neurons.
Methods and Materials
Animals
Male C57BL/6J mice (C57) were purchased from the Jackson Laboratory (Bar Harbor, ME). Mice were 4–5 weeks old on arrival and tested when they were 8–9 weeks old for siRNA experiments (electrophysiology and drinking behavior, Fig 3-5). Mice used for high Ca++ and isoflurane experiments (Fig 1), to identify the presence of KCNK13 protein (Fig 2A) and for mRNA expression after acute ethanol treatment (Fig 2B) were between 4 to 6 weeks old. All mice were treated in strict accordance with the NIH Guide for the Care and Use of Laboratory Animals, and all experimental methods were approved by the Animal Care Committee of the University of Illinois at Chicago.
Figure 3. In vivo downregulation of Kcnk13 with lentiviral siRNA.
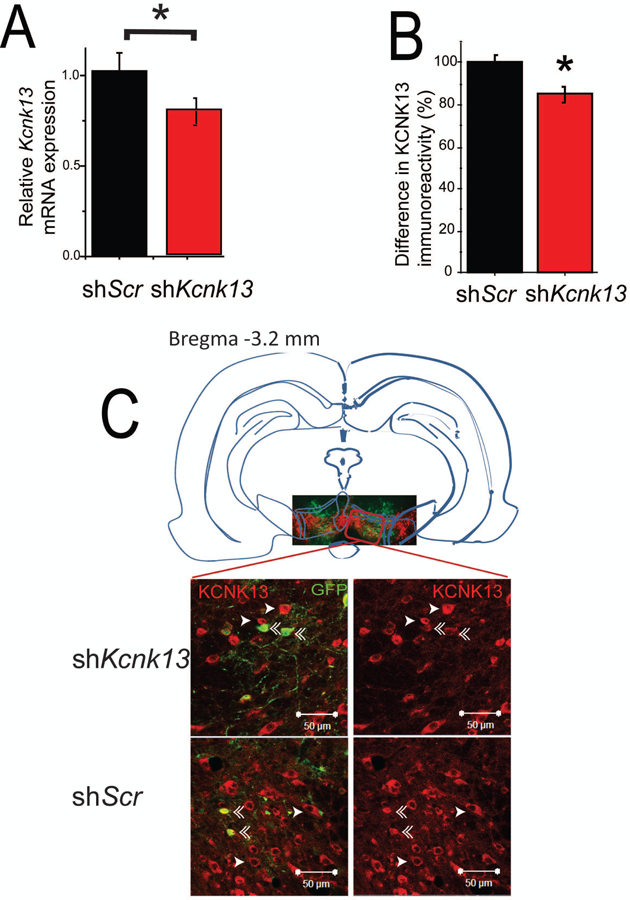
A. Reduced expression of Kcnk13 mRNA in the VTA of mice expressing shKcnk13 compared with mice expressing the control shRNA, shScr. Effectiveness of Kcnk13 shRNA in the VTA was assessed by qPCR of RNA from dissected VTA three weeks after lentivirus infection. Kcnk13 was reduced by 22.2% (unpaired t-test, t-statistic= 1.92,* p< 0.05, n=6).
B, C. Reduced expression of KCNK13 protein in the VTA of mice expressing shKcnk13. Immunohistochemistry was performed three weeks after lentiviral infection using antibodies to KCNK13 and GFP. KCNK13 immunoreactivity was compared between GFP-positive neurons and GFP-negative neurons.
B. GFP-positive neurons in mice expressing shKcnk13 expressed 15.1± 3.4% less KCNK13 immunoreactivity than GFP-negative neurons (paired t-test, t= −4.88, p<0.001, n=18). In mice expressing shScr in the VTA, GFP-positive neurons expressed 0.64 ± 3.4% more KCNK13 immunoreactivity compared to GFP-negative neurons (paired t-test, t= 0.305, p>0.05, n=15). Asterisk (*) indicates significant difference between GFP-positive and GFP-negative neurons.
C. Illustration of the typical location of microinjection sites (inset picture showing VTA region immunostained for GFP and TH) and immunostaining of KCNK13 and GFP in mice expressing shKcnk13 and shScr (four larger pictures). Single arrowhead indicates KCNK13-immunoreactive cells, double arrowhead indicates cells immunoreactive for both KCNK13 and GFP. Note the reduced KCNK13 immunoreactivity in the GFP-immunoreactive cells.
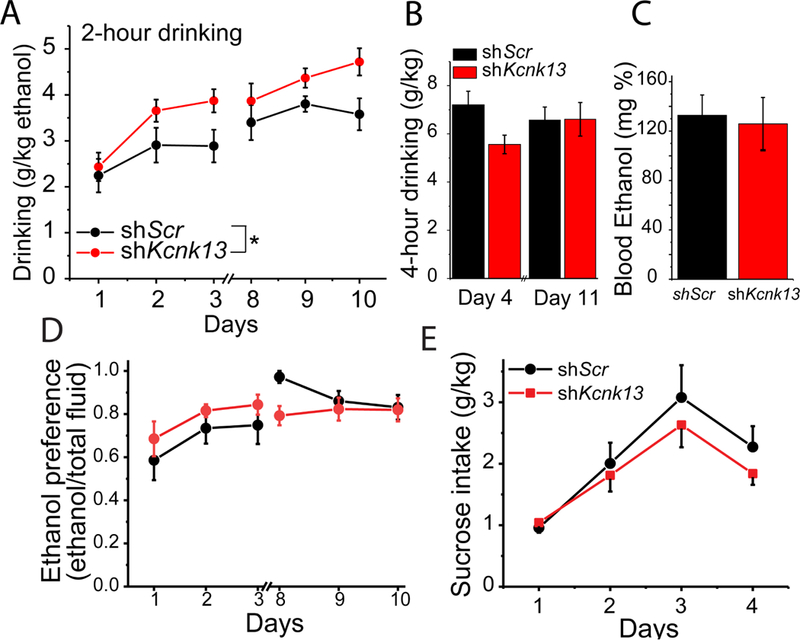
Figure 5. Knockdown of Kcnk13 increases binge-like ethanol consumption.
A. Lentivirus expressing shRNA targeting Kcnk13 (shKcnk13) or a non-targeting shRNA (shScr) was microinjected into the mouse VTA. Three weeks after injection, mice were tested for bingelike alcohol drinking. Mice injected with shKcnk13 (n=10) drank significantly more during the two-hour drinking sessions than mice injected with shScr (n=6) (two-way ANOVA, effect of shRNA, F1, 84=13.5, p<0.001).
B. On day 4 and day 11 of the Drinking in the Dark protocol, mice were given access to ethanol solutions for 4 hours instead of 2 hours. There was no significant difference between the groups in the amount of ethanol consumed in 4 hours.
C. Blood ethanol concentrations were assessed after the four-hour drinking session on day 11. No significant difference in blood alcohol levels between the groups was observed.
D. Ethanol preference (assessed during the drinking in the dark test), is calculated as the percent of ethanol consumed over total fluid consumption. There was no significant difference in ethanol preference, but as both groups preferred ethanol to a great extent (~80%), there may have been a ceiling effect; that is, as both the shKcnk13 (n=10) and the control (n=6) groups preferred ethanol to such a great extent, there was little room to distinguish between the groups (two-way RM ANOVA, F1, 4=0.811, p>0.05).
E. Following two weeks of ethanol drinking, mice that had received injections of either lentiviral-delivered Kcnk13 shRNA (shKcnk13, n=6) or scrambled control (shScr, n=6) were tested for sucrose consumption for four days, for two hours per day. There was no significant difference between the groups (two-way ANOVA, F1, 40=0.26, p>0.05).
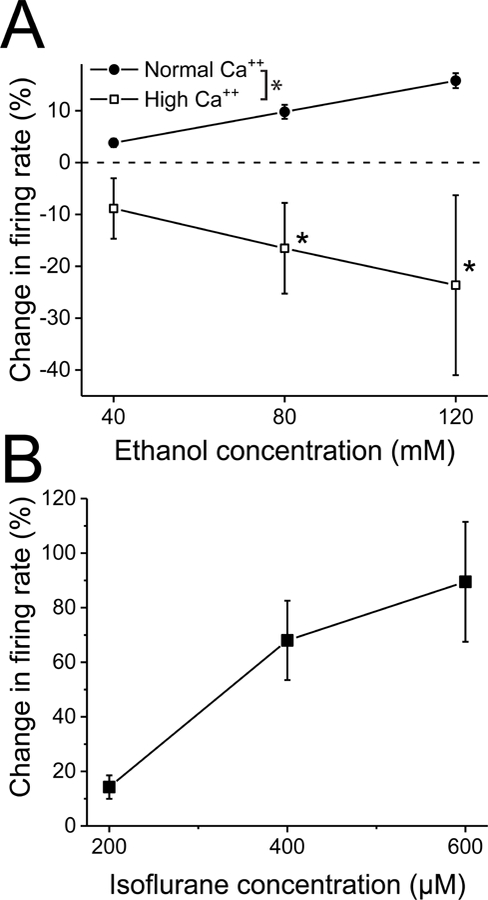
Figure 1. High extracellular calcium blocks, and isoflurane mimics, ethanol excitation of VTA neurons.
A. High Ca++ medium blocks ethanol excitation. Ethanol was tested in the presence and absence of high Ca++ external medium. Increases in Ca++ concentration from normal (2.5 mM) were made by substituting Ca++ for sodium in the external medium. In normal medium, ethanol (40, 80, 120 mM) produced excitation of 3.8 ± 0.8, 9.8 ± 1.4, and 15.8 ± 1.4, respectively. High Ca++ was adjusted to maintain a regular firing rate. Despite this precaution, changing to high Ca++ medium (ranging from 5.8 to 12.5 mM total Ca++) significantly reduced the baseline firing from 2.6 ± 0.21 to 1.9 ± 0.25 Hz (paired t test, t=3.07, df=7, p<0.02, n=8 from 6 mice). In high Ca++ medium, ethanol produced changes in firing rate of −8.9 ± 5.8% (40 mM), −16.5 ± 8.7% (80 mM), and −23.6 ± 17.3% (120 mM). Ethanol excitation was significantly reduced with a significant interaction between concentration and the effect of high Ca++ (two-way RM ANOVA, F1, 7= 6.4, p<0.04 for effect of high Ca++, Tukey post-hoc comparison, * p<0.05; n=8).
B. Isoflurane increases the firing rate of VTA neurons. Isoflurane (200–600 µM) was added to the superfusate for 5 minutes, and the change in firing rate was measured. Isoflurane produced a significant concentration-dependent increase in firing (one-way ANOVA, F2, 7= 10.58, p<0.002, n=8 from 4 mice).
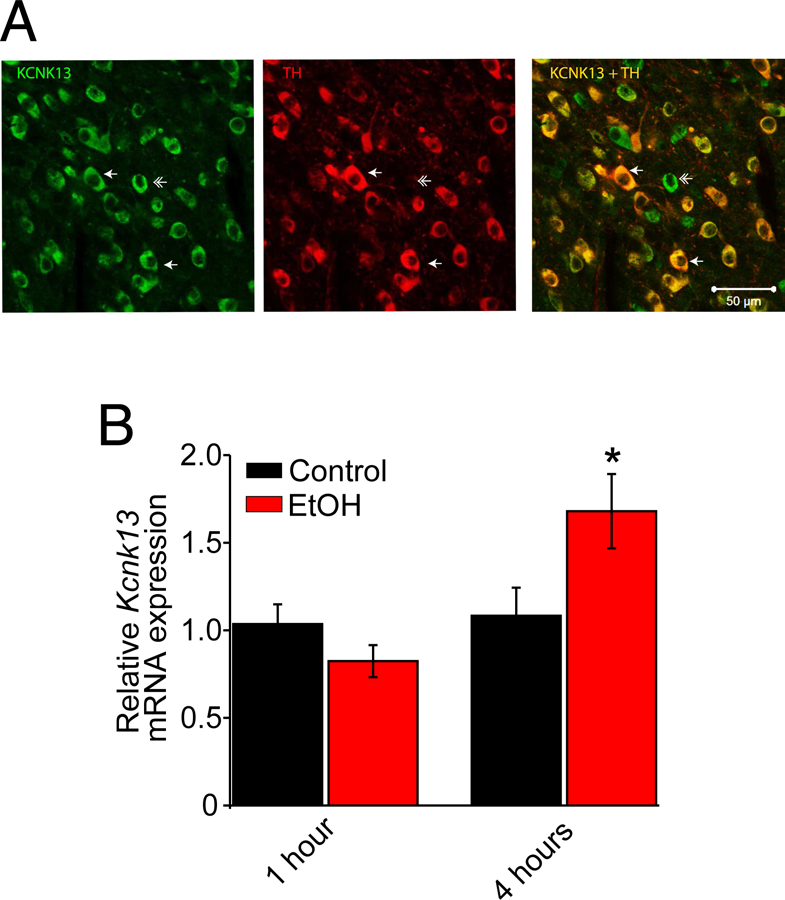
Figure 2. KCNK13 is expressed in dopamine and non-dopamine neurons in the VTA and Kcnk13 expression is induced by acute alcohol.
A. Immunohistochemistry was performed using antibodies to KCNK13 and tyrosine hydroxylase (TH). Left panel: Neurons labeled green are KCNK13-positive. Middle panel: Neurons labeled red are TH-positive. Right panel: Merging the images shows that some neurons (single arrow) are stained for both TH and KCNK13, and other neurons are TH negative but KCNK13 positive (double arrow).
B. Quantitative PCR was performed in tissue from mice given a systemic injection of ethanol (EtOH, 3 g/kg, IP) or saline (Control) one hour or four hours before sacrifice. There was no change in Kcnk13 mRNA one hour after ethanol administration; four hours after ethanol treatment, there was a significant increase in Kcnk13 mRNA (unpaired t test, t = −2.25, p<0.05. n=9).
Electrophysiology
The brain slice preparation technique has been described previously (Appel et al., 2006). Briefly, the brain was removed rapidly; coronal sections (400 µm thick) containing the VTA were cut and were placed immediately in the recording chamber in which artificial cerebrospinal fluid (aCSF) was flowing at 2 ml/min at 35º C. The composition of the aCSF in these experiments was (in mM): NaCl 126, KCl 2.5, NaH2PO4 1.24, CaCl2 2.4, MgSO4 1.3, NaHCO3 26, glucose 11. The composition of the cutting solution was (in mM): KCl 2.5, CaCl2 2.4, MgSO4 1.3, NaHCO3 26, glucose 11, and sucrose 220. All solutions were saturated with 95% O2/5% CO2 (pH=7.4).
Recording electrodes were placed in the VTA under visual control. Only those neurons which were located within the lateral VTA and which conformed to the criteria for DA neurons established in the literature and in this laboratory (Brodie et al., 1988; Lacey et al., 1989; Mueller and Brodie, 1989) were studied. Not all VTA neurons in this study were characterized pharmacologically as dopamine-containing neurons (Margolis et al., 2006; Margolis et al., 2012), but all of the neurons conforming to these electrophysiological criteria that have been tested with baclofen (0.1–1 µM) in our laboratory have been inhibited; sensitivity to inhibition by baclofen is a property of DA VTA neurons but not GABAergic VTA neurons (Margolis et al., 2012). The VTA neurons in this study (32 cells from 14 mice) had a mean firing rate of 2.28 ± 0.16 Hz with a mean interspike interval of 0.54 ± 0.05 msec. The mean coefficient of variation (CV) for the cells reported below was 0.13 ± 0.02. Note that a low CV does not distinguish between DA and non-DA neurons (Margolis et al., 2012).
Drugs were added to the aCSF using a calibrated infusion pump from stock solutions 100 to 1000 times the desired final concentrations. Final concentrations were calculated from aCSF flow rate, pump infusion rate and concentration of drug stock solution. The small volume chamber (about 300 µl) used in these studies permitted the rapid application and washout of drug solutions. Ethanol (95%; 190 proof) and most of the salts used to prepare the extracellular media were purchased from Sigma (St. Louis, MO). Isoflurane was from Henry Schein Animal Health (Dublin, OH).
Extracellular recording electrodes were made from 1.5 mm diameter glass tubing (Sutter Instruments, Novato, CA) with filament and were filled with 0.9% NaCl. Tip resistance of all microelectrodes ranged from 2 – 4 MΩ. A high-gain extracellular amplifier (x-Cell, FHC, Inc., Bowdoin, ME) was used in conjunction with an IBM-PC-based data acquisition system (ADInstruments, Inc.). Firing rate was determined before and during drug application, and was calculated over one minute intervals prior to administration of drugs and during the drug effects. The change in firing rate is expressed as a percentage of the initial firing rate, which controls for slight changes in firing rate that may occur over time.
Short hairpin RNA (shRNA)
Three different shRNAs were designed to target mouse Kcnk13 using the siDirect 2.0 design tool (Naito and Ui-Tei, 2012). The three 21-nucleotide targeting sequences were incorporated into hairpins and the oligonucleotides cloned into the lentiviral vector pLL3.7 (Lasek et al., 2007). The control shRNA (shScr) sequence has been described previously and encodes a nonspecific hairpin, predicted to not target any gene in the mouse genome. Sequences and in vitro testing of the Kcnk13 shRNA are shown in Supplementary Fig S1. Lentivirus was produced in 293FT cells as previously described (Dutton et al., 2016).
Stereotaxic surgery and lentiviral injections
Mice were anesthetized with ketamine/xylazine, placed in a stereotaxic alignment system (David Kopf Instruments) and injected with 1.0 µl of virus solution bilaterally. VTA coordinates were: AP −3.2 mm, ML +/− 0.5 mm, DV −4.7 mm. Viral titers were 3 × 107 pg p24 gag antigen/ml. Mice recovered for 3 weeks prior to testing ethanol consumption or electrophysiological studies. Viral infection was confirmed by immunohistochemistry after the completion of drinking in the dark (DID) testing using anti-GFP and anti-TH antibodies (see Figure 3C small image for example).
Immunofluorescent staining of brain sections
Mice were euthanized with pentobarbital followed by transcardial perfusion with cold phosphate-buffered saline (PBS) and 4% paraformaldehyde. Brains were processed as described previously (Kharazia et al., 2003). Primary antibodies for TH and KCNK13 detection (Fig 1) were mouse anti-TH (EMD Millipore, Burlington, MA, catalog number MAB318) and rabbit anti-KCNK13 (Novus, Littleton, CO, catalog number NBP2–41132). Primary antibodies for GFP and tyrosine hydroxylase (TH) detection (Fig 3C, small image) were rabbit anti-TH (Millipore, Temecula, CA, catalog number AB152) and mouse anti-GFP (Thermo-Fisher, Asheville, NC, catalog number A-11120). The primary antibodies for KCNK13 and GFP double staining (Fig 3C, larger 4 images) were rabbit anti-KCNK13 (Novus, Littleton, CO) and mouse anti-GFP (Thermo-Fisher, Asheville, NC). Images of lentiviral infection in the VTA (TH and GFP) were captured using an EVOS fl inverted fluorescence microscope with a 4X objective lens (Life Technologies) and images of KCNK13 expression in VTA (KCNK13 and TH, KCNK13 and GFP) were captured using a Zeiss LSM 710 confocal microscope (Carl Zeiss, Thornwood, NY). For quantification of knockdown of KCNK13 in the VTA after lentiviral infection, the intensity of KCNK13 staining in the infected VTA was quantified from 2–10 cells (GFP+, and GFP-) per section, 3 sections per mouse and 5–6 mice per group (shScr and shKcnk13) 3 weeks after lentiviral infection. Intensity of KCNK13 immunofluorescence was measured using the National Institutes of Health ImageJ software, and compared between GFP+ and GFP− cells. As comparisons were made between GFP+ and GFP− cells in each slice, a paired t-test was used to establish statistical significance for differences between the groups.
Drinking in the dark (DID)
The 4-day ethanol drinking test was a two-bottle variation of the standard DID test as described previously (Rhodes et al., 2005). Briefly, mice were individually housed in a 12-hour reversed light/dark cycle room (lights off 10 am to 10 pm) for 2 weeks prior to behavioral testing. The DID test was performed by replacing the water bottle 3 hours into the dark cycle with a pair of sipper tubes, one containing water and the other containing 20% ethanol in water. Locations of the alcohol and water tubes were alternated daily. On the first 3 days of each week (day 1, 2, 3 and day 8, 9, 10), mice were given access to the ethanol solution for 2 hours. On the fourth day of each week (day 4 and day 11), mice were given access to the ethanol solution for 4 hours. We did not test at the two hour time point on days 4 and 11, as we were concerned that disruption at the two hour time point would alter drinking over the 4 hour session in the groups differentially. Mice had no access to alcohol on Days 5, 6 and 7. Blood samples (20 µl) were collected immediately after the 4-hour drinking session on day 11 to measure blood ethanol concentrations (BECs). Blood was collected in heparinized capillary tubes via tail vein puncture. BECs were determined using a nicotinamide adenine dinucleotide-alcohol dehydrogenase enzymatic assay (Zapata et al., 2006). To test sucrose consumption, mice underwent a test identical to the ethanol consumption test, except one of the provided sipper tubes contained 2% sucrose instead of 20% ethanol.
Quantitative real-time PCR (qPCR)
Mice were sacrificed one or four hour(s) after intraperitoneal injection of 3g/kg ethanol or saline. Brains were rapidly harvested after sacrifice, and the VTA was dissected using RNase-free conditions. RNA was isolated from VTA tissue using the RNeasy mini kit (Qiagen, Valencia, CA) and subjected to first strand cDNA synthesis using reverse transcriptase (Thermo Fisher, Waltham, MA). Quantitative real-time PCR was used to determine the mRNA level of Kcnk13. Relative expression was calculated using the delta Cq method. Details of the methods used for qPCR to examine the mRNA expression of Kcnk13 in the VTA after acute ethanol exposure is provided in Supplementary Information. The results are expressed as fold change in reference to Gapdh expression.
Statistical Analysis
Statistical analyses were performed with Origin (Originlab, Northampton, MA) or GraphPad Prism version 6.05 (GraphPad Software, Inc., La Jolla, CA). Comparison of differences in baseline firing rate (Figure 1A, 4C), and differences in protein expression (Figure 3B), were assessed with paired t tests (within subjects); differences in mRNA expression (Figure 2B and3A) and BECs (Figure 5C) were assessed with unpaired t tests (between subjects). One-way ANOVA was used to assess significance of excitation induced by isoflurane (Figure 1B) and 4 hour drinking (Figure 5B). Two-way ANOVAs were used to compare ethanol concentrationresponses or 2-hour drinking between groups (Figure 4C and 5A) or concentration-responses between groups (Figure 1A). In the case of the ANOVAs, Tukey’s post-hoc tests were used for multiple comparisons testing as appropriate.
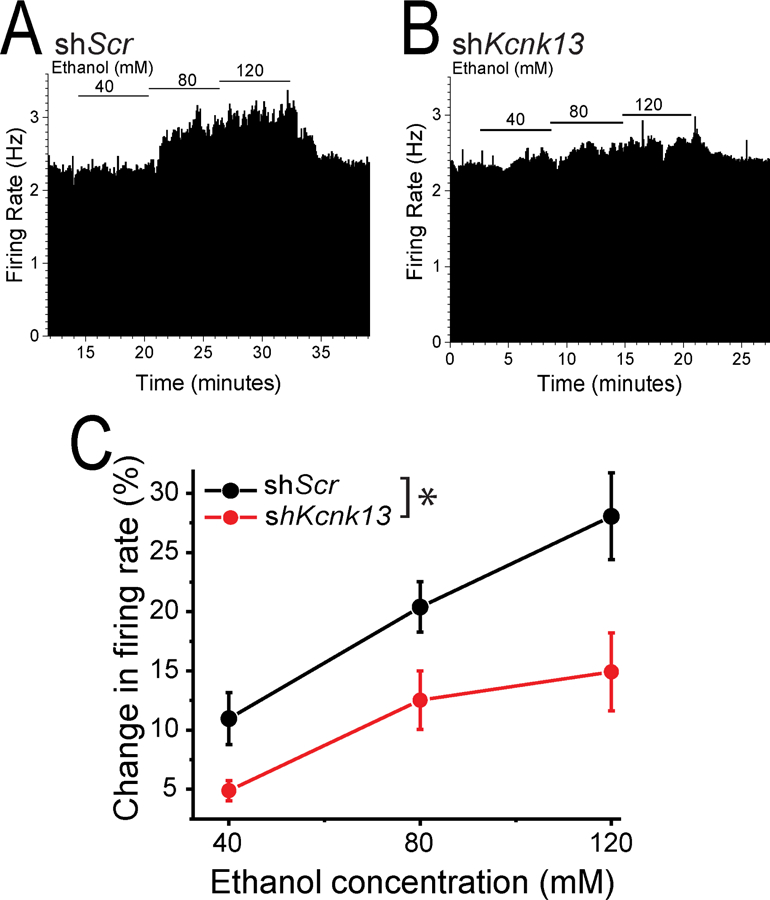
Figure 4. Ethanol excitation is attenuated with in vivo downregulation of Kcnk13.
A, B Ratemeter graphs of the responses to ethanol of single neurons after VTA infection with lentivirus expressing either shScr (A) or shKcnk13 (B). Ethanol (40, 80, 120 mM) produced excitation of (A) 1.85, 26.5, and 37.7 %, and (B) 5.5, 9.9, and 12.8 %.
C. Pooled results from extracellular recordings of VTA neurons in brain slices from mice expressing either shKcnk13 or shScr. The effect of ethanol (40, 80 120 mM) on firing rate was tested. Ethanol excitation was significantly decreased in VTA neurons from shKcnk13 mice compared with control (two-way RM ANOVA, F1, 45= 17.34, p<0.006 for effect of shRNA, n=8). Mean baseline firing between the treatment groups was not significantly different (shScr 1.90 ± 0.46 Hz, shKcnk13, 2.50 ± 0.46 Hz; unpaired t-test, t=1.08, p > 0.05, n=8)
Results
The leak potassium channel KCNK13 is potentially involved in ethanol excitation in the VTA.
A number of conditions interfere with leak potassium channel function, including external quinine, quinidine, low external pH, temperature, volatile anesthetics like isoflurane (Enyedi and Czirjak, 2010; Goldstein et al., 2001), and high external calcium ion (Ca++) (Goldstein et al., 2001; Rajan et al., 2000). We demonstrated previously that quinine (Nimitvilai et al., 2016) and quinidine (Appel et al., 2003) block ethanol excitation, pointing to the possibility that a specific leak potassium channel might underlie ethanol-induced excitation in the VTA. In initial experiments to try to identify the quinidine-sensitive target of alcohol, we increased the extracellular Ca++ concentration, which blocked the ethanol-induced excitation (Figure 1A). Initial firing rate of neurons in this experiment was 1.9 ± 0.2 Hz with a CV of 0.15 ± 0.08. In the presence of high extracellular Ca++, ethanol produced a concentration-dependent decrease in firing. One possibility for this inhibition could be the effect of ethanol to increase the activity of inhibitory barium-sensitive potassium channels (McDaid et al., 2008; Okamoto et al., 2006). In the absence of the excitatory action of ethanol, this inhibitory action of ethanol might become apparent (You et al., 2018). As Ca++ has a variety of membrane and intracellular effects (on protein kinase C and calcium calmodulin kinase, for example), extensive additional studies will be needed to determine the mechanism of the ethanol-induced inhibition in the presence of high Ca++.
As leak potassium channels are sensitive to volatile anesthetics, we tested the effect of isoflurane on neuronal firing. Initial firing rate of neurons in this experiment was 2.6 ± 0.2 Hz with a CV of 0.16 ± 0.04. Isoflurane (200 – 600 µM) mimicked the excitatory effect of ethanol (Figure 1B). Isoflurane-induced excitation is consistent with role of THIK channels (KCNK12 and KCNK13) in controlling VTA neuronal firing rate and possibly mediating ethanol excitation of VTA neurons, as other leak channels are activated by volatile anesthetics. As KCNK12 is largely localized to the endoplasmic reticulum (Chatelain et al., 2013; Renigunta et al., 2014), we focused our investigation of ethanol excitation and drinking behavior on KCNK13.
Despite an early report using in situ hybridization that indicated low expression of Kcnk13 mRNA in the VTA (Rajan et al., 2001), our immunohistochemical examination of the VTA indicates that KCNK13 immunoreactivity is observed in VTA neurons (Figure 2A). Both TH+ and TH− VTA neurons demonstrated KCNK13 immunoreactivity, indicating that KCNK13 is not expressed solely in DA VTA neurons. The presence of these channels in the VTA reinforced the possibility that KCNK13 on VTA neurons may modulate their activity.
Kcnk13 mRNA is upregulated by acute ethanol exposure in the VTA
To determine whether ethanol can alter the expression of Kcnk13, we measured Kcnk13 mRNA levels by qPCR at two time points, one hour and four hours, after an acute injection of ethanol (3 g/kg, i.p.). We observed a significant increase in Kcnk13 mRNA levels in the VTA of the mice four hours, but not one hour, after ethanol treatment (unpaired t test, t = −2.25, p<0.05 for the 4 hour group, n=9 from 9 mice per group) (Figure 2B), indicating that Kcnk13 may be an ethanol-regulated gene or ethanol may interfere with the degradation of Kcnk13 mRNA. Consistent with this observation, at the 4 h time point at which there is an increase in Kcnk13 mRNA levels in the VTA, there is also an increase in the potency of ethanol to excite VTA neurons (Supplementary Figure S2).
Knockdown of Kcnk13 decreases ethanol-induced excitation of VTA neurons
To demonstrate an involvement of the specific leak potassium channel KCNK13 in ethanol-induced excitation in the VTA, we stereotaxically injected the VTA of mice with lentivirus expressing shRNA targeting Kcnk13 (Figure 3). A prior in vitro validation experiment demonstrated that Kcnk13 was reduced in Neuro-2a cells by this shRNA by ~50% when compared with the control shRNA (Supplemental Figure S1). Three weeks after injection of the lentivirus expressing the shRNA into the VTA, Kcnk13 mRNA expression was significantly reduced (by 22.2%; unpaired t-test, t= 1.92, n=6 mice per group) (Figure 3A). Additional experiments using quantitative immunohistochemistry demonstrated that KCNK13 protein expression was reduced by 15.1 ± 3.4% (paired t-test, t= −4.88, p<0.001, n=18 from 6 mice) (Figure 3B, C). We next tested for the excitatory effect of ethanol on VTA neurons from mice expressing Kcnk13 shRNA compared with mice expressing a control shRNA (shScr). In this study, the initial firing rate of shScr neurons (8 cells from 2 mice) was 1.9 ± 0.3 Hz with a CV of 0.09 ± 0.02 and initial firing rate of shKcnk13 neurons (8 cells from 2 mice) was 2.5 ± 0.5 Hz with a CV of 0.13 ± 0.04. Neither the initial firing rates (unpaired t-test, t=−1.08, p>0.05) nor CVs (unpaired t-test, t=−0.83, p>0.05) were different between the groups. Compared to the shScr groups, ethanol-induced excitation of VTA neurons from shKcnk13 mice was significantly lower (two-way RM ANOVA, F1, 45= 17.34, p<0.006 for effect of shRNA, n=8 per group) (Figure 4). Although KCNK13 protein expression was decreased by only about 15%, ethanol excitation was reduced by close to 50% at each concentration (Figure 4C). These results demonstrate that reduction of Kcnk13 expression diminished the excitatory effect of ethanol, supporting the idea that KCNK13 contributes to ethanol excitation of VTA neurons.
KCNK13 regulates binge-like ethanol consumption
Since KCNK13 regulates ethanol-induced excitation of VTA neurons, we hypothesized that it may also play a role in ethanol drinking. To test this, we microinjected lentivirus expressing shKcnk13 or shScr into the VTA and measured binge-like ethanol consumption three weeks after injection using a two-bottle variation of the DID test (Rhodes et al., 2005). Mice expressing shKcnk13 in the VTA drank more ethanol than controls during the two-hour sessions (two-way ANOVA, effect of shRNA, F1, 84=13.5, p<0.001) (Figure 5A), indicating an important role of KCNK13 in this model of binge alcohol drinking.
In contrast to the effect on drinking during two-hour drinking sessions, ethanol consumption during four-hour drinking sessions was not different between the groups (one-way ANOVA, F3,28= 1.42, p>0.05) (Figure 5B), possibly indicating a compensatory upregulation (see Discussion below). Blood ethanol levels achieved on the last day of drinking were 132.8 ± 16.5 mg % and 125.9 ± 21.3 mg%, in the mice that received lentivirus expressing shScr and shKcnk13 (unpaired t-test, t-statistic= −0.23, DF=14, p>0.05; n=16 total, 6 shScr and 10 shKcnk13), respectively, indicating that both groups of mice were achieving similar and pharmacologically relevant BECs (Figure 5C). The groups did not differ in ethanol preference (Figure 5D), possibly due to the overall high ethanol preference by both groups (i.e., a possible ceiling effect). Knockdown of Kcnk13 had no significant effect on sucrose drinking (Figure 5E), indicating a selectivity of the effect of KCNK13 knockdown on ethanol intake. The similar sucrose intake between shScr and shKcnk13 groups also suggests that KCNK13 knockdown did not have a general effect on motor performance and that the effect on binge drinking did not generalize to all substances.
Discussion
Using pharmacological, molecular and behavioral methods, our results support a significant role for KCNK13 channels in ethanol excitation of VTA neurons. This effect of ethanol on KCNK13 in the VTA is likely to be behaviorally relevant, as Kcnk13 knockdown in the VTA increased ethanol consumption. Ethanol excites DA neurons of the VTA in the absence of synaptic connections, indicating that ethanol-induced excitation is mediated by ethanolsensitive elements on the membrane of these neurons (Brodie et al., 1999). Among those cellular elements (You et al., 2018), KCNK13 channels should be considered as an important ethanolsensitive ion channel on VTA neurons.
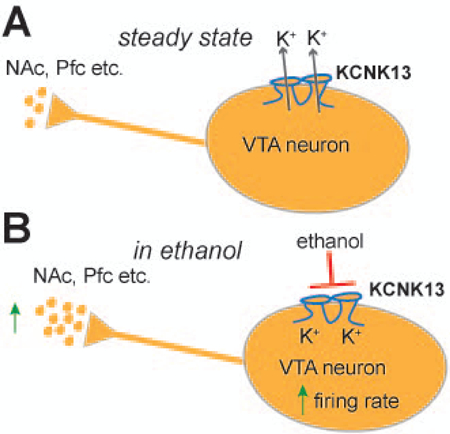
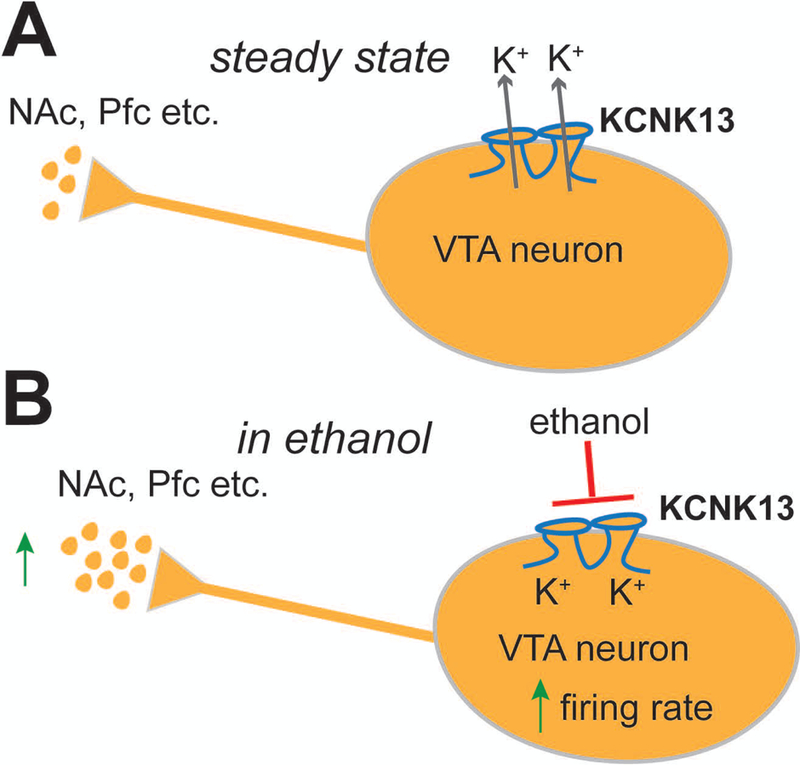
A model of the role of KCNK13 channels on VTA neuronal activity is presented in Figure 6. In general, flow of potassium ions through KCNK13 and other leak channels maintains a negative intracellular membrane potential, controlling the spontaneous firing rate and the steady-state level of neurotransmitters released onto the targets of these neurons (Figure 6A). Ethanol blocks KCNK13 channels and reduces this potassium current, depolarizing the membrane and increasing the firing rate, resulting in an increase in neurotransmitter release onto the targets of VTA projections (Figure 6B). In the case of the shRNA experiments, reducing the number of KCNK13 channels shifts the ethanol concentration-response curve to the right, thus we would predict that this reduction would increase the amount of ethanol needed to produce an equivalent release of neurotransmitters, including dopamine, in VTA target regions.
Figure 6. Model for the role of KCNK13 in the VTA and in the response to ethanol.
A. Under normal conditions, KCNK13 channels and other leak potassium channels are open, helping to maintain the negative resting membrane potential of VTA neurons. At rest there is spontaneous action potential firing due to pacemaker activity in the neuron, and this causes baseline release of neurotransmitters in target regions like the nucleus accumbens and prefrontal cortex.
B. With the addition of ethanol, KCNK13 channels are blocked, causing a depolarization of the membrane and increased firing rate. The increase in the firing rate of the DA VTA neuron results in increased release of neurotransmitter in target regions.
Downregulation of Kcnk13 with shRNA did not result in a significant increase in baseline firing rate, possibly due to a compensatory upregulation of other, ethanol-insensitive leak potassium channels. Note the reduction of mRNA and protein for KCNK13 channels was relatively modest (~22% and ~15% repectively), and it is unknown what proportion of the overall leak channel population is comprised of KCNK13 channels. In our experiments, we confirmed that knockdown of Kcnk13 in the VTA reduced the ability of ethanol to excite VTA neurons and increased binge-like ethanol consumption. Together, these results support a role for KCNK13 in the VTA in alcohol intake, demonstrating that reducing KCNK13 decreases ethanol-induced excitation of VTA neurons and causes an increase in binge drinking.
Mice expressing shKcnk13 in the VTA drank more ethanol than control mice in the twohour sessions, indicating that KCNK13 plays an important role in regulating binge-like alcohol drinking. We hypothesize that due to a reduction of the alcohol-sensitive target KCNK13 in the VTA, more alcohol was required in the shKcnk13-expressing mice to reach the same level of VTA activation as shScr-expressing mice (Figure 6). There was no effect of Kcnk13 knockdown on sucrose consumption, indicating that Kcnk13 is not generally involved in consumption of rewarding sweet solutions. We did not test for the effect of Kcnk13 knockdown on the consumption of a bitter solution such as quinine, therefore it is still a possibility that the increase in ethanol consumption in mice expressing shKcnk13 is due to decreased sensitivity to the bitter taste of ethanol.
Interestingly, we found that the amount of ethanol consumed, and the BECs achieved, after 4 hours of drinking were similar in mice expressing the Kcnk13 shRNA in the VTA and shScr controls. This result might be explained by an increase in Kcnk13 expression four hours after ethanol treatment, as demonstrated in Figure 2. Upregulation of Kcnk13 over four hours could compensate for ethanol-induced inhibition of KCNK13, and overcome the modest (1522%) knockdown by shKcnk13. A major additional study would be needed to accurately assess the time-course of changes in Kcnk13 mRNA and KCNK13 protein expression over a two-hour or four-hour drinking session, and over one to four days of DID. Alcohol intake over the twohour sessions suggests the possibility that intrinsically lower levels of KCNK13 may be a risk factor for binge drinking. More extensive studies of the interaction of Kcnk13 expression with alcohol consumption, and the use of a conditional Kcnk13 knockout specifically in VTA neuronal subtypes, will be necessary to understand the specifics of dynamic changes in KCNK13 on VTA neurons that may drive binge drinking.
In addition to KCNK13, other neurotransmitters and ion channels are likely to play roles in the regulation of ethanol-induced excitation. We and others have demonstrated several actions of ethanol on VTA neurons, including reducing M-current (Koyama et al., 2007), and increasing both h-current (Brodie and Appel, 1998; Okamoto et al., 2006) and a barium-sensitive potassium conductance (Herman et al., 2015; McDaid et al., 2008); these effects would alter the magnitude of ethanol excitation by affecting the passive electrical membrane properties of the neuron. Increasing or decreasing the resistance of the membrane, for example, would make the cells more or less excited by ethanol action on KCNK13 channels. Likewise, the efficiency of KCNK13-mediated excitation could be altered by a variety of agents, including h-channel blockers (McDaid et al., 2008; Okamoto et al., 2006), knockout of G protein-coupled potassium channels (Herman et al., 2015), and pharmacological agents that affect cholinergic (Ericson et al., 2008; Larsson et al., 2002), GABAergic (Steffensen et al., 2009; Theile et al., 2011) or glutamatergic (Steffensen et al., 2000) neurotransmission. Unlike these other agents, pharmacological conditions that block KCNK13 (e.g., quinine (Nimitvilai et al., 2016), high extracellular Ca++ (Figure 1)), also reduce the excitatory effect of ethanol, suggesting that KCNK13 is a primary target of ethanol in the VTA. The results presented here indicate a novel and important role of KCNK13 in ethanol actions on VTA neurons.
While both TH-immunopositive and -immunonegative cells expressed KCNK13, ethanol-induced excitation of neurons with electrophysiological characteristics of DA neurons was reduced by treatments that decreased expression of KCNK13, although as noted above, nondopamine neurons may have the same electrophysiological profile (Margolis et al., 2006). The VTA contains primarily GABAergic and DA neurons; DA neurons are excited by ethanol (Brodie et al., 1990), whereas ethanol inhibits GABAergic neurons (Gallegos et al., 1999). We did not examine the response of neurons adhering to the electrophysiological profile for GABAergic neurons, so we do not know whether there are additional membrane targets that might attenuate the excitatory effect of ethanol on KCNK13 on those GABAergic neurons. If KCNK13 channels play a significant role in regulating excitability of GABAergic neurons of the VTA, blockade of those channels by ethanol would be expected to excite the GABA neurons as well. Extensive studies with selective knockout models or upregulation of KCNK13 in GABAergic and DAergic neurons would be helpful in developing a better understanding the role of KCNK13 in the VTA.
Four hours of access to alcohol in the drinking in the dark paradigm resulted in blood ethanol concentrations of greater than 125 mg/dl (> 27 mM; Figure 5C), while 0.08 g/dl (80 mg%, legal intoxication in many US states) is about 17 mM. One confound noted in the past has been that the concentrations of ethanol needed to activate DA-like VTA neurons in rodent brain slices (40–120 mM, but also see (Avegno et al., 2016; Mrejeru et al., 2015)) are higher than those likely to be achieved during normal ethanol intake in humans, and are higher than ethanol concentrations that cause the increased dopamine release observed with in vivo microdialysis (Vena and Gonzales, 2015) or in vivo voltammetry (Jones et al., 2006; Signs et al., 1987). This reduced ethanol potency in vitro may be partially explained by the loss of connections to VTA neurons from other brain areas that is a consequence of the brain slice method. However, Ungless et al. demonstrated a significant decrease in a leak potassium current after axotomy in Aplysia (Ungless et al., 2002), although the specific leak channel in these experiments was not identified. Downregulation of leak potassium channels in response to axotomy has been established in mammalian tissue as well; TREK2 (Acosta et al., 2014) and TRASK (Tulleuda et al., 2011) are reduced after axotomy in rat nociceptors, for example. As axotomy is a normal consequence of in vitro neuronal preparations, it is possible that rapid downregulation of channels like KCNK13 occurs as a consequence of brain slice preparation, resulting in a decrease in the magnitude of ethanol excitation. Additional studies will be necessary to support this speculative hypothesis, but, if true, this would further support the role of KCNK13 in ethanol excitation, and, in addition, explain why ethanol is less potent in stimulating DA VTA neurons in vitro than in vivo.
Ethanol activation of neurons of the VTA was first observed in vivo (Gessa et al., 1985) and in vitro (Brodie et al., 1990) decades ago, and several targets for ethanol on VTA neurons have been identified. The results presented here demonstrate a significant role for KCNK13 channels in mediation of excitation of VTA neurons by ethanol. Furthermore, we have provided a link between KCNK13 in the VTA and binge-like drinking. The studies shown here provide initial evidence of a role of KCNK13 channels in the regulation of VTA neuronal excitation in response to ethanol. As VTA neurons are fundamentally implicated in the etiology of symptoms of substance use disorders, pharmacological targeting of KCNK13 channels could be an effective way to treat alcoholism. If a decrease in KCNK13 channels increases drinking behavior, a blocker of ethanol action on KCNK13 channels, or an enhancer of KCNK13 activity, may decrease ethanol seeking behavior. In addition, our drinking studies indicate that an innate deficit in KCNK13 prior to alcohol experience may be a risk factor for high levels of binge drinking, increasing the clinical relevance of investigating KCNK13. More work is needed to determine how KCNK13 levels are regulated in the VTA, whether KCNK13 channels in other brain areas control pre- or postsynaptic alcohol effects, and whether reduced levels of KCNK13 confer an increased innate risk for high alcohol intake. Ultimately, KCNK13 may be an important target of ethanol that regulates many effects of alcohol in the brain.
Supplementary Material
Highlights:
The ventral tegmental area (VTA) is important in neurobiological processes related to the development of alcoholism
Pharmacological evidence implicated two-pore potassium channels in ethanol-induced excitation of VTA neurons
Downregulation of KCNK13 (THIK-1) using shRNA decreased ethanol-induced neuronal excitation and increased binge-like drinking
KCNK13 may be an important target of ethanol in the VTA and other central nervous system regions
Acknowledgments:
The authors gratefully acknowledge that this work was supported by PHS Grant R01AA05846 (MSB), U01 AA020912 (AWL) and P50AA022538 (SCP; AWL; MSB). Contributions by Dr. Sarah B. Appel to initial work that served as a prelude to the studies reported here is acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Data:
Additional information (Supplementary Methods and Figure S1 and S2) is available in the Supplementary Materials section.
Financial Disclosures:
All authors reported no related financial interests or potential conflicts of interest.
References
- Acosta C, Djouhri L, Watkins R, Berry C, Bromage K, Lawson SN, 2014. TREK2 expressed selectively in IB4-binding C-fiber nociceptors hyperpolarizes their membrane potentials and limits spontaneous pain. J Neurosci 34, 1494–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel SB, Liu Z, McElvain MA, Brodie MS, 2003. Ethanol excitation of dopaminergic ventral tegmental area neurons is blocked by quinidine. J. Pharmacol. Exp. Ther 306, 437–446. [DOI] [PubMed] [Google Scholar]
- Appel SB, Wise L, McDaid J, Koyama S, McElvain MA, Brodie MS, 2006. The effects of long chain length n-alcohols on the firing frequency of dopaminergic neurons of the ventral tegmental area. J. Pharmacol. Exp. Ther [DOI] [PubMed]
- Avegno EM, Salling MC, Borgkvist A, Mrejeru A, Whitebirch AC, Margolis EB, Sulzer D, Harrison NL, 2016. Voluntary adolescent drinking enhances excitation by low levels of alcohol in a subset of dopaminergic neurons in the ventral tegmental area. Neuropharmacology 110, 386–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie MS, Appel SB, 1998. The effects of ethanol on dopaminergic neurons of the ventral tegmental area studied with intracellular recording in brain slices. Alcohol Clin. Exp. Res 22, 236–244. [PubMed] [Google Scholar]
- Brodie MS, Pesold C, Appel SB, 1999. Ethanol directly excites dopaminergic ventral tegmental area reward neurons. Alcohol Clin. Exper. Res 23, 1848–1852. [PubMed] [Google Scholar]
- Brodie MS, Shefner SA, Dunwiddie TV, 1988. Ethanol increases the firing of dopamine neurons of the ventral tegmental area in vitro. Alcohol Clin. Exper. Res 12, 323. [DOI] [PubMed] [Google Scholar]
- Brodie MS, Shefner SA, Dunwiddie TV, 1990. Ethanol increases the firing rate of dopamine neurons of the rat ventral tegmental area in vitro. Brain. Res 508, 65–69. [DOI] [PubMed] [Google Scholar]
- Canavier CC, Oprisan SA, Callaway JC, Ji H, Shepard PD, 2007. Computational model predicts a role for ERG current in repolarizing plateau potentials in dopamine neurons: implications for modulation of neuronal activity. J Neurophysiol 98, 3006–3022. [DOI] [PubMed] [Google Scholar]
- Chatelain FC, Bichet D, Feliciangeli S, Larroque MM, Braud VM, Douguet D, Lesage F, 2013. Silencing of the tandem pore domain halothane-inhibited K+ channel 2 (THIK2) relies on combined intracellular retention and low intrinsic activity at the plasma membrane. J Biol Chem 288, 35081–35092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng C, Li KY, Zhou C, Ye JH, 2009. Ethanol enhances glutamate transmission by retrograde dopamine signaling in a postsynaptic neuron/synaptic bouton preparation from the ventral tegmental area. Neuropsychopharmacology 34, 1233–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutton JW 3rd, Chen H, You C, Brodie MS, Lasek AW, 2016. Anaplastic lymphoma kinase regulates binge-like drinking and dopamine receptor sensitivity in the ventral tegmental area. Addict Biol [DOI] [PMC free article] [PubMed]
- Enyedi P, Czirjak G, 2010. Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol Rev 90, 559–605. [DOI] [PubMed] [Google Scholar]
- Ericson M, Lof E, Stomberg R, Chau P, Soderpalm B, 2008. Nicotinic acetylcholine receptors in the anterior, but not posterior, ventral tegmental area mediate ethanol-induced elevation of accumbal dopamine levels. J Pharmacol Exp Ther 326, 76–82. [DOI] [PubMed] [Google Scholar]
- Gallegos RA, Lee RS, Criado JR, Henriksen SJ, Steffensen SC, 1999. Adaptive responses of gamma-aminobutyric acid neurons in the ventral tegmental area to chronic ethanol. J. Pharmacol. Exp. Ther 291, 1045–1053. [PubMed] [Google Scholar]
- Gessa GL, Muntoni F, Collu M, Vargiu L, Mereu G, 1985. Low doses of ethanol activate dopaminergic neurons in the ventral tegmental area. Brain. Res 348, 201–203. [DOI] [PubMed] [Google Scholar]
- Goldstein SA, Bockenhauer D, O’Kelly I, Zilberberg N, 2001. Potassium leak channels and the KCNK family of two-P-domain subunits. Nat Rev Neurosci 2, 175–184. [DOI] [PubMed] [Google Scholar]
- Guan Y, Xiao C, Krnjevic K, Xie G, Zuo W, Ye JH, 2012. GABAergic actions mediate opposite ethanol effects on dopaminergic neurons in the anterior and posterior ventral tegmental area. J Pharmacol Exp Ther 341, 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman MA, Sidhu H, Stouffer DG, Kreifeldt M, Le D, Cates-Gatto C, Munoz MB, Roberts AJ, Parsons LH, Roberto M, Wickman K, Slesinger PA, Contet C, 2015. GIRK3 gates activation of the mesolimbic dopaminergic pathway by ethanol. Proc Natl Acad Sci U S A 112, 7091–7096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SR, Mathews TA, Budygin EA, 2006. Effect of moderate ethanol dose on dopamine uptake in rat nucleus accumbens in vivo. Synapse 60, 251–255. [DOI] [PubMed] [Google Scholar]
- Kharazia VN, Jacobs KM, Prince DA, 2003. Light microscopic study of GluR1 and calbindin expression in interneurons of neocortical microgyral malformations. Neuroscience 120, 207–218. [DOI] [PubMed] [Google Scholar]
- Koob GF, Volkow ND, 2010. Neurocircuitry of addiction. Neuropsychopharmacology 35, 217–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkova TM, Ponomarenko AA, Brown RE, Haas HL, 2004. Functional diversity of ventral midbrain dopamine and GABAergic neurons. Mol Neurobiol 29, 243–259. [DOI] [PubMed] [Google Scholar]
- Koyama S, Brodie MS, Appel SB, 2007. Ethanol inhibition of m-current and ethanolinduced direct excitation of ventral tegmental area dopamine neurons. J. Neurophysiol 97, 19771985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey MG, Mercuri NB, North RA, 1989. Two cell types in rat substantia nigra zona compacta distinguished by membrane properties and the actions of dopamine and opioids. J. Neurosci 9, 1233–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson A, Svensson L, Soderpalm B, Engel JA, 2002. Role of different nicotinic acetylcholine receptors in mediating behavioral and neurochemical effects of ethanol in mice. Alcohol 28, 157–167. [DOI] [PubMed] [Google Scholar]
- Lasek AW, Janak PH, He L, Whistler JL, Heberlein U, 2007. Downregulation of mu opioid receptor by RNA interference in the ventral tegmental area reduces ethanol consumption in mice. Genes Brain Behav 6, 728–735. [DOI] [PubMed] [Google Scholar]
- Margolis EB, Lock H, Hjelmstad GO, Fields HL, 2006. The ventral tegmental area revisited: is there an electrophysiological marker for dopaminergic neurons? J. Physiol 577, 907924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Toy B, Himmels P, Morales M, Fields HL, 2012. Identification of rat ventral tegmental area GABAergic neurons. PLoS One 7, e42365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaid J, McElvain MA, Brodie MS, 2008. Ethanol effects on dopaminergic ventral tegmental area neurons during block of Ih: involvement of barium-sensitive potassium currents. J. Neurophysiol 100, 1202–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrejeru A, Marti-Prats L, Avegno EM, Harrison NL, Sulzer D, 2015. A subset of ventral tegmental area dopamine neurons responds to acute ethanol. Neuroscience 290, 649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller AL, Brodie MS, 1989. Intracellular recording from putative dopamine-containing neurons in the ventral tegmental area of Tsai in a brain slice preparation. J. Neurosci. Methods 28, 15–22. [DOI] [PubMed] [Google Scholar]
- Naito Y, Ui-Tei K, 2012. siRNA Design Software for a Target Gene-Specific RNA Interference. Front Genet 3, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimitvilai S, You C, Arora DS, McElvain MA, Vandegrift BJ, Brodie MS, Woodward JJ, 2016. Differential Effects of Toluene and Ethanol on Dopaminergic Neurons of the Ventral Tegmental Area. Front Neurosci 10, 434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto T, Harnett MT, Morikawa H, 2006. Hyperpolarization-activated cation current (Ih) is an ethanol target in midbrain dopamine neurons of mice. J. Neurophysiol 95, 619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan S, Wischmeyer E, Karschin C, Preisig-Muller R, Grzeschik KH, Daut J, Karschin A, Derst C, 2001. THIK-1 and THIK-2, a novel subfamily of tandem pore domain K+ channels. J Biol Chem 276, 7302–7311. [DOI] [PubMed] [Google Scholar]
- Rajan S, Wischmeyer E, Xin Liu G., Preisig-Muller R, Daut J, Karschin A, Derst C, 2000. TASK-3, a novel tandem pore domain acid-sensitive K+ channel. An extracellular histiding as pH sensor. J Biol Chem 275, 16650–16657. [DOI] [PubMed] [Google Scholar]
- Renigunta V, Zou X, Kling S, Schlichthorl G, Daut J, 2014. Breaking the silence: functional expression of the two-pore-domain potassium channel THIK-2. Pflugers Arch 466, 1735–1745. [DOI] [PubMed] [Google Scholar]
- Rhodes JS, Best K, Belknap JK, Finn DA, Crabbe JC, 2005. Evaluation of a simple model of ethanol drinking to intoxication in C57BL/6J mice. Physiol Behav 84, 53–63. [DOI] [PubMed] [Google Scholar]
- Rivera-Meza M, Quintanilla ME, Bustamante D, Delgado R, Buscaglia M, HerreraMarschitz M, 2014. Overexpression of hyperpolarization-activated cyclic nucleotide-gated channels into the ventral tegmental area increases the rewarding effects of ethanol in UChB drinking rats. Alcohol Clin Exp Res 38, 911–920. [DOI] [PubMed] [Google Scholar]
- Signs SA, Yamamoto BK, Schechter MD, 1987. In vivo electrochemical determination of extracellular dopamine in the caudate of freely-moving rats after a low dose of ethanol. Neuropharmacology 26, 1653–1656. [DOI] [PubMed] [Google Scholar]
- Steffensen SC, Nie Z, Criado JR, Siggins GR, 2000. Ethanol inhibition of N-methyl-Daspartate responses involves presynaptic gamma-aminobutyric acid(B) receptors. J. Pharmacol. Exp. Ther 294, 637–647. [PubMed] [Google Scholar]
- Steffensen SC, Walton CH, Hansen DM, Yorgason JT, Gallegos RA, Criado JR, 2009. Contingent and non-contingent effects of low-dose ethanol on GABA neuron activity in the ventral tegmental area. Pharmacol. Biochem. Behav 92, 68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theile JW, Morikawa H, Gonzales RA, Morrisett RA, 2011. GABAergic transmission modulates ethanol excitation of ventral tegmental area dopamine neurons. Neuroscience 172, 94103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulleuda A, Cokic B, Callejo G, Saiani B, Serra J, Gasull X, 2011. TRESK channel contribution to nociceptive sensory neurons excitability: modulation by nerve injury. Mol Pain 7, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungless MA, Gasull X, Walters ET, 2002. Long-term alteration of S-type potassium current and passive membrane properties in aplysia sensory neurons following axotomy. J Neurophysiol 87, 2408–2420. [DOI] [PubMed] [Google Scholar]
- Vena AA, Gonzales RA, 2015. Temporal profiles dissociate regional extracellular ethanol versus dopamine concentrations. ACS Chem Neurosci 6, 37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C, Shao XM, Olive MF, Griffin WC III, Li KY, Krnjevic K, Zhou C, Ye JH, 2009. Ethanol facilitates glutamatergic transmission to dopamine neurons in the ventral tegmental area. Neuropsychopharmacology 34, 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You C, Vandegrift B, Brodie MS, 2018. Ethanol actions on the ventral tegmental area: novel potential targets on reward pathway neurons. Psychopharmacology (Berl) 235, 1711–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapata A, Gonzales RA, Shippenberg TS, 2006. Repeated ethanol intoxication induces behavioral sensitization in the absence of a sensitized accumbens dopamine response in C57BL/6J and DBA/2J mice. Neuropsychopharmacology 31, 396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.