

Abstract
Post-translational protein modification controls the function of Tau as a scaffold protein linking a variety of molecular partners. This is most studied in the context of microtubules, where Tau regulates their stability as well as the distribution of cellular components to defined compartments. However, Tau is also located in the cell nucleus; and is found to protect DNA. Quantitative assessment of Tau modification in the nucleus when compared to the cytosol may elucidate how subcellular distribution and function of Tau is regulated. We undertook an unbiased approach by combing bimolecular fluorescent complementation and mass spectrometry in order to show that Tau phosphorylation at specific residues is increased in the nucleus of proliferating pluripotent neuronal C17.2 and neuroblastoma SY5Y cells. These findings were validated with the use of nuclear targeted Tau and subcellular fractionation, in particular for the phosphorylation at T181, T212 and S404. We also report that the DNA damaging drug Etoposide increases the translocation of Tau to the nucleus whilst reducing its phosphorylation. We propose that overt phosphorylation of Tau, a hallmark of neurodegenerative disorders defined as tauopathies, may negatively regulate the function of nuclear Tau in protecting against DNA damage.
Introduction
Proteinopathies represent a large spectrum of human disorders caused by proteins with a cytotoxic gain of function or the failure to perform a normal activity, both due to abnormal conformation and modification1. In their pathogenic forms, these proteins hold the predisposition to self-assemble into toxic soluble oligomers or insoluble aggregates. Given that cellular protein clearance is less efficient for multimeric or aggregated protein assemblies, a gradual accumulation and the formation of large deposits such as those typical for progressive neurodegenerative disorders may occur2. This is further accelerated by aging, which correlates with proteostasis defects, mostly due to a decline in protein clearance3. Other liabilities are increased protein production, abnormal post-translational modification, or changes in the amino acid sequence of the protein in genetic variants causing hereditary disease forms4. The aging brain may be concerned by the co-existence of distinct proteinopathies such as those involving Tau in neurofibrillar tangles and β-amyloid plaques in Alzheimer’s disease, or α-synuclein in Lewi bodies of Parkinson’s disease5,6. Then again, distinct proteinopathies may cause clinically similar disorders as it is the case for the deposition of aberrant forms of Tau, FUS or TDP-43 in the ALS/FTD disease spectrum7. Tauopathies, both in sporadic and familial forms of frontotemporal dementia with parkinsonism-17 caused by mutations in the Tau gene (MAPT), are characterized by Tau assembled in highly ordered paired helical filaments within neuronal cells8,9.
Tau was originally isolated as a microtubule-associated protein10. Its primary structure covers domains with distinct characteristics typical of a scaffold protein: an amino-terminal projection sequence followed by a proline-rich sequence domain, a microtubule-binding region and a carboxy-terminal tail. In its unbound state, Tau is described as highly soluble, heath-stable, unfolded protein. Tau binding to microtubules leads to a conformational switch with the negatively charged projection domain dissociating from the positively charged microtubule-binding domain. Tau links microtubules to other binding partner such as motor complexes or cellular membranes11, a process that is regulated by post-translational modifications9,12. Tau is post-translationally modified by proteolysis, acetylation, methylation, glycosylation and phosphorylation. This latter is the prevalent modification with more than eighty potential sites at serines, threonines and tyrosines. About twenty sites contribute to the normal function of Tau, but increased phosphorylation at these or at additional sites occurs during early development and is present in pathological lesions13. Phosphorylation within or outside the microtubule domain affects Tau association to microtubules. This may disturb microtubule-mediated axonal transport or increase free Tau, the probability of fibril formation14 or its secretion as a critical event for cell-to-cell spreading of Tau lesions in the brain15,16. Microtubule-dissociated Tau may also locate to other cellular compartments11,17,18. Toxic insults induce abnormal Tau distribution, as it is the case for β-amyloid-mediated re-location of Tau from the axonal to the somatodendritic compartment of neurons19. Distinct cellular locations of Tau may be associated to different post-translational modifications9,12,20 and different post-translational modifications of Tau may be associated to different functions, e.g. at the neuronal synapse21,22. Intriguingly, Tau is also found in the nucleus in vitro23–27 and in vivo28. Tau interacts with RNA and DNA29–32 and appears to protect DNA from denaturation and radicals33–36. Binding of DNA by Tau is linked to its dephosphorylated form37.
In our study, we revisited the hypothesis that nuclear Tau is characterized by a distinct post-translational modification. Using a set of parallel approaches to characterize Tau modification in the nucleus, we found increased phosphorylation at distinct sites for nuclear localized Tau and identified pharmacological modulators that differentially affect the subcellular location and modification of Tau.
Results
Cellular distribution of Tau by bimolecular fluorescent complementation
Bimolecular fluorescence complementation (biFC) represents a relatively simple technology to reveal subcellular protein location38. We adapted biFC to investigate the presence of Tau in specific sublocations within cultured cells. Here, we generated GFP1–10 sensors targeted to the nucleus, to the lumen of the endoplasmic reticulum (ER) or to the mitochondrial outer membrane (OMM). For the nuclear targeting sequence fused at the amino-terminus of GFP1–10 (nucGFP1–10), we took advantage of a well-described artificial targeting peptide carrying three times a nuclear targeting sequence derived from the SV40 large T antigen39. For the erGFP1–10 sensor, we fused the signal peptide of human calreticulin at the amino-terminus of the sensor and added the ER retention signal KDEL at its carboxy-terminus (Fig. 1a). The ommGFP1–10 sensor was generated as described18,40. The correct cellular distribution of the sensors was then assessed by immune fluorescent staining of transiently transfected mouse pluripotent neuronal C17.2 cells using an antibody against GFP. Cells were counterstained for calnexin located in the ER and the nuclear envelop and with the dye DAPI binding to the nuclear DNA. This procedure revealed the ubiquitous (i.e. cytosolic and nuclear) distribution of the untargeted GFP1–10 sensor, the nuclear localization of the nucGFP1–10 sensor that overlapped with the DAPI staining, and the co-localization of the ER-marker calnexin with the erGFP1–10 sensor but not with the ommGFP1–10 sensor associated to mitochondria40 (Fig. 1a). The specificity of α-GFP staining was highlighted by the absence of signal in the surrounding untransfected cells, which were positive only for calnexin and DAPI (Fig. 1a). A subcellular distribution consistent to that observed in C17.2 cells was obtained in human kidney HEK293 cells and in human SH-SY5Y neuroblastoma cells (Supplementary Fig. 1).
Figure 1.

Targeted GFP1–10 sensors reveal subcellular pools of Tau. (a) The subcellular distribution of the indicated GFP1–10 sensors in transiently transfected mouse C17.2 cells is shown by confocal microscopy upon immune staining of PFA-fixed cells with an anti-GFP antibody (upper row, in red). The cells are counter-stained with the ER-marker calnexin (middle row, in cyan) and the nuclear stain DAPI (shown in the merged images, bottom row, in blue). (b) The GFP1–10 sensors were then co-transfected with S11-Tau, which reveals by biFC biofluorescence (upper row, in green) microtubule-associated Tau with GFP1–10, nuclear Tau with nucGFP1–10, and mitochondria-associated Tau with ommGFP1–10. No biFC is obtained with the erGFP1–10 sensor, unless Tau is targeted to the ER lumen (erTau-S11, column on the far right). The cells are counter-stained for total human Tau with the Tau13 antibody (middle row, in red) and DAPI (merged images, bottom row, in blue).
In order to reconstitute GFP fluorescence, we then co-transfected C17.2 cells with the longest – 441 amino-acid long – isoform of human Tau fused at the amino-terminus with the eleventh β-strand of GFP (S11). S11 complements the GFP1–10 sensor for reconstituting a biofluorescent GFP. Co-location of S11-Tau and GFP1–10, resulted in strong cytosolic reconstituted GFP-fluorescence (biFC) (Fig. 1b). Here, cells were counter-stained with a human-specific Tau antibody, which revealed accurate co-localization of biFC with the expected cellular distribution of Tau along microtubules. The use of the nucGFP1–10 sensor revealed the presence of Tau within the cell nucleus, whereas ommGFP1–10 demonstrated mitochondria-associated Tau. Thus, although the main pool of Tau was again detected in the cytosol by the α-Tau antibody, the two targeted sensors visualized minor pools of Tau within the nucleus or bound to mitochondria (Fig. 1b). Microtubule-associated Tau was better visualized with a β-tubulin antibody when the cells were fixed in ice-cold methanol38, on the other hand methanol fixation was less suited to reveal the GFP biofluorescent signal and protein localization within the nucleus. No biFC was obtained with the erGFP1–10 sensor (Fig. 1b) and for that matter also with a GFP1–10 sensor targeted to the mitochondrial matrix (Supplementary Fig. 2)18 indicating the absence of detectable Tau protein levels within the secretory pathway or mitochondria. In order to show the functionality of the erGFP1–10 sensor, we generated a construct encoding for a Tau carrying at its amino-terminus the signal peptide of influenza hemagglutinin followed by a consensus sequence for amino-glycosylation, whereas the S11-peptide was added at the carboxy-terminus (erTau-S11). Tau targeted to the lumen of the ER complemented the erGFP1–10 sensor and generated a biFC signal (Fig. 1b) that co-localized with the ER-maker calnexin. Additional evidence for the presence of erTau-S11 in the secretory pathway was its significantly slower migration on SDS PAGE due to plausible glycosylation (see below Fig. 2b, compare lanes 7 and 4) and robust secretion.
Figure 2.

Tau and nuclear targeted Tau are phosphorylated in C17.2 cells. (a) Cell extracts (10 µg total protein) obtained from C17.2 cells transiently transfected with Tau (Tau441) or nuclear targeted Tau (nucTau) are analysed by western blot with the human-specific Tau13 antibody in the absence or presence of λ-phosphatase treatment (λ-phosph). Bacterial recombinant Tau was mixed with a mock transfected cell lysate (recTau441). GAPDH served as a loading control. (b) Confocal microscopy images of C17.2 cells demonstrate nuclear targeting of nucTau (Tau13 staining, in red). Cells are counterstained for microtubules (α-tubulin, in cyan) and the nucleus (DAPI, in blue in the merged image).
The aim of this study was to assess whether distinct subcellular location of Tau may affect its post-translational modification. So, the next step was to first examine whether C17.2 cells actively modified Tau. For this, we analysed Tau on SDS polyacrylamide gel electrophoresis followed by western blot with the Tau13 monoclonal antibody (Fig. 2a, lane 4; Supplementary Fig. 3). This analysis revealed that human Tau in C17.2 cells displayed a heterogeneous apparent molecular weight, as known for phosphorylated Tau. Consistent with this interpretation, the majority of the Tau forms on the western blot had a larger apparent molecular mass than that of the unmodified recombinant Tau441 expressed in bacteria (Fig. 2a, lane 2; Supplementary Fig. 3). Treatment of cell lysates with λ-phosphatase reduced heterogeneity and apparent molecular mass of Tau expressed by C17.2 cells, comparably to that of recombinant Tau (Fig. 2a, lane 3; Supplementary Fig. 3). Similar migration pattern and sensitivity to λ-phosphatase was observed also for endogenous human Tau and for overexpressed Tau441 in human SH-SY5Y cells, for overexpressed Tau441 in human HEK293 cells and for S11-Tau in mouse C17.2 cells (Supplementary Fig. 4). Moreover, Tau protein forced into the nucleus by the same nuclear import signal used for the sensor (nucTau, Fig. 2b) showed multiple complex post-translational modification and sensitivity to λ-phosphatase (Fig. 2a, lanes 6 and 7; Supplementary Fig. 3). Specificity of the human-specific mouse monoclonal Tau13 antibody was emphasized by the absence of immune positive signals in mock-transfected mouse C17.2 cells, despite the presence of endogenous mouse Tau (Fig. 2a, lane 1; Supplementary Fig. 3). Reduced GAPDH signal, used as loading control in this experiment, indicated partial unspecific protein degradation in the cell lysates treated with λ-phosphatase when compared to the untreated control (Fig. 2a, compare lanes 3 and 4 or lanes 6 and 7; Supplementary Fig. 3), which may explain the presence of a putative proteolytic fragment of Tau migrating slightly faster than the main Tau band in the λ-phosphatase-treated lysates (Fig. 2a, lane 3; Supplementary Fig. 3).
Mass Spectrometry reveals subcellular-specific Tau phosphorylation
Having established the presence of Tau within the nucleus of C17.2 cells (Fig. 1b) and its post-translational modification by phosphorylation (Fig. 2a), next we optimized conditions for the isolation of S11-Tau using the GFP1–10 sensors as immune isolation baits, in order to characterize Tau modification depending on its cellular distribution38. First, we prepared detergent-free extract obtained from C17.2 cells co-transfected with S11-Tau and GFP1–10 and demonstrated the presence of the two biFC partners by western blot (Fig. 3a, lane 5). Immune isolation of the GFP-containing biFC complex was performed by single-step affinity purification with anti-GFP VHH antibody coupled to magnetic agarose beads (GFP-Trap). This procedure, led to efficient co-purification of S11-Tau from extract obtained from cells co-transfected with S11-Tau and GFP1–10 (Fig. 3a, lane 10). In order to evaluate the specificity of the immune isolation procedure, the negative controls included cell extracts obtained from mock transfected cells (Fig. 3a, lane 1), cells transfected with either one of the two proteins without the respective biFC partner (Fig. 3a, lanes 2, 3, 7, 8), or replacing the GFP1–10 sensor with intact GFP (Fig. 3a, lanes 4 and 9). Tau immune reactivity in the GFP-bound fractions was very faint, i.e. at unspecific background levels, in the samples obtained from cells lacking GFP1–10 or co-expressing intact GFP instead of GFP1–10. Moreover, mixing cell extracts obtained from cells expressing separately S11-Tau or GFP1–10 before the immune isolation did not result in co-isolation of Tau (Fig. 3a, lanes 6 and 11). This showed lack of post-extraction GFP reconstitution, which reduced the liability of co-isolating Tau protein assembled post-extraction with the GFP1–10 sensor when the two binding partners did not co-localized in intact cells. β-actin immune blotting was used as loading control for the unprocessed cell extracts, and its absence in the GFP-immune isolates was a further evidence of the specificity of the enrichment procedure. Furthermore, based on the results obtained with standard immune precipitation with the αGFP rabbit polyclonal ab290 coupled to protein G-agarose beads, it should be noted that GFP-Trap inefficiently isolated unreconstituted GFP1–10. Based on these data, we predicted isolation of subcellular Tau pools from cells expressing different targeted GFP1–10 sensors by GFP-Trap. This procedure would consent the identification of subcellular location-specific post-translational modifications of Tau, which we did by LC-MS/MS analysis.
Figure 3.
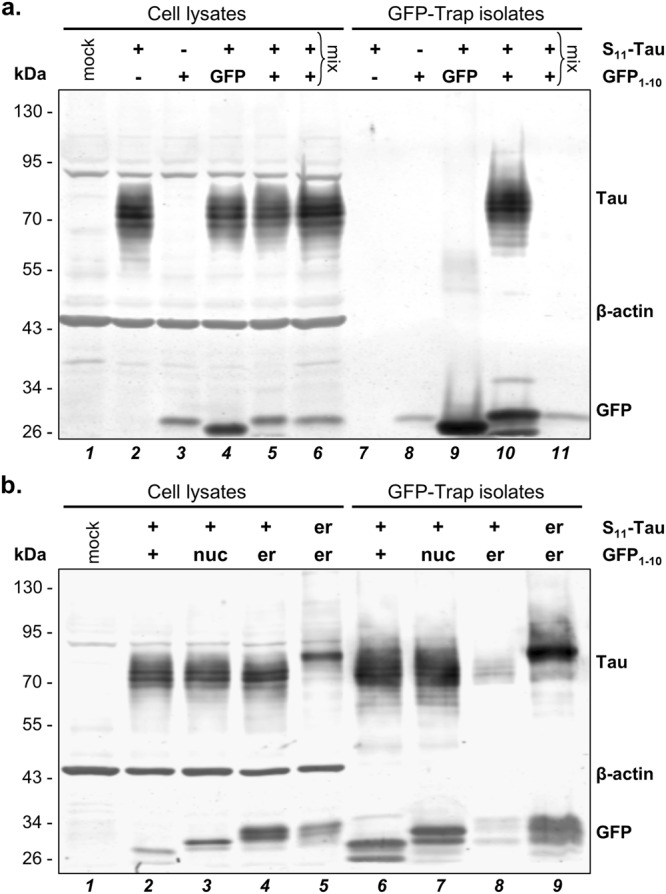
Immune isolation of Tau by mean of the GFP1–10 sensors. (a) Cell lysates from C17.2 cells transfected as indicated are analysed by western blot before (cell lysates) or after immune isolation on anti-GFP magnetic beads (GFP-Trap isolates). This shows specific isolation of S11-Tau when co-transfected with GFP1–10, but not in the negative controls, including post-lysis mixing of lysates obtained from cells transfected with either S11-Tau or GFP1–10 (lanes labelled with “mix”). β-actin is the loading control for the cell lysates, whereas the presence of the sensor is verified with an anti-GFP antibody. (b) Cell lysates and GFP-immune isolates obtained from C17.2 cells transiently transfected with S11-Tau and the indicated sensors are analysed by western blot as described for (a). Molecular weight markers are given on the left of the blots, full blots are shown scanned by dual infrared imaging.
We then generated scaled-up extracts from C17.2 cells transfected with plasmids encoding S11-Tau and the untargeted GFP1–10 or the nucGFP1–10 sensors (Fig. 3b, lanes 2 and 3) and isolated the biFC complex with GFP-Trap (Fig. 3b, lanes 6 and 7). The specificity of the affinity purification for reconstituted GFP formed before cell extraction was confirmed with the erGFP1–10 sensor. Consistent with the biFC data by microscopy (Fig. 1b), Tau-affinity purification by means of erGFP1–10 sensor occurred from cells expressing ER-targeted erTau-S11 but not when expressing S11-Tau (Fig. 3b, lanes 4, 5, 8, 9). The same procedure was also applied to isolate ommGFP1–10-bound Tau (Supplementary Fig. 5). Next, GFP-affinity isolated Tau was digested with Lys-C/trypsin and the resulting peptides were analysed by LC-MS/MS. Sequence coverage resulting from this analysis reached 81% of the Tau polypeptide (Fig. 4). Search for post-translationally modified human Tau-derived peptides revealed multiple phospho-threonine and -serine residues (Fig. 4 and Table 1). No other classes of post-translational modifications were detected among those searched, i.e. lysine methylation, acetylation, ubiquitination and nitration. Out of the thirteen phosphopeptides detected, two exhibited double-phosphorylation. The first included two threonines (T175 and T181; numbering according to the Tau441 isoform), and the other a threonine and a serine (T231 and S235). For all phosphosites detected, the corresponding unmodified residue was also identified, indicating that the sites are partly phosphorylated in C17.2 cells under normal culturing conditions. All samples were analysed twice and we calculated mean peptide intensity from the two measurements, each normalized for the total intensities for all human Tau-derived peptides. We then compared the relative intensities of the phospho-Tau peptides isolated by means of the GFP1–10, nucGFP1–10 or the ommGFP1–10 sensors (Table 1). Significantly increased phosphorylation of peptides embedding T181, one of the three S198/199/202 (the exact position was not resolved), T212 and the double-phosphorylation at T231/S235 was detected in samples isolated with the nucGFP1–10 sensor when compared to GFP1–10 or ommGFP1–10. A significant increase in the abundance of the phosphopeptide embedding S404 was found for nucGFP1–10-Tau when compared to GFP1–10-Tau, but not for ommGFP1–10-Tau, possibly suggesting a differential Tau modification at mitochondria when compared to the cytosol as recently discussed18, and also supported by increased pS356 and reduced pT231/pS235 for ommGFP1–10-Tau when compared to GFP1–10-Tau. For all other phosphosites no differences between the sensors were found. In order to consolidate these results, in a second analysis, we probed biological pentaplicates for the GFP1–10 and nucGFP1–10 sensors. The overall recovery of peptides in this analysis was lower than the first one, with seven distinct phosphosites detected (Table 2). Here, nucGFP1–10-bound Tau was significantly more phosphorylated at T181 and T231, whereas phosphorylation at the other sites was not significantly different. Overall, the mass spectrometric analysis revealed an increased phosphorylation for Tau isolated with the nucGFP1–10 sensor compared to that of the ubiquitously distributed GFP1–10 or ommGFP1–10, at least at three threonine and two serine residues out of the thirteen phosphosites measured. These findings were unforeseen considering previous work performed by immune staining with the dephosphorylated specific Tau1 antibody37. Therefore, we challenged our data by undertaking two independent approaches based on western blot analysis with commercial phospho-specific antibodies.
Figure 4.

LC-MS/MS reveals Tau phosphorylation. Primary sequence of human Tau441 with highlighted sequence coverage (81%; letters in black) and phosphorylation sites (letters in red) revealed by LC-MS/MS. The microtubule binding domain of Tau is underlined.
Table 1.
First LC-MS/MS mass spectrometric analysis of Tau phosphorylation isolated with different GFP1–10 sensors. Tryptic peptides obtained from the indicated GFP1–10-Tau immune isolates were analysed twice and the mean peptide intensities, each normalized for the total abundance of all Tau peptides, are given as the relative mean percent for each phosphosite (human Tau441 numbering).
| Ctrl: GFP1–10 | nucGFP1–10 | ommGFP1–10 | nuc vs ctrl | nuc vs omm | omm vs ctrl | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n = 3 | SD | n = 2 | SD | n = 3 | SD | p | ratio | p | ratio | p | ratio | |
| pT181 | 2.49% | 0.22% | 3.11% | 0.25% | 2.13% | 0.34% | 0.004 | 1.2 | 6 × 10 −6 | 1.5 | ns | 0.9 |
| pT175 + pT181 | 0.05% | 0.03% | 0.04% | 0.02% | 0.01% | 0.01% | ns | 0.8 | ns | 3.3 | ns | 0.2 |
| pS198/199/202 | 2.55% | 0.10% | 3.42% | 0.37% | 2.19% | 0.09% | 5 × 10−5 | 1.3 | <10−6 | 1.6 | ns | 0.9 |
| pT212 | 0.71% | 0.11% | 1.31% | 0.08% | 0.41% | 0.07% | 0.006 | 1.9 | 3 × 10−5 | 3.2 | ns | 0.6 |
| pS214 | 0.25% | 0.03% | 0.17% | 0.01% | 0.15% | 0.06% | ns | 0.7 | ns | 1.2 | ns | 0.6 |
| pT217 | 0.10% | 0.02% | 0.08% | 0.05% | 0.07% | 0.01% | ns | 0.8 | ns | 1.2 | ns | 0.7 |
| pT231 | 0.65% | 0.08% | 0.75% | 0.11% | 0.63% | 0.15% | ns | 1.2 | ns | 1.2 | ns | 1.0 |
| pT231 + pS235 | 3.16% | 0.48% | 3.65% | 0.81% | 1.91% | 0.37% | 0.032 | 1.2 | <10−6 | 1.9 | <10−6 | 0.6 |
| pS262 | 0.01% | 0.01% | 0.02% | 0.01% | 0.04% | 0.03% | ns | 2.5 | ns | 0.5 | ns | 5.5 |
| pS356 | 0.16% | 0.02% | 0.17% | 0.13% | 0.57% | 0.55% | ns | 1.1 | ns | 0.3 | 0.047 | 3.5 |
| pS396 | 0.02% | 0.01% | 0.01% | 0.01% | 0.01% | 0.01% | ns | 0.7 | ns | 1.0 | ns | 0.7 |
| pS404 | 0.28% | 0.02% | 0.78% | 0.19% | 0.53% | 0.02% | 0.027 | 2.7 | ns | 1.5 | ns | 1.9 |
| pS416 | 0.19% | 0.04% | 0.11% | 0.03% | 0.07% | 0.06% | ns | 0.6 | ns | 1.5 | ns | 0.4 |
2way ANOVA and Tukey’s multiple comparison test (alpha 0.05).
Table 2.
Second LC-MS/MS mass spectrometric analysis of Tau phosphorylation isolated with GFP1–10 or nucGFP1–10. Tryptic peptide intensities, each normalized for the total abundance of all Tau peptides, are given as relative percent for each phosphosite (human Tau441 numbering).
| GFP1–10 | nucGFP1–10 | p | ratio | |||
|---|---|---|---|---|---|---|
| mean | SD | mean | SD | |||
| pT181 | 0.28% | 0.31% | 0.91% | 0.32% | 0.012 | 3.3 |
| pS198/199/202 | 2.16% | 0.80% | 2.33% | 0.39% | ns | 1.1 |
| pT212 | 0.04% | 0.03% | 0.30% | 0.24% | ns | 7.6 |
| pT231 | 0.50% | 0.26% | 1.16% | 0.43% | 0.009 | 2.3 |
| pT231 + pS235 | 0.21% | 0.19% | 0.63% | 0.46% | ns | 3.1 |
| pS262 | 0.05% | 0.03% | 0.11% | 0.07% | ns | 2.3 |
| pS404 | 2.71% | 1.07% | 2.89% | 0.55% | ns | 1.1 |
| pS416 | 0.14% | 0.13% | 0.04% | 0.05% | ns | 0.3 |
2way ANOVA and Bonferroni’s multiple comparison test (n = 5, alpha 0.05).
Nuclear targeting of Tau modifies its phosphorylation
For the first approach, C17.2 cells were transfected with untagged human Tau441 or nuclear targeted nucTau (Fig. 2b). One day later, total cell lysates were analysed by western blot, whereby cell lysates were loaded in a way to obtain similar amounts of Tau (Fig. 5a, Supplementary Fig. 6). Relative phosphorylation, after normalization for Tau measured with the Tau13 pan-antibody, was determined for biological triplicates, and calculated as ratio of the value for nucTau over that of wild-type Tau (Fig. 5b). Phosphorylation was increased for nucTau at the three sites examined, i.e. T181 by 3.8 ± 0.4 fold the value of wild-type Tau (p 1 × 10−9, 2way ANOVA, alpha 0.05), T212 by 4.0 ± 0.2 fold (p 6 × 10−10) and S404 by 2.4 ± 0.2 fold (p 9 × 10−5). Notably, nucTau detection was also increased for the Tau1 antibody with 2.6 ± 0.2 fold (p 1 × 10−5), specific for non-phosphorylated Ser195/198/199/20241. All phosphorylation dependent sites were also increased for nucTau in transiently transfected human SH-SY5Y cells (Fig. 5c, Supplementary Fig. 7): T181 by 1.8 ± 0.1 (p 4 × 10−7), T212 by 4.0 ± 0.4 (p 1 × 10−15), S404 by 1.5 ± 0.1 (p 0.004) and Tau1 by 1.8 ± 0.3 (p 8 × 10−7).
Figure 5.

Nuclear targeting of Tau modifies its phosphorylation. (a) Cell lysates obtained from C17.2 cells transiently transfected with Tau or nucTau are analysed by western blot with pan-Tau antibody Tau13 or antibodies against the indicated phosphosites or dephosphorylated Ser195/198/199/202 (Tau1). Molecular weight markers are shown on the left. (b and c) Quantification of Tau and nucTau and their modifications in C17.2 (n = 3) or SH-SY5Y (n = 5) cells. Values are mean ± SD. 2way ANOVA and Bonferroni’s multiple comparison test, **p < 0.01, ***p < 0.001.
Analysis of wild-type Tau phosphorylation in a nuclear fraction
For the second approach, we took advantage of the availability in the laboratory of a mouse C17.2 cell line with tetracycline-inducible expression of human Tau441 in order to isolate enriched cytosolic and nuclear fractions by a differential centrifugation procedure. Purity of the fractions was assessed by immune detection with the cytosolic GAPDH marker and the nuclear H3 marker histone 3 (Fig. 6a, Supplementary Fig. 8). Quantification of the signals obtained from biological triplicates demonstrated for the nuclear fraction a presence of GAPDH reaching 2.0 ± 1.6% for GAPDH, 99.6 ± 5.4% for H3 and 17.3 ± 2.5% for Tau (Fig. 6b). In order to analyse same amounts of Tau for both fractions, the nuclear samples were concentrated by 3-chloroacetic acid (TCA) precipitation before detecting phosphorylated Tau by western blot (Fig. 6c, Supplementary Fig. 9). Again, nuclear Tau displayed increased phosphorylation at T181 by 2.2 ± 0.2 fold the value of normal Tau (p 3 × 10−5, 2way ANOVA, alpha 0.05.), S404 (2.4 ± 0.1 fold; p 0.043) and decreased phosphorylation at the Tau1 epitope (1.6 ± 0.2 fold; p 0.0001) (Fig. 6d). Also T212 was found more phosphorylated in the nuclear fraction when compared to the cytosolic fraction, but this did not reach significance (3.2 ± 2.1%; p 0.9), possibly because the detection of pT212 in both fractions was only slightly above background determined with samples obtained from untransfected C17.2 cells, resulting in high variation between replicates.
Figure 6.

Subcellular fractionation reveals location-specific Tau modification. (a) Cell lysates, cytosolic and nuclear fractions obtained from biological triplicates of C17.2 cells induced for human Tau expression for 24 hr are analysed by western blot with antibodies for Tau (Tau13), for the cytosolic GAPDH marker and for the nuclear histone 3 (H3) marker. Molecular weight markers are shown on the left. Full blots are presented in Supplementary Fig. 4 online. (b) Quantification is plotted as mean percent ± SD recovered in the two subcellular fractions. (c) Cytosolic and TCA-precipated nuclear fractions are analysed by western blot with antibodies for pan-Tau, against the indicated phosphosites or for dephosphorylated Ser195/198/199/202 (biological triplicates). (d) Quantification of modified Tau normalized for the amount of total Tau detected in each sample. To facilitate the comparison between sites, each modified site is further normalized with the respective values measured in the cytosolic fraction. Values are mean ± SD, n = 3. 2way ANOVA and Bonferroni’s multiple comparison test, *p < 0.05, ***p < 0.001.
DNA damage modifies subcellular location and modification of Tau
Having established that Tau phosphorylation depends on its subcellular location, we analysed whether compounds expected to affect cellular Tau distribution may influence Tau modification. We focused our attention on treatments known to cause a nuclear Tau translocation following DNA damage, or to interfere with the Tau-microtubule interaction. Microscopic immune-fluorescent analysis of C17.2 cells with induced human Tau expression confirmed that treatment with the topoisomerase II inhibitor Etoposide induced activation of the DNA damage response, visualized with phospho-specific antibodies against γH2A-X (Fig. 7a) or against four nuclear phosphorylated kinases (Fig. 7b). Quantification of the fluorescent signal by confocal microscopy in the DAPI-stained nuclei demonstrated that activation of the early DNA damage response kinases in the presence of Etoposide, when compared to the untreated controls, reached 173 ± 3% for ATM (mean ± sem, n = 218 nuclei, p 10−15, 2-tailed unpaired Mann-Whitney test), 165 ± 3% for ATR (n = 212–225, p 10−15,), 470 ± 17% for Chk1 (n = 208–214, p 10−15,) and 378 ± 14% for Chk2 (n = 200–222, p 10−15,). The nuclear signal for Tau in the presence of Etoposide reached 132 ± 2% (n = 857–859, p 10−15,) that obtained for control conditions, confirming that the DNA damage caused translocation of Tau to the nuclear compartment (Fig. 7c). Vinblastine caused the collapse of the microtubule network as shown by the presence of brightly fluorescent microtubule fragments in the perinuclear region visualized with a β-tubulin antibody (Fig. 7a) but did not induce nuclear Tau translocation (Supplementary Fig. 10).
Figure 7.

DNA damage induces de-phosphorylation of nuclear Tau. (a) The effect of Etoposide and Vinblastine treatment on mouse C17.2 cells is shown by confocal microscopy upon immune staining of PFA-fixed cells with antibodies against the microtubule marker β-tubulin (in cyan) or the DNA damage marker γH2A-X (in red). (b) Confocal microscopic quantification of the activated kinases in the nucleus (DAPI mask). Mean percent ± sem relative to the respective controls. 2-tailed unpaired Mann-Whitney test, ****p < 0.0001. (c) Confocal microscopy images of C17.2 cells with induced Tau441 expression treated in the absence or presence of 60 µM Etoposide for 5 hr and stained for with the human Tau antibody (Tau13). Quantification of immune fluorescent detection of human Tau in DAPI-stained nuclei. Mean percent ± sem relative to the control. 2-tailed unpaired Mann-Whitney test, ****p < 0.0001. (d) Cytosolic and nuclear fractions obtained from biological replicates of C17.2 cells with induced Tau expression and treated as indicated are analysed by western blot with antibodies for the two phosphosites pT181 and pS404 and for the dephosphorylated Tau1 epitope are quantified upon normalization for total Tau. Data represent mean percent ± SD relative to the untreated controls. Non-parametric Kruskal-Wallis test, *p < 0.05, **p < 0.01.
In order to evaluate a possible effect of the drug treatments, Tau phosphorylation was analysed by western blot in total cell lysates as well as in cytosolic and nuclear fractions (Fig. 7d). When compared to the control conditions, Etoposide treatment reduced pT181 in total lysates to 42 ± 18% (mean ± SD, n = 6, p 0.01, non-parametric Kruskal-Wallis test), in the cytosol to 46 ± 18% (p 0.005) and, notably, even more in the nucleus to 28 ± 14% (p 0.001). An increase of the de-phosphorylated dependent Tau1 epitope to 155 ± 26% (p 0.024) was found in the nuclear fraction, but no significant changes were observed in the cytosolic fraction or in total cell lysates. Etoposide treatment did not affect significantly pS404. In contrast, Vinblastine-treated cells displayed reduced pS404 to 64 ± 9% (p 0.046, n = 3) in the cytosol and substantially also in total lysates (70 ± 7%, not significant) but not in the nuclear fraction (102 ± 21%). On the other hand, we found increased Tau1 signal in the presence of Vinblastine in total cell lysates to 211 ± 51% (p 0.019, n = 3) and in the nuclear fraction to 182 ± 31% (p 0.024) but this did not reach significance in the cytosolic fraction (174 ± 18%). We also tested the effect of the nuclear export inhibitor Leptomycin b and found a significant increase in the nuclear staining for Tau reaching 144 ± 4% (mean ± sem, n = 154–178 nuclei, p 10−15) but this effect on the retention of Tau in the nucleus did not affect any of the phosphosites of Tau analysed in the nuclear fraction, in the cytosolic fraction or in total cell lysates.
In summary, our data show that the phosphorylation of Tau differs depending on its subcellular location and that phosphorylation of nuclear and cytosolic Tau is regulated by different cellular pathways.
Discussion
With a set of cellular models and analytical methods, we investigated nuclear Tau modification and obtained evidence for different phosphorylation at defined sites in this cellular compartment. First evidence of subcellular location-dependent modification was produced by mass spectrometry of S11-Tau isolated with GFP1–10 sensors targeted to distinct cellular compartments. In this analysis, phosphorylation of nuclear sensor-bound Tau, when compared to Tau captured with the untargeted sensor, was increased at five out of thirteen Tau phosphopeptides detected by mass spectrometry, namely those including T181, S198/199/202, T212, S404 and the double phosphorylated T231/S235. Reinforcing these data, these residues, with the exception of S404, were also more phosphorylated for the nuclear sensor when compared to Tau isolated by means of the sensor targeted to the outer mitochondrial membrane. On the other hand, association to mitochondria reduced pT231/pS235 but increased pS356, confirming the possibility of a mitochondria-localized pool of Tau with unique functions18. Previously published work claimed reduced nuclear Tau phosphorylation by immune staining with the dephosphorylated Ser195/198/199/202-specific antibody Tau137. Data from our laboratory indicate that the analysis of (de)phosphorylation of endogenous Tau by immune staining of cultured cells needs confirmation with a proper negative control. In fact, using Tau1 and some phospho-Tau antibodies used in this study on human SH-SY5Y cells, wild-type or with destroyed MAPT gene, resulted in a nucleus-localized staining that was not dependent on the presence or the absence of endogenous or overexpressed Tau. Nevertheless, because of this and the fact that a second mass spectrometric analysis showed increased nuclear Tau phosphorylation only at residues T181 and T231/S235, we continued our study with two different cellular systems. First, we studied the effect of forced nuclear targeting of Tau (nucTau) by western blot with phospho-specific antibodies. Quantifiable signals were obtained for pT181, pT212, pS404 and for Tau1 both in transfected mouse C17.2 and human SH-SY5Y cells. For both cell lines, increased immune detection at the same four epitopes was found for nucTau. Since the utilization of the nuclear targeted sensor and of nucTau may influence normal cellular Tau distribution, we then investigated Tau modification in isolated nuclei of C17.2 cells expressing human wild-type Tau. This confirmed the presence of Tau in the nucleus at levels higher than those of the potential cytosolic contaminant GAPDH. When compared to the cytosolic fraction, also Tau present in the nuclear fraction displayed increased detection of three out of four epitopes analysed. Our data describe the phosphorylation status of nuclear Tau in proliferative pluripotent neuronal C17.2 and neuroblastoma SY5Y cells and this may not apply in differentiated cells such as neurons.
The presence of Tau in the nucleus was documented previously in vitro23–27 and in vivo28 where a diffuse distribution in the nucleoplasm or association to nucleoli in its dephosphorylated, Tau1 antibody-positive form was reported24,26,27,42. Within the nucleus, Tau interacts with RNA and DNA29–32 and appears to protect the neuron from age-related insults. In vitro, Tau raises the melting temperature of DNA43. Tau binds and bends DNA when its proline-rich and microtubule-binding domains associate to the AT-rich minor DNA groove independently of the nucleotide sequence33,36,44. These data suggest that Tau may shuttle between cytoplasm and nucleus, as canonical heat-shock (HS) proteins do when they induce the HS response45,46. Indeed, rescue from HS-induced DNA damage is observed when nuclear-targeted Tau is expressed in Tau-deficient cells or mouse brain37,47. Linking the role of Tau in DNA metabolism with disease, cells expressing FTDP-17 mutations display more chromosomal defects that those expressing normal Tau48. Here, we show that treatment with the DNA damaging drug Etoposide also causes increased nuclear location of Tau, whereby this is accompanied by decreased phosphorylation at T181 and at the Tau1 epitope in the nuclear fraction, without affecting pS404. Decreased phosphorylation of Tau in the presence of Etoposide is remarkable in view of the many kinases that are activated during the DNA damage response. Considering the role of phosphorylation in regulating protein-protein interaction and the scaffold nature of Tau as protein linking different binding partner, it is tempting to consider that our data underlie a possible mechanism regulating different functions of Tau. In this regards de-phosphorylation of Tau during the DNA damage response at residue T181 and at the Tau1 epitope, i.e. within the proline-reach domain that has been implicated in dephosphorylated Tau-DNA association44, may offer a molecular mechanism for the DNA protective role of Tau. As a matter of fact, pT181 is the epitope commonly used to show increased phospho-Tau as a cerebrospinal fluid biomarker for an ongoing neurodegenerative process. Our data may indicate that a neurodegeneration-linked increase in pT181 could weaken the DNA protecting role of Tau. Interestingly, treatment of the cells with Vinblastine did not increased nuclear Tau localization and did not change pT181, but reduced phosphorylation of S404 in the cytoplasm but not in the nucleus, suggesting that the carboxy-terminal tail of Tau may have a predominant cytoplasmic function. Increased nuclear Tau retention in the presence of Leptomycin did not affect phosphorylation of Tau at the sites analysed in this study. Notably, increased nuclear Tau was found for the 1N4R isoform of Tau49, whereas a pathological form of Tau was shown to interfere with nuclear import and export50, indicating that additional mechanisms regulating nuclear Tau translocation and modification exist.
Tau alters the integrity of cytoplasmic and nuclear RNA47 and may regulate RNA metabolism when present in stress granules, which protect RNA from degradation during cellular stress51. Nuclear localization and a role of Tau in DNA and RNA homeostasis are reminiscent to TDP-43 and FUS, two other proteins involved in frontotemporal dementia (FTD)52,53. These are RNA binding proteins that play key roles in transcription, mRNA splicing, stability and transport. Their subcellular location is affected by autosomal dominant FTD mutations52,53. It is tempting to consider a common pathway for Tau/TDP/FUS-linked FTD. Whilst FUS is clearly involved in the DNA damage response54–56, a role of TDP-43 and Tau appears less evident. Further knowledge on nuclear Tau may provide clues in the understanding of FTD involving TDP-43 and FUS proteins. Due to the wide distribution in the CNS and periphery, Tau is potentially a global player for genome surveillance, in particular against DNA insults accumulating as a function of aging.
Materials and Methods
Expression plasmids
cDNAs were inserted in the constitutive expression vector pcDNA3 or in pcDNA5 for inducible expression. For nucGFP1–10, the GFP1–10 plasmid38 was modified at the amino-terminus with a nuclear targeting sequence consisting of an artificial peptide carrying three tandem sequence derived from the SV40 large T antigen (MDIDPKKKRKVDPKKKRKVDPKKKRKVD)39. To insert this sequence, we used as template GFP1–10 for a PCR with the three overlapping forward primers TCGGATCCATGGACCCGAAGAAGAAACGCAAGGTCGATCCGAAGAAG, CAAGGTCGATCCGAAGAAGAAGCGGAAGGTCGATCCAAAGAAAAAAAGG and CGATCCAAAGAAA AAAAGGAAGGTGTCCAAAGGAGAAGAACTG and the reverse primer TTCACTCGAGCTATGTTCCTTTTTCATTTGG. This inserted also the restriction sites BamHI and XhoI (in bold) for subcloning the amplified fragment in pcDNA3, whereby the initial methionine of GFP was eliminated. The signal peptide at the amino-terminus of erGFP1–10 (MLLSVPLLLGLLGLAVA) was that of human calreticulin and was obtained with the forward primer CACGCAGGAATTCATGCTGCTATCCGTGCCGCTGCTGCTCGGCCTCCTCGGCCTGGCCGTCGCCTCCAAAGGAGAAGAACTG (the initial methionine of GFP was eliminated) and the reverse primer ACTTCTCACTCGAGTTACAGCTCGTCCTTTGTTCCTTTTTCATTTGGATC, which also inserted a KDEL sequence for retention in the ER before the stop codon of GFP1–1040.
Tau expression plasmids encoded the 441 amino acid-splice variant 2N4R of human Tau. The plasmid encoding for untagged Tau, S11-Tau, and Tau-S11 were generated as described38. The nuclear targeting sequence of nucTau was the same used for nucGFP1–10 and obtained with forward primer ATATAAGCTTACCATGGATATCGACCCGAAG and reverse primer TTAAGGATCCATCCACCTTCCTTTTTTTCTTTGG. The amplified fragment was then inserted before the Tau cDNA at the restriction sites HindIII and BamHI (in bold) to obtain the desired polypeptide with the nuclear targeting sequence followed by a short linker (GST) and the sequence of human Tau including the initial methionine. The plasmid encoding for erTau-S11, was generated using as template Tau-S11, and the primers TTCGGATCCATGAAGACCATCATTGCTTTGAGCTACATTTTCTGTCT GGCTCTCGGCCAAGACCTTCCAGGAAATGACAACAGCACAGCAGCTGAGCCCCGCCAGG and TTCACTCGAGTCATGTGATGCCGGCGGCGTTC followed by subcloning with BamHI and XhoI (in bold) in pcDNA3. The inserted sequence corresponded to MKTIIALSYIFCLALGQDLPGNDNSTA, which covered the signal peptide and the amino-glycosylation consensus sequence (underlined) of influenza A hemagglutinin without the initial methionine of Tau.
Cell culturing, cell lines and plasmid transfections
Mouse multipotent neural progenitor C17.2 cells and human neuroblastoma SH-SY5Y cells were cultured as described previuosly38. Inducible C17.2 cell lines were generated with the Flp-In T-Rex tetracycline-inducible cell system according to the manufacturer instructions (Invitrogen, K650001) and were selected and maintained in the presence of 150 µg/mL Hygromycin B and 15 µg/ml Blasticidin S. Protein expression was induced by one day-incubation with 30 ng/mL Tetracycline, a treatment time and dose that were sufficient to reach steady-state levels of expression. Plasmid transfections were performed with Lipofectamine 3000 (Invitrogen, L-3000-008) as described38.
Drug treatments
During the last 5 h of tetracycline incubation, C17.2 cells with inducible Tau expression were treated with 60 µM Etoposide (Abcam, ab120227; 100 mM stock in DMSO), 3 µM Vinblastine (Sigma-Aldrich, V1377; 11 mM stock in DMSO), or 60 nM Leptomycin B (Sigma-Aldrich, L2913; 10.3 µM in 70% ethanol). At the end of the drug treatment, cells were fixed and processed for fluorescence microscopy. For the immune fluorescence quantification of nuclear Tau and activated kinases, a DAPI mask on ImageJ was used.
Fluorescence microscopy
For immune fluorescence microscopy, cells were routinely grown on 8-well slides (Ibidi, 80826-IBI) coated with poly-D-lysine. One day after plasmid transfection, cells were fixed either in paraformaldehyde (PFA) or in methanol. For PFA, a 4% solution in PBS pre-warmed at 37 °C was added 1:1 (V:V) to the culture medium for 10 min at 37 °C, followed by a second 5 min fixation in 4% PFA at room temperature, 5 min quenching in 100 μM glycine and three washes with PBS. For methanol, the culture medium was replaced with methanol chilled at −20 °C followed by 10 min incubation at −20 °C. Then, the methanol solution was removed by aspiration and the fixed cell layer gently washed three times with PBS. For the immune staining procedure, all steps were performed at room temperature. The fixed cells were blocked with 5% normal goat serum, 0.3% Triton X-100 in PBS for 1 h followed by PBS washes. The antibodies were diluted in a working solution composed of 0.5% normal goat serum, 0.3% Triton X-100 in PBS. Primary antibodies, usually incubated for 1 h at 37 °C, were specific for human Tau (Tau13, Santa Cruz, sc-21796, used at 1 μg/mL), GFP (Proteintech Europe, 66002–1-Ig, 7 μg/mL), calnexin (kind gift of Prof. Maurizio Molinari, IRB, Bellinzona, Switzerland, diluted 1:1,000), α-tubulin (Abcam, ab1825, 0.5 μg/mL or Cell Signaling, DM1A, diluted 1:500), pS129-H2A.X (Santa Cruz, sc-517348, 0.5 μg/mL), pATM (Cell Signaling, S1981, diluted 1:500), pChk1 (Cell Signaling, S345, diluted 1:500), pChk2 (Cell Signaling, T68, diluted 1:500), pATR (Cell Signaling, S428, diluted 1:500). Secondary antibodies (2 μg/mL for 1 h at room temperature in the dark) were anti-mouse IgG (Thermo Fisher Scientific, Alexa594, A-11032, or Alexa 488, A-11001) or anti-rabbit IgG (Thermo Fisher Scientific, Alexa594, A-11037, or Alexa488 A-11034). Nuclei were counterstained with DAPI (0.5 μg/mL). After the staining, cells were kept in 0.05% sodium azide in PBS in the fridge before analysis with a confocal microscope (Nikon C2). Images were taken with a line by line scan using a sequence of excitation with the 405 nm laser and emission filter 464/40–700/100 nm, 488 nm laser and emission filter 525/50 nm, and 561 nm laser and 561/LP nm emission filter.
Cell lysis and western blotting
Cells were rinsed once with PBS and collected by scraping and low speed centrifugation. Cell lysates were prepared in 100 μL ice-cold RIPA supplemented with inhibitor cocktails for proteases (Sigma-Aldrich, S8820) and phosphatases (Sigma-Aldrich, 04906845001). All samples were then maintained on ice or at 4 °C. Cell lysates were shaken for 30 min at 1,400 rpm (Eppendorf, Thermomixer 5355) and RIPA extracts collected after centrifugation for 10 min at 20,000 g. Total protein concentration was determined by Bradford (Pierce, BCA Protein Assay Kit 23227). For phosphatase treatment, RIPA extracts (10 μg protein), prepared without phosphatase inhibitors, were incubated in the absence or presence of 20 U/μL Lambda PP (BioLabs, P0753S) for 1 h at 30 °C as described by the supplier. Total cell lysates were prepared by direct solubilisation of cell pellets in 100 μL sample buffer: 1.5% SDS, 8.3% glycerol, 0.005% Bromophenol blue, 1.6% β-mercaptoethanol and 62.5 mM Tris-HCl pH 6.8.
SDS-polyacrylamide gel electrophoresis was performed on hand-casted 10% gels at constant 90–110 V (Bio-Rad, Mini-PROTEAN 1658033FC). Protein was transferred on PVDF membranes (Merck-Millipore, Immobilon-FL IPFL00010) using a Trans-Blot Turbo Transfer System (Bio-Rad, 1704150). After the transfer, the membranes were incubated in blocking buffer (Licor Biosciences Odyssey, 927–50000) for 1 h at room temperature. Bound primary antibodies were revealed by the two secondary antibodies anti-mouse IgG coupled to IRDye RD 680 or anti-rabbit IgG coupled to IRDye 800CW (Licor Biosciences, 926–68070 and 926–32211) on a dual infrared imaging scanner (Licor Biosciences, Odyssey CLx 9140) and quantified with the software provided (Licor Biosciences, Image Studio V5.0.21, 9140–500). Antibodies used for western blot were Tau13 (0.2 μg/mL), Tau1 (Merck-Millipore, MAB-3420, 0.5 μg/mL), phospho-Tau T181 (AT270, Invitrogen, MN1050, 0.2 μg/mL), T212 (Abcam, ab4842, 1:1,000), S404 (Invitrogen, 44–758 G, 1:1,000), GAPDH (Abcam, ab181602, 1:2,500), histone 3 (Abcam, ab176842, 1:10,000), β-actin (Sigma-Aldrich, A1978, 0.084 μg/mL), and GFP (Abcam, ab5449, 1:5,000).
Samples preparation for LC-MS/MS
Detergent-free extracts were prepared in PBS supplemented with protease and phosphatase inhibitors by three freeze/thaw cycles in liquid nitrogen followed by centrifugation for 10 min at 20,000 g. Immune isolation of complemented GFP was performed on magnetic beads. For this, cell extracts obtained by pooling biological triplicate (at least 600 μg total protein) were brought to 500 μL with PBS, supplemented with 5 μL of 50% magnetic bead slurry GFP-Trap (ChromoTek, gtma-20) and incubated on an orbital rotator (Labnet, 096621) for 2 h at 4 °C. With the aid of a magnet, the beads were washed twice with PBS and once with water. A small portion of the beads was eluted with sample buffer to analyse the immune isolates by western blot.
Proteolytic peptides were obtained by double digestion. For this, GFP-trap bead-bound immune isolates were resuspended in 50 µL 50 mM NH4HCO3 and 8 M urea and reduced in 2 µM dithiotreitol for 1 h at 37 °C with shaking at 800 rpm. Proteins were then alkylated in 4.5 mM iodoacetamide for 45 min in the dark at room temperature. Sequential digestion was performed for 3 h with Lys-C (WAKO, 125–05061; 20 ng/µL), and then overnight with trypsin (Promega, V5113; 2.5 ng/µL) by adding 144 µL 50 mM NH4HCO3. The digestion was stopped by acidification to pH < 3 with 50% formic acid. The peptide mixtures were loaded on C18 spin tips (Pierce, 84850), and desalted twice with 0.1% formic acid by centrifugation for 1 min at 100 g. Peptides were eluted twice with 80% acetonitrile, lyophilized (Eppendorf, Concentrator plus 5305), solubilized in 0.1% formic acid and analysed by mass spectrometry.
LC-MS/MS
Samples were measured on a nanoLC (Thermo Fisher, EASY-nLC 1000) coupled to a mass spectrometer (Thermo Fisher, Q Exactive Plus). Peptides were separated on a 40 cm × 0.75 μm column packed in-house with reversed-phase resin (ReproSil-Pur C18- AQ 1.9 μm, Dr. Maisch). Peptides were eluted for 60 min using a segmented linear 5–40% gradient (solvent A: 0.1% formic acid; solvent B: 99.9% acetonitrile, 0.1% formic acid) at 300 nL/min. Survey full-scan mass spectra were acquired with mass range 350–1,500 m/z, at a resolution of 70,000 at 200 m/z and the 20 most intense ions above an intensity of 3.6e4 were sequentially isolated, fragmented (HCD at 25 NCE) and measured at a resolution of 17,500 at 200 m/z. Peptides with a charge of + 1 or with unassigned charge state were excluded from fragmentation for MS2, and a dynamic exclusion of 30 s was applied. Ions were accumulated to a target value of 3e6 for MS1 and of 1e5 for MS2.
In Sequest HT, mass spectra were searched against a mouse database (Uniprot, downloaded July 8th, 2016; supplemented with sequences of human Tau). Carbamidomethylation of cysteine was set as a fixed modification and oxidation of methionine and phosphorylation of serine, threonine and tyrosine were allowed as a variable modification. Precursor mass tolerance was 10 ppm and fragment mass tolerance 0.02 Da. Trypsin was set as enzyme allowing for maximally two missed cleavages. Percolator was used to filter the results to 1% FDR at peptide level and phosphoRS 3.0 for phosphorylation-site localization. Skyline 3 was used to extract the monoisotopic and the first and second isotope precursor intensity traces of all identified Tau peptides.
Cytosolic and nuclear fractionation
The protocol was derived from an online method (Abcam). All samples were maintained on ice and all centrifugations were performed at 4 °C. C17.2 cells were washed once with PBS and scraped in hypotonic buffer: 250 mM sucrose, 10 mM KCl, 2 mM CaCl2, 1.5 mM MgCl2, 20 mM HEPES pH 7.4, supplemented with protease and phosphatase inhibitors. Cells were broken by passing the cell suspension four times through a 25-gauge needle. An aliquot of this sample was collected and labelled as “total”. The cytosolic fraction was obtained as supernatant of a sequential centrifugation for 5 min at 1,000 g and for 10 min at 20,000 g. For the nuclear fraction, the pellet from the first centrifugation was washed twice by sequential resuspension in hypotonic buffer, ten-time passage through a 25-gauge needle and centrifugation for 10 min at 1,000 g. The final pellet was resuspended in RIPA supplemented with 1.25 mM MgCl2, protease and phosphatase inhibitor, treated with Benzonase nuclease (Merck-Millipore, 70746–4, 0.56 U/µL) for 15 min at 37 °C and collected as the supernatant of a final centrifugation for 10 min at 20,000 g.
In order to concentrate the nuclear fraction, this was mixed with 1 volume of 20% TCA, incubated for 30 min on ice and centrifuged 20 min at 20,000 g. The pellet was washed with −20 °C cold 100% acetone, followed by centrifugation for 20 min at 20,000 g. After supernatant removal, residual acetone was evaporated at room temperature and the samples resuspended in SDS PAGE sample buffer and neutralization with 1 M Tris-HCl pH 10.8
Statistical analysis
Statistical analysis was performed with the aid of GraphPad Prism version 7.02 as indicated in the results section and figure legends.
Electronic supplementary material
Acknowledgements
We thank the whole laboratory for support and advice during this study. We thank Prof. Hilal Lashuel, EPFL, Switzerland for his generous supply of recombinant Tau and Prof. Maurizio Molinari, IRB, Switzerland for his generous supply of α-calnexin antibody. PPa is supported by grants from the Gelu Foundation, the Donatella e Ilaria Merlini Foundation and from the Swiss National Science Foundation, grant 31003A_166612. TC is supported by grants from the Ministry of University and Research (Bando SIR 2014 No. RBSI14C65Z to T.C) and from the Università degli Studi di Padova (Progetto Giovani 2012 No. GRIC128SP0 and Progetto di Ateneo 2016 No. CALI_SID16_01).
Author Contributions
G.U. and A.S. designed, performed and quantified experiments and co-wrote the manuscript. P.B. performed proteomic measurements and analysed the data. T.C. generated expression plasmids, proposed and designed experiments; C.F. and M.S. designed and performed experiments. PPi supervised proteomic analysis, reviewed the manuscript. S.P. designed and supervised experiments. P.P.a. conceived the study, designed and supervised the whole study, wrote the manuscript and provided funding in support of this study.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-36374-4.
References
- 1.Luheshi LM, Crowther DC, Dobson CM. Protein misfolding and disease: from the test tube to the organism. Curr. Opin. Chem. Biol. 2008;12:25–31. doi: 10.1016/j.cbpa.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 2.Villemagne VL, Dore V, Burnham SC, Masters CL, Rowe CC. Imaging tau and amyloid-beta proteinopathies in Alzheimer disease and other conditions. Nat. Rev. Neurol. 2018;14:225–236. doi: 10.1038/nrneurol.2018.9. [DOI] [PubMed] [Google Scholar]
- 3.Clayton KA, Van Enoo AA, Ikezu T. Alzheimer’s Disease: The Role of Microglia in Brain Homeostasis and Proteopathy. Front. Neurosci. 2017;11:680. doi: 10.3389/fnins.2017.00680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaushik S, Cuervo AM. Proteostasis and aging. Nat. Med. 2015;21:1406–1415. doi: 10.1038/nm.4001. [DOI] [PubMed] [Google Scholar]
- 5.Clarimon J, et al. Early-onset familial lewy body dementia with extensive tauopathy: a clinical, genetic, and neuropathological study. J. Neuropathol. Exp. Neurol. 2009;68:73–82. doi: 10.1097/NEN.0b013e3181927577. [DOI] [PubMed] [Google Scholar]
- 6.Clavaguera F, et al. Peripheral administration of tau aggregates triggers intracerebral tauopathy in transgenic mice. Acta Neuropathol. 2014;127:299–301. doi: 10.1007/s00401-013-1231-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hock EM, Polymenidou M. Prion-like propagation as a pathogenic principle in frontotemporal dementia. J. Neurochem. 2016;138(Suppl 1):163–183. doi: 10.1111/jnc.13668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kovacs G. G. Tauopathies. Handb. Clin. Neurol. 2017;145:355–368. doi: 10.1016/B978-0-12-802395-2.00025-0. [DOI] [PubMed] [Google Scholar]
- 9.Li C, Gotz J. Tau-based therapies in neurodegeneration: opportunities and challenges. Nat. Rev. Drug. Discov. 2017;16:863–883. doi: 10.1038/nrd.2017.155. [DOI] [PubMed] [Google Scholar]
- 10.Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. 1975;72:1858–1862. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ait-Bouziad N, et al. Discovery and characterization of stable and toxic Tau/phospholipid oligomeric complexes. Nat. Commun. 2017;8:1678. doi: 10.1038/s41467-017-01575-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morris M, et al. Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat. Neurosci. 2015;18:1183–1189. doi: 10.1038/nn.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee G, Leugers CJ. Tau and tauopathies. Prog. Mol. Biol. Transl. Sci. 2012;107:263–293. doi: 10.1016/B978-0-12-385883-2.00004-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jeganathan S, von Bergen M, Brutlach H, Steinhoff HJ, Mandelkow E. Global hairpin folding of tau in solution. Biochemistry. 2006;45:2283–2293. doi: 10.1021/bi0521543. [DOI] [PubMed] [Google Scholar]
- 15.Fuster-Matanzo Almudena, Hernández Félix, Ávila Jesús. Tau Spreading Mechanisms; Implications for Dysfunctional Tauopathies. International Journal of Molecular Sciences. 2018;19(3):645. doi: 10.3390/ijms19030645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Katsinelos T, et al. Unconventional Secretion Mediates the Trans-cellular Spreading of Tau. Cell Rep. 2018;23:2039–2055. doi: 10.1016/j.celrep.2018.04.056. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Mandelkow E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016;17:5–21. doi: 10.1038/nrn.2015.1. [DOI] [PubMed] [Google Scholar]
- 18.Cieri Domenico, Vicario Mattia, Vallese Francesca, D'Orsi Beatrice, Berto Paola, Grinzato Alessandro, Catoni Cristina, De Stefani Diego, Rizzuto Rosario, Brini Marisa, Calì Tito. Tau localises within mitochondrial sub-compartments and its caspase cleavage affects ER-mitochondria interactions and cellular Ca2+ handling. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2018;1864(10):3247–3256. doi: 10.1016/j.bbadis.2018.07.011. [DOI] [PubMed] [Google Scholar]
- 19.Zempel H, Mandelkow EM. Tau missorting and spastin-induced microtubule disruption in neurodegeneration: Alzheimer Disease and Hereditary Spastic Paraplegia. Mol. Neurodegener. 2015;10:68. doi: 10.1186/s13024-015-0064-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Q, et al. The cellular distribution and Ser262 phosphorylation of tau protein are regulated by BDNF in vitro. PloS one. 2014;9:e91793. doi: 10.1371/journal.pone.0091793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frandemiche ML, et al. Activity-dependent tau protein translocation to excitatory synapse is disrupted by exposure to amyloid-beta oligomers. J. Neurosci. 2014;34:6084–6097. doi: 10.1523/JNEUROSCI.4261-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Regan P, et al. Tau phosphorylation at serine 396 residue is required for hippocampal LTD. J. Neurosci. 2015;35:4804–4812. doi: 10.1523/JNEUROSCI.2842-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cross D, Tapia L, Garrido J, Maccioni RB. Tau-like proteins associated with centrosomes in cultured cells. Exp. Cell Res. 1996;229:378–387. doi: 10.1006/excr.1996.0383. [DOI] [PubMed] [Google Scholar]
- 24.Greenwood JA, Johnson GV. Localization and in situ phosphorylation state of nuclear tau. Exp. Cell Res. 1995;220:332–337. doi: 10.1006/excr.1995.1323. [DOI] [PubMed] [Google Scholar]
- 25.Lambert MP, Sabo S, Zhang C, Enam SA, Klein WL. Constitutive Alzheimer’s-type tau epitopes in a neuritogenic rat CNS cell line. Neurobiol. Aging. 1995;16:583–589. doi: 10.1016/0197-4580(95)00042-D. [DOI] [PubMed] [Google Scholar]
- 26.Loomis PA, Howard TH, Castleberry RP, Binder LI. Identification of nuclear tau isoforms in human neuroblastoma cells. Proc. Natl. Acad. Sci. USA. 1990;87:8422–8426. doi: 10.1073/pnas.87.21.8422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thurston VC, Zinkowski RP, Binder LI. Tau as a nucleolar protein in human nonneural cells in vitro and in vivo. Chromosoma. 1996;105:20–30. doi: 10.1007/BF02510035. [DOI] [PubMed] [Google Scholar]
- 28.Brady RM, Zinkowski RP, Binder LI. Presence of tau in isolated nuclei from human brain. Neurobiol. Aging. 1995;16:479–486. doi: 10.1016/0197-4580(94)00170-6. [DOI] [PubMed] [Google Scholar]
- 29.Bryan JB, Nagle BW, Doenges KH. Inhibition of tubulin assembly by RNA and other polyanions: evidence for a required protein. Proc. Natl. Acad. Sci. USA. 1975;72:3570–3574. doi: 10.1073/pnas.72.9.3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Corces VG, et al. Effects of DNA on microtubule assembly. Eur. J. Biochem. 1980;105:7–16. doi: 10.1111/j.1432-1033.1980.tb04468.x. [DOI] [PubMed] [Google Scholar]
- 31.Corces VG, Salas J, Salas ML, Avila J. Binding of microtubule proteins to DNA: specificity of the interaction. Eur. J. Biochem. 1978;86:473–479. doi: 10.1111/j.1432-1033.1978.tb12330.x. [DOI] [PubMed] [Google Scholar]
- 32.Kampers T, Friedhoff P, Biernat J, Mandelkow EM, Mandelkow E. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS lett. 1996;399:344–349. doi: 10.1016/S0014-5793(96)01386-5. [DOI] [PubMed] [Google Scholar]
- 33.Camero S, et al. Tau protein provides DNA with thermodynamic and structural features which are similar to those found in histone-DNA complex. J. Alzheimers Dis. 2014;39:649–660. doi: 10.3233/JAD-131415. [DOI] [PubMed] [Google Scholar]
- 34.Hua Q, et al. Microtubule associated protein tau binds to double-stranded but not single-stranded DNA. Cell. Mol. Life. Sci. 2003;60:413–421. doi: 10.1007/s000180300034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu Y, et al. Hyperphosphorylation results in tau dysfunction in DNA folding and protection. J. Alzheimers Dis. 2013;37:551–563. doi: 10.3233/JAD-130602. [DOI] [PubMed] [Google Scholar]
- 36.Wei Y, et al. Binding to the minor groove of the double-strand, tau protein prevents DNA from damage by peroxidation. PloS one. 2008;3:e2600. doi: 10.1371/journal.pone.0002600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sultan A, et al. Nuclear tau, a key player in neuronal DNA protection. J. Biol. Chem. 2011;286:4566–4575. doi: 10.1074/jbc.M110.199976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Foglieni C, et al. Split GFP technologies to structurally characterize and quantify functional biomolecular interactions of FTD-related proteins. Sci. Rep. 2017;7:14013. doi: 10.1038/srep41598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ding Y, et al. Ratiometric biosensors based on dimerization-dependent fluorescent protein exchange. Nat. methods. 2015;12:195–198. doi: 10.1038/nmeth.3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cali T, Ottolini D, Soriano ME, Brini M. A new split-GFP-based probe reveals DJ-1 translocation into the mitochondrial matrix to sustain ATP synthesis upon nutrient deprivation. Hum. Mol. Genet. 2015;24:1045–1060. doi: 10.1093/hmg/ddu519. [DOI] [PubMed] [Google Scholar]
- 41.Petry FR, et al. Specificity of anti-tau antibodies when analyzing mice models of Alzheimer’s disease: problems and solutions. PloS one. 2014;9:e94251. doi: 10.1371/journal.pone.0094251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Y, Loomis PA, Zinkowski RP, Binder LI. A novel tau transcript in cultured human neuroblastoma cells expressing nuclear tau. J. Cell. Biol. 1993;121:257–267. doi: 10.1083/jcb.121.2.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hua Q, He R. Human neuronal tau promoting the melting temperature of DNA. Chinese Science Bulletin. 2000;45:999–1002. doi: 10.1007/BF02884979. [DOI] [Google Scholar]
- 44.Qi H, et al. Nuclear magnetic resonance spectroscopy characterization of interaction of Tau with DNA and its regulation by phosphorylation. Biochemistry. 2015;54:1525–1533. doi: 10.1021/bi5014613. [DOI] [PubMed] [Google Scholar]
- 45.Niu P, et al. Overexpressed heat shock protein 70 protects cells against DNA damage caused by ultraviolet C in a dose-dependent manner. Cell stress chaperones. 2006;11:162–169. doi: 10.1379/CSC-175R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang D, et al. Nuclear heat shock protein 72 as a negative regulator of oxidative stress (hydrogen peroxide)-induced HMGB1 cytoplasmic translocation and release. J. Immunol. 2007;178:7376–7384. doi: 10.4049/jimmunol.178.11.7376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Violet M, et al. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front. Cell. Neurosci. 2014;8:84. doi: 10.3389/fncel.2014.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rossi G, et al. A new function of microtubule-associated protein tau: involvement in chromosome stability. Cell cycle. 2008;7:1788–1794. doi: 10.4161/cc.7.12.6012. [DOI] [PubMed] [Google Scholar]
- 49.Vanderweyde T, et al. Interaction of tau with the RNA-Binding Protein TIA1 Regulates tau Pathophysiology and Toxicity. Cell Rep. 2016;15:1455–1466. doi: 10.1016/j.celrep.2016.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu C, Götz J. Profiling murine tau with 0N, 1N and 2N isoform-specific antibodies in brain and peripheral organs reveals distinct subcellular localization, with the 1N isoform being enriched in the nucleus. PLoS one. 2013;8:e84849. doi: 10.1371/journal.pone.0084849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eftekharzadeh B, et al. Tau Protein Disrupts Nucleocytoplasmic Transport in Alzheimer’s Disease. Neuron. 2018;99:925–940. doi: 10.1016/j.neuron.2018.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79:416–438. doi: 10.1016/j.neuron.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thomas M, Alegre-Abarrategui J, Wade-Martins R. RNA dysfunction and aggrephagy at the centre of an amyotrophic lateral sclerosis/frontotemporal dementia disease continuum. Brain. 2013;136:1345–1360. doi: 10.1093/brain/awt030. [DOI] [PubMed] [Google Scholar]
- 54.Deng Q, et al. FUS is phosphorylated by DNA-PK and accumulates in the cytoplasm after DNA damage. J. Neurosci. 2014;34:7802–7813. doi: 10.1523/JNEUROSCI.0172-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang WY, et al. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci. 2013;16:1383–1391. doi: 10.1038/nn.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang Liuqing, Gal Jozsef, Chen Jing, Zhu Haining. Self-assembled FUS binds active chromatin and regulates gene transcription. Proceedings of the National Academy of Sciences. 2014;111(50):17809–17814. doi: 10.1073/pnas.1414004111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.