

Abstract
Biodegradable polymeric nanoparticles (NPs) have demonstrated significant potential to improve the systemic delivery of RNA interference (RNAi) therapeutics, such as small interfering RNA (siRNA), for cancer therapy. However, the slow and inefficient siRNA release inside tumor cells generally observed for most biodegradable polymeric NPs may result in compromised gene silencing efficacy. Herein, a biodegradable and redox-responsive NP platform, composed of a solid poly(disulfide amide) (PDSA)/cationic lipid core and a lipid–poly(ethylene glycol) (lipid–PEG) shell for systemic siRNA delivery to tumor cells, is developed. This newly generated NP platform can efficiently encapsulate siRNA under extracellular environments and can respond to the highly concentrated glutathione (GSH) in the cytoplasm to induce fast intracellular siRNA release. By screening a library of PDSA polymers with different structures and chain lengths, the optimized NP platform shows the unique features of i) long blood circulation, ii) high tumor accumulation, iii) fast GSH-triggered intracellular siRNA release, and iv) exceptionally effective gene silencing. Together with the facile polymer synthesis technique and robust NP formulation enabling scale-up, this new redox-responsive NP platform may become an effective tool for RNAi-based cancer therapy.
Keywords: biodegradable nanoparticle, cancer therapy, redox-responsive, siRNA delivery
1. Introduction
Bioresponsive nanoparticles (NPs) that can respond to biological signals or pathological abnormalities, such as pH, enzyme, redox, and hypoxia,[1] have become appealing delivery platforms for the development of next-generation nanomedicines.[2] In particular, owing to the huge difference of reductive agent glutathione (GSH) concentration in the cytoplasm (≈2–10 × 10−3 m) versus extracellular fluids (≈2–10 × 10−6 m),[3] redox-responsive NPs have emerged as a fascinating tool for active intracellular delivery of various therapeutics, especially the bio-macro-molecules that need to be delivered and released into the cytoplasm for therapeutic effects.
In the past decade, a variety of redox-responsive polymers have been successfully developed for in vivo delivery of small molecular drugs to achieve a better therapeutic effect.[3d,4] One of the representative polymers is the disulfide bond–containing polymer, which can be rapidly degraded intracellularly by reductive agents such as GSH.[4c] This rapidly bioresponsive degradation behavior (from minutes to hours)[5] is distinct from the hydrolytically degradable polymers such as aliphatic polyesters and polycarbonates,[6] which usually show gradual degradation kinetics in body tissues with degradation period ranging from days to weeks even months.[4b,7] Despite the advantages of rapid degradation and fast intracellular cargo release, moderate effort has been made to construct redox-responsive NPs for systemic delivery of therapeutic nucleic acids such as small interfering RNA (siRNA), which represents a novel therapeutic modality for cancer treatment by silencing the expression of target gene(s), especially those that encode “undruggable” proteins.[8] Currently, redox-responsive polymers with the characteristic of GSH-triggered de-PEGylation[9] or de-crosslinking[4b,c,10] have been used to formulate NPs for systemic siRNA delivery, which can simultaneously achieve long blood circulation and fast intracellular siRNA release. Nevertheless, the applications of these polymers are subject to several limitations, such as complicated synthesis strategy and incomplete biodegradation issue, which may introduce difficulty in scale-up of therapeutic NP formulations.
We recently developed a facile and one-pot approach to synthesize a completely biodegradable and rapidly redox-responsive L-cysteine-based poly(disulfide amide) (PDSA) polymer.[11] The physiochemical properties of this multidisulfide bond–containing polymer (e.g., structure, molecular weight, and hydrophobicity) can be finely tuned by adjusting the structures and numbers of the repeat units in the polymeric chain. Inspired by this encouraging result, we speculate that the rapid redox response of the PDSA polymer may promote intracellular siRNA delivery and thus enhance the silencing of cancer-associated genes to achieve a better anticancer effect. To this end, we further prepare a library of PDSA polymers (Scheme 1A) in this work and systemically investigated the utility of these polymers in NPs for in vivo siRNA delivery. When mixing these polymers with cationic lipid, siRNA, and lipid–poly(ethylene glycol) (lipid–PEG), a new redox-responsive NP platform composed of a cationic lipid/siRNA complex–containing hydrophobic PDSA core and a lipid–PEG shell can be generated (Scheme 1B). With this new NP platform, we chose kinesin family member 11 (KIF11) and MYC as two proof-of-concept targets, and systematically evaluated our redox-responsive PDSA NPs for KIF11 (siKIF11) and MYC (siMYC) siRNA delivery and their anticancer efficacy. As an important member of the kinesin-like protein family, KIF11 controls mitosis, migration, and intracellular transport through interaction with microtubules, and is involved the development of malignant cancers and angiogenesis.[12] At present, KIF11 is regarded as one of the most promising new targets for antimitotic drugs and several small molecule inhibitors have entered Phase I and II clinical trials for solid tumors and hematological malignancies.[12c] MYC is a multifunctional, nuclear phosphoprotein that plays a role in cell cycle progression, apoptosis, and cellular transformation.[13] MYC overexpression is a common feature of many cancers including prostate adenocarcinoma, where increased MYC levels are detected at all stages of disease initiation and progression.[14] Moreover, it has recently been demonstrated that MYC overexpression is directly correlated with drug (e.g., cisplatin) resistance.[15] Our in vivo results show that systemic delivery of siKIF11 and siMYC with the redox-responsive PDSA NP platform can efficiently silence KIF11 and MYC expression in tumor cells and significantly inhibit prostate cancer (PCa) tumor growth.
Scheme 1.
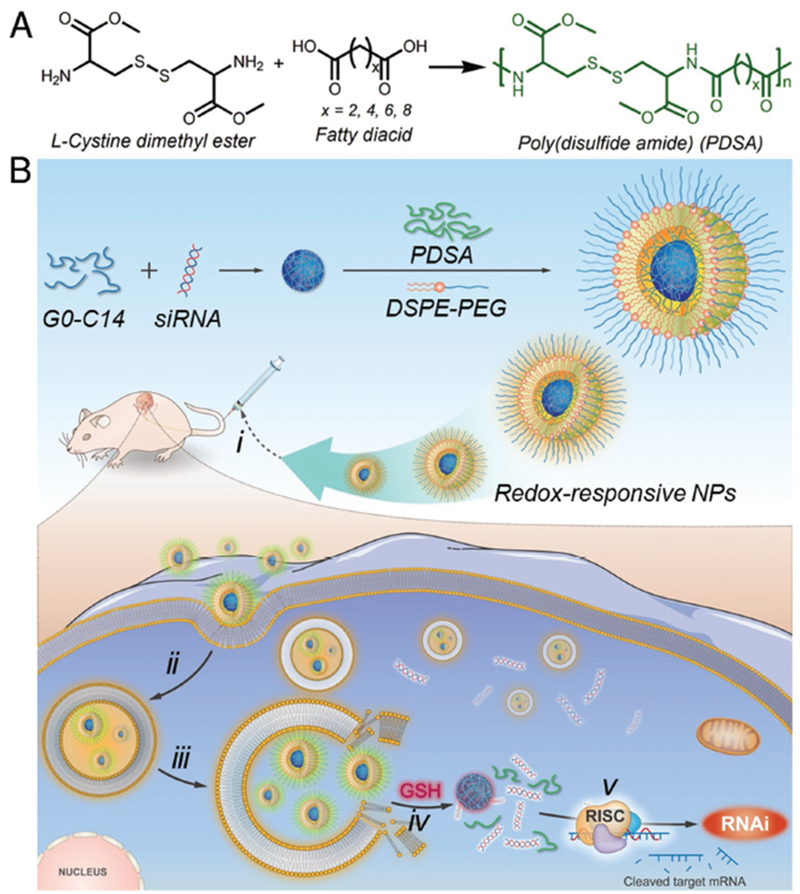
A) Synthesis scheme of the redox-responsive PDSA polymer. B) Schematic illustration of the redox-responsive PDSA NP platform for systemic siRNA delivery and cancer therapy. The PDSA polymer can coassemble with cationic lipid (G0-C14), siRNA, and DSPE–PEG to form stable NPs with G0-C14/siRNA complexes embedded in the hydrophobic PDSA core and DSPE–PEG covering on surface. i) After intravenous administration, the siRNA-loaded NPs can extravasate from leaky tumor vasculature and accumulate at the tumor site. After uptake by ii) tumor cells and iii) endosomal escape, the highly concentrated GSH in the cytoplasm can break the multiple disulfide bonds in the PDSA polymer and iv) induce the NP disassembly to rapidly release siRNA, thus v) resulting in efficient gene silencing to inhibit tumor growth.
2. Results and Discussion
2.1. Synthesis and Characterization of PDSA Polymers and RNA Interference (RNAi) NPs
Starting from the commercially available L-cystine dimethyl ester and fatty diacids with a different number of methylene groups (x = 2, 4, 6, and 8), hydrophobic PDSA polymers with different chemical structures (denoted PDSAx) were synthesized via one-step polycondensation reaction (Scheme 1A). In addition, we also chose sebacic acid (x = 8) and changed the feed composition as well as reaction time to obtain another three PDSA polymers with different molecular weights (denoted PDSA8a, PDSA8b, and PDSA8c). Their molecular weights and chemical structures were examined by gel permeation chromatography (GPC, Figure 1A) and nuclear magnetic resonance (NMR, Figure S1, Supporting Information), respectively, to confirm successful synthesis. Due to the hydrophobic nature of these PDSA polymers, they can form hydrophobic interactions with amphiphilic cationic lipid, which have been widely used for complexing with siRNA.[8c,16] Therefore, a new RNA interference (RNAi) NP platform can be formulated through the self-assembly nanoprecipitation method by mixing the siRNA aqueous solution with the dimethylformamide (DMF) mixture of PDSA polymer, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (poly ethylene glycol)-3000] (DSPE-PEG3k), and amphiphilic cationic lipid G0-C14 (Figure S2, Supporting Information) that we had previously developed.[17] In this procedure, the cationic lipid can first complex the siRNA in the DMF solution via electrostatic interaction with the hydrophobic C14 chains positioned on the surface of the cationic lipid/siRNA complexes, which are subsequently encapsulated by the hydrophobic PDSA polymer via hydrophobic interaction to form PDSA cores when adding the DMF mixture to aqueous solution. Simultaneously, the amphiphilic DSPE-PEG3k molecules will be coated on the surface of the PDSA cores to form stable siRNA-loaded NPs. We designed and synthesized a library of PDSA polymers with different chemical structures and molecular weights to adjust the physiochemical properties of the siRNA-loaded NPs. As shown in Figure 1A, as the PDSA polymeric chain is increasing, the siRNA encapsulation efficiency (EE%) increases but the size and zeta potential (ζ) of the resulting NPs decrease, possibly because the increased PDSA length leads to an increased hydrophobicity that can condense the cationic lipid/siRNA complexes to form much more compact hydrophobic core with improved siRNA loading ability and decreased zeta potential. Notably, due to the relatively low siRNA encapsulation efficiency (<20%) and large size (>200 nm) of the PDSA2 NPs, their redox response and gene silencing efficacy were not further evaluated in the subsequent experiments.
Figure 1.
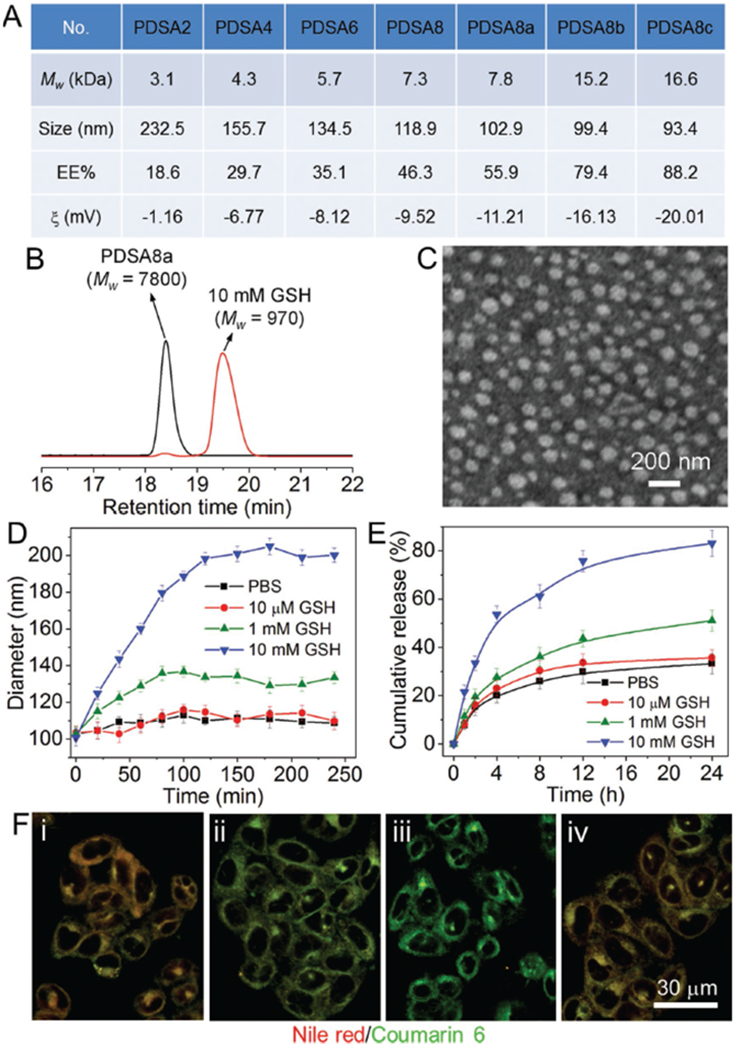
A) Molecular weight of the PDSA polymers and size, siRNA encapsulation efficiency (EE%), and zeta potential (ζ) of the siRNA-loaded NPs made with these PDSA polymers. B) GPC profile of the PDSA8a polymer incubated with 10 × 10−3 m GSH for 1 h. C) TEM image of the siLuc-loaded PDSA8a NPs. D) Size change of the siLuc-loaded PDSA8a NPs incubated with GSH at different concentrations. E) In vitro siRNA release profile of the DY547-siRNA-loaded PDSA8a NPs incubated with GSH at different concentrations. F) Fluorescent images of the Luc-HeLa cells incubated with the PDSA8a NPs loaded with coumarin 6 (green fluorescence) and Nile red (red fluorescence) for i) 1 h, ii) 2 h, and iii) 4 h, and iv) the cells first treated with 50 × 10−6 m NEM for 1 h followed by the PDSA8a NPs loaded with coumarin 6 and Nile red for 4 h.
We chose PDSA8a polymer to evaluate its redox response. Figure 1B shows the molecular weight change of this polymer upon the addition of GSH. After incubating with 10 × 10−3 m GSH for 1 h, the molecular weight (Mw) of polymer decreases from 7800 to 970 g mol−1, proving the redox-responsive characteristic of the PDSA8a polymer. With this GSH-triggered degradation, the morphology of the luciferase siRNA (siLuc)-loaded PDSA8a NPs changes from spherical shape (Figure 1C) into amorphous aggregates (Figure S3, Supporting Information) after 4 h incubation with 10 × 10−3 m GSH. However, the NPs maintain their spherical morphology when incubated with 10 × 10−6 m GSH for 24 h (Figure S3, Supporting Information). This result is consistent with the size change of the NPs upon the addition of GSH. As shown in Figure 1D, the NPs are stable, and no obvious size change can be observed when incubating the NPs with phosphate buffered saline (PBS) or GSH at a low concentration (e.g., 10 × 10−6 m). In contrast, due to the degradation and aggregation triggered by the highly concentrated GSH (e.g., 10 × 10−3 m), there is a significant increase in the NP size. This characteristic allows the NPs to show a redox-dependent release of DY547-labeled siLuc (DY547-siRNA). As shown in Figure 1E, more than 80% of the loaded siRNA has been released from the NPs incubated with 10 × 10−3 m GSH for 24 h, which is much higher than that of the NPs incubated with PBS (≈33%), 10 × 10−6 m GSH (≈35%), or 1 × 10−3 m GSH (≈51%).
We next investigated the intracellular redox response of the PDSA NPs. Forster resonance energy transfer (FRET) pairs of hydrophobic coumarin 6 (400Ex/510Em) and Nile red (520Ex/590Em) were encapsulated into the PDSA8a NPs and then incubated with the Luc-expressing HeLa (Luc-HeLa) cells. In this system, upon excitation of the donor dye (coumarin 6), its emitted energy can be transferred to the acceptor dye (Nile red) due to its close proximity (<10 nm), and thus red fluorescence corresponding to the Nile red can be observed.[11,18] After the NP disassembly triggered by the intracellular GSH, the FRET pair can be separated, and green fluorescence corresponding to coumarin 6 can be observable under an excitation of 400 nm. Figure 1F shows the fluorescent images of the Luc-HeLa cells incubated with the PDSA8a NPs loaded with the FRET pair for different times. After 1 h of incubation, obvious NP uptake and FRET effect can be observed (Figure 1Fi). Two hours later, the red fluorescence resulting from the FRET decreases, whereas the green fluorescence corresponding to coumarin 6 increases (Figure 1Fii). After 4 h incubation, cells show a dominant green fluorescence and nearly no red fluorescence can be detected (Figure 1Fiii). However, if cells are pretreated with N-ethylmaleimide (NEM, 50 × 10−6 m) that can react with thiols and thus reduce the effective concentration of intracellular GSH,[19] the FRET effect still maintains (Figure 1Fiv) despite the fact that some internalized NPs have entered the cytoplasm (Figure S4, Supporting Information). All these data indicate that the PDSA polymers can respond to the intracellular redox environment, leading to fast degradation of the polymeric chains and disassembly of their NPs.
2.2. In Vitro Gene Silencing
Having confirmed the redox response of the PDSA NPs, we then evaluated their gene silencing efficacy using Luc-HeLa cells. The siLuc was used to selectively suppress Luc expression. As shown in Figure 2A, all siRNA-loaded NPs can efficiently suppress Luc expression and ≥80% knockdown in Luc expression can be achieved at a 10 × 10−9 m siRNA dose. In particular, the NPs made with PDSA8a polymer can suppress the Luc expression by ≈95% without obvious cytotoxicity (Figure S5A, Supporting Information). The possible reason is that the PDSA8a NPs show moderate zeta potential (e.g., higher than PDSA8b and 8c NPs) and siRNA loading ability (e.g., stronger than PDSA4, 6, and 8 NPs). Therefore, PDSA8a NPs were selected for the following experiments. To verify the contribution of the redox response to efficient gene silencing of PDSA NPs, we used commercially available poly(lactic-co-glycolic acid) (PLGA) with a similar molecular weight (viscosity, 0.1–0.25 dL g−1) as that of the PDSA8a polymer to prepare non-responsive PLGA NPs, then examined their gene silencing efficacy. Without redox response, the PLGA NPs show much lower gene silencing efficacy compared to the PDSA8a NPs, and more than 40% Luc can still be expressed in the treated cells (Figure 2A), which is similar to the Luc expression in the cells first treated by 50 × 10−6 m NEM for 1 h followed by transfection with siLuc-loaded PDSA8a NPs. This result strongly demonstrates the importance of redox response to the efficient gene silencing rendered by the PDSA NPs.
Figure 2.

A) Luc expression in the Luc-HeLa cells treated with the siLuc-loaded PDSA NPs, PLGA NPs, and 50 × 10−6 m NEM for 1 h followed by the siLuc-loaded PDSA NPs at a 10 × 10−9 m siRNA dose. B) Western blot analysis of KIF11 expression in PC3 cells treated with the siKIF11-loaded PDSA8a NPs. C) Immunofluorescence staining analysis of KIF11 expression in PC3 cells treated with i) control NPs and ii) siKIF11-loaded PDSA8a NPs at a 5 × 10−9 m siRNA dose. D) Western blot analysis of MYC expression in PC3 cells treated with the siMYC-loaded PDSA8a NPs. E) Immunofluorescence staining analysis of MYC expression in PC3 cells treated with i) control NPs and ii) siMYC-loaded PDSA8a NPs at a 25 × 10−9 m siRNA dose. F,G) Proliferation profiles of PC3 cells treated with F) siKIF11- or G) siMYC-loaded PDSA8a NPs. siLuc-loaded PDSA8a NPs were used as the control in these experiments.
After screening the NP platform with optimal silencing efficacy (PDSA8a NPs) and low cytotoxicity (Figure S5B, Supporting Information), we then examined whether this RNAi NP platform can be used to silence KIF11 in PCa cells. KIF11 is a motor protein that belongs to the kinesin-like protein family and is involved the development of malignant cancers including PCa.[12] As an attractive anticancer target, inhibition of KIF11 by small molecules has demonstrated the ability to induce mitotic arrest and apoptosis of multiple cancers.[12c] The prostate cancer (PC3) cells were used a model cell line to examine the KIF11 silencing efficacy of the PDSA8a NPs. Figure 2B shows the western blot analysis of KIF11 expression in the PC3 cells. The siKIF11-loaded NPs can dramatically suppress the KIF11 expression and nearly no KIF11 can be detected at a very low siRNA dose (e.g., 5 × 10−9 m). The immunofluorescence staining analysis (Figure 2C) further demonstrates efficient KIF11 silencing by the PDSA8a NPs. After the treatment with siKIF11-loaded NPs at a 5 × 10−9 m siRNA dose, red fluorescence corresponding to the KIF11 is absent (Figure 2Cii), indicating almost 100% knockdown of the KIF11 expression. To further extend the application of the PDSA8a NPs for gene silencing and cancer treatment, we also evaluated their ability to downregulate the expression of MYC, a cancer-associated protein overexpressed in the earliest phase of PCa and metastatic PCa.[14] As shown in Figure 2D, the siMYC-loaded NPs can efficiently suppress MYC expression (≈60% knockdown) at a 15 × 10−9 m siRNA dose. At a 25 × 10−9 m siRNA dose, MYC expression is nearly extinguished (<2%; Figure S6, Supporting Information). Similar results can also be found in the immunofluorescence staining analysis (Figure 2Eii). With this suppressed KIF11 or MYC expression, the proliferation rate of PC3 cells is significantly inhibited (Figure 2F,G). Particularly, only 25% of the cells treated with the siKIF11-loaded NPs for 24 h at a 10 × 10−9 m siRNA dose are alive after 8 days (Figure 2F), while there is about tenfold increase in the number of cells treated with the siLuc-loaded PDSA8a NPs (Control). Notably, because the very low expression of KIF11 and MYC in the normal human prostate epithelial cells (HPrEc), the silencing of KIF11 or MYC does not affect the fate of HPrEc cells significantly (Figure S7, Supporting Information).
2.3. Pharmacokinetics and Biodistribution
After validating the efficient gene silencing of the PDSA8a NPs in vitro, we next assessed their pharmacokinetics (PK) and biodistribution (BioD). PK was examined by intravenous injection of DY677-siRNA-loaded NPs to normal adult Balb/c mice (1 nmol siRNA dose per mouse, n = 3). As shown in Figure 3A, the NPs show long blood circulation with a half-life (t1/2) of ≈4.92 h. In contrast, the naked siRNA is rapidly cleared from the blood and its blood t1/2 is less than 10 min. This prolonged blood circulation is mainly due to the protection of PEG outer layer.[20] The BioD was evaluated by intravenously injecting DY677-siRNA-loaded NPs into athymic nude mice bearing PC3 xenograft tumors. Figure 3B shows the fluorescent image of the mice at 24 h post injection. Given the long blood circulation characteristic of the PDSA8a NPs, they show a much higher tumor accumulation than the naked siRNA (Figure 3C). The tumors and major organs were harvested at 24 h post injection (Figure 3D,E), and the quantification of BioD is shown in Figure 3F. The siRNA-loaded NPs show about sixfold higher accumulation in tumors than the naked siRNA.
Figure 3.

A) Pharmacokinetics of naked DY677-siRNA and DY677-siRNA-loaded PDSA8a NPs. B,C) Overlaid fluorescent image of the PC3 xenograft tumor-bearing nude mice at 24 h post injection of B) DY677-siRNA-loaded PDSA8a NPs and C) naked DY677-siRNA. Tumors are indicated by ellipses. D,E) Overlaid fluorescent image of the tumors and main organs of the PC3 xenograft tumor-bearing nude mice sacrificed at 24 h post injection of the D) DY677-siRNA-loaded PDSA8a NPs and E) naked DY677-siRNA. F) Biodistribution of the NPs quantified from panels (D) and (E).
2.4. In Vivo Gene Silencing and Antitumor Efficacy
With the promising in vitro and PK/BioD results described above, we further evaluated whether our redox-responsive siRNA delivery platform can silence KIF11 and MYC expressions in vivo and show anticancer effects. To this end, the siRNA-loaded PDSA8a NPs were intravenously injected into the PC3 xenograft tumor-bearing athymic nude mice (1 nmol siRNA dose per mouse, n = 3) for three consecutive days. As shown in Figure 4A–C, the siKIF11- or siMYC-loaded NPs lead to ≈85% knockdown in KIF11 protein expression (Figure 4A) or ≈55% knockdown in MYC protein expression (Figure 4B) compared to the control NPs. A similar decrease of KIF11 and MYC expressions was also observed by immunohistochemical (IHC) analysis (Figure S8, Supporting Information). To confirm whether the NP-mediated KIF11 and MYC silencing has an anticancer effect, the siRNA-loaded NPs were intravenously injected into the PC3 xenograft tumor-bearing mice once every 2 days at a 1 nmol siRNA dose per mouse (n = 5). After four consecutive injections of the siKIF11-loaded NPs, the tumor growth was significantly inhibited compared to the mice treated with PBS, naked siKIF11, or control NPs loaded with siLuc (Figure 4D,E). Within a long evaluation period of 40 days, there is less than twofold increase (from ≈l78 to ≈155 mm3) in tumor size of the mice treated with the siKIF11-loaded NPs (Figure 4D). However, the tumor size in the three control groups is ≈6-fold larger than that of the mice treated with siKIF11-loaded NPs. The NP-mediated MYC silencing also shows an impressive anticancer effect. As shown in Figure 4F,G, after five intravenous injections of the siMYC-loaded NPs into PC3 xenograft tumor-bearing mice, the tumor growth rate is efficiently inhibited. A 3.5-fold increase in tumor size (from ≈70 to 278 mm3) was observed at day 22 in mice that received siMYC-loaded NPs, which is a much smaller increase than that seen in mice treated with PBS naked siMYC or siLuc-loaded NPs (≈8-fold increase in tumor size). It is important to note that the siKIF11-loaded NPs show neglectable in vivo side effects, and there is no obvious influence on mouse body weight (Figure S9, Supporting Information). To further evaluate the potential in vivo side effects, the PDSA8a NPs loaded with siKIF11 or siMYC were intravenously injected to normal adult mice (1 nmol siRNA dose per mouse, n = 3). Blood serum analysis showed that tumor necrosis alpha (TNF-α), interferon gamma (IFN-γ), interlukin-6 (IL-6), and interlukin-12 (IL-12) levels were in the normal range at 24 h post injection of the siKIF11- or siMYC-loaded NPs (Figure S10, Supporting Information). After three daily injections, no noticeable histological changes were noticed in the tissues from heart, liver, spleen, lung, or kidney (Figure S11, Supporting Information). All of these results indicate the good biocompatibility of this redox-responsive NP platform.
Figure 4.

A–C) Western blot analysis of A) KIF11 and B) MYC expression in the PC3 xenograft tumor tissue after systemic treatment by control NPs and siKIF11- or siMYC-loaded PDSA8a NPs. D) Tumor size of the PC3 xenograft tumor-bearing nude mice (n = 5) after systemic treatment by PBS, naked siKIF11, control NPs, and siKIF11-loaded PDSA8a NPs. The intravenous injections are indicated by the arrows. E) Photograph of the harvested PC3 xenograft tumors after 40 days of evaluation of the mice in panel (D). F) Tumor size of the PC3 xenograft tumor-bearing nude mice (n = 5) after systemic treatment by PBS, naked siMYC, control NPs, and siMYC-loaded PDSA8a NPs. The intravenous injections are indicated by the arrows. G) Photograph of the harvested PC3 xenograft tumors after 22 day evaluation of the mice in panel (F). siLuc-loaded PDSA8a NPs were used as control NPs.
3. Conclusion
In summary, we have designed and developed a new-generation redox-responsive NP platform for systemic siRNA delivery and effective cancer therapy. This new NP platform is comprised of a cationic lipid/siRNA complex-containing hydrophobic PDSA core and a lipid–PEG shell. The physiochemical properties of this nanoplatform can be finely tuned by adjusting the two biocompatible repeat units in the PDSA polymeric chain. In vitro results show that the PDSA NP platform can respond to the cytosolic GSH to achieve fast intracellular siRNA release, leading to efficient silencing of two target proteins (KIF11 and MYC) in PCa cells. In vivo results show that this NP platform with long blood circulation has a high tumor accumulation, can efficiently suppress the KIF11 and MYC expression in tumor tissue, and can significantly inhibit PCa tumor growth with negligible toxicities. Combined with the facile polymer synthesis technique and robust NP formulation enabling scale-up, we expect this long-circulating, biodegradable and fast redox-responsive RNAi NP platform to be of high interest for the treatment of a wide range of solid tumors.
4. Experimental Section
Preparation of the PDSA NPs:
The PDSA polymer was dissolved in DMF to form a homogeneous solution with a concentration of 20 mg mL−1. To prepare the siRNA-loaded NPs, a mixture of 1 nmol siRNA (0.1 nmol μL−1 aqueous solution) and 50 μL of G0-C14 (5 mg mL−1 in DMF) in a N/P molar ratio of 1:20 was prepared and added to the mixture of 200 μL of PDSA polymer solution and 140 μL of DSPE-PEG3k solution (20 mg mL−1 in DMF). Under vigorously stirring (1000 rpm), the mixture was added dropwise to 5 mL of deionized water. The formed NP dispersion was transferred to an ultrafiltration device (EMD Millipore, MWCO 100 K), and centrifuged to remove the organic solvent and free compounds. After washing with deionized water (3 × 5 mL), the siRNA-loaded NPs were dispersed in 1 mL of pH 7.4 PBS buffer. To prepare the NPs loaded with FRET pair of coumarin 6 and Nile red, a mixture of 2.5 μL of coumarin 6 (20 × 10−3 m in DMF) and 12 μL of Nile red (50 × 10−3 m in DMF) in a molar ratio of 1:12 was added to the mixture of 200 μL of PDSA polymer and 140 μL of DSPE-PEG3k solution (20 mg mL−1 in DMF). Under vigorously stirring (1000 rpm), the mixture was added dropwise to 5 mL of deionized water. The formed NPs were washed according to the method described above and finally dispersed in 1 mL of pH 7.4 PBS buffer.
Characterizations of the PDSA NPs:
Size and zeta potential of the NPs were determined by dynamic light scattering (DLS, Brookhaven Instruments Corporation). The morphology of NPs was visualized on a Tecnai G2 Spirit BioTWIN transmission electron microscope (TEM). Before observation, the sample was stained with 1% uranyl acetate and dried under air. To determine siRNA encapsulation efficiency (EE%), DY547-labeled siLuc (DY547-siRNA) loaded NPs were prepared according to the method aforementioned. A small volume (5 μL) of the NP solution was withdrawn and mixed with 20-fold dimethyl sulfoxide (DMSO). The fluorescence intensity of DY547-siRNA was measured using a Synergy HT multi-mode microplate reader. The amount of loaded siRNA in the NPs was calculated according to the standard curve.
Evaluation of the Reduction Response of the PDSA NPs:
The siLuc-loaded PDSA NPs were prepared according to the method described above and then dispersed in 1 mL of pH 7.4 PBS buffer containing GSH at different concentrations. At a predetermined interval, the size of the NPs was examined by DLS, and the morphology of the NPs was observed by TEM. To examine the influence of reduction response on the siRNA release, DY547-labeled siLuc-loaded NPs were prepared according to the method as described above. Subsequently, the NPs were dispersed in 1 mL of pH 7.4 PBS buffer and then transferred to a Float-a-lyzer G2 dialysis device (MWCO = 100 kDa, Spectrum) that was immersed in PBS buffer containing GSH at 37 °C. At a predetermined interval, 5 μL of the NP solution was withdrawn and mixed with 20-fold DMSO. The fluorescence intensity of DY547-labeled siRNA was determined using a microplate reader.
Cell Culture:
Luc-HeLa and PC3 cells were incubated in Roswell Park Memorial Institute (RPMI) 1640 medium with 10% fetal bovine serum (FBS) at 37 °C in a humidified atmosphere containing 5% CO2.
Confocal Laser Scanning Microscope (CLSM):
Luc-HeLa (50 000 cells) were seeded in disks and incubated in 2 mL of RPMI 1640 medium containing 10% FBS for 24 h. After replacing the medium with 2 mL of fresh medium, DY547-labeled siLuc-loaded NPs were added, and the cells were allowed to incubate for different times. After removing the medium and subsequently washing with PBS buffer thrice, lysotracker green and Hoechst 33342 were added to stain the endosomes and nuclei, respectively. The cells were finally viewed under a FV1000 CLSM (Olympus).
Evaluation of Intracellular Reduction Response of the PDSA NPs:
Luc-HeLa (50 000 cells) were seeded in disks and incubated in 2 mL of RPMI 1640 medium containing 10% FBS for 24 h. After replacing the medium with 2 mL of fresh medium, NPs loaded with FRET pair of coumarin 6 and Nile red were added, and the cells were allowed to incubate for different times. After removing the medium and subsequently washing with PBS buffer thrice, the cells were fixed with 4% paraformaldehyde and finally viewed under CLSM.
Luc Silencing:
Luc-HeLa cells were seeded in 96-well plates (5000 cells per well) and incubated in 0.1 mL of RPMI 1640 medium) with 10% FBS for 24 h. Thereafter, the medium was replaced by fresh medium, and siLuc-loaded NPs were added. After 24 h incubation, the cells were washed with PBS buffer and allowed to incubate in fresh medium for another 48 h. The Luc expression was determined using Steady-Glo luciferase assay kits. Cytotoxicity was measured using AlamarBlue assay according to the manufacturer’s protocol. The luminescence or fluorescence intensity was measured using a microplate reader, and the average value of five independent experiments was collected.
In Vitro KIF11 and MYC Silencing:
PC3 cells were seeded in 6-well plates (50 000 cells per well) and incubated in 2 mL of RPMI 1640 medium containing 10% FBS for 24 h. Subsequently, the medium was replaced by fresh medium, and then siKIF11- or siMYC-loaded NPs were added. After incubation for 24 h, the cells were washed with pH 7.4 PBS buffer and further incubated in fresh medium for another 48 h. For the cells treated with siKIF11-loaded NPs, they were digested by trypsin and the proteins were extracted using modified radioimmunoprecipitation assay lysis buffer (50 × 10−3 m Tris-HCl pH 7.4, 150 × 10−3 m NaCl, 1% NP-40 substitute, 0.25% sodium deoxycholate, 1 × 10−3 m sodium fluoride, 1 × 10−3 m Na3VO4, 1 × 10−3 m ethylenediaminetetraacetic acid (EDTA)), supplemented with protease inhibitor cocktail and 1 × 10−3 m phenylmethanesulfonyl fluoride (PMSF). For the cells treated with siMYC-loaded NPs, the cells were incubated with the medium containing 10 × 10−6 m lactacystin and 10 μg mL−1 calpain inhibitor I to prevent MYC degradation for 1 h and then digested by trypsin. Subsequently, the proteins were extracted according to the protocol described above. The expressions of KIF11 and MYC were examined using western blot described below.
Western Blot:
Equal amount of protein, as determined with a bicinchoninic acid (BCA) protein assay kit (Pierce/Thermo Scientific) according to the manufacturer’s instruction, was added to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels and separated by gel electrophoresis. After transferring the protein from gel to polyvinylidene difluoride membrane, the blot was blocked with 3% bovine serum albumin (BSA) in tris buffered saline with Tween 20 (TBST) (50 × 10−3 m Tris-HCl pH 7.4, 150 × 10−3 m NaCl, and 0.1% Tween 20), and then incubated with a mixture of primary rabbit antibody (polyclonal anti-KIF11 (Abcam, ab61199), monoclonal anti-MYC (Abcam, ab32072), and monoclonal anti-cMYC (Cell Signaling, D84C12), and polyclonal anti-β-actin (Cell Signaling, 13E5)). The expression of KIF11 and MYC was detected with horseradish peroxidase (HRP)-conjugated secondary antibody (anti-rabbit immunoglobulin G (IgG) HRP-linked antibody, Cell Signaling) and an enhanced chemiluminescence (ECL) detection system (Pierce).
Immunofluorescence Staining:
PC3 cells (50 000 cells) were seeded in disks and incubated in 2 mL of RPMI 1640 medium containing 10% FBS for 24 h. After replacing the medium with fresh medium, siKIF11- or siMYC-loaded NPs were added, and the cells were allowed to incubate with the siRNA-loaded NPs for 24 h. Subsequently, the cells were washed with pH 7.4 PBS buffer thrice and then incubated with fresh medium for another 48 h. Thereafter, the cells were fixed with 4% paraformaldehyde, and then permeabilized by incubation in 0.2% Triton X-100 in pH 7.4 PBS buffer for 5 min, followed by washing with pH 7.4 PBS buffer (3 × 5 min). Subsequently, the cells were blocked with blocking buffer (2% normal goat serum, 2% BSA, and 0.2% gelatin in pH 7.4 PBS buffer) at room temperature for 1 h. After washing the cells with pH 7.4 PBS buffer (3 × 5 min), KIF11 or MYC rabbit antibody diluted in 1% BSA solution was added and the cells were incubated for 1 h. Subsequently, the cells were with pH 7.4 PBS buffer (3 × 5 min), and then further incubated with Alex Fluor 647-linked secondary antibody (Life Technologies, A-11011) and Alex Fluor 488-conjugated phalloidin (Life Technologies, A12379) for another 1 h. After washing with pH 7.4 PBS buffer (3 × 5 min), the cells were viewed under a FV1000 CLSM.
In Vitro Cell Proliferation:
PC3 cells were seeded in 6-well plates (20 000 cells per well) and incubated in 2 mL of RPMI 1640 medium containing 10% FBS for 24 h. After replacing the medium with fresh medium, siKIF11- or siMYC-loaded NPs were added and the cells were allowed to incubate with the siRNA-loaded NPs for 24 h. Thereafter, the cells were washed with pH 7.4 PBS buffer thrice and fresh medium was added for further incubation. At predetermined intervals, the cytotoxicity was measured by AlamarBlue assay according to the manufacturer’s protocol. After each measurement, the AlamarBlue agent was removed and 2 mL of fresh medium was added for further incubation.
Animals:
Healthy male BALB/c mice and athymic nude mice (4–5 weeks old) were purchased from Charles River Laboratories. All in vivo studies were performed in accordance with National Institutes of Health animal care guidelines and in strict pathogen-free conditions in the animal facility of Brigham and Women’s Hospital and University of Maryland. Animal protocol was approved by the Institutional Animal Care and Use Committees on animal care (Harvard Medical School and the University of Maryland Baltimore County).
Pharmacokinetics Study:
Healthy male BALB/c mice were randomly divided into two groups (n = 3) and given an intravenous injection of either i) DY677-labeled siLuc-loaded NPs or ii) DY677-labeled naked siLuc at a 1 nmol siRNA dose per mouse. At a predetermined time interval, orbital vein blood (20 μL) was withdrawn using a tube containing heparin, and the wound was pressed for several seconds to stop the bleeding. The fluorescence intensity of DY677-labeled siRNA in the blood was determined by a microplate reader.
PC3 Xenograft Tumor Model:
The PC3 xenograft tumor model was constructed by subcutaneous injection with 200 μL of PC3 cell suspension (a mixture of RPMI 1640 medium and Matrigel in 1:1 volume ratio) with a density 2 × 106 cells mL−1 into the back region of healthy male athymic nude mice. When the tumor volume reached 50–70 mm3, the mice were used for the following in vivo experiments.
Biodistribution:
PC3 xenograft tumor-bearing mice were randomly divided into two groups (n = 3) and given an intravenous injection of either i) DY677-labeled naked siLuc or ii) DY677-labeled siLuc-loaded NPs at a 1 nmol siRNA dose per mouse. Twenty-four hours after the injection, the mice were imaged using the Maestro 2 In-Vivo Imaging System (Cri Inc). Main organs and tumors were then harvested and imaged. To quantify the accumulation of siRNA-loaded NPs in the tumors and main organs, the fluorescence intensity of each tissue was quantified by Image-J.
In Vivo KIF11 Silencing:
PC3 xenograft tumor-bearing mice were randomly divided into two groups (n = 3) and intravenously injected with i) siLuc-loaded NPs or ii) siKIF11-loaded NPs for three consecutive days. Twenty-four hours after the final injection, mice were sacrificed and tumors were harvested for western blot analysis and immunohistochemistry staining. For the western blot analysis, the proteins in the tumor were extracted using modified radioimmunoprecipitation assay lysis buffer (50 × 10−3 m Tris-HCl pH 7.4, 150 × 10−3 m NaCl, 1% NP-40 substitute, 0.25% sodium deoxycholate, 1 × 10−3 m sodium fluoride, 1 × 10−3 m Na3VO4, 1 × 10−3 m EDTA), supplemented with protease inhibitor cocktail and 1 × 10−3 m PMSF. Western blot was performed according to the method described above.
IHC Staining:
IHC staining was performed on formalin-fixed paraffin-embedded tumor sections. Briefly, tumor slides were first heated to 60 °C for 1 h, desparaffinized with xylene (3 × 5 min), and washed with different concentrations of alcohol. After retrieval of antigen using DAKO target retrieval solution at 95–99 °C for 40 min, followed by washing, the slides were blocked with peroxidase blocking buffer (DAKO Company) for 5 min. After washing buffer (DAKO Company), the slides were incubated with KIF11 rabbit antibody diluted in DAKO antibody solution for 1 h. The slides were then washed and incubated with peroxidase-labeled polymer for 30 min. After washing and staining with DAB+ substrate–chromogen solution and hematoxylin, the slides that remounted and viewed under a MVX10 MacroView Dissecting scope equipped with an Olympus DP80 camera.
In Vivo MYC Silencing:
PC3 xenograft tumor-bearing mice were randomly divided into two groups (n = 3) and intravenously injected with i) siLuc-loaded NPs or ii) siMYC-loaded NPs for three consecutive days. Twenty-four hours after the final injection, 40 μL of PBS containing 10 × 10−6 m lactacystin and 10 μg mL−1 calpain inhibitor I was administrated by intratumoral injection to prevent MYC degradation. One hour later, the mice were sacrificed and tumors were harvested for western blot analysis and immunohistochemistry staining according to the method described above.
Immune Response:
Healthy male BALB/c mice were randomly divided into five groups (n = 3) and given an intravenous injection of either i) PBS, ii) naked siKIF11, iii) naked siMYC, iv) siKIF11- or v) siMYC-loaded NPs at a 1 nmol siRNA dose per mouse. Twenty-four hours after injection, blood was collected and serum was isolated for measurements of representative cytokines (TNF-α, IL-6, IL-12, and IFN-γ by enzyme-linked immunosorbent assay (ELISA, PBL Biomedical Laboratories and BD Biosciences) according to the manufacturer’s instruction.
Histology:
Healthy male BALB/c mice were randomly divided into five groups (n = 3) and administered daily intravenous injections of either i) PBS, ii) naked siKIF11, iii) naked siMYC, iv) siKIF11- or v) siMYC-loaded NPs at a 1 nmol siRNA dose per mouse. After three consecutive injections, the main organs were collected 24 h post the final injection, fixed with 4% paraformaldehyde, and embedded in paraffin. Tissue sections were stained with hematoxylin–eosin (H&E) and then viewed under an optical microscope.
Inhibition of Tumor Growth:
PC3 xenograft tumor-bearing mice were randomly divided into four groups (n = 5) and intravenously injected with i) PBS, ii) naked si KI F11 or siMYC, iii) siLuc-loaded NPs, or iv) siKIF11 or siMYC-loaded NPs at a 1 nmol siRNA dose per mouse once every 2 days. The tumor growth was monitored every 2 days by measuring perpendicular diameters using a caliper, and tumor volume was calculated as follows
| (1) |
where W and L are the shortest and longest diameters, respectively.
Statistical Analysis:
Statistical significance was determined by a two-tailed Student’s t-test assuming equal variance. A p-value of <0.05 is considered statistically significant.
Supplementary Material
Acknowledgements
X.X., J.W., and S.L. contributed equally to this work. This work was supported in part by the Department of Defense Prostate Cancer Research Program Synergistic Idea Development Award (W81XWH-15-1-0728); the National Institutes of Health grants (HL127464, CA200900); the David H. Koch-Prostate Cancer Foundation (PCF) Program in Cancer Nanotherapeutics and the Movember-PCF Challenge Award; the Thousand Talents Program for Distinguished Young Scholars; and the Science and Technology Planning Project of Guangdong Province (2016A010103015) and Shenzhen City (JCYJ20170307141438157). O.C.F. has financial interest in Selecta Biosciences, Tarveda Therapeutics, and Placon Therapeutics.
Footnotes
The ORCID identification number(s) for the author(s) of this article can be found under https://doi.org/10.1002/smll.201802565.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
Contributor Information
Dr. Xiaoding Xu, Center for Nanomedicine and Department of Anesthesiology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, USA; Guangdong Provincial Key Laboratory of Malignant, Tumor Epigenetics and Gene Regulation, Medical Research Center, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou 510120, China
Dr. Jun Wu, Center for Nanomedicine and Department of Anesthesiology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, USA; School of Biomedical Engineering, Sun Yat-Sen University, Guangzhou 510006, China
Shuaishuai Liu, Department of Biological Sciences, University of Maryland, Baltimore County, Baltimore, MD 21250, USA, bieberic@umbc.edu.
Dr. Phei Er Saw, Center for Nanomedicine and Department of Anesthesiology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, USA; Guangdong Provincial Key Laboratory of Malignant, Tumor Epigenetics and Gene Regulation, Medical Research Center, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou 510120, China
Dr. Wei Tao, Center for Nanomedicine and Department of Anesthesiology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, USA
Yujing Li, Center for Nanomedicine and Department of Anesthesiology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, USA.
Lisa Krygsman, Center for Nanomedicine and Department of Anesthesiology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, USA.
Prof. Srinivasan Yegnasubramanian, Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA
Prof. Angelo M. De Marzo, Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA; Department of Pathology, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA
Prof. Jinjun Shi, Center for Nanomedicine and Department of Anesthesiology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, USA, jshi@bwh.harvard.edu
Prof. Charles J. Bieberich, Department of Biological Sciences, University of Maryland, Baltimore County, Baltimore, MD 21250, USA, bieberic@umbc.edu
Prof. Omid C. Farokhzad, Center for Nanomedicine and Department of Anesthesiology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, USA; King Abdulaziz University, Jeddah 21589, Saudi Arabia, ofarokhzad@bwh.harvard.edu
References
- [1].a) Wang S, Huang P, Chen X, Adv. Mater 2016, 28, 7340; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang S, Huang P, Chen X, ACS Nano 2016, 10, 2991; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lee Y, Lee S, Lee DY, Yu B, Miao W, Jon S, Angew. Chem., Int. Ed 2016, 55, 10676; [DOI] [PubMed] [Google Scholar]; d) Kwon EJ, Lo JH, Bhatia SN, Proc. Natl. Acad. Sci. USA 2015, 112, 14460; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Zhu L, Wang T, Perche F, Taigind A, Torchilin VP, Proc. Natl. Acad. Sci. USA 2013, 110, 17047; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Xu X, Saw PE, Tao W, Li Y, Ji X, Bhasin S, Liu Y, Ayyash D, Rasmussen J, Huo M, Shi J, Farokhzad OC, Adv. Mater 2017, 29, 1700141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Shi J, Kantoff PW, Wooster R, Farokhzad OC, Nat. Rev. Cancer 2017, 17, 20; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lu Y, Aimetti AA, Langer R, Gu Z, Nat. Rev. Mater 2017, 2, 16075. [Google Scholar]
- [3].a) Castellani P, Balza E, Rubartelli A, Antioxid. Redox Signaling 2014, 20, 1086; [DOI] [PubMed] [Google Scholar]; b) Schafer FQ, Buettner GR, Free Radicals Biol. Med 2001, 30, 1191; [DOI] [PubMed] [Google Scholar]; c) Wu G, Fang YZ, Yang S, Lupton JR, Turner ND, J. Nutr 2004, 134, 489; [DOI] [PubMed] [Google Scholar]; d) Sun H, Meng F, Cheng R, Deng C, Zhong Z, Antioxid. Redox Signaling 2014, 21, 755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Sun H, Meng F, Cheng R, Deng C, Zhong Z, Expert Opin. Drug Delivery 2013, 10, 1109; [DOI] [PubMed] [Google Scholar]; b) Meng F, Hennink WE, Zhong Z, Biomaterials 2009, 30, 2180; [DOI] [PubMed] [Google Scholar]; c) Cheng R, Feng F, Meng F, Deng C, Feijen J, Zhong Z, J. Controlled Release 2011, 152, 2. [DOI] [PubMed] [Google Scholar]
- [5].a) Gilbert HF, Methods Enzymol 1995, 251, 8; [DOI] [PubMed] [Google Scholar]; b) Raina S, Missiakas D, Annu. Rev. Microbiol 1997, 51, 179. [DOI] [PubMed] [Google Scholar]
- [6].a) Yang C, Panwar N, Wang Y, Zhang B, Liu M, Toh H, Yoon HS, Tjin SC, Chong PHJ, Law W-C, Chen C-K, Yong K-T, Nanoscale 2016, 8, 9405; [DOI] [PubMed] [Google Scholar]; b) Lin G, Chen C-K, Yin F, Yang C, Tian J, Chen T, Xu G, He C, Lin MC-M, Wang J, Lu F, Wang X, Yong K-T, J. Mater. Chem B 2017, 5, 3327. [DOI] [PubMed] [Google Scholar]
- [7].a) Ali SAM, Zhong SP, Doherty PJ, Williams DF, Biomaterials 1993, 14, 648; [DOI] [PubMed] [Google Scholar]; b) Pitt GG, Gratzl MM, Kimmel GL, Surles J, Sohindler A, Biomaterials 1981, 2, 215; [DOI] [PubMed] [Google Scholar]; c) Sun H, Mei L, Song C, Cui X, Wang P, Biomaterials 2006, 27, 1735. [DOI] [PubMed] [Google Scholar]
- [8].a) Zuckerman JE, Davis ME, Nat. Rev. Drug Discovery 2015, 14, 843; [DOI] [PubMed] [Google Scholar]; b) Kanasty R, Dorkin JR, Vegas A, Anderson D, Nat. Mater 2013, 12, 967; [DOI] [PubMed] [Google Scholar]; c) Tseng Y-C, Mozumdar S, Huang L, Adv. Drug Delivery Rev 2009, 61, 721; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Davis ME, Zuckerman JE, Choi CHJ, Seligson D, Tolcher A, Alabi CA, Yen Y, Heidel JD, Ribas A, Nature 2010, 464, 1067; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Xu X, Wu J, Liu Y, Yu M, Zhao L, Zhu X, Bhasin S, Li Q, Ha E, Shi J, Farokhzad OC, Angew. Chem., Int. Ed 2016, 55, 7091; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Xu X, Wu J, Liu Y, Saw PE, Tao W, Yu M, Zope H, Si M, Victorious A, Rasmussen J, Ayyash D, Farokhzad OC, Shi J, ACS Nano 2017, 11, 2618; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Yao YD, Sun TM, Huang SY, Dou S, Lin L, Chen JN, Ruan JB, Mao CQ, Y Yu F, Zeng MS, Zang JY, Liu Q, Su FX, Zhang P, Lieberman J, Wang J, Song E, Sci. Transl. Med 2012, 4, 130ra48. [DOI] [PubMed] [Google Scholar]
- [9].a) Kim HJ, Oba M, Pittella F, Nomoto T, Cabral H, Matsumoto Y, Miyata K, Nishiyama N, Kataoka K, J. Drug Targeting 2012, 20, 33; [DOI] [PubMed] [Google Scholar]; b) Wang K, Hu Q, Zhu W, Zhao M, Ping Y, Tang G, Adv. Funct. Mater 2015, 25, 3380; [Google Scholar]; c) An S, He D, Wagner D, Jiang C, Small 2015, 11, 5142. [DOI] [PubMed] [Google Scholar]
- [10].a) Li J, Yu X, Wang Y, Yuan Y, Xiao H, Cheng D, Shuai X, Adv. Mater 2014, 26, 8217; [DOI] [PubMed] [Google Scholar]; b) Zou Y, Zheng M, Yang W, Meng F, Miyata K, Kim HJ, Kataoka K, Zhong Z, Adv Mater 2017, 29, 1703285. [DOI] [PubMed] [Google Scholar]
- [11].Wu J, Zhao L, Xu X, Bertrand N, Choi WI, Yameen B, Shi J, Shah V, Mulvale M, MacLean JL, Farokhzad OC, Angew. Chem., Int. Ed 2015, 54, 9218. [DOI] [PubMed] [Google Scholar]
- [12].a) Liu X, Gong H, Huang K, Cancer Sci 2013, 104, 651; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Exertier P, Javerzat S, Wang B, Franco M, Herbert J, Platonova N, Winandy M, Pujol N, Nivelles O, Ormenese S, Godard V, Becker J, Bicknell R, Pineau R, Wilting J, Bikfalvi A, Hagedorn M, Oncotarget 2013, 4, 2302; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Rath O, Kozielski F, Nat. Rev. Cancer 2012, 12, 527; [DOI] [PubMed] [Google Scholar]; d) Venere M, Horbinski CM, Crish JF, Jin X, Vasanji A, Major J, Burrows A, Chang C, Prokop J, Wu Q, Sims PA, Canoll P, Summers MK, Rosenfeld SS, Rich JN, Sci. Transl. Med 2015, 7, 304ra143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Miller DM, Thomas SD, Islam A, Muench D, Sedoris K, Clin. Cancer Res 2012, 18, 5546; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nilsson JA, Cleveland JL, Oncogene 2003, 22, 9007; [DOI] [PubMed] [Google Scholar]; c) Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV, Cancer Discovery 2015, 5, 1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Koh CM, Bieberich CJ, Dang CV, Nelson WG, Yegnasubramanian S, De Marzo AM, Genes Cancer 2010, 1, 617; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hubbard GK, Mutton LN, Khalili M, McMullin RP, Hicks JL, Bianchi-Frias D, Horn LA, Kulac I, Moubarek MS, Nelson PS, Yegnasubramanian S, De Marzo AM, Bieberich CJ, Cancer Res 2016, 76, 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Casinelli G, LaRosa J, Sharma M, Cherok E, Banerjee S, Branca M, Edmunds L, Wang Y, Sims-Lucas S, Churley L, Kelly S, Sun M, Stolz D, Graves JA, Cell Death Discovery 2016, 2, 16082; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pyndiah S, Tanida S, Ahmed KM, Cassimere EK, Choe C, Sakamuro D, Sci. Signaling 2011, 4, ra19. [DOI] [PubMed] [Google Scholar]
- [16].Zhu X, Xu Y, Solis LM, Tao W, Wang L, Behrens C, Xu X, Zhao L, Liu D, Wu J, Zhang N, Wistuba II, Farokhzad OC, Zetter BR, Shi J, Proc. Natl. Acad. Sci. USA 2015, 112, 7779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Xu X, Xie K, Zhang X-Q, Pridgen EM, Park GY, Cui DS, Shi J, Wu J, Kantoff PW, Lippard SJ, Langer R, Walker GC, Farokhzad OC, Proc. Natl. Acad. Sci. USA 2013, 110, 18638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yang Z, Cao J, He Y, Yang JH, Kim T, Peng X, Kim JS, Chem. Soc. Rev 2014, 43, 4563. [DOI] [PubMed] [Google Scholar]
- [19].a) Gregory JD, J. Am. Chem. Soc 1955, 77, 3922; [Google Scholar]; b) Bhuniya S, Maiti S, Kim EJ, Lee H, Sessler JL, Hong KS, Kim JS, Angew. Chem., Int. Ed 2014, 53, 4469; [DOI] [PubMed] [Google Scholar]; c) Xu XD, Cheng YJ, Wu J, Cheng H, Cheng SX, Zhuo RX, Zhang XZ, Biomaterials 2016, 76, 238. [DOI] [PubMed] [Google Scholar]
- [20].a) Knop K, Hoogenboom R, Fischer D, Schubert US, Angew. Chem., Int. Ed 2010, 49, 6288; [DOI] [PubMed] [Google Scholar]; b) Xu X, Saw PE, Tao W, Li Y, Ji X, Yu M, Mahmoudi M, Rasmussen J, Ayyash D, Zhou Y, Farokhzad OC, Shi J, Nano Lett 2017, 17, 4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.