

Abstract
The structure of heparin and heparan sulfate (Hep/HS) oligosaccharides, as determined by the length and the pattern of sulfation, acetylation, and uronic acid epimerization, dictates their biological function through modulating interactions with protein targets. But fine structural determination is a very challenging task due to the lability of the sulfate modifications and difficulties in separating isomeric HS chains. Previously, we reported a strategy for chemical derivatization involving permethylation, desulfation, and trideuteroperacetylation, combined with standard reverse phase LC-MS/MS that enables the structural sequencing for heparin/HS oligosaccharides of sizes up to dodecasaccharide by positionally replacing all sulfates with more stable trideuteroacetyl groups, allowing for robust MS/MS sequencing. However, isomeric oligosaccharides that contain both N-sulfation and N-acetylation become isotopomers after labeling, differing only in the sites of deuteration. This prevents chromatographic separation of these different mixed domain sequences post-derivatization, and makes sequencing by MS/MS difficult due to co-fragmentation of the isotopomers leading to chimeric product ion spectra. In order to improve chromatographic separation of mixed domain oligosaccharides, we have introduced a propionylation step in place of trideuteroacetylation for labeling of sites of sulfation. HS standard disaccharides have been used to evaluate the efficiency of this improved chemical derivatization. The results show that we can quantitatively replace sulfation with propionyl groups with the same high efficiency as the previously reported trideuteroacetylation. After derivatization, we demonstrate the ability to chromatographically separate two mixed domain tetrasaccharide isomers differing solely by the order of N-sulfation and N-acetylation, allowing for full sequencing of each by MS/MS. These results represent a marked improvement in the ability of our previously reported derivatization strategy to analyze complex mixtures of Hep/HS oligosaccharides without a decrease in sensitivity.
Keywords: Heparin/Heparan sulfate, Glycosaminoglycans, Chemical derivatization, LC-MS/MS
Graphical abstract
1. Introduction
Glycosaminoglycans (GAGs) are linear polysaccharides consisting of repeating disaccharide units, most commonly found in vivo conjugated with a core protein to form a proteoglycan. GAGs play important biological roles in many aspects of development and physiology of multicellular organisms[1–5], and are found on the surface of virtually all mammalian cells. With the exception of hyaluronic acid, GAGs are heavily modified by deacetylation, N- and O-sulfation, and epimerization at the C5 position of uronic acid. Modification patterns are introduced in the Golgi, and are not based on a template. Consequently, GAG structures display substantial heterogeneity within tissues, and an enormous molecular diversity across different tissues and over developmental time[6]. Heparan sulfate (HS) is the most heterogeneous class in GAGs with variability in O-sulfation positions, amine group modifications, and uronic acid epimerization[7, 8], all of which mediate a wide range of key biochemical and developmental processes[9–11], have important biological activities in angiogenesis, blood coagulation, cell adhesion and tumor metastasis[12–14], as well as mediating host-pathogen interactions[15, 16].
Structure elucidation of heparin/HS oligosaccharide is important for structure–function studies. Mass spectrometry is a useful tool for the structural analysis of heparin/HS oligosaccharides. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) using electrospray ionization (ESI) offers high sensitivity, accuracy, and throughput[17]. However, due to the instability of sulfate groups and the high structural heterogeneity leading to considerable difficulties in separating isomeric structures, only a few function specific heparin/HS motifs have been elucidated, such as the heparin pentasaccharide responsible for interactions with antithrombin III and coagulation inhibition[18], and such elucidations often require the use of alternative techniques other than LC-MS/MS sequencing. Disaccharide compositional profiling of HS/heparin disaccharides generated by depolymerization of the polysaccharides is useful and allows for quantitative analysis[19–22]. While this method provides compositional information, it does not reveal the sequence information on oligosaccharide domains of the polysaccharide chains. As an alternative strategy, a chemical derivatization scheme was developed that employs the most widely used LC-MS/MS platform for separation and sequencing of heparin/HS oligosaccharides. This scheme involves the position-specific replacement of labile, hydrophilic groups of the heparin/HS oligosaccharide with stable hydrophobic groups, a modification and repurposing of a chemical derivatization scheme that was initially reported by Dell et al. for GAG analysis by Fast Atom Bombarment (FAB) ionization [23].
In our previous report, we have presented a related approach involving sequential permethylation, desulfation, and pertrideuteroacetylation to modify native HS oligosaccharides for online separation and structural analysis of mixtures of HS oligosaccharides. Using this technology, we observed sufficient sequential glycosidic bond cleavage fragments to enable accurate sequencing using LC-CID-MS/MS[24, 25], and this technology was sufficient for us to make the first identification of a Robo1-specific Hep/HS oligosaccharide sequence[26, 27]. While this method has demonstrated success, a significant weakness was identified: in a sequencing experiment of HS tetrasaccharides from natural sources, we were unable to distinguish between d-HexA2S-GlcNS-GlcA-GlcNAc and d-HexA2S-GlcNAc-GlcA-GlcNS. Similarly, d-HexA-GlcNAc6S-GlcA-GlcNS and d-HexA-GlcNS6S-GlcA-GlcNAc also co-eluted and co-fragmented, preventing us from being able to reliably relatively quantitate the two isomers[28]. In both of these cases, this difficulty arose because after derivatization, the two derivatized tetrasaccharides became isotopomers, differing only in the location of three deuterium atoms. Using trideuteroacetylation to mark sites of sulfation resulted in the inability to chromatographically separate oligosaccharides with mixed NS and NAc groups, that differed only by the site of N-sulfation/acetylation. In order to remedy this, we have replaced the trideuteroacetylation with a propionylation step. We observed propionylation to work efficiently with HS disaccharides, showing that we can quantitatively replace sulfation with propionyl groups. We have also derivatized two synthetic oligosaccharide isomeric standards that differ only by the site of NAc/NS. After derivatization, we can chromatographically separate these analytes and cleanly sequence each analyte separately using solely glycosidic bond fragments.
2. Results and discussion
2.1 Chemical derivatization scheme to differentiate mixed GlcN-modification domains
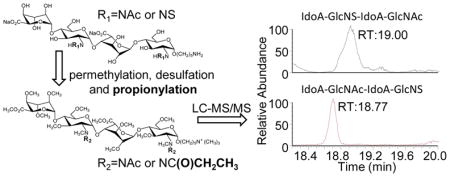
As shown in Figure S-1 (Supporting Information), the Hep/HS disaccharide has the basic structure ΔUA-GlcN, where the amine group can either be an acetylated amine, a sulfated amine, or rarely a free amine. In our previous work[29], the sequencing protocol replaces sulfates with trideuteroacetyl groups. HS disaccharides with GlcNS and GlcNAc residue can be easy differentiated from each other after derivatization by MS analysis, due to the mass difference of N-acetyl group and trideuteroacetyl group is 3Da. But, in oligosaccharides that contain a mixture of GlcNS and GlcNAc, isomeric analytes that differ solely by the position of GlcNS and GlcNAc become isotopomers after derivatization, differing solely by the position of deuterium atoms. For example, as shown in Figure 1, two tetrasaccharides (dp4s) with one N-acetyl group and one N-sulfo group have a different sequence, i.e. dp4-a: IdoA-GlcNS-IdoA-GlcNAc and dp 4-b: IdoA-GlcNAc-IdoA-GlcNS. After permethylation, desulfation and tri-deuteroacetylation, the N-sulfo group near to the nonreducing end of dp4-a was replaced by trideuteroacetyl group (Figure 1-A). The N-sulfation site near to the reducing end of dp4-b has also been labeled during derivatization with a trideuteroacetyl group (Figure 1-B). They both have an exact mass of 1005.5184, and the isotopomers will not separate by reverse phase LC. In order to remedy this, we have replaced the trideuteroacetylation with a propionylation step. The use of a propionyl group will now cause mixed domain isomers to differ by the position of one or more methyl groups (due to the N-acetyl to N-propionyl change), which should be much easier to separate by reverse phase LC.
Figure 1.

Tetrasaccharides with one N-acetyl group and one N-sulfo group before and after chemical derivatization using the previously reported trideuteroactylation method. Dp4-a and Dp4-b have a different sequence, but after derivatization differ only by the location of three deuterium atoms. These isotopomers are impractical to chromatographically separate, resulting in ambiguous chimeric MS/MS spectra.
2.2 Chemical derivatizations of HS disaccharide standards
The improved derivatization protocol used for the modification of the heparin/HS disaccharides is outlined in Figure 2. To evaluate the reaction efficiency of propionylation, we selected HS disaccharides (dp2) I-S: ΔUA2S-GlcNS6S and I-A: ΔUA2S-GlcNAc6S to use as examples. Previous experience has shown that the difficulty in achieving complete derivatization increases as a function of charge density; therefore, we chose the most highly sulfated commercially available GlcNS and GlcNAc disaccharides to test our protocol. After derivatization, the two disaccharides were analyzed by LC-MS. LC-MS analysis of the I-S derivatization product gave one major peak and two minor peaks (Figure 3A). The major peak was observed at m/z 576.253, corresponding to the protonated molecule [M+H]+ from ΔUA2S-GlcNS6S after derivatization (theoretical m/z 576.258). The extracted ion chromatogram (EIC) of 576.253 shows two peaks with a retention time (RT) of 25.07 min and 26.16 min, respectively (Figure 3B), representing the α and β anomers at the reducing end. The EIC of the two minor mass peaks (m/z 544.225 and 512.200) shown in Figure 3C and D clearly indicate that they have the same RT as m/z 576.253, indicating that these two minor masses are source induced fragmentation products; namely, loss of one or two methanols.
Figure 2.

Improved chemical derivatization scheme for differentiating mixed domain isomers. Permethylation protects the unsulfated groups (magenta), as well as allows the assignment of sulfation sites within the GlcN based on the effects of sulfation on the extent of permethylation. Sulfates (blue) are then gently removed by solvolysis, and the sites of sulfation are labeled with propionyl group (red).
Figure 3.
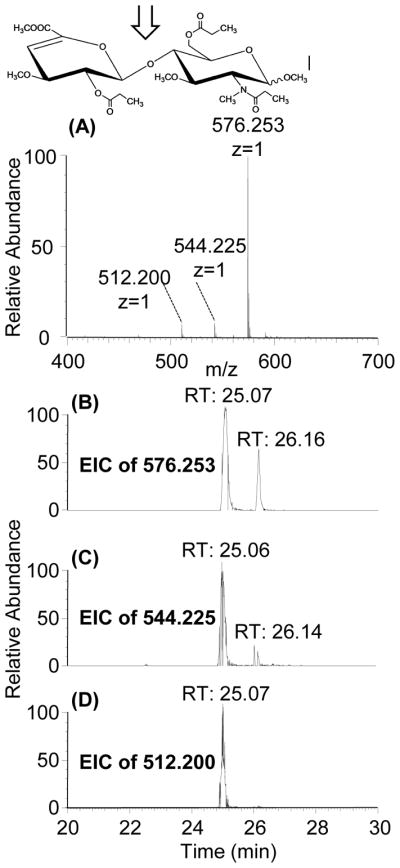
(A) MS spectrum and (B) EIC profiles for derivatized I-S: ΔUA2S-GlcNS6S (m/z 576.253). The two peaks in the EIC profile represent reducing end anomerization. The EIC profiles of two detected minor mass peaks, m/z 544.225 and 512.200 (C and D, respectively) show the same retention time as m/z 576.253 (B), indicating they are both source fragmentation products.
Mass spectra of the I-A derivatization product (Figure 4A) revealed a major peak at m/z 562.250, which corresponds to the expected mass of I-A after derivatization (theoretical m/z is 562.242). The EIC of 562.250 also showed two peaks (Figure 4B), consistent with anomerization at the reducing end. Two minor mass peaks (m/z 520.238 and 530.222) are also detected (Figure 4A) and appear to be source-induced loss of methanol (Figure 4C) and ethenone from the N-acetyl group (Figure 4D). We did not detect side reaction products from derivatization of disaccharides using the new propionylation protocol, consistent with previous reports using trideuteroacetylation [28, 29].
Figure 4.

(A) MS spectrum and (B) EIC profile for derivatized I-A: ΔUA2S-GlcNAc6S. Derivatized I-A (m/z 562.250) yielded two EIC peaks, due to anomerization at the reducing end. The EIC profiles of two detected minor mass peaks, m/z 530.222 and 520.238 (C and D, respectively) show the same RT as m/z 562.25 (B), indicating they are both source fragmentation products.
2.3 Tetrasaccharide standards propionylation reverse phase LC-MS
To evaluate the application of our method to longer HS oligosaccharides with mixed NS and NAc groups, we performed chemical derivatizations and LC-MS/MS analysis of two synthetic dp4 isomeric standards that differ only by the site of NAc/NS. As shown in Figure 5A and 5B, a set of two isomeric synthetic dp4s with an alkyl linker (named T1: IdoA-GlcNS-IdoA-GlcNAc-O(CH2)5-NH2 and T2: IdoA-GlcNAc-IdoA-GlcNS-O(CH2)5-NH2) were used as a synthetic mixture. The primary amine-bearing linker eliminated anomerization, and after permethylation introduced a quaternary amine with a fixed positive charge. After chemical derivatization of the mixture, LC-MS was performed for the initial analysis. The MS spectrum of the derivatized mixture is shown in Figure 5C. Underpermethylation can be observed, as can permethylation-induced β-elimination products as previously reported [26–29]; however, no under-propionylation products can be detected in the LC-MS spectrum as shown in Figure 5C. Discussion of the mechanism of underpermethylation and steps to improve permethylation efficiency and product stability are beyond the scope of this manuscript, but will be reported in future manuscripts on the topic.
Figure 5.

Structures of the synthetic tetrasaccharide T1: IdoA-GlcNS-IdoA-GlcNAc-O(CH2)5-NH2 (A) and T2: IdoA-GlcNAc-IdoA-GlcNS-O(CH2)5-NH2 (B), before and after chemical derivatization. The primary amine on the reducing end linker is converted to a quaternary amine during the permethylation, providing a permanent fixed charge on the reducing end. An averaged MS spectrum taken from the elution window of the tetrasaccharides and their under-derivatized versions showed an abundant peak for the fully derivatized tetrasaccharide. β-elimination products were detected (not shown), as well as underpermethylation and in-source fragmentation products. However, no evidence of underpropionylation was observed.
LC-MS showed that replacement of pertrideuteroacetylation with perproprionylation resulted in derivatization efficiency comparable to that previously reported [26–29]. LC-MS/MS analysis of the derivatized mixture of the two tetrasaccharides results in the identification of two peaks of m/z 1100.563, separated by 23 seconds. While the two isomeric tetrasaccharides retain the same mass after derivatization, we can differentiate between the two based on their MS/MS spectra, using any product ion that cleaves between the GlcNS and the GlcNAc residues, as shown in Figure 6. Focusing on the Y2 product ion of the precursor ion [M]+ (m/z 1100.563), the product ion of m/z 609.360 is diagnostic of IdoA-GlcNS-IdoA-GlcNAc-O(CH2)5-NH2, while the product ion of m/z 623.376 is diagnostic of IdoA-GlcNAc-IdoA-GlcNS-O(CH2)5-NH2. EICs of m/z 609.360 and m/z 623.376 from the CID MS/MS spectra of m/z 1100.563 are presented in Figure 6D. Based on these product ion traces, we can clearly identify that IdoA-GlcNS-IdoA-GlcNAc-O(CH2)5-NH2 eluted at 19.00 min, while IdoA-GlcNAc-IdoA-GlcNS-O(CH2)5-NH2 eluted at 18.77 min. The two isomers were clearly separated from each other, and could be readily identified solely by glycosidic bond cleavages after derivatization.
Figure 6.

LC-MS/MS spectra of the derivatized mixture of the two tetrasaccharide isomers. (A) EIC of derivatized dp4 (m/z 1100.563) from the LC-MS experiment. CID MS/MS spectra of the precursor ion [M]+ (m/z 1100.563) resulted in two qualitatively different product ion spectra: (B) one dominated by the 609.360 product ion, and (C) one dominated by the 623.376 product ion. (D) Theoretical fragmentation of the two derivatized isomers indicate that the Y2 product ion would differentiate between the two isomers, and EICs of the two Y2 ions show clear chromatographic separation of the isomeric mixture, with the product ion of m/z 609.360 diagnostic for T1: IdoA-GlcNS-IdoA-GlcNAc-O(CH2)5-NH2, and the product ion of m/z 623.376 is diagnostic for T2: IdoA-GlcNAc-IdoA-GlcNS-O(CH2)5-NH2.
3. Conclusion
The replacement of trideuteroacetylation with a propionylation step was achieved for both model disaccharides and synthetic tetrasaccharides with no decrease in derivatization efficiency, lower cost for derivatization reagents (due to the elimination of hexadeuteroacetic anhydride), and no increase in derivatization time. The resulting derivatized products have increased retention on a C18 reverse phase column, and the fragmentation patterns are highly similar to those previously reported [28, 29]. Even more notably, the replacement of trideuteroacetylation with propionylation eliminates the problem previously observed with mixed domain GlcNS/GlcNAc isomers; namely, the conversion of positional isomers to isotopomers after derivatization, preventing chromatographic separation and making confident identification challenging. We observed propionylation to efficiently differentiate two isomeric synthetic oligosaccharide isomeric standards that differ only by the site of GlcNAc/GlcNS (IdoA-GlcNAc-IdoA-GlcNS-O(CH2)5-NH2 and IdoA-GlcNS-IdoA-GlcNAc-O(CH2)5-NH2). We were able to clearly resolve these two isomers. Previous work has shown that even partial resolution of isomers is sufficient to differentiate them based on product ion alignment [28]; the level of separation we were able to achieve from these isomers is more than sufficient. One unusual case remains a theoretical problem for the propionylation method. Currently, we can differentiate GlcNS3S from GlcNAc6S based on the ability of the 3-O sulfation to deactivate the hydrogen of the N-sulfated amine, resulting in a single underpermethylation[29]. This causes the GlcNS3S to have the same mass as GlcNAc6S after derivatization using the propionylation method presented here. While this theoretical problem exists, it can be easily remedied by replacing propionic anhydride with decadeuteropropionic anhydride, shifting the mass of the GlcNS3S by 5 Da relative to GlcNAc6S. Combined with future improvements in permethylation efficiency, propionylation will allow us to extend this methodology to more complex mixed domain Hep/HS oligosaccharides for more thorough and accurate analysis of these complex mixtures of modified oligosaccharides.
4. Experimental
4.1 Materials and reagents
Unsaturated heparin disaccharides I-S: ΔUA,2S-GlcNS,6S (exact mass: 576.9713) and I-A: ΔUA,2S-GlcNAc,6S (exact mass: 539.0251) were purchased from Iduron (Cheshire, UK). Two tetrasaccharides with alkyl linker (T1: IdoA-GlcNS-IdoA-GlcNAc-O(CH2)5-NH2 and T2: IdoA-GlcNAc-IdoA-GlcNS-O(CH2)5-NH2) were synthesized and purified as previously described[27]. A more description of the analytical characterization of these two disaccharides by NMR and high resolution mass spectrometry is given in the Supporting Information, Figures S-2 through S-5. Sep-Pak C18 Plus Short Cartridge (360 mg Sorbent per Cartridge, 55–105 μm Particle Size, 50/pk) were purchased from Waters (Milford, MA, USA). Unless otherwise noted, all chemical reagents used in the chemical derivatization scheme were purchased from Sigma-Aldrich Inc (St. Louis, MO).
4.2 Chemical Derisvatization of HS Oligosaccharides
A series of chemical derivatizations were performed to replace the labile and strongly acidic sulfate groups with much more stable and hydrophobic propionyl groups, which enable the HS oligosaccharides to be retained well and separated by C18 reverse phase liquid chromatography and fragmented by MS/MS without losing the information about the sulfation modifications. Detailed procedures have been described in our previous work for structural analysis of synthetic HS oligosaccharides[29]. First, Hep/HS oligosaccharides (50 μg each) were dissolved in 1.0 mL water, loaded onto a Dowex 50WX8 (hydrogen form resin) column in 10% triethylamine where they were converted to triethylammonium (TEA) salts in order to increase their solubility in dimethyl sulfoxide (DMSO)[30]. In the TEA salt preparation section, Milli-Q ultra pure water was used to flush the hydrogen form resin, in order to ensure there are no metal ions. After adding 10% TEA, the pH of the solution passed through the hydrogen form resin was higher than pH 11. It is important that sodium salt GAGs completely exchange to the TEA salt before adding the DMSO/base solution. Also, permethylation is critically influenced by water, so anhydrous chemical reagents are preferred for permethylation unless otherwise noted. 100 μL sodium hydroxide solution (50% in water) was suspended in 4 mL dimethyl sulfoxide (DMSO, anhydrous, ≥99.9%), thoroughly vortexed, then centrifuged at 4500 rpm 5 minutes. Excess DMSO was removed, and additional DMSO was added, with this process repeated five times. The final base was resuspended in 1 mL DMSO. TEA salts of Hep/HS oligosaccharides (25–50 μg) were resuspended in 200 μL DMSO/base solution, followed by addition of 100 μL of iodomethane. After 5 min of vortexing and 10 min of sonicating, the reaction was stopped by adding 2 mL of water and sparged with nitrogen to remove iodomethane, followed by desalting using a C18 Sep-Pak cartridge. The dried permethylated products were then converted to their pyridinium salts by ion exchange over Dowex 50WX8, then resuspended in 20 μL of DMSO containing 10% methanol and incubated for 4 h at 95 °C to quantitatively remove all sulfate groups [29]. After removing the sulfate groups, the dried samples were resuspended in 175 μL of pyridine and 25 μL of propionic anhydride and incubated at 55 °C overnight. The solvents were then removed by using a Speed-Vac concentrator, and the samples were resuspended in 20% acetonitrile/water at a concentration of 0.2 μg/μL for later LC-MS/MS analysis.
4.3 LC-MS/MS Analysis
All samples were analyzed on a Thermo Orbitrap Fusion Tribrid (Thermo Fisher Scientific) coupled with an Ultimate 3000 Nano LC system (Dionex). Reverse-phase separation was accomplished using a 0.3 mm i.d.×15 mm C18 PepMap 100 trap column with 5 μm particle size (Thermo Fisher Scientific) coupled with a 75μm ×15cm Acclaim PepMap 100 C18 analytical column with 2 μm particle size (Thermo Fisher Scientific). Mobile-phase solvent A consisted of 0.1% formic acid in water, and mobile-phase B consisted of 0.1% formic acid in acetonitrile. The flow rate was set to 300 nL/min. A 25 min gradient (2% to 95% buffer B) was used to elute unsaturated disaccharides, and 45 min gradient (20% to 50% buffer B) was used for the synthetic tetrasacchrides. Nanoelectrospray voltage was set to 2.6 kV, positive ion. Full MS scan range was set to 150–1500 m/z at a resolution of 60,000, RF lens was 6%, and the automatic gain control (AGC) target was set to 2.0 × 105. For the MS/MS scans, the resolution was set to 50,000, the precursor isolation width was 3 m/z units, and ions were fragmented by collision-induced dissociation (CID) at a normalized collision energy of 50%.
Supplementary Material
Highlights.
Site-specific replacement of sulfates in Hep/HS with propionyl groups with equal efficiency to previously reported trideuteroacetylation
After derivatization, achieved chromatographic separation of isomeric Hep/HS oligosaccharides differing solely by the position of GlcNAc and GlcNS monosaccharides
Achieved efficient sequencing of a defined mixture of isomeric Hep/HS synthetic tetrasaccharides by LC-MS/MS
Acknowledgments
This work was supported by the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) through the Research Resource for Integrated Glycotechnology (P41GM103390).
References
- 1.Bishop JR, Schuksz M, Esko JD. Nature. 2007;446:1030. doi: 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
- 2.Coulson-Thomas VJ. Int J Exp Pathol. 2016;97:213–229. doi: 10.1111/iep.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Poulain FE, Yost HJ. Development. 2015;142:3456–3467. doi: 10.1242/dev.098178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saied-Santiago K, Bülow HE. Dev Dyn. 2018;247:54–74. doi: 10.1002/dvdy.24555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith PD, Coulson-Thomas VJ, Foscarin S, Kwok JCF, Fawcett JW. Exp Neurol. 2015;274:100–114. doi: 10.1016/j.expneurol.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 6.Townley RA, Bülow HE. Curr Opin Struct Biol. 2018;50:144–154. doi: 10.1016/j.sbi.2018.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gandhi Neha S, Mancera Ricardo L. Chem Biol Drug Des. 2008;72:455–482. doi: 10.1111/j.1747-0285.2008.00741.x. [DOI] [PubMed] [Google Scholar]
- 8.Rabenstein DL. Nat Prod Rep. 2002;19:312–331. doi: 10.1039/b100916h. [DOI] [PubMed] [Google Scholar]
- 9.Couchman JR. Nature Reviews Molecular Cell Biology. 2003;4:926. doi: 10.1038/nrm1257. [DOI] [PubMed] [Google Scholar]
- 10.Fannon M, Forsten KE, Nugent MA. Biochemistry. 2000;39:1434–1445. doi: 10.1021/bi991895z. [DOI] [PubMed] [Google Scholar]
- 11.Wu ZL, Zhang L, Yabe T, Kuberan B, Beeler DL, Love A, et al. J Biol Chem. 2003;278:17121–17129. doi: 10.1074/jbc.M212590200. [DOI] [PubMed] [Google Scholar]
- 12.Liu D, Shriver Z, Venkataraman G, El Shabrawi Y, Sasisekharan R. Proceedings of the National Academy of Sciences. 2002;99:568–573. doi: 10.1073/pnas.012578299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu J, Thorp Suzanne C. Med Res Rev. 2001;22:1–25. doi: 10.1002/med.1026. [DOI] [PubMed] [Google Scholar]
- 14.Perrimon N, Bernfield M. Nature. 2000;404:725. doi: 10.1038/35008000. [DOI] [PubMed] [Google Scholar]
- 15.GÖTte M. The FASEB Journal. 2003;17:575–591. doi: 10.1096/fj.02-0739rev. [DOI] [PubMed] [Google Scholar]
- 16.Perrimon N, Bernfield M. Semin Cell Dev Biol. 2001;12:65–67. doi: 10.1006/scdb.2000.0237. [DOI] [PubMed] [Google Scholar]
- 17.Guerrini M, Guglieri S, Casu B, Torri G, Mourier P, Boudier C, et al. J Biol Chem. 2008;283:26662–26675. doi: 10.1074/jbc.M801102200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Toyoda H, Kinoshita-Toyoda A, Selleck SB. J Biol Chem. 2000;275:2269–2275. doi: 10.1074/jbc.275.4.2269. [DOI] [PubMed] [Google Scholar]
- 19.Desaire H, Leary JA. J Am Soc Mass Spectrom. 2000;11:916–920. doi: 10.1016/S1044-0305(00)00168-9. [DOI] [PubMed] [Google Scholar]
- 20.Wei W, Niñonuevo MR, Sharma A, Danan-Leon LM, Leary JA. Anal Chem. 2011;83:3703–3708. doi: 10.1021/ac2001077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang B, Chang Y, Weyers AM, Sterner E, Linhardt RJ. J Chromatogr. 2012;1225:91–98. doi: 10.1016/j.chroma.2011.12.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gill VL, Aich U, Rao S, Pohl C, Zaia J. Anal Chem. 2013;85:1138–1145. doi: 10.1021/ac3030448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zaia J. Mol Cell Proteomics. 2013;12:885–892. doi: 10.1074/mcp.R112.026294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dell A, Rogers ME, Thomas-Oates JE, Huckerby TN, Sanderson PN, Nieduszynski IA. Carbohydr Res. 1988;179:7–19. [Google Scholar]
- 25.Zaia J. Mass Spectrom Rev. 2004;23:161–227. doi: 10.1002/mas.10073. [DOI] [PubMed] [Google Scholar]
- 26.Li Z, Moniz H, Wang S, Ramiah A, Zhang F, Moremen KW, et al. J Biol Chem. 2015;290:10729–10740. doi: 10.1074/jbc.M115.648410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zong C, Huang R, Condac E, Chiu Y, Xiao W, Li X, et al. J Am Chem Soc. 2016;138:13059–13067. doi: 10.1021/jacs.6b08161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang R, Zong C, Venot A, Chiu Y, Zhou D, Boons G-J, et al. Anal Chem. 2016;88:5299–5307. doi: 10.1021/acs.analchem.6b00519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang R, Liu J, Sharp JS. Anal Chem. 2013;85:5787–5795. doi: 10.1021/ac400439a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heiss C, Wang Z, Azadi P. Rapid Commun Mass Spectrom. 2011;25:774–778. doi: 10.1002/rcm.4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.