

Abstract
In vitro whole-organism screens of T. brucei with representative examples of brain-penetrant microtubule (MT)-stabilizing agents identified lethal triazolopyrimidines and phenylpyrimidines with sub-μM potency. In mammalian cells, these anti-proliferative compounds disrupt MT-integrity and reduce total tubulin levels. Their parasiticidal potency, combined with their generally favorable pharmacokinetic properties, which include oral bioavailability and brain-penetration, suggest that these compounds are potential leads against African trypanosomiasis.
Keywords: Triazolopyrimidine, Microtubules, Human African Trypanosomiasis, Trypanosoma brucei
Graphical Abstract
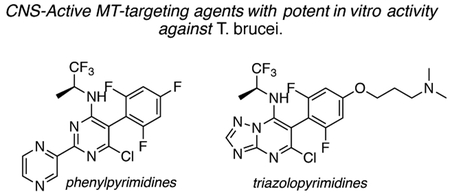
In vitro whole-organism screens of T. brucei with examples of different classes of small molecules known to modulate mammalian MT dynamics and structure identified a number of triazolopyrimidines and phenylpyrimidines with sub-μM parasiticidal activity. Compounds of this type may be considered as potential leads against African trypanosomiasis.
Human African Trypanosomiasis (HAT; Sleeping Sickness) is an insect-borne parasitic disease caused by Trypanosoma brucei that threatens populations in sub-Saharan Africa. The disease-causing pathogen replicates at the tsetse fly bite site before disseminating from the skin through the hemolymphatic system where it replicates in the spleen, liver, lymph nodes, skin, eyes, endocrine system and heart (Stage 1 infection). The parasites that cross the blood-brain barrier (BBB) proliferate within the central nervous system (CNS) causing CNS disease (Stage 2). The East African (Rhodesian; rHAT) form of the disease follows a more acute clinical progression than the West African (Gambian; gHAT) infection. However, both forms generate symptoms that include personality changes, psychosis, sensory disorders, tremor, ataxia, daytime somnolence and convulsions.[1] Death is inevitable if the infection is left untreated or is inadequately treated. [1–2]
Current treatment options for HAT rely mainly on drugs that are often associated with severe or life-threatening adverse events.[1b] For Stage 1 gHAT, the aromatic diamidine, pentamidine (1, Figure 1), is employed but is less effective against rHAT, in which case, suramin (2, Figure 1) is used. For Stage 2 gHAT, the regimen of choice is a combination of the injectable nitrofuran derivative, nifurtimox (3, Figure 1), and the oral ornithine decarboxylase inhibitor, eflornithine (4, Figure 1). For Stage 2 rHAT, therapy relies on the trivalent arsenical, melarsoprol (5, Figure 1) that can cause a potentially fatal reactive encephalopathy. [1b, 3] Cross-resistance to 1 and 5 has been documented [4] and treatment failures with 2 have also been recorded. [5] Although two new promising drug candidates, the oxaborole, SCYX-7158 (6, Figure 1), and the nitroimidazole, fexinidazole (7, Figure 1), are in different stages of clinical development,[6] the search for new/alternative treatment options remains a priority.
Figure 1.

The structures of current anti-trypanosomal drugs and drug candidates.
Microtubule (MT)-stabilizing drugs form a cornerstone of cancer chemotherapy;[7] however, several studies suggest that compounds of this type have the potential to treat other human diseases, including certain diseases of the CNS.[8] Tubulin/MTs have also been proposed as a valuable drug target to treat parasitic infections.[9] For example, low nM concentrations of the MT-stabilizing drug, paclitaxel (8, Table 1), inhibit the replication of T. brucei [10] and a related parasite, Trypanosoma cruzi.[11] Although 8 and most other MT-stabilizing natural products cannot be considered for CNS indications due to limited BBB permeability,[12] there has been considerable progress in the identification of brain-penetrant examples. Indeed, selected compounds from different classes of natural products (e.g., epothilones[12a] and dictyostatin[13]), as well as non-naturally occurring compounds, such as triazolopyrimidines and related heterocycles (Figure 2),[14] have demonstrated generally good CNS-uptake. The availability of these compounds provides an opportunity to explore their potential to treat Stage 2 HAT and, possibly, other neuroparasitic infections.
Table 1.
Representative examples from different scaffolds of MT-active compounds and their IC50 values vs. T. b. brucei Lister 427 in vitro.
| Compound | Structure |
T. brucei IC50 (nM)[a] |
1h B/P[b] |
|---|---|---|---|
| 1 | 48.17 ± 25 | ND | |
| 8 |  |
3.2 ± 3.0 | 0.07# |
| 9 | 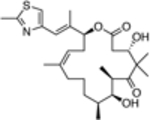 |
163.9 ± 30 | 7.6# |
| 10 |  |
>4,000 | 2.2 |
| 11 |  |
>4,000 | ND |
| 12 | 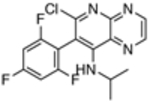 |
>4,000 | ND |
| 13 | 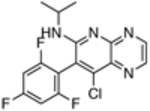 |
>4,000 | ND |
| 14 | 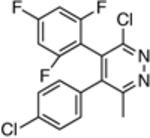 |
3,327 ± 1821 | ND |
| 15 | 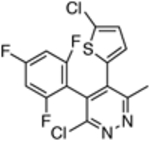 |
2,501 ± 1839 | ND |
| 16 | 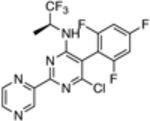 |
109.5 ± 46.9 | 0.58 |
| 17 | 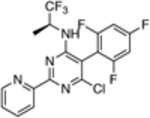 |
66.1 ± 18 | 0.34 |
| 18 |  |
10.9 ± 6.8 | 0.35 |
| 19 |  |
2.5 ± 2.4 | ND |
| 20 |  |
>4,000 | 0.8 |
| 21 |  |
81.3 ± 15 | 0.037 |
| 22 |  |
263 ± 43 | 0.49 |
| 23 |  |
145.8 ± 14.9 | 0.27 |
Assays were conducted 3 times each in duplicate; the mean and SD for each IC50 value across 6 data points are shown;
brain-to-plasma (B/P) compound concentration ratio 1 h after an intra-peritoneal (ip) injection of 5 mg/kg of test compound; brain-to-plasma compound concentration ratios are from published studies; [12a, 14]
B/P 2 h after an ip injection of 5 mg/kg of test compound.
ND = not determined.
Figure 2.

Representative structures of non-naturally occurring MT-active small molecules.
Interestingly, such compounds are known to act on mammalian tubulin/MTs through different modes of action. For example, the natural product, epothilone D (9, Table 1), binds to the paclitaxel binding site on β-tubulin and promotes MT-stabilization and bundling in a manner similar to paclitaxel.[15] By contrast, the non-naturally occurring triazolopyrimidines and related heterocycles are believed to interact within the binding site of the vinca alkaloids.[16] Moreover, depending on the nature of the heterocyclic core and the substitution pattern, these molecules can either preserve or disrupt MT integrity.[17] The possible significance of these different modes of action in the context of parasitic tubulin/MTs and, ultimately, parasite growth inhibition activity has yet to be evaluated. Thus, the primary goal of the present study was to assess and compare the anti-parasitic activity of several representative compounds. To this end, we evaluated selected MT-active compounds (10–23, Table 1) that had been synthesized and characterized previously as part of an Alzheimer’s disease drug discovery program.[14, 17]
The in vitro assay employed to evaluate our selection of test compounds was a previously described SYBR Green-based whole-cell assay developed for T. b. brucei Lister 427, [18] but modified to a 96-well format. As summarized in Table 1, consistent with prior reports, 8 exhibited a single digit nM IC50 value, whereas the brain-penetrant MT-stabilizing natural product, 9, was weaker but still significantly anti-trypanosomal. For other examples of the triazolopyrimidine series and related classes of MT-targeting compounds, a relatively wide range of anti-parasitic activities was measured. Compounds from the imidazole (10 and 11), pyridopyrazine (12 and 13), as well as the pyridazine series (14 and 15), which are all reported to possess both MT-stabilizing and anti-fungal properties,[19] possessed either no or limited activity in the T. brucei assay. Conversely, different examples of the phenylpyrimidine[14a, 20] and triazolopyrimidine[14a, 21] series were significantly parasiticidal. In particular, the phenylpyrimidines, 16 and 17, generated IC50 values that were comparable with the current HAT drug, pentamidine (1; Table 1 and Figure 3), whereas the related congeners, 18 and 19, were significantly more potent with IC50 values in the low nM range. Among the triazolopyrimidines, although 20 did not produce 50% growth inhibition at the highest concentration tested (4 μM), other closely related congeners bearing an alkoxy side chain in the fluorinated ring (i.e., 21–23) generated nM IC50 values. Taken together, these results indicate that among the non-naturally occurring compounds that interact with mammalian tubulin at the vinblastine binding site, only the phenylpyrimidines and selected triazolopyrimidines exhibit promising anti-parasitic activity in vitro.
Figure 3.

Inhibition of T. b. brucei Lister 427 growth by representative MT-stabilizers. T. brucei assays employed the SYBR Green DNA-binding agent.[18] Assays were conducted 3 times each in duplicate; the means and SD values across 6 data points are shown.
Mode of action studies in mammalian cells reveal that, in general, all MT-active phenylpyrimidines, as well as the triazolopyrimidines that contain the side chain at the para position of the fluorinated phenyl group (e.g., 21–23), disrupt MT-integrity. This disruption is often associated with a proteasome-dependent reduction in total tubulin levels.[17] Conversely, MT-active triazolopyrimidines which lack the alkoxy side chain (e.g., 20) promote MT-stabilization without impacting total tubulin levels or MT-integrity in mammalian cells.[17] These observations led to the hypothesis that, depending on the particular substitution pattern, different triazolopyrimidine congeners may exhibit different binding modes within the vinca site, ultimately producing different effects on MT structure and function.[22] Studies are needed to determine whether triazolopyrimidines and phenylpyrimidines act on parasite tubulin/MT in a manner similar to that described for mammalian cells. However, the observation that potent (i.e., sub-μM) anti-parasitic activity is observed only for triazolopyrimidines containing the alkoxy side chain (cf., 20, 21–23) and for all tested phenylpyrimidines (16–19) suggests that the more potent trypanocides are those congeners that interfere with MT-integrity and reduce total tubulin levels in mammalian cells. Such compounds, like most MT-targeting agents, are known to exert anti-mitotic activity in rapidly dividing cells, including mammalian cells.[20–21] Yet, it is possible that these molecules or their analogues may prove therapeutically useful to treat Stage 2 HAT provided that they can reach brain concentrations sufficient to kill CNS-resident parasites upon administration of tolerable doses. Studies are underway in our laboratories to evaluate whether acute treatments based on these CNS-active MT-targeting agents are effective against both peripheral and/or brain T. brucei infections in a mouse model of HAT.
In summary, in vitro whole-organism screens of T. brucei with examples of different classes of small molecules known to modulate mammalian MT dynamics and structure identified a number of triazolopyrimidines and phenylpyrimidines with sub-μM parasiticidal activity. Given their potent in vitro anti-parasitic activity and their favorable PK properties,[14a, 21] including oral bioavailability and brain penetration, these compounds hold promise as potential leads for treatment of stage 2 HAT.
Experimental Section
The test compounds evaluated in the T. b. brucei Lister 427 assay were synthesized as described previously.[14, 17] Compounds in 100% DMSO stocks were diluted in 100 μL of modified HMI-9 medium in 96-well plates (Corning 3903) such that the final DMSO concentration was 0.5%. Bloodstream form trypanosomes in log-phase growth were then diluted to 2×105 trypanosomes/mL in modified HMI-9 medium, and dispensed into the previously prepared 96-well plates at 100 μL/well. After incubation, trypanosomes were lysed by addition of 50 μL of lysis buffer containing 1x SYBR Green.
Acknowledgements
Financial support for this work was provided by NIH Grants AG044332 (NIA) and R21AI133394 (NIAID).
References:
- [1].a) Barrett MP, Burchmore RJ, Stich A, Lazzari JO, Frasch AC, Cazzulo JJ, Krishna S, Lancet 2003, 362, 1469–1480; [DOI] [PubMed] [Google Scholar]; b) Sanjeev KS, A., Clinical manifestations, diagnosis, and treatment of African trypanosomiasis. http://www.uptodate.com/contents/clinical-manifestations-diagnosis-and-treatment-of-african-trypanosomiasis-H1, 2016.
- [2].Kennedy PG, J. Clin. Invest 2004, 113, 496–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Braakman HM, van de Molengraft FJ, Hubert WW, Boerman DH, Neurology 2006, 66, 1094–1096; [DOI] [PubMed] [Google Scholar]; b) Balasegaram M, Harris S, Checchi F, Ghorashian S, Hamel C, Karunakara U, Bull. World Health Organ. 2006, 84, 783–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Legros D, Evans S, Maiso F, Enyaru JC, Mbulamberi D, Trans. R. Soc. Trop. Med. Hyg 1999, 93, 439–442; [DOI] [PubMed] [Google Scholar]; b) de Koning HP, Trends Parasitol. 2008, 24, 345–349. [DOI] [PubMed] [Google Scholar]
- [5].Pepin J, Milord F, Adv Parasitol. 1994, 33, 1–47. [DOI] [PubMed] [Google Scholar]
- [6].a) Matthews KR, Mol. Biochem. Parasitol 2015, 200, 30–40; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mesu V, Kalonji WM, Bardonneau C, Mordt OV, Blesson S, Simon F, Delhomme S, Bernhard S, Kuziena W, Lubaki JF, Vuvu SL, Ngima PN, Mbembo HM, Ilunga M, Bonama AK, Heradi JA, Solomo JLL, Mandula G, Badibabi LK, Dama FR, Lukula PK, Tete DN, Lumbala C, Scherrer B, Strub-Wourgaft N, Tarral A, Lancet 2018, 391, 144–154. [DOI] [PubMed] [Google Scholar]
- [7].Jordan MA, Wilson L, Nat. Rev. Cancer 2004, 4, 253–265. [DOI] [PubMed] [Google Scholar]
- [8].Brunden KR, Trojanowski JQ, Smith AB III, Lee VM, Ballatore C, Bioorg. Med. Chem 2013, 22, 5040–5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Werbovetz KA, Mini-Rev. Med. Chem 2002, 2, 519–529; [DOI] [PubMed] [Google Scholar]; b) Ochola DO, Prichard RK, Lubega GW, J. Parasitol 2002, 88, 600–604; [DOI] [PubMed] [Google Scholar]; c) Dostál V, Libusová L, Protoplasma 2014, 251, 991–1005; [DOI] [PubMed] [Google Scholar]; d) Fennell B, Naughton J, Barlow J, Brennan G, Fairweather I, Hoey E, McFerran N, Trudgett A, Bell A, Expert Opin. Drug Discov. 2008, 3, 501–518. [DOI] [PubMed] [Google Scholar]
- [10].Lama R, Sandhu R, Zhong B, Li B, Su B, Bioorg. Med. Chem. Lett 2012, 22, 5508–5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Baum SG, Wittner M, Nadler JP, Horwitz SB, Dennis JE, Schiff PB, Tanowitz HB, Proc. Natl. Acad. Sci. U S A 1981, 78, 4571–4575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Brunden KR, Yao Y, Potuzak JS, Ferrer NI, Ballatore C, James MJ, Hogan AM, Trojanowski JQ, Smith AB III, Lee VM, Pharmacol. Res 2011, 63, 341–351; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fellner S, Bauer B, Miller DS, Schaffrik M, Fankhanel M, Spruss T, Bernhardt G, Graeff C, Farber L, Gschaidmeier H, Buschauer A, Fricker G, J. Clin. Invest 2002, 110, 1309–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Brunden KR, Gardner NM, James MJ, Yao Y, Trojanowski JQ, Lee VMY, Paterson I, Ballatore C, Smith AB III, ACS Med. Chem. Lett 2013, 4, 886–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Lou K, Yao Y, Hoye AT, James MJ, Cornec AS, Hyde E, Gay B, Lee VM, Trojanowski JQ, Smith AB III, Brunden KR, Ballatore C J. Med. Chem 2014, 57, 6116–6127; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cornec AS, Monti L, Kovalevich J, Makani V, James MJ, Vijayendran KG, Oukoloff K, Yao Y, Lee VM, Trojanowski JQ, Smith AB Iii, Brunden KR, Ballatore C, J. Med. Chem 2017, 60, 5120–5145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bollag DM, McQueney PA, Zhu J, Hensens O, Koupal L, Liesch J, Goetz M, Lazarides E, Woods CM, Cancer Res. 1995, 55, 2325–2333. [PubMed] [Google Scholar]
- [16].Sáez-Calvo G, Sharma A, Balaguer F. d. A., Barasoain I, Rodríguez-Salarichs J, Olieric N, Muñoz-Hernández H, Berbís MÁ, Wendeborn S, Peñalva MA, Matesanz R, Canales Á, Prota AE, Jímenez-Barbero J, Andreu JM, Lamberth C, Steinmetz MO, Díaz JF, Cell Chemical Biology 2017, 24, 737–750 e736. [DOI] [PubMed] [Google Scholar]
- [17].Kovalevich J, Cornec AS, Yao Y, James M, Crowe A, Lee VM, Trojanowski JQ, Smith AB III, Ballatore C, Brunden KR, J. Pharmacol. Exp. Ther 2016, 357, 432–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Faria J, Moraes CB, Song R, Pascoalino BS, Lee N, Siqueira-Neto JL, Cruz DJ, Parkinson T, Ioset JR, Cordeiro-da-Silva A, Freitas-Junior LH, J. Biomol. Screen 2015, 20, 70–81. [DOI] [PubMed] [Google Scholar]
- [19].a) Crowley PJ, Lamberth C, Müller U, Wendeborn S, Nebel K, Williams J, Sageot O-A, Carter N, Mathie T, Kempf H-J, Godwin J, Schneiter P, Dobler MR, Pest. Manag. Sci 2010, 66, 178–185; [DOI] [PubMed] [Google Scholar]; b) Lamberth C, Trah S, Wendeborn S, Dumeunier R, Courbot M, Godwin J, Schneiter P, Bioorg. Med. Chem 2012, 20, 2803–2810; [DOI] [PubMed] [Google Scholar]; c) Lamberth C, Dumeunier R, Trah S, Wendeborn S, Godwin J, Schneiter P, Corran A, Bioorg. Med. Chem 2013, 21, 127–134. [DOI] [PubMed] [Google Scholar]
- [20].Zhang N, Ayral-Kaloustian S, Nguyen T, Hernandez R, Lucas J, Discafani C, Beyer C, Bioorg. Med. Chem 2009, 17, 111–118. [DOI] [PubMed] [Google Scholar]
- [21].Zhang N, Ayral-Kaloustian S, Nguyen T, Afragola J, Hernandez R, Lucas J, Gibbons J, Beyer C, J. Med. Chem 2007, 50, 319–327. [DOI] [PubMed] [Google Scholar]
- [22].Oukoloff K, Kovalevich J, Cornec AS, Yao Y, Owyang ZA, James M, Trojanowski JQ, Lee VM, Smith AB III, Brunden KR, Ballatore C, Bioorg. Med. Chem. Lett 2018, 28, 2180–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]