

Abstract
Salvianolic acid B (SalB), a natural polyphenolic compound extracted from the herb of Salvia miltiorrhiza, possesses antioxidant and neuroprotective properties and has been shown to be beneficial for diseases that affect vasculature and cognitive function. Here we investigated the protective effects of SalB against subarachnoid hemorrhage (SAH)-induced oxidative damage, and the involvement of underlying molecular mechanisms. In a rat model of SAH, SalB inhibited SAH-induced oxidative damage. The reduction in oxidative damage was associated with suppressed reactive oxygen species generation; decreased lipid peroxidation; and increased glutathione peroxidase, glutathione, and superoxide dismutase activities. Concomitant with the suppressed oxidative stress, SalB significantly reduced neurologic impairment, brain edema, and neural cell apoptosis after SAH. Moreover, SalB dramatically induced nuclear factor-erythroid 2-related factor 2 (Nrf2) nuclear translocation and increased expression of heme oxygenase-1 and NADPH: quinine oxidoreductase-1. In a mouse model of SAH, Nrf2 knockout significantly reversed the antioxidant effects of SalB against SAH. Additionally, SalB activated sirtuin 1 (SIRT1) expression, whereas SIRT1-specific inhibitor sirtinol pretreatment significantly suppressed SalB-induced SIRT1 activation and Nrf2 expression. Sirtinol pretreatment also reversed the antioxidant and neuroprotective effects of SalB. In primary cultured cortical neurons, SalB suppressed oxidative damage, alleviated neuronal degeneration, and improved cell viability. These beneficial effects were associated with activation of the SIRT1 and Nrf2 signaling pathway and were reversed by sirtinol treatment. Taken together, these in vivo and in vitro findings suggest that SalB provides protection against SAH-triggered oxidative damage by upregulating the Nrf2 antioxidant signaling pathway, which may be modulated by SIRT1 activation.
Keywords: Salvianolic acid B, Subarachnoid hemorrhage, Oxidative damage, Nrf2, Sirtuin 1
1. Introduction
Worldwide, subarachnoid hemorrhage (SAH) causes neurologic injury with high morbidity and mortality [1]. However, effective therapeutic strategies to improve SAH outcomes remain limited. Recent advances have indicated that early brain injury (EBI) after SAH is the main determinant of outcomes [2–4]. Although the exact pathophysiology of EBI is not fully elucidated, accumulating evidence indicates that oxidative damage contributes greatly to the development of EBI after SAH [5–7]. Theoretically, pharmacologically reducing oxidative damage would be a reasonable approach for the treatment of SAH.
Salvianolic acid B (SalB), a polyphenolic compound extracted from the traditional Chinese herb Salvia miltiorrhiza, has been used to treat cardiovascular and cerebral vascular diseases [8]. Multiple studies have shown that SalB possesses antioxidative and neuroprotective activities in vivo and in vitro [8–11]. For instance, recent research showed that SalB effectively attenuates oxidative stress-related disorders, such as myocardial ischemia and reperfusion, cerebral ischemia, and diabetes [12–14]. In addition, SalB can easily pass the blood-brain barrier and act directly on the central nervous system. Studies have shown that SalB has neuroprotective activity in traumatic brain injury, stroke, Alzheimer’s disease, and Parkinson’s disease [12,15–17]. However, no study has investigated whether SalB is beneficial in experimental SAH.
Nuclear factor-erythroid 2-related factor 2 (Nrf2) is an essential transcription factor that controls the intrinsic antioxidative system to reduce the progression of various oxidative stress-related disorders [18–21]. After stimulation, Nrf2 translocates into the nucleus and binds to the antioxidant response element (ARE), thereby promoting transcription of several antioxidant and detoxification genes, such as heme oxygenase-1 (HO-1), NADPH: quinine oxidoreductase-1 (NQO-1), and superoxide dismutase (SOD) [18–21]. HO-1, one of the cytoprotective enzymes activated by the Nrf2 pathway, plays a critical role in maintaining cellular homeostasis by degrading heme [22,23]. Notably, HO-1 may exert beneficial or detrimental effects in the brain after different types and stages of injuries [22–25]. Of course, in addition to HO-1, other antioxidant enzymes are also critical for maintaining cellular redox homeostasis against oxidative insults. Intriguingly, SalB was shown to induce activation of the Nrf2 signaling pathway in a variety of disease models [15,26,27]. Additionally, SalB is a potent activator of sirtuin 1 (SIRT1) [12,28,29], a nicotinamide adenine dinucleotide-dependent deacetylase implicated in a wide range of cellular functions. Mounting evidence indicates that SIRT1 activation ameliorates brain injury after SAH and plays a key role in regulating the Nrf2 signaling pathway [2,30–32]. However, it is not clear whether SalB can activate the Nrf2 signaling pathway by modulating SIRT1 activation after SAH. Therefore, we evaluated whether SalB has a beneficial effect against SAH and investigated the relationship between Nrf2 signaling and SIRT1 signaling in this model.
2. Material and methods
2.1. Chemicals and reagents
SalB (purity > 97%), sirtinol, and dimethylsulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Neurobasal medium, B27, glutamine, HEPES, penicillin and streptomycin were purchased from Gibco (Carlsbad, CA, USA). Poly-d-lysine (cat# ST508) and Cresyl violet were purchased from Beyotime Biotechnology (Shanghai, China). 4,6-diamidino-2-phenylindole (DAPI, cat# ab104139) was purchased from Abcam (Cambridge, MA, USA).
2.2. Animal preparation
All experimental protocols in this study were approved by the Animal Care and Use Committee of Nanjing University and conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. Adult male Sprague-Dawley rats (250–300 g) were purchased from the Animal Center of Nanjing University. Eight- to 10-week-old male C57BL/6 (wild-type, WT) mice were purchased from Jackson Laboratory. Nrf2 gene knockout (KO) mice (on C57BL/6 background, 8–10 weeks old), originally generated by Dr. Masayuki Yamamoto, were bred in-house. Both rats and mice were acclimated to a 12-h light/dark cycle with free access to food and water. Primary cortical neurons were prepared from the pups of pregnant C57BL/6 mice at 15–18 days’ gestation. The website Randomization.com (http://www.randomization.com) was used to assign animals into different study groups [25,33].
2.3. In vivo models of SAH
SAH was induced in rats by a single blood injection, as described previously with some modification [4,34]. Briefly, rats were anesthetized with 10% chloral hydrate (0.35 mL/100 g). The chloral hydrate dose and administration route were chosen according to previous studies for stereotaxic neurosurgery [2,35–37]. Rats were then positioned prone in a stereotactic frame, and a burr hole was drilled into the skull 7.5 mm anterior to the bregma at the midline. Bone wax was applied to the burr hole to prevent loss of cerebrospinal fluid and bleeding from the midline vessels. A needle with a rounded tip was inserted into the burr hole at a 45 degree angle to the sagittal plane. The needle was advanced until the tip reached the base of the skull, 2–3 mm anterior to the chiasma (approximately 10–12 mm from the brain surface) and then retracted 0.5 mm. A total of 0.35 mL nonheparinized fresh autologous arterial blood from the femoral artery was slowly (over 20 s) injected into the drill hole with a syringe pump under aseptic conditions. The needle was then kept in this position for 2 min to prevent blood backflow and cerebrospinal fluid leakage. Sham animals underwent the same procedure but were injected with 0.35 mL of physiologic saline instead of blood. After they recovered from the anesthesia, rats were returned to their normal housing area. The brain regions taken for evaluation are shown in Fig. 1A.
Fig. 1.
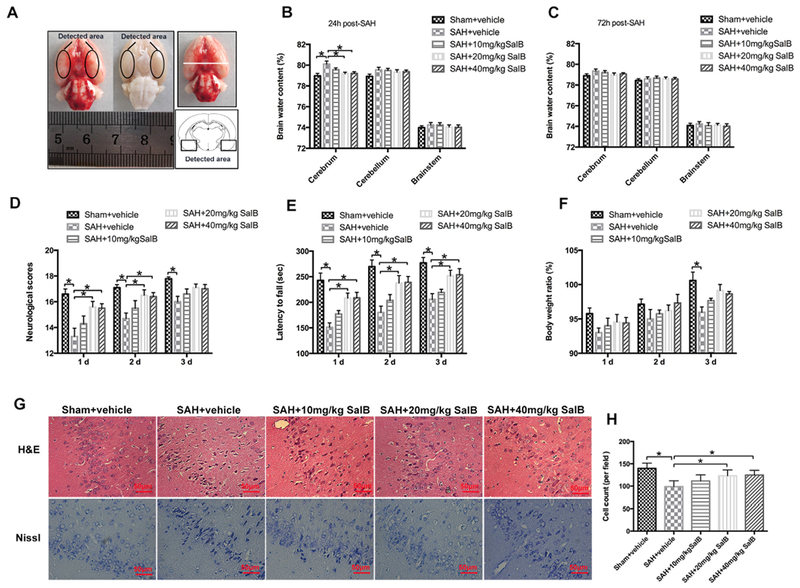
Dose-response effects of salvianolic acid B (SalB) on subarachnoid hemorrhage (SAH) in rats. (A) Schematic representation of the cortex area sampled for analysis. (B, C) Effects of three SalB doses on brain water content at 24 h (B) and 72 h (C) after SAH. (D-F) Effects of three SalB doses on neurologic scores (D), rotarod performance (E), and body weight loss (F) at 24 h, 48 h, and 72 h after SAH. (G) Representative photomicrographs of H&E and Nissl staining at 72 h after SAH. (H) Quantification of the proportion of surviving neurons at 72 h after SAH. Bars represent the mean ± SEM. *P < 0.05.
We used mice to further confirm the potential beneficial effects of SalB observed in rats. In addition, we employed Nrf2 KO mice to investigate whether the neuroprotective effects of SalB occur via Nrf2 signaling. Mice were anesthetized with isoflurane (4.0% for induction, 2.0% for maintenance) and ventilated with oxygen-enriched air (20%:80%) via a nose cone, as in our previous studies [38,39]. After anesthetization, a burr hole was drilled into the skull 4.5 mm anterior to the bregma. A needle with a round tip was tilted 45 degrees in the sagittal plane and inserted into the burr hole. The needle was lowered until the tip reached the base of the skull (approximately 5 mm from the brain surface) and retracted 0.5 mm. Bone wax was applied to the burr hole before needle insertion to prevent loss of cerebrospinal fluid and bleeding from midline vessels. Ten C57BL/6 WT mice were used as blood donors. We collected 50 μL of nonheparinized arterial blood from the heart of a donor for aseptic injection into the burr hole of an anesthetized mouse. After recovering from anesthesia, mice were returned to their normal housing area. The severity of SAH was evaluated by an 18-point scoring system reported by Sugawara et al. [40]. Animals with SAH grades less than 8 were excluded.
2.4. In vivo study design
In the first set of experiments, 159 rats (180 rats were used, 21 rats died) were randomly [33] divided into the following groups: sham + vehicle, sham + 20 mg/kg SalB, SAH + vehicle, SAH + 10 mg/kg SalB, SAH + 20 mg/kg SalB, and SAH + 40 mg/kg SalB. Rats were killed at 24 and 72 h after SAH. Post-assessments included neurologic scores, rotarod performance, body weight ratio, brain water content, western blot, biochemical analysis, real-time PCR, and histopathologic study.
In the second set of experiments, 26 WT mice and 31 Nrf2 KO mice underwent SAH and then were randomly assigned to receive vehicle or 20 mg/kg SalB. Animals were killed at 24 h after SAH. Post-assessments included neurologic scores, rotarod performance, biochemical analysis, and histopathologic study.
In the third set of experiments, 64 rats were randomly assigned into SAH + vehicle group, SAH + sirtinol group, SAH + 20 mg/kg SalB group, and SAH + 20 mg/kg SalB + sirtinol group. Rats were killed at 24 h after SAH. Post-assessments included neurologic scores, rotarod performance, brain water content, western blot, biochemical analysis, and histopathologic study.
2.5. Primary neuronal culture
Primary cortical neurons were cultured as previously reported [4,20,41]. In brief, cerebral cortex was isolated from brains of fetal mice. After the blood vessels and meninges were carefully removed, the cortex was digested with 0.25% trypsin for 5 min at 37 °C. Then the suspensions were filtered through a 22-μm filter and centrifuged at 1500 rpm for 5 min. The remaining cells were distributed in poly-D-lysine-coated plates and suspended in neurobasal medium supplemented with B27, glutamate, HEPES, penicillin, and streptomycin. Half of the medium was replaced with fresh medium every 2 days. Neurons that had been in culture for 8–10 days were used for in vitro studies.
For the in vitro SAH model, primary cortical neurons were incubated for 24 h with oxyhemoglobin (oxyHb, Ruibio, 07109) dissolved in culture medium at a final concentration of 10 μM. This concentration of oxyHb was based on our previous studies [4]. The primary cortical neurons were randomly assigned into six groups: control, oxyHb, oxyHb + SalB (5 μM, 20 μM, and 40 μM), and oxyHb + SalB + sirtinol. The neurons were collected for western blot analysis, histopathology, and cell viability analysis.
2.6. Drug administration
For in vivo experiments, SalB was dissolved in physiologic saline at concentrations of 10 mg/mL, 20 mg/mL, or 40 mg/mL. After SAH induction, different doses of SalB or an equal volume of vehicle was administered intraperitoneally at 1 h after insults and then once daily until euthanasia. Sirtinol, a SIRT1-specific inhibitor, was diluted in DMSO to a concentration of 2 mmol/L. Sirtinol (2 mmol/L, 30 μL/kg) or vehicle (30 μL/kg) was administrated into the left lateral ventricle (0.8 mm posterior and 1.5 mm lateral to the bregma, and 3.7 mm below the dural layer) through a syringe 2 h before SAH was induced. For in vitro experiments, SalB was dissolved in culture medium to reach final concentrations of 5 μmol/L, 20 μmol/L, and 40 μmol/L. Sirtinol was dissolved in DMSO and then added to culture medium to reach a final concentration of 20 μmol/L. The doses of SalB and sirtinol used in vivo and in vitro were selected according to previous studies [2,8,27].
2.7. Brain water content
Brain water content was evaluated at 24 and 72 h after SAH. The brains were separated into cerebrum, cerebellum, and brainstem. Each part was weighed immediately after removal (wet weight) and again after being dried at 80 °C for 72 h (dry weight). Brain water content was calculated as [(wet weight − dry weight)/wet weight] × 100% [2,42].
2.8. Behavioral analysis and body weight
Neurologic functions at 24, 48, and 72 h post-SAH were evaluated with an 18-point scoring system (Table 1) reported by Sugawara et al. [40]. Body weight change was expressed as a ratio of body weight after surgery to body weight before surgery [34,38].
Table 1.
Definitions of neurological scores.
| Test | Score | |||
|---|---|---|---|---|
| 0 | 1 | 2 | 3 | |
| Spontaneous activity (in cage for 5 min) | No movement | Barely moves position | Moves but does not approach at least three sides of cage | Moves and approaches at least three sides of cage |
| Spontaneous movements of all limbs | No movement | Slight movement of limbs | Moves all limbs but slowly | Moves all limbs the same as pre-SAH |
| Movements of forelimbs (outstretching while held by tail) | No outreaching | Slight outreaching | Outreach is limited and less than pre-SAH | Outreach is the same as pre-SAH |
| Climbing wall of wire cage | NA | Fails to climb | Climbs weakly | Normal climbing |
| Reaction to touch on both sides of trunk | NA | No response | Weak response | Normal response |
| Response to vibrissae touch | NA | No response | Weak response | Normal response |
SAH, subarachnoid hemorrhage; NA, not available.
An accelerating rotarod was used to assess motor deficits [25,43]. Animals were adapted to the rotarod for a period of 3 days before randomization. Then the testing was performed at 1, 2, and 3 days after surgery. The rotating speed was gradually increased from 4 to 40 rpm over a 5-min period. The time spent on the rotarod was recorded. Three trials were performed, and the individual times from these trials were averaged.
2.9. Western blot analysis
Whole cell protein extraction, cytosolic protein extraction, and nuclear protein extraction were carried out according to established protocols [44,45]. Equal protein amounts were separated by polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane. The membrane was blocked in 5% skim milk for 2 h at room temperature and then incubated with primary antibodies against Bcl2 (1:200, Santa Cruz, Dallas, TX, USA), Bax (1:200, cat# sc-493, Santa Cruz), caspase-3 (1:400, cat# 9661, Cell Signaling), Nrf2 (1:1000, cat# ab31163, Abcam), HO-1 (1:1000, cat# ab13243, Abcam), NQO-1 (1:1000, cat# ab34173, Abcam), SIRT1 (1:200, cat# SC-15404, Santa Cruz), ac-FoxO1 (1:200, cat# sc-49437, Santa Cruz), ac-p53 (1:400, cat# 2570, Cell Signaling), β-actin (1:3000, cat# AP0060, Bioworld Technology, Minneapolis, MN, USA), and Histone H3 (1:3000, cat# BS7416, Bioworld Technology) overnight at 4 °C. Subsequently, the membrane was incubated with horseradish peroxidase (HRP)-conjugated IgG for 2 h at room temperature. Protein bands were detected by enhanced chemiluminescence solution (Thermo Fisher Scientific, Waltham, MA, USA). Band density was quantified with UN-Scan-It 6.1 software (Silk Scientific Inc., Orem, UT, USA).
2.10. Biochemical estimation
The levels of intracellular malondialdehyde (MDA), glutathione peroxidase (GSH-Px), glutathione (GSH), and SOD were determined with commercially available kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) in accordance with the manufacturer’s instructions. The H2O2 levels in the fresh brain samples were measured with the hydrogen peroxide assay kit (Abcam) according to the manufacturer’s instructions. Protein concentrations were determined with the BCA protein assay kit (Thermo Fisher Scientific).
2.11. RT-PCR
Based on our established protocol [43,46], brain samples were homogenized in TRIzol reagent (Invitrogen, Grand Island, NY, USA). Equal amounts of mRNA were reverse transcribed to cDNA with the PrimeScript RT reagent kit (Thermo Fisher Scientific). The primers were synthesized by ShineGene Biotechnology (Shanghai, China) and are shown in Table 2. Quantitative real-time PCR analysis was tested on a Stratagene Mx3000P RT-PCR system (Agilent Technologies, USA) with real-time SYBR Green PCR technology (ComWin Biotechnology, China) [47]. Total RNA concentrations from each sample were normalized by quantity of glyceraldehyde 3-phosphate dehydrogenase mRNA.
Table 2.
Real-time PCR primers used in this study.
| Gene | Forward primer (5′ to 3′) | Reverse primer (3′ to 5′) |
|---|---|---|
| HO-1 | GCGAAACAAGCAGAACCCA | ACCCTCCATGAGTAGGACTCG |
| NQO-1 | GCGTCTGGAGACTGTCTGGG | CGTTCAGGTAAGGTCGGC |
| GAPDH | AAGAAGGTGGTGAAGCAGGC | TCCACCACCCTGTTGCTGTA |
2.12. Hematoxylin and eosin (H&E) and Nissl staining
In brief, brain sections were stained with H&E or Cresyl violet solution according to standard procedures [33,44,48–50], and then mounted onto microscope slides with Permount (Thermo Fisher Scientific). Staining was visualized under a light microscope.
2.13. Immunofluorescence staining
Immunofluorescence staining was performed according to our previously published protocols [44,50]. Briefly, brain sections (6 μm) or cultured cells were incubated with primary antibodies against 8-hydroxyguanosine (8-OHdG, Abcam), Nrf2 (1:100, Abcam), SIRT1 (1:50, Santa Cruz), caspase-3 (1:100, Cell Signaling), and NeuN (1:200, cat# MAB377, EMD Millipore, USA) overnight at 4 °C. Brain sections were then incubated with corresponding secondary antibodies Alexa Fluor 488 and/or Alexa Fluor 594 (Jackson ImmunoResearch Incorporation, West Grove, PA, USA). Fluorescence was examined under a ZEISS HB050 inverted microscope system. The fluorescently stained cells were analyzed by Image J software.
2.14. TUNEL staining
A terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) detection kit (Roche Inc., Indianapolis, USA) was used for TUNEL staining according to the manufacturer’s instructions [25,51]. Specifically, brain sections or cultured neurons were incubated with TUNEL reaction mixture for 1 h at room temperature. After three washes with phosphate-buffered saline, they were stained with DAPI to show total nuclei. Samples were examined under a ZEISS HB050 inverted microscope system. The fluorescently stained cells were analyzed by Image J software.
2.15. Cell counting
Because the inferior basal temporal lobe always contained blood and differed histologically from control rat brain, we evaluated mainly the basal temporal lobe adjacent to the clotted blood [6]. Two investigators blinded to sample group counted cells in six randomly selected high-power fields (400 ×) in each coronal section. The mean percentage of positive cells per section was calculated [44,49], and the final average number of positive cells was determined in four sections per animal.
2.16. Detection of DCFH-DA staining
In brief, primary cultured neurons were incubated with 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA, Sigma) for 10 min at 37 °C. DCFH fluorescence was measured under an inverted fluorescence microscope (IX53, Olympus, Japan), with equal exposure times for each group. The mean relative fluorescence intensity for each group was measured with the Image-Pro Plus system.
2.17. Cell viability analysis
Viability of primary cultured neurons was evaluated by measuring lactate dehydrogenase (LDH) activity [4,52]. LDH activity was determined with an assay kit according to the manufacturer’s protocol (Beyotime Biotechnology). Briefly, neuronal cells were incubated with an LDH release agent. After incubation, the supernatant was collected and the OD value at 490 nm was measured by a spectrophotometer.
2.18. Statistical analysis
All data are presented as mean ± SEM. Differences among multiple groups were compared by one-way or two-way analysis of variance (ANOVA) with Bonferroni post hoc test. Statistical significance was inferred at P < 0.05.
3. Results
3.1. Mortality rate and SAH grade
No animals died in the sham + vehicle or sham + 20 mg/kg SalB group. The mortality rate of the rats was 19.2% (10 of 52) in the SAH + vehicle group; 16.3% (17 of 104) in the SAH + SalB group; 33.3% (6 of 18) in the SAH + sirtinol group; and 25% (4 of 16) in the SAH + SalB + sirtinol group. The mortality rate of the mice was 7.7% (1 of 13) in the WT SAH + vehicle group; 7.7% (1 of 13) in the WT SAH + SalB group; 25% (4 of 16) in the Nrf2 KO SAH + vehicle group; and 20% (3 of 15) in the Nrf2 KO SAH + SalB group. No significant differences in the SAH grade were observed among experimental groups at 24 h after SAH (Supplementary Fig. 1).
3.2. Dose-response effects of SalB on SAH
In a dose-response study, SalB was administered to rats after SAH at 10, 20, and 40 mg/kg. Doses of 20 mg/kg and 40 mg/kg, but not 10 mg/kg, significantly alleviated brain edema at 24 h after SAH (Fig. 1B). No significant differences in brain edema were detected among the experimental groups at 72 h after SAH (Fig. 1C). In addition, SalB at 20 and 40 mg/kg markedly improved neurologic scores and rotarod performance after SAH. However, 10 mg/kg SalB did not reduce neurologic impairment after SAH (Fig. 1D, E). Body weight ratio results showed that SAH caused greater body weight loss than the sham procedure after 3 days. However, body weight loss did not differ significantly among SAH + vehicle and SAH + SalB groups (Fig. 1F). Furthermore, H&E and Nissl staining showed that SalB treatment at 20 and 40 mg/kg significantly improved neuronal survival after SAH, with no statistical difference between the two concentrations (Fig. 1G, H). Because 20 mg/kg was the lowest dose to provide maximal effect, we used this dose for the remaining experiments.
3.3. Influence of SalB treatment on oxidative stress at 24 h post-SAH
Results showed that SAH significantly induced oxidative damage, as evidenced by increases in lipid peroxidation and H2O2 generation and decreases in GSH-Px, GSH, and SOD activities, when compared with those of the sham + vehicle group (Fig. 2A-E). In contrast, SalB treatment at 20 mg/kg and 40 mg/kg, but not 10 mg/kg, markedly reduced MDA and H2O2 content, but upregulated the activity of GSH-Px, GSH, and SOD (Fig. 2A-E). 8-OHdG immunofluorescence staining also revealed that SalB treatment suppressed oxidative DNA damage in the basal temporal lobe adjacent to the clotted blood (Fig. 2F, G). These data suggest that SalB can effectively reduce the oxidative damage induced by SAH.
Fig. 2.
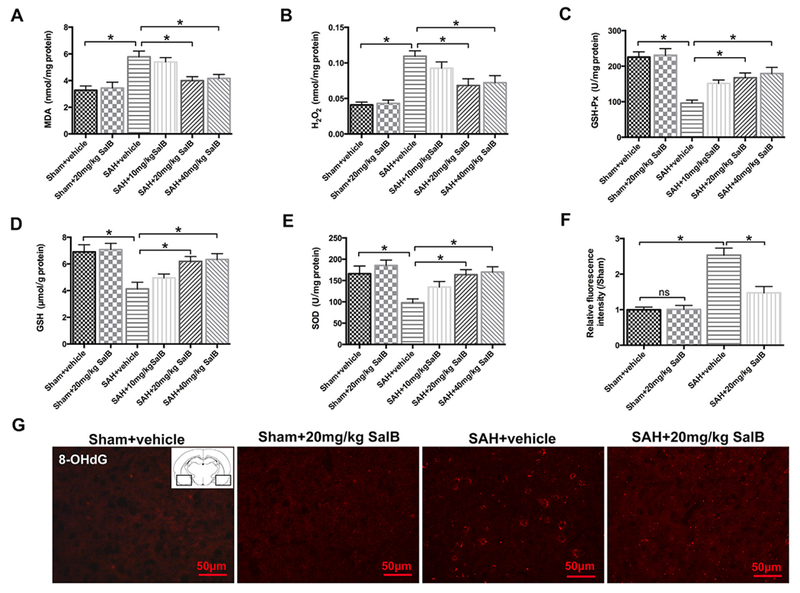
Effects of SalB on oxidative damage in rats at 24h after SAH. (A-E) Quantification of malondialdehyde (MDA, A), H2O2 (B), glutathione peroxidase (GSH-Px, C), glutathione (GSH, D), and superoxide dismutase (SOD, E) activities in different groups. (F) Quantitative analysis of 8-hydroxyguanosine (8-OHdG) immunofluorescence staining. (G) Representative photomicrographs of immunofluorescence staining for 8-OHdG. Bars represent the mean ± SEM. *P < 0.05; ns, not significant.
3.4. Effects of SalB on brain water content, neuronal death, and neurologic function after SAH
To better explore the neuroprotective effects of SalB on SAH injury, we evaluated brain edema, neurologic scores, and neural cell death. SAH caused significant brain edema and neurologic impairment that was alleviated by SalB treatment (Fig. 3A-C). In addition, rats with SAH had greater numbers of caspase-3- and TUNEL-positive neurons than did sham-operated rats (Fig. 3D-G). Likewise, western blot results showed that SAH increased apoptosis at 24 h, compared with that in the sham + vehicle group, as evidenced by enhanced levels of cleaved caspase-3 and Bax, and decreased levels of Bcl2 (Fig. 3H, I). These alterations were reversed by SalB treatment (Fig. 3H, I).
Fig. 3.
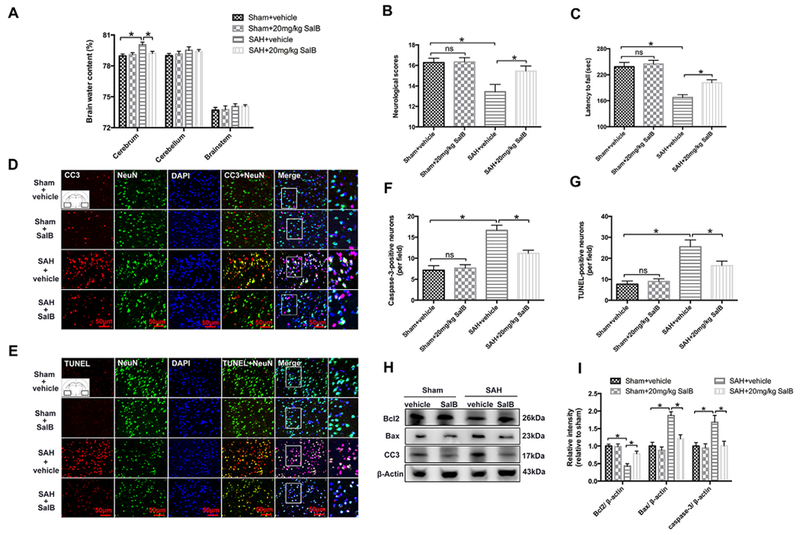
Effects of SalB on brain edema, neurologic function, and neuronal death in rats at 24 h post-SAH. (A-C) Effects of SAEi and SalB on water content (A), neurologic function (B), and latency to fall in the rotarod test (C). (D, E) Representative photomicrographs of immunofluorescence staining for caspase-3 (CC3, D) and TUNEL staining (E) in the indicated experimental groups. (F, G) Semi-quantitative analysis of caspase-3 immunofluorescence staining and TUNEL staining. (H) Western blot analysis for Bcl2, Bax, and caspase-3. (I) Quantification of Bcl2, Bax, and caspase-3. Bars represent the mean ± SEM. *P < 0.05; ns, not significant.
3.5. Effects of SalB on the Nrf2 signaling pathway after SAH
Western blot analysis showed that total and nuclear expression levels of Nrf2 protein increased after SAH (Fig. 4A, B). No significant difference in the cytosolic expression levels of Nrf2 protein between SAH and sham-operated groups was detected (Fig. 4A, B). SalB further induced the nuclear and total expression of Nrf2 protein as well as the nuclear/cytosolic Nrf2 ratio after SAH (Fig. 4A, B). In addition, both protein and mRNA levels of HO-1 and NQO-1, the downstream targets of Nrf2, were significantly increased after SalB treatment (Fig. 4C, D). Double immunofluorescent staining confirmed the Western blot results, showing that SalB enhanced nuclear translocation of Nrf2 after SAH (Fig. 4E, F).
Fig. 4.
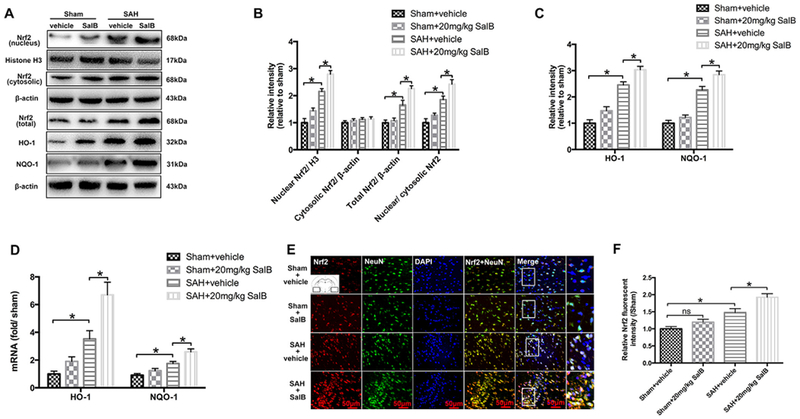
Effects of SalB treatment on the Nrf2 signaling pathway in rats at 24 h post-SAH. (A) Western blot analysis for the expression of Nrf2, HO-1, and NQO-1 in the indicated groups. (B, C) Quantitative analyses of Nrf2, HO-1, and NQO-1 in each group. (D) Real-time PCR analysis for HO-1 and NQO-1 expression. (E) Representative photomicrographs of double immunofluorescence staining for Nrf2 and NeuN. (F) Quantification of Nrf2 immunofluorescence staining. Bars represent the mean ± SEM. *P < 0.05; ns, not significant.
3.6. Effects of SalB on oxidative damage in Nrf2 KO mice after SAH
To further confirm whether the neuroprotective effect of SalB after SAH occurs via Nrf2 signaling, we employed Nrf2 KO mice. Consistent with the results in rats, SalB treatment significantly improved neurologic function and reduced oxidative damage in WT mice after SAH. In contrast, the neuroprotective and antioxidative effects of SalB against SAH were absent in Nrf2 KO mice (Fig. 5A-H). These data suggest that Nrf2 signaling plays a key role in defending against oxidative damage after SAH, and that SalB enhances the Nrf2 signaling pathway. However, we noted that SalB still reduced the number of TUNEL-positive neurons in Nrf2 KO mice after SAH (Fig. 5F, G). This finding suggests that the beneficial effects of SalB against SAH might not be solely dependent on Nrf2 activation.
Fig. 5.
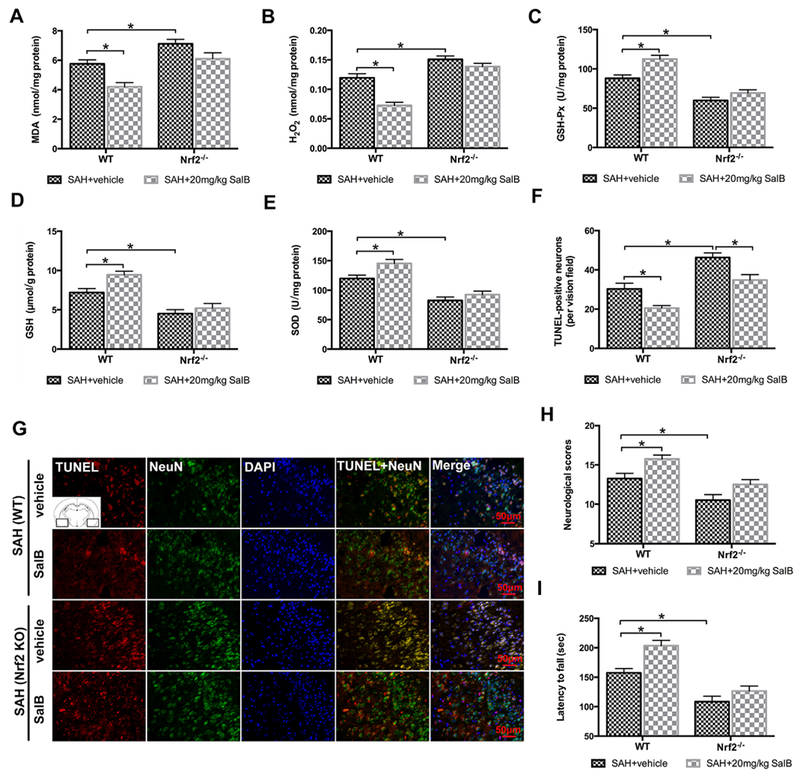
Effects of SalB on SAH injury at 24 h in wild-type (WT) and Nrf2 knockout (KO) mice. (A-E) Quantification of MDA, H2O2, GSH-Px, GSH, and SOD activities in WT and Nrf2 KO mice treated with vehicle or 20 mg/kg SalB. (F) Quantitative analysis of the number of TUNEL-positive neurons in each group. (G) Representative photomicrographs of TUNEL staining. (H, I) Effects of SAH and SalB on neurologic function (H) and latency to fall in the rotarod test (I). Bars represent the mean ± SEM. *P < 0.05.
3.7. Effects of SalB on SIRT1 signaling after SAH
Western blot analysis showed that SalB treatment significantly increased expression of SIRT1 and Nrf2, and decreased levels of acetyl-FoxO1 and acetyl-p53. These changes were all reversed by SIRT1-specific inhibitor sirtinol (Fig. 6A, B). In support of our western blot findings, double immunofluorescence staining showed that SalB upregulated SIRT1 expression and Nrf2 nuclear translocation in neurons and that these effects were abolished by sirtinol (Fig. 6C-F).
Fig. 6.
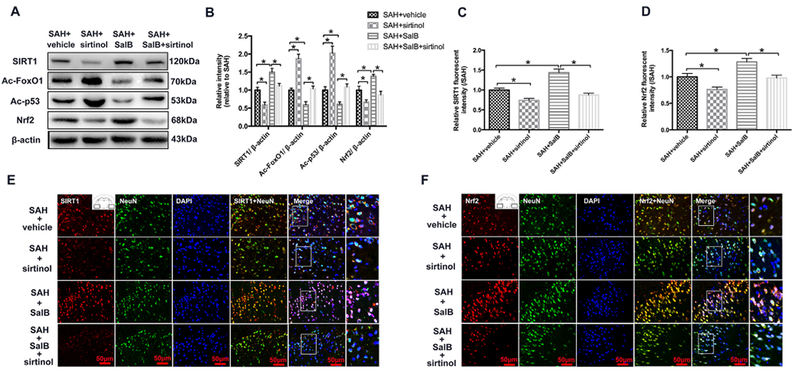
Effects of SalB on sirtuin 1 (SIRT1) activation in rats after SAH. (A, B) Representative western blot (A) and quantification (B) of expression of SIRT1, acetyl (ac)-FoxO1, ac-P53, and Nrf2. (C, D) Quantification of SIRT1 (C) and Nrf2 (D) immunofluorescence staining. (E, F) Representative photomicrographs show double immunofluorescence staining of SIRT1 and NeuN (E) and Nrf2 and NeuN (F) in the indicated experimental groups. Bars represent the mean ± SEM. *P < 0.05.
3.8. SIRT1 inhibition reverses the neuroprotective effects of SalB after SAH
SIRT1 inhibition by sirtinol further exacerbated oxidative damage after SAH and significantly abolished the antioxidative effects of SalB, as evidenced by increased lipid peroxidation and H2O2 generation, and decreased GSH-Px and SOD activities (Fig. 7A-D). Moreover, sirtinol reversed the SalB-induced decrease in numbers of caspase-3- and TUNEL-positive neurons and neurological impairment (Fig. 7E-J).
Fig. 7.
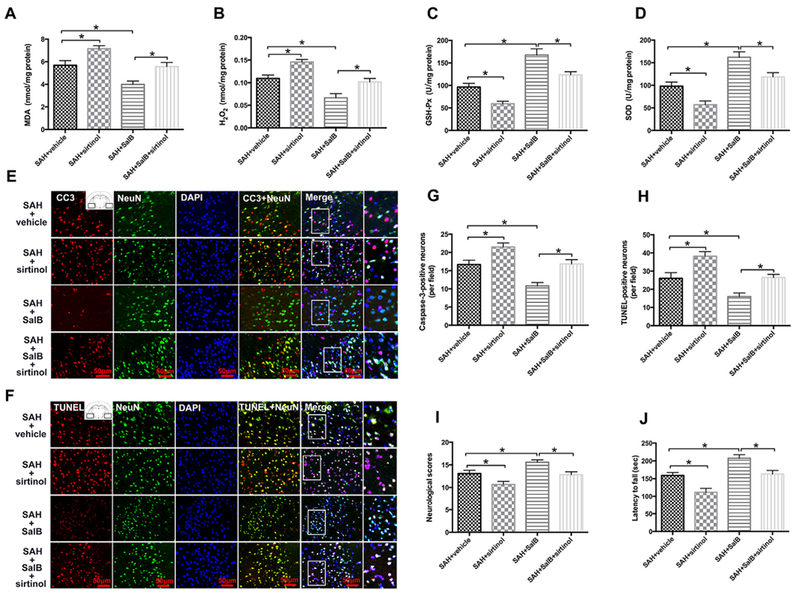
Effects of SIRT1 inhibition on SAH injury in rats. (A-D) Quantification of MDA, H2O2, GSH-Px, and SOD activities in different groups. (E, F) Representative photomicrographs show immunofluorescence staining for caspase-3 (CC3) and TUNEL staining in the indicated experimental groups. (G, H) Semi-quantitative analysis of caspase-3 immunofluorescence staining and TUNEL staining. (I, J) Effects of SIRT1 inhibition on SalB-induced improvements in neurologic function (I) and latency to fall in the rotarod test (J).
3.9. Effects of SalB on SIRT1 and Nrf2 signaling in primary cortical neurons
Consistent with the results in vivo, western blot analysis showed that SalB treatment could significantly and dose-dependently increase the expression of SIRT1, Nrf2, HO-1, and NQO-1 in primary cortical neurons. These increases were reversed by SIRT1-specific inhibitor sirtinol (Fig. 8A, B). Double immunofluorescence staining confirmed the western blot results (Fig. 8C-F).
Fig. 8.
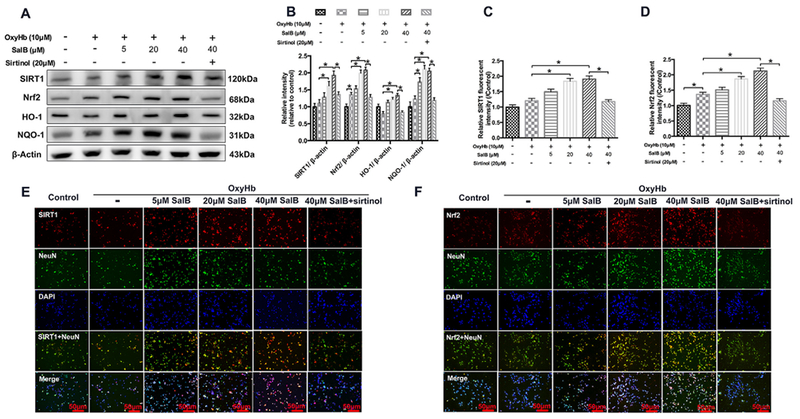
Effects of SalB treatment on the SIRT1 and Nrf2 signaling pathway in primary neurons exposed to oxyHb. (A, B) Representative western blots (A) and quantification of relative protein expression (B) for SIRT1, Nrf2, HO-1, and NQO-1. (C, D) Quantification of SIRT1 (C) and Nrf2 (D) immunofluorescence staining. (E, F) Representative photomicrographs of double immunofluorescence staining for SIRT1 and NeuN (E) and Nrf2 and NeuN (F) in the indicated experimental groups. Bars represent the mean ± SEM. *P < 0.05.
3.10. Effects of SalB on primary cortical neurons exposed to oxyHb
We further evaluated the therapeutic effects of SalB on neuronal damage in primary cortical neurons exposed to oxyHb. As shown in Fig. 9A, oxyHb aggravated neuronal degeneration as evidenced by swollen cell bodies and loss of synapses. SalB dose-dependently ameliorated the oxyHb-induced neuronal degeneration, but the improvement was reversed by SIRT1 inhibition. In addition, exposure of neurons to oxyHb induced a marked increase in reactive oxygen species (ROS) generation that was dose-dependently reversed by SalB (Fig. 9B, C). Sirtinol abolished the SalB-induced reduction in ROS generation. We then investigated the influence of different doses of SalB on TUNEL staining and LDH release. Our results indicated that SalB dose-dependently reduced the number of TUNEL-positive neurons and restored cell viability in primary cortical neurons (Fig. 9D–F). These findings suggest that SalB might protect against neuronal injury in an in vitro SAH model.
Fig. 9.
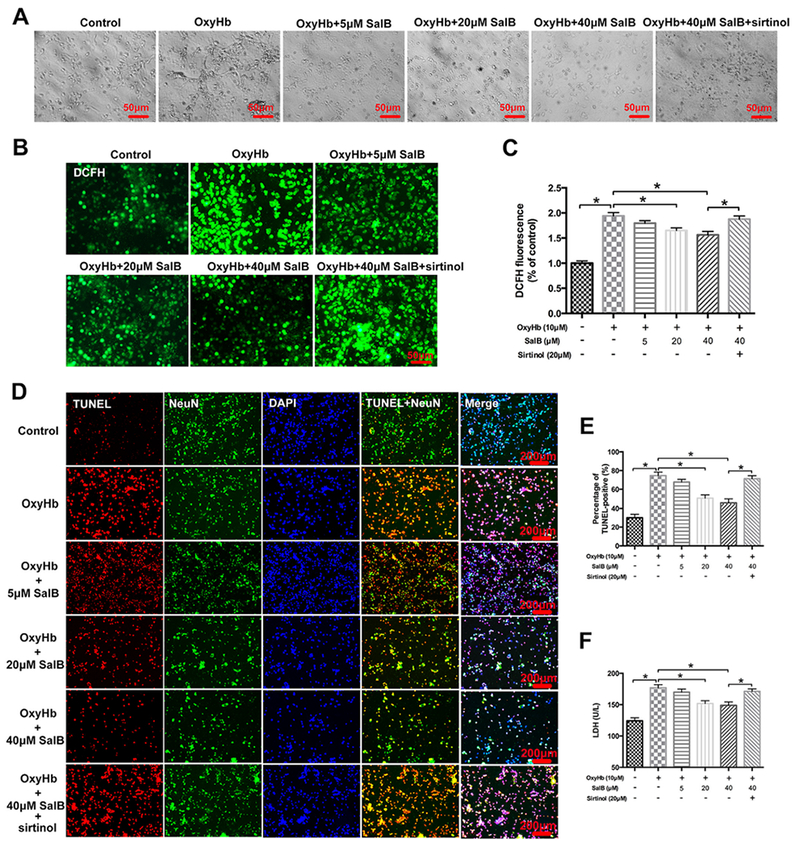
Effects of SalB on ROS production, TUNEL staining, and cell viability in primary cortical neurons. (A) Representative photomicrographs of cultured neurons obtained by bright field microscopy. (B, C) Representative photomicrographs (B) and quantification (C) of 2,7-dichlorodihdrofluorescein (DCFH) immunofluorescence in primary cortical neurons. (D) Representative photomicrographs of TUNEL staining in primary cortical neurons. (E) Quantification of the number of TUNEL-positive neurons in all experimental groups. (F) Quantitative analysis of lactate dehydrogenase (LDE1) activity in the indicated groups. Bars represent the mean ± SEM. *P < 0.05.
4. Discussion
Substantial evidence indicates that oxidative stress is closely associated with EBI after hemorrhagic stroke, particularly SAH [2,5–7,53–55]. Excessive free radicals, including hydroxyl radical, superoxide anion, and H2O2, are generated early and consume intrinsic antioxidant systems such as SOD, GSH, and GSH-px. These free radicals lead to neuronal damage by promoting lipid peroxidation, protein breakdown, and DNA damage that in turn induces cellular apoptosis, endothelial damage, and blood-brain barrier disruption [53–56]. Importantly, brain is an organ with a high content of polyunsaturated fatty acids, which makes it vulnerable to free radical attacks and lipid peroxidation. Hence, pharmacologic inhibition of post-SAH oxidative damage may be a successful strategy.
SalB has been shown to have potent antioxidant effects and to prevent various disorders involving free-radical toxicity [11,15,57]. In addition, numerous studies have reported that SalB is neuroprotective in various central nervous system diseases [8,12,15]. However, to our knowledge, no study has yet investigated the potential benefits in experimental SAH. In our study, we first evaluated the antioxidant effects of SalB after SAH. Consistent with previous studies [5,58], ROS production was markedly induced in the early period after SAH. The overproduction of ROS triggered severe damage related to increased MDA levels and decreased antioxidant enzyme activity. MDA is a reliable marker of oxidative stress and indicates the presence of damage and destruction to cell membranes [6,52]. SOD, GSH, and GSH-px are essential antioxidant enzymes that inhibit the overproduction of ROS [32]. SalB administration not only dramatically inhibited the overproduction of ROS induced by SAH, but also enhanced SOD and GSH-px activity and GSH content, suggesting that SalB can ameliorate oxidative stress after SAH. Concomitant with the decreased oxidative damage, SalB significantly ameliorated brain edema, neural cell apoptosis, and neurologic impairment after SAH. However, the underlying molecular mechanisms of SalB’s beneficial actions remain obscure.
The Nrf2 signaling pathway, a primary host defense, plays an important role in the maintenance of cellular homeostasis [18,19,32]. Under normal conditions, Nrf2 is located mainly in the cytoplasm and anchored by the Kelch-like ECH-associated protein 1 (Keapl). Once the cell encounters stimulation, such as oxidative stress, Nrf2 dissociates from Keapl, translocates into the nucleus, and binds to the ARE, subsequently inducing antioxidant enzyme expression, including NQO-1, HO-1, and SOD [25,32]. Mounting evidence from preclinical studies indicates that Nrf2 signaling activation can ameliorate SAH injury and that genetic elimination of Nrf2 aggravates SAH-induced secondary complications [59,60]. In fact, recent studies in other research areas have shown that SalB upregulates Nrf2 and HO-1 protein expression and enhances the ability of potential antioxidants [61,62]. To investigate whether the protective effect of SalB against oxidative damage is associated with Nrf2 activation, we examined the effects of SalB on the Nrf2/ARE antioxidant pathway and the regulation of antioxidant enzymes. We found that SalB treatment significantly induced Nrf2 nuclear translocation, as well as HO-1 and NQO-1 upregulation in the brain after SAH. Moreover, knockout of Nrf2 blocked SalB’s anti-oxidative effects on SAH-induced brain injury. These findings suggest that Nrf2 plays a key role in defending against SAH-induced oxidative damage and that, by enhancing the Nrf2 signaling pathway, SalB has the potential to enhance this defense. Nonetheless, the ability of SalB to ameliorate SAH-induced apoptosis in Nrf2−/− mice without significantly reversing neurologic impairment suggests that the benefits of SalB might not be solely dependent on Nrf2 activation.
How SalB regulates Nrf2 activation remains unclear. A probable mechanism might involve SIRT1, which regulates a variety of cellular biochemical processes, including oxidative stress, apoptosis, metabolism, and senescence [2]. SIRT1 deacetylates various substrates, such as FoxOs, P53, PPARr, and PCGla, which all are potentially involved in neuronal survival [63]. For example, SIRT1 might ameliorate oxidative stress through the post-translational deacetylation of FoxO1. In addition, SIRT1 deacetylates P53 and prevents P53-mediated transcriptional activity to block apoptosis [64]. Moreover, numerous recent studies have shown that SIRT1 exerts potent antioxidant effects by enhancing the expression and transcriptional activity of Nrf2 [30,32]. More importantly, extensive research in other fields has shown that SalB is a potent SIRT1 activator [12,29]. However, it is unknown whether SalB modulates SIRT1 activation to enhance Nrf2 signaling after SAH. In the present study, we provide evidence that SalB can strongly upregulate SIRT1 and inhibit FoxO1 and P53 acetylation to reduce brain injury after SAH. SalB treatment significantly increased Nrf2 expression after SAH. Interestingly, SIRT1 specific inhibitor sirtinol effectively abolished SalB-induced SIRT1 activation and Nrf2 expression, and thereby aggravated SAH-induced oxidative damage and brain injury. These results are consistent with those of other studies, which suggested that SalB-mediated Nrf2 antioxidant signaling pathways may be regulated by the activation of SIRT1 [30,32]. Based on these outcomes, we further evaluated the influence of SalB on primary cortical neurons exposed to oxyHb. Consistent with the results in vivo, SalB dose-dependently ameliorated ROS production and neuronal damage. These data further confirm that SalB is a potent antioxidant agent. As in our in vivo studies, the beneficial effects of SalB in vitro were associated with SIRT1 upregulation and Nrf2 signaling activation, and were reversed by sirtinol. Taken together, these results indicate that the neuroprotective effects of SalB might be attributed to its ability to enhance SIRT1 expression and thereby activate the Nrf2 signaling pathway.
Our study has a few limitations. First, we used two different anesthetic drugs—chloral hydrate and isoflurane—to anesthetize the animals. Previous studies reported that isoflurane might reduce brain inflammation and delay the development of brain injury after SAH [65,66]. However, isoflurane is a very common and safe anesthetic drug and is widely used for mouse anesthesia. To avoid the possible effects of isoflurane on the progression of SAH, we used chloral hydrate to anesthetize rats to confirm the beneficial effects of SalB after SAH. Although chloral hydrate can be used to anesthetize small animals [2,35,36], it has weaknesses. Accumulating evidence indicates that chloral hydrate does not provide enough analgesia and produces marked respiratory depression at doses required for surgical anesthesia [67]. In future studies, we will utilize other anesthetic drugs such as phenobarbitone or avertin to confirm the beneficial effects of SalB after SAH. Second, we cannot exclude the possibility that other functions and signaling pathways might also contribute to the beneficial effects of SalB on EBI. For instance, SalB can inhibit microglial activation, suppress the toll-like receptor 4 signaling pathway, and enhance the phosphoinositide 3-kinase (PI3K) pathway [8,68,69], which also has been implicated in the pathophysiology of EBI after SAH or intracerebral hemorrhage [70,71]. Third, how SalB modulates SIRT1 and Nrf2 activation is not fully elucidated. It is known that adenosine monophosphate-activated protein kinase (AMPK) and PI3K, which serve as upstream protein kinases of Nrf2, also play key roles in the regulation of Nrf2 [18]. Previous studies have reported that SIRT1 deacetylates LKB1 and secondarily affects AMPK, and that SalB can directly affect AMPK and PI3K [68,72,73]. Whether AMPK and PI3K are involved in the activation of Nrf2 signaling by SalB still needs to be investigated. Fourth, the exact relationship between SIRT1 and Nrf2 signaling is not clear. Although we observed that sirtinol can inhibit both SIRT1 and Nrf2 expression, how SIRT1 regulates Nrf2 signaling remains obscure. Mounting evidence suggests that SIRT1 can regulate Nrf2 expression and activity to modulate the transcription of antioxidant enzymes [32,72]. However, Nrf2 also can positively regulate SIRT1 expression and deacetylase activity, suggesting a positive feedback loop between Nrf2 and SIRT1 signaling [74,75]. Additionally, some researchers have shown that SIRT1 can mediate Nrf2 deacetylation level to terminate the nuclear Nrf2 translocation and that overexpression of SIRT1 can disturb cellular redox balance by inhibiting Nrf2-induced antioxidant defense [76]. These reports suggest a complex relationship between SIRT1 and Nrf2 signaling. Therefore, additional studies are required to further decipher these important mechanistic questions.
In summary, we provide evidence that SalB can ameliorate SAH-induced oxidative damage. The effects of SalB were largely dependent on upregulation of the Nrf2 signaling pathway, in part by enhancing SIRT1 activation. Although more work is required, this study suggests that SalB might be a promising treatment option for SAH and other oxidative stress-related injuries.
Supplementary Material
Acknowledgments
Funding
This work was supported by grants from The National Natural Science Foundation of China (No. 81471183 to XZ, 81501022 to XSZ, 81771292 to CHH, and U1704166 to XMC), the National Institutes of Health (R01AT007317, R01NS078026, R56NS096549, R21NS101614, R21NS102899, and UG3NS106937 to JW), and a Stimulating and Advancing ACCM Research (StAAR) grant from the Department of Anesthesiology and Critical Care Medicine, Johns Hopkins University. Xiangsheng Zhang is the recipient of the China Scholarship Council Joint Ph.D. training award.
Abbreviations:
- 8-OHdG
8-hydroxyguanosine
- AMPK
adenosine monophosphate-activated protein kinase
- ARE
antioxidant response element
- DCFH-DA
2,7-dichlorodihdrofluorescein diacetate
- DMSO
dimethylsulfoxide
- EBI
early brain injury
- GSH
glutathione
- GSH-Px
glutathione peroxidase
- HO-1
heme oxygenate-1
- LDH
lactate dehydrogenase
- MDA
malondialdehyde
- NQO-1
NADPH: quinine oxidoreductase-1
- Nrf2
nuclear factor-erythroid 2-related factor 2
- PI3K
phosphoinositide 3-kinase
- SAH
subarachnoid hemorrhage
- SalB
Salvianolic acid B
- SIRT1
sirtuin 1
- SOD
superoxide dismutase
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling
Footnotes
Appendix A. Supplementary material
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.freeradbiomed.2018.06.035.
Conflict of interest
The authors declare no conflict of interest.
The authors declare no financial or other conflict of interest.
References
- [1].Macdonald RL, Schweizer TA, Spontaneous subarachnoid haemorrhage, Lancet 389 (2017) 655–666. [DOI] [PubMed] [Google Scholar]
- [2].Zhang XS, Wu Q, Wu LY, Ye ZN, Jiang TW, Li W, Zhuang Z, Zhou ML, Zhang X, Hang CH, Sirtuin 1 activation protects against early brain injury after experimental subarachnoid hemorrhage in rats, Cell Death Dis 7 (2016) e2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Sehba FA, Hou J, Pluta RM, Zhang JH, The importance of early brain injury after subarachnoid hemorrhage, Prog. Neurobiol 97 (2012) 14–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Zhang X, Wu Q, Zhang Q, Lu Y, Liu J, Li W, Lv S, Zhou M, Zhang X, Hang C, Resveratrol attenuates early brain injury after experimental subarachnoid hemorrhage via inhibition of NLRP3 inflammasome activation, Front. Neurosci 11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zhang L, Li Z, Feng D, Shen H, Tian X, Li H, Wang Z, Chen G, Involvement of Nox2 and Nox4 NADPH oxidases in early brain injury after subarachnoid hemorrhage, Free Radic. Res 51 (2017) 316–328. [DOI] [PubMed] [Google Scholar]
- [6].Zhang XS, Zhang X, Zhou ML, Zhou XM, Li N, Li W, Cong ZX, Sun Q, Zhuang Z, Wang CX, Shi JX, Amelioration of oxidative stress and protection against early brain injury by astaxanthin after experimental subarachnoid hemorrhage, J. Neurosurg 121 (2014) 42–54. [DOI] [PubMed] [Google Scholar]
- [7].Zhan Y, Chen C, Suzuki H, Hu Q, Zhi X, Zhang JH, Hydrogen gas ameliorates oxidative stress in early brain injury after subarachnoid hemorrhage in rats, Crit. Care Med 40 (2012) 1291–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhang J, Xie X, Tang M, Zhang J, Zhang B, Zhao Q, Han Y, Yan W, Peng C, You Z, Salvianolic acid B promotes microglial M2-polarization and rescues neurogenesis in stress-exposed mice, Brain Behav. Immun 66 (2017) 111–124. [DOI] [PubMed] [Google Scholar]
- [9].Kim DH, Park SJ, Kim JM, Jeon SJ, Kim DH, Cho YW, Son KH, Lee HJ, Moon JH, Cheong JH, Ko KH, Ryu JH, Cognitive dysfunctions induced by a cholinergic blockade and Abeta 25-35 peptide are attenuated by salvianolic acid B, Neuropharmacology 61 (2011) 1432–1440. [DOI] [PubMed] [Google Scholar]
- [10].Zhang JQ, Wu XH, Feng Y, Xie XF, Fan YH, Yan S, Zhao QY, Peng C, You ZL, Salvianolic acid B ameliorates depressive-like behaviors in chronic mild stress-treated mice: involvement of the neuroinflammatory pathway, Acta Pharmacol. Sin 37 (2016) 1141–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tang Y, Jacobi A, Vater C, Zou X, Stiehler M, Salvianolic acid B protects human endothelial progenitor cells against oxidative stress-mediated dysfunction by modulating Akt/mTOR/4EBP1, p38 MAPK/ATF2, and ERK1/2 signaling pathways, Biochem. Pharmacol 90 (2014) 34–49. [DOI] [PubMed] [Google Scholar]
- [12].Lv H, Wang L, Shen J, Hao S, Ming A, Wang X, Su F, Zhang Z, Salvianolic acid B attenuates apoptosis and inflammation via SIRT1 activation in experimental stroke rats, Brain Res. Bull 115 (2015) 30–36. [DOI] [PubMed] [Google Scholar]
- [13].Deng Y, Yang M, Xu F, Zhang Q, Zhao Q, Yu H, Li D, Zhang G, Lu A, Cho K, Teng F, Wu P, Wang L, Wu W, Liu X, Guo DA, Jiang B, Combined salvianolic acid B and ginsenoside Rg1 Exerts cardioprotection against ischemia/reperfusion injury in rats, PLoS One 10 (2015) e0135435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ren Y, Tao S, Zheng S, Zhao M, Zhu Y, Yang J, Wu Y, Salvianolic acid B improves vascular endothelial function in diabetic rats with blood glucose fluctuations via suppression of endothelial cell apoptosis, Eur. J. Pharmacol 791 (2016) 308–315. [DOI] [PubMed] [Google Scholar]
- [15].Zhang XS, Zhang X, Wu Q, Li W, Zhang QR, Wang CX, Zhou XM, Li H, Shi JX, Zhou ML, Astaxanthin alleviates early brain injury following subarachnoid hemorrhage in rats: possible involvement of Akt/bad signaling, Mar. Drugs 12 (2014) 4291–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chen T, Liu W, Chao X, Zhang L, Qu Y, Huo J, Fei Z, Salvianolic acid B attenuates brain damage and inflammation after traumatic brain injury in mice, Brain Res. Bull 84 (2011) 163–168. [DOI] [PubMed] [Google Scholar]
- [17].Lee YW, Kim DH, Jeon SJ, Park SJ, Kim JM, Jung JM, Lee HE, Bae SG, Oh HK, Son KH, Ryu JH, Neuroprotective effects of salvianolic acid B on an Abeta 25-35 peptide-induced mouse model of Alzheimer’s disease, Eur. J. Pharmacol 704 (2013) 70–77. [DOI] [PubMed] [Google Scholar]
- [18].Lv H, Liu Q, Zhou J, Tan G, Deng X, Ci X, Daphnetin-mediated Nrf2 antioxidant signaling pathways ameliorate tert-butyl hydroperoxide (t-BHP)-induced mitochondrial dysfunction and cell death, Free Radic. Biol. Med 106 (2017) 38–52. [DOI] [PubMed] [Google Scholar]
- [19].Wang J, Fields J, Zhao C, Langer J, Thimmulappa RK, Kensler TW, Yamamoto M, Biswal S, Dore S, Role of Nrf2 in protection against intracerebral hemorrhage injury in mice, Free Radic. Biol. Med 43 (2007) 408–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chang CF, Cho S, Wang J, Epicatechin protects hemorrhagic brain via synergistic Nrf2 pathways, Ann. Clin. Transl. Neurol 1 (2014) 258–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lan X, Han X, Li Q, Wang J, Epicatechin, a natural flavonoid compound, protects astrocytes against hemoglobin toxicity via Nrf2 and AP-1 signaling pathways, Mol. Neurobiol 54 (2017) 7898–7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wang J, Dore S, Heme oxygenase-1 exacerbates early brain injury after intracerebral haemorrhage, Brain 130 (2007) 1643–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhang Z, Song Y, Zhang Z, Li D, Zhu H, Liang R, Gu Y, Pang Y, Qi J, Wu H, Wang J, Distinct role of heme oxygenase-1 in early- and late-stage intracerebral hemorrhage in 12-month-old mice, J. Cereb. Blood Flow. Metab 37 (2017) 25–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Schallner N, Pandit R, LeBlanc R 3rd, Thomas AJ, Ogilvy CS, Zuckerbraun BS, Gallo D, Otterbein LE, Hanafy KA, Microglia regulate blood clearance in subarachnoid hemorrhage by heme oxygenase-1, J. Clin. Investig 125 (2015) 2609–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Cheng T, Wang W, Li Q, Han X, Xing J, Qi C, Lan X, Wan J, Potts A, Guan F, Wang J, Cerebroprotection of flavanol (−)-epicatechin after traumatic brain injury via Nrf2-dependent and -independent pathways, Free Radic. Biol. Med 92 (2016) 15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tongqiang L, Shaopeng L, Xiaofang Y, Nana S, Xialian X, Jiachang H, Ting Z, Xiaoqiang D, Salvianolic Acid B prevents iodinated contrast media-induced acute renal injury in rats via the PI3K/Akt/Nrf2 pathway, Oxid. Med. Cell Longev 2016 (2016) 7079487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Liu X, Xavier C, Jann J, Wu H, Salvianolic acid B (Sal B) protects retinal pigment epithelial cells from oxidative stress-induced cell death by activating glutaredoxin 1 (Grx1), Int. J. Mol. Sci 17 (2016) e1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zeng W, Shan W, Gao L, Gao D, Hu Y, Wang G, Zhang N, Li Z, Tian X, Xu W, Peng J, Ma X, Yao J, Inhibition of HMGB1 release via salvianolic acid B-mediated SIRT1 up-regulation protects rats against non-alcoholic fatty liver disease, Sci. Rep 5 (2015) 16013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Li M, Lu Y, Hu Y, Zhai X, Xu W, Jing H, Tian X, Lin Y, Gao D, Yao J, Salvianolic acid B protects against acute ethanol-induced liver injury through SIRT1-mediated deacetylation of p53 in rats, Toxicol. Lett 228 (2014) 67–74. [DOI] [PubMed] [Google Scholar]
- [30].Do MT, Kim HG, Choi JH, Jeong HG, Metformin induces microRNA-34a to downregulate the Sirt1/Pgc-1alpha/Nrf2 pathway, leading to increased susceptibility of wild-type p53 cancer cells to oxidative stress and therapeutic agents, Free Radic. Biol. Med 74 (2014) 21–34. [DOI] [PubMed] [Google Scholar]
- [31].Feng J, Li S, Chen H, Tanshinone IIA inhibits myocardial remodeling induced by pressure overload via suppressing oxidative stress and inflammation: possible role of silent information regulator 1, Eur. J. Pharmacol 791 (2016) 632–639. [DOI] [PubMed] [Google Scholar]
- [32].Zhang Y, Tao X, Yin L, Xu L, Xu Y, Qi Y, Han X, Song S, Zhao Y, Lin Y, Liu K, Peng J, Protective effects of dioscin against cisplatin-induced nephrotoxicity via the microRNA-34a/sirtuin 1 signalling pathway, Br. J. Pharmacol 174 (2017) 2512–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Han X, Zhao X, Lan X, Li Q, Gao Y, Liu X, Wan J, Yang Z, Chen X, Zang W, Guo AM, Falck JR, Koehler RC, Wang J, 20-HETE synthesis inhibition promotes cerebral protection after intracerebral hemorrhage without inhibiting angiogenesis, J. Cereb. Blood Flow. Metab (2018) (271678×18762645). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wu Q, Qi L, Li H, Mao L, Yang M, Xie R, Yang X, Wang J, Zhang Z, Kong J, Sun B, Roflumilast reduces cerebral inflammation in a rat model of experimental subarachnoid hemorrhage, Inflammation 40 (2017) 1245–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sammut S, Dec A, Mitchell D, Linardakis J, Ortiguela M, West AR, Phasic dopaminergic transmission increases NO efflux in the rat dorsal striatum via a neuronal NOS and a dopamine D(1/5) receptor-dependent mechanism, Neuropsychopharmacology 31 (2006) 493–505. [DOI] [PubMed] [Google Scholar]
- [36].Shen H, Liu C, Zhang D, Yao X, Zhang K, Li H, Chen G, Role for RIP1 in mediating necroptosis in experimental intracerebral hemorrhage model both in vivo and in vitro, Cell Death Dis 8 (2017) e2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Li Q, Wan J, Lan X, Han X, Wang Z, Wang J, Neuroprotection of brain-permeable iron chelator VK-28 against intracerebral hemorrhage in mice, J. Cereb. Blood Flow. Metab 37 (2017) 3110–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wu H, Wu T, Xu X, Wang J, Wang J, Iron toxicity in mice with collagenase-induced intracerebral hemorrhage, J. Cereb. Blood Flow. Metab 31 (2011) 1243–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lan X, Han X, Li Q, Li Q, Gao Y, Cheng T, Wan J, Zhu W, Wang J, Pinocembrin protects hemorrhagic brain primarily by inhibiting toll-like receptor 4 and reducing M1 phenotype microglia, Brain Behav. Immun 61 (2017) 326–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sugawara T, Ayer R, Jadhav V, Zhang JH, A new grading system evaluating bleeding scale in filament perforation subarachnoid hemorrhage rat model, J. Neurosci. Methods 167 (2008) 327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wang J, Zhuang H, Dore S, Heme oxygenase 2 is neuroprotective against intracerebral hemorrhage, Neurobiol. Dis 22 (2006) 473–476. [DOI] [PubMed] [Google Scholar]
- [42].Yang J, Li Q, Wang Z, Qi C, Han X, Lan X, Wan J, Wang W, Zhao X, Hou Z, Gao C, Carhuapoma JR, Mori S, Zhang J, Wang J, Multimodality MRI assessment of grey and white matter injury and blood-brain barrier disruption after intracerebral haemorrhage in mice, Sci. Rep 7 (2017) 40358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Li Q, Han X, Lan X, Hong X, Li Q, Gao Y, Luo T, Yang Q, Koehler RC, Zhai Y, Zhou J, Wang J, Inhibition of tPA-induced hemorrhagic transformation involves adenosine A2b receptor activation after cerebral ischemia, Neurobiol. Dis 108 (2017) 173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zhang XS, Li W, Wu Q, Wu LY, Ye ZN, Liu JP, Zhuang Z, Zhou ML, Zhang X, Hang CH, Resveratrol Attenuates acute inflammatory injury in experimental subarachnoid hemorrhage in rats via inhibition of TLR4 pathway, Int. J. Mol. Sci 17 (2016) e1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Pan LN, Zhu W, Li Y, Xu XL, Guo LJ, Lu Q, Wang J, Astrocytic Toll-like receptor 3 is associated with ischemic preconditioning-induced protection against brain ischemia in rodents, PLoS One 9 (2014) e99526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wang X, Ma S, Yang B, Huang T, Meng N, Xu L, Xing Q, Zhang Y, Zhang K, Li Q, Zhang T, Wu J, Yang GL, Guan F, Wang J, Resveratrol promotes hUCMSCs engraftment and neural repair in a mouse model of Alzheimer’s disease, Behav. Brain Res 339 (2018) 297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wu Q, Zhang XS, Wang HD, Zhang X, Yu Q, Li W, Zhou ML, Wang XL, Astaxanthin activates nuclear factor erythroid-related factor 2 and the antioxidant responsive element (Nrf2-ARE) pathway in the brain after subarachnoid hemorrhage in rats and attenuates early brain injury, Mar. Drugs 12 (2014) 6125–6141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zhu W, Gao Y, Wan J, Lan X, Han X, Zhu S, Zang W, Chen X, Ziai W, Hanley DF, Russo SJ, Jorge RE, Wang J, Changes in motor function, cognition, and emotion-related behavior after right hemispheric intracerebral hemorrhage in various brain regions of mouse, Brain Behav. Immun 69 (2018) 568–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zhao D, Xu X, Pan L, Zhu W, Fu X, Guo L, Lu Q, Wang J, Pharmacologic activation of cholinergic alpha7 nicotinic receptors mitigates depressive-like behavior in a mouse model of chronic stress, J. Neuroinflamm 14 (2017) 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Han X, Lan X, Li Q, Gao Y, Zhu W, Cheng T, Maruyama T, Wang J, Inhibition of prostaglandin E2 receptor EP3 mitigates thrombin-induced brain injury, J. Cereb. Blood Flow. Metab 36 (2016) 1059–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Zhang Z, Liu J, Fan C, Mao L, Xie R, Wang S, Yang M, Yuan H, Yang X, Sun J, Wang J, Kong J, Huang S, Sun B, The GluN1/GluN2B NMDA receptor and metabotropic glutamate receptor 1 negative allosteric modulator has enhanced neuroprotection in a rat subarachnoid hemorrhage model, Exp. Neurol 301 (2018) 13–25. [DOI] [PubMed] [Google Scholar]
- [52].Li Q, Han X, Lan X, Gao Y, Wan J, Durham F, Cheng T, Yang J, Wang Z, Jiang C, Ying M, Koehler RC, Stockwell BR, Wang J, Inhibition of neuronal ferroptosis protects hemorrhagic brain, JCI Insight 2 (2017) e90777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].A RE a.Zhang JH, Oxidative stress in subarachnoid haemorrhage: significance in acute brain injury and vasospasm, Acta Neurochir. Suppl 104 (2008) 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wu H, Zhang Z, Hu X, Zhao R, Song Y, Ban X, Qi J, Wang J, Dynamic changes of inflammatory markers in brain after hemorrhagic stroke in humans: a postmortem study, Brain Res 1342 (2010) 111–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Wang J, Tsirka SE, Contribution of extracellular proteolysis and microglia to intracerebral hemorrhage, Neurocrit Care 3 (2005) 77–85. [DOI] [PubMed] [Google Scholar]
- [56].Wang W, Li M, Chen Q, Wang J, Hemorrhagic transformation after tissue plasminogen activator reperfusion therapy for ischemic stroke: mechanisms, models, and biomarkers, Mol. Neurobiol 52 (2015) 1572–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Fu J, Fan HB, Guo Z, Wang Z, Li XD, Li J, Pei GX, Salvianolic acid B attenuates spinal cord ischemia-reperfusion-induced neuronal injury and oxidative stress by activating the extracellular signal-regulated kinase pathway in rats, J. Surg. Res 188 (2014) 222–230. [DOI] [PubMed] [Google Scholar]
- [58].Li M, Wang W, Mai H, Zhang X, Wang J, Gao Y, Wang Y, Deng G, Gao L, Zhou S, Chen Q, Wang X, Methazolamide improves neurological behavior by inhibition of neuron apoptosis in subarachnoid hemorrhage mice, Sci. Rep 6 (2016) 35055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Liu Y, Qiu J, Wang Z, You W, Wu L, Ji C, Chen G, Dimethylfumarate alleviates early brain injury and secondary cognitive deficits after experimental subarachnoid hemorrhage via activation of Keap1-Nrf2-ARE system, J. Neurosurg 123 (2015) 915–923. [DOI] [PubMed] [Google Scholar]
- [60].Li T, Wang H, Ding Y, Zhou M, Zhou X, Zhang X, Ding K, He J, Lu X, Xu J, Wei W, Genetic elimination of Nrf2 aggravates secondary complications except for vasospasm after experimental subarachnoid hemorrhage in mice, Brain Res 1558 (2014) 90–99. [DOI] [PubMed] [Google Scholar]
- [61].Lin M, Zhai X, Wang G, Tian X, Gao D, Shi L, Wu H, Fan Q, Peng J, Liu K, Yao J, Salvianolic acid B protects against acetaminophen hepatotoxicity by inducing Nrf2 and phase II detoxification gene expression via activation of the PI3K and PKC signaling pathways, J. Pharmacol. Sci 127 (2015) 203–210. [DOI] [PubMed] [Google Scholar]
- [62].Lee HJ, Seo M, Lee EJ, Salvianolic acid B inhibits atherogenesis of vascular cells through induction of Nrf2-dependent heme oxygenase-1, Curr. Med. Chem 21 (2014) 3095–3106. [DOI] [PubMed] [Google Scholar]
- [63].Zhang F, Wang S, Gan L, Vosler PS, Gao Y, Zigmond MJ, Chen J, Protective effects and mechanisms of sirtuins in the nervous system, Prog. Neurobiol 95 (2011) 373–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Chong ZZ, Shang YC, Wang S, Maiese K, SIRT1: new avenues of discovery for disorders of oxidative stress, Expert Opin. Ther. Targets 16 (2012) 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Altay O, Hasegawa Y, Sherchan P, Suzuki H, Khatibi NH, Tang J, Zhang JH, Isoflurane delays the development of early brain injury after subarachnoid hemorrhage through sphingosine-related pathway activation in mice, Crit. Care Med 40 (2012) 1908–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Altay O, Suzuki H, Hasegawa Y, Caner B, Krafft PR, Fujii M, Tang J, Zhang JH, Isoflurane attenuates blood-brain barrier disruption in ipsilateral hemisphere after subarachnoid hemorrhage in mice, Stroke 43 (2012) 2513–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Baxter MG, Murphy KL, Taylor PM, Wolfensohn SE, Chloral hydrate is not acceptable for anesthesia or euthanasia of small animals, Anesthesiology 111 (209) (2009) 209–210 (author reply). [DOI] [PubMed] [Google Scholar]
- [68].Liu CL, Xie LX, Li M, Durairajan SS, Goto S, Huang JD, Salvianolic acid B inhibits hydrogen peroxide-induced endothelial cell apoptosis through regulating PI3K/Akt signaling, PLoS One 2 (2007) e1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Sun A, Liu H, Wang S, Shi D, Xu L, Cheng Y, Wang K, Chen K, Zou Y, Ge J, Salvianolic acid B suppresses maturation of human monocyte-derived dendritic cells by activating PPARgamma, Br. J. Pharmacol 164 (2011) 2042–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Lan X, Han X, Li Q, Yang QW, Wang J, Modulators of microglial activation and polarization after intracerebral haemorrhage, Nat. Rev. Neurol 13 (2017) 420–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Zhang Z, Zhang Z, Lu H, Yang Q, Wu H, Wang J, Microglial polarization and inflammatory mediators after intracerebral hemorrhage, Mol. Neurobiol 54 (2017) 1874–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Chen J, Lai J, Yang L, Ruan G, Chaugai S, Ning Q, Chen C, Wang DW, Trimetazidine prevents macrophage-mediated septic myocardial dysfunction via activation of the histone deacetylase sirtuin 1, Br. J. Pharmacol 173 (2016) 545–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Huang MQ, Zhou CJ, Zhang YP, Zhang XQ, Xu W, Lin J, Wang PJ, Salvianolic Acid B, Ameliorates hyperglycemia and dyslipidemia in db/db mice through the AMPK pathway, Cell Physiol. Biochem 40 (2016) 933–943. [DOI] [PubMed] [Google Scholar]
- [74].Huang K, Gao X, Wei W, The crosstalk between Sirt1 and Keap1/Nrf2/ARE antioxidative pathway forms a positive feedback loop to inhibit FN and TGF-beta1 expressions in rat glomerular mesangial cells, Exp. Cell Res 361 (2017) 63–72. [DOI] [PubMed] [Google Scholar]
- [75].Song NY, Lee YH, Na HK, Baek JH, Surh YJ, Leptin induces SIRT1 expression through activation of NF-E2-related factor 2: implications for obesity-associated colon carcinogenesis, Biochem. Pharmacol 153 (2018) 282–291. [DOI] [PubMed] [Google Scholar]
- [76].Kawai Y, Garduno L, Theodore M, Yang J, Arinze IJ, Acetylation-deacetylation of the transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2) regulates its transcriptional activity and nucleocytoplasmic localization, J. Biol. Chem 286 (2011) 7629–7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.