

Abstract
Oxidative stress is closely linked to the toxic responses of various cell types in normal and pathophysiological conditions. Deoxynivalenol (DON), an inducer of stress responses in the ribosome and the endoplasmic reticulum (ER), causes mitochondrial dysfunction and mitochondria-dependent apoptosis through oxidative stress in humans and animals. The NF-κB pathway, which is closely linked to oxidative stress, is hypothesized to be a critical signaling pathway for DON-induced toxicity and is a potential target for intervention. The present study was conducted to explore the protective effects of pyrrolidine dithiocarbamate (PDTC) from the toxic effects of DON in rat anterior pituitary GH3 cells. Our results showed that DON activated the NF-κB transcription factors and induced cellular oxidative stress, mitochondrial dysfunction, and apoptosis. Morphological studies using transmission electron microscopy (TEM) and cell apoptosis analyses suggested that PDTC prevented DON-induced mitochondrial dysfunction and apoptosis, probably by preventing the DON-induced translocation of NF-κB p65 into the nucleus, and by inhibiting DON-induced iNOS expression. This led to the blocking of the NF-κB pathway and inhibition of iNOS activity.
1. Introduction
Oxidative stress is closely linked to toxic responses of various cell types in normal and pathophysiological conditions. Deoxynivalenol (DON), produced by the Fusarium graminearum and F. culmorum species, is an inducer of stress responses in the ribosome and the endoplasmic reticulum (ER). It causes mitochondrial dysfunction and mitochondria-dependent apoptosis through oxidative stress [1, 2]. The consumption of DON-contaminated products causes a wide range of disorders in animals and humans, affecting the gastrointestinal, reproductive, neuroendocrine, and immune systems [3–5].
The main cellular targets of DON are the ribosome and the ER [6, 7]. However, studies have indicated that DON-induced toxicity also induced oxidative stress and endocrine imbalance [8]. DON targets the mitochondria and causes the mitochondrial membrane potential (ΔΨm) to decrease, leading to the deformation of the mitochondria and the subsequent release of cytochrome c into the cytoplasm [9–11]. Mitochondrial impairment occurred in the livers of fetuses when their mothers consumed DON [12]. Moreover, DON reduced intracellular hormone levels, including those of insulin, leptin, insulin-like growth factor 1 (IGF-1), and IGF acid-labile subunit (IGFALS), which could potentially cause DON-induced growth retardation [13, 14]. We recently discovered that DON inhibited the synthesis of growth hormone (Gh1) in rat GH3 cells, by reducing the cell viability and by inducing apoptosis [15]. Thus, we hypothesized that protecting cells from DON-induced cytotoxicity would prevent growth retardation.
Previous studies have identified that the NF-κB signaling pathway, which occurs downstream of MAPK signaling, can be widely activated after DON treatment in the human Caco-2 and HT-29 cell lines [16, 17]. NF-κB is activated by cytokines, such as TNFα and interleukin (IL), and regulates downstream effects on cell function [18, 19]. It regulates downstream antioxidant and prooxidant genes such as inducible nitric oxide synthase (iNOS), neuronal nitric oxide synthase, superoxide dismutase, catalase, heme oxygenase-1, xanthine oxidoreductase, NADPH : quinone oxidoreductase, and cyclooxygenase-2 [20]. We previously found that the T-2 toxin induced the transcription of Nfkbil1 and Nfrkb in GH3 cells [15], suggesting that the NF-κB signaling pathway was critical to mycotoxin-induced toxicity.
The rat GH3 cell line is a clonal strain of rat pituitary tumor that can synthesize and secrete prolactin and growth hormone. Trichothecenes induce considerable toxicity in endocrine GH3 cells by causing mitochondrial dysfunction, growth hormone synthesis inhibition, cell apoptosis, and inflammation [15, 21]. Therefore, we used an in vitro model of GH3 cells to study the effects of the NF-κB inhibitor, pyrrolidine dithiocarbamate (PDTC), on DON-induced mitochondrial dysfunction and apoptosis. We discovered the mechanisms of DON-induced cytotoxicity in relation to nitric oxide (NO) generation, oxidant-antioxidant balance, and NF-κB activation. The morphological changes in DON-treated cells were determined using flow cytometry and transmission electron microscopy (TEM). The effect of PDTC on DON-induced cytotoxicity was evaluated by TEM, with particular focus on phosphoryl-NF-κB p65 nuclear localization, iNOS expression, and mitochondrial injury. Protection from apoptosis was monitored by flow cytometry.
2. Materials and Methods
2.1. Reagents and Chemicals
DON was obtained from Sigma-Aldrich (St. Louis, MO, USA). PDTC was obtained from Beyotime (Shanghai, P.R. China). Anti-iNOS (ab15323) and anti-actin (ab1801) antibodies were purchased from Abcam (Cambridge, MA, USA). Anti-phospho-IκBα (Ser32/36; 5A5), anti-phosphoryl-NF-κB p65 (Ser536; 93H1), and peroxidase-coupled goat anti-rabbit and mouse IgG (H + L) secondary antibodies were obtained from CST (Danvers, MA, USA). The apoptosis detection kit (Annexin V-FITC) was purchased from BestBio (Shanghai, China).
2.2. Cell Culture
The cells were cultivated as previously reported [15, 21]. Briefly, the GH3 cells from passages 5 to 15 were cultivated in high-glucose DMEM (HyClone Laboratories, Inc., Logan, Utah, USA) with 10% heat-inactivated fetal bovine serum (FBS; Gibco BRL, Gaithersburg, MD, USA) and 1% penicillin-streptomycin (HyClone Laboratories, Inc., Logan, Utah, USA) at 37°C, in the presence of 5% CO2. After 24 hours of incubation, the culture medium was changed to high-glucose DMEM, and the cells were incubated with or without the test reagents (DON and PDTC) for the indicated time intervals.
For cytotoxicity analysis, the cells were seeded in a 96-well plate (at a density of 1 × 104 cells per well) and treated with 0, 300, 600, or 1200 mg/L of DON [15]. For quantitative real-time PCR, western blot, and chemoimmunological assays, GH3 cells were seeded in a six-well plate (at a density of 1 × 105 cells per well) with 2 mL of medium. For TEM, the cells were seeded in a 75 cm2 flask (at a density of 5 × 105 cells per flask) with 12 mL of medium. In some experiments, the cells were treated with inhibitors for 45 min to an hour, then exposed to DON for 12 hours. All experiments were performed in triplicate on at least three independent occasions.
2.3. Quantitative Real-Time PCR (qRT-PCR)
Total RNA was extracted using TRIzol (Invitrogen, Breda, The Netherlands) [15] and analyzed by quantitative real-time RT-PCR using the iNOS gene specific primers (S: 5′CCTCAGGCTTGGGTCTTGTTA3′; AS: 5′ATCCTGTGTTGTTGGGCTGG3′) as previously described. Fold changes in mRNA expression levels were calculated using the 2−ΔΔCt method [11].
2.4. Western Blotting
Total protein was extracted, quantified, separated on a 12% SDS-PAGE gel, and transferred onto a polyvinylidene fluoride membrane (Millipore, Billerica, MA, USA) as previously described [15]. Membranes were incubated with anti-actin (ab1801), anti-phospho-NF-κB p65 (Ser536; 93H1; diluted 1 : 1000), anti-phospho-IκBα (Ser32/36; 5A5; diluted 1 : 1000), and anti-iNOS (ab15323; diluted 1 : 500) antibodies overnight at 4°C, according to the manufacturer's instructions.
2.5. Oxidative Stress Indices
The CAT activity, malondialdehyde (MDA), SOD, and glutathione peroxidase (GSH-Px) levels were assessed using commercial kits (Nanjing Jiancheng BioEngineering Institute Co. Ltd., Nanjing, China).
2.6. Chemoimmunology of Phosphoryl-NF-κB p65
Immunofluorescence was used to determine phosphoryl-NF-κB p65 localization. GH3 cells were fixed with paraformaldehyde (v/v, 1/25) at 37°C for 10 minutes. They were then permeabilized with cold acetone at −20°C for 3 minutes. After a PBS wash (0.1 mM, pH 7.4), cells were saturated with 3% BSA in PBS for 30 minutes, and incubated with the anti-phosphoryl-NF-κB p65 antibody (diluted 1 : 100) at 4°C overnight. After another PBS wash (0.1 mM, pH 7.4), the cells were incubated with the secondary antibody for 30 minutes at room temperature. Coverslips were washed twice with PBS (0.1 mM, pH 7.4), incubated with the goat anti-mouse IgG antibody conjugated with Alexa Fluor 555 (Cell Signaling Technology, Danvers, MA, USA) for 30 minutes in the dark, incubated in 5 μM DAPI staining solution (Invitrogen) for 5 minutes, and then washed in PBS. The fluorescence was monitored using an UltraVIEW VoX confocal system (PerkinElmer, Co., Norwalk, CT, USA).
2.7. GH3 Cell Morphology by Transmission Electron Microscopy (TEM)
The morphological variation in mitochondria was investigated as described earlier [21]. Briefly, the cells were fixed with glutaraldehyde (v/v, 2.5/100), postfixed in osmium tetroxide (w/v, 1/100), dehydrated in absolute ethanol, then embedded stepwise by polymerization at 45°C for 12 hours and at 60°C for 36–48 hours. The 70 nm ultrathin slices were stained with lead citrate for 10 minutes and with uranyl acetate for 30 minutes. Finally, they were washed thrice with ddH2O and dried. The slices were viewed with the H-7650 TEM (Hitachi, Japan).
2.8. Cell Apoptosis
The cells were harvested, washed, and centrifuged (2000 ×g, 4°C, 10 min). Then, they were suspended in Annexin V-FITC binding buffer at a density of 1 × 106 cells per mL. They were then incubated with 10 μL of propidium iodide (PI) solution (BD BioScience, San Jose, CA, USA) in the dark for 15 minutes. Apoptosis was measured using CyAn ADP as described previously [11].
2.9. Statistical Analyses
Data were analyzed by performing a two-way analysis of variance using the SPSS software (SPSS Inc., version 17.0, Chicago, IL, USA). P values < 0.05 indicated statistical significance.
3. Results and Discussion
Oxidative stress plays a major role in the mediation of cellular damage and dysfunction. It is inseparably linked to mitochondrial dysfunction and cell apoptosis [22, 23]. Free radicals contribute to the development of mycotoxicosis by inducing lipid peroxidation and changes in antioxidant status, and by causing the loss of cellular mitochondrial membrane potential [24, 25]. In this study, we found that after DON treatment, the activity of MDA and antioxidant enzymes such as CAT and SOD significantly increased, whereas the activity of GSH-Px significantly decreased (Table 1). This result corroborates other studies [26, 27] in which T-2 toxin exposure was associated with significant decreases in GSH-Px activity in granulosa cells from rats and in hepatic cells from chicken. However, a significant increase in GSH-Px activity was observed in DON-treated HT-29 cells [16]. Because GSH-Px functions as a scavenger of lipid peroxides and is induced by reactive oxygen species and hydroxyl free radicals in cells, the reduction in GSH-Px activity indicates a serious oxidant-antioxidant imbalance in cells. This implies that the rat GH3 cell line is likely more sensitive to DON toxicity than the human HT-29 cell line.
Table 1.
Activities of GSH-Px, GST, CAT, and SOD in GH3 cells.
| Group (ng/mL) | CAT activity (U/mg protein) | SOD (U/mL) | GSH-Px (U/mg protein) | MDA (μmol/mg protein) |
|---|---|---|---|---|
| 0 | 0.407 ± 0.008 | 1.907 ± 0.224 | 150.262 ± 0.001 | 1.476 ± 0.023 |
| 300 | 0.949 ± 0.012∗ | 2.938 ± 0.426∗ | 134.592 ± 0.034∗ | 1.736 ± 0.032∗ |
| 600 | 1.129 ± 0.006∗ | 4.630 ± 0.075∗∗ | 85.149 ± 0.217∗∗ | 1.698 ± 0.038∗ |
| 1200 | 2.326 ± 0.005∗∗ | 5.798 ± 0.257∗∗ | 53.719 ± 0.421∗∗ | 1.777 ± 0.042∗ |
Note: CAT 1 U = the amount of enzyme that consumes 1 nmole H2O2/min. GST 1 U = the amount of enzyme that conjugates 1 μmole CDNB/min. GSH-Px 1 U = the amount of enzyme that converts 1 μM GSH to GSSG in the presence of H2O2/min. SOD 1 U = the amount of enzyme required for 50% inhibition of pyrogallol autooxidation. Data are shown as means ± SD (n = 3) from three separate experiments performed in triplicate. ∗ indicates P values < 0.05, and ∗∗ indicates P values < 0.01.
The NF-κB transcription factors control many processes such as immunity, oxidative stress, and apoptosis. Phosphorylation of p65 NF-κB at serine 536 is mediated by multiple protein kinases, including the IκB kinase [28]. We found that DON induced the phosphorylation of IκBα kinase as well as the phosphorylation and nuclear translocation of the p65 proteins (Figure 1). The pretreatment of cells with PDTC before DON treatment resulted in reduced p65 phosphorylation and translocation (Figure 2). In the nucleus, NF-κB p65 binds to the iNOS gene promoter and upregulates its gene expression [29]. Therefore, we also investigated the iNOS mRNA and protein levels. The quantitative RT-PCR showed that iNOS gene expression increased after DON treatment, but decreased significantly with PDTC pretreatment (Figure 3(a)). The immunoblotting analysis showed similar patterns for the protein levels (Figure 3(b)). Reactive nitrogen species (RNS) and ROS are free radicals that cause oxidative stress. ROS generation did not significantly increase after DON treatment in human HT-29 cells [30] and RAW264.7 cells [31], whereas it increased significantly in HepG-2 cells [24]. At doses of 250 and 500 ng/mL, DON resulted in increased ROS and RNS production in human HT-29 cells [16]. Taken together, the PDTC appears to act as an antioxidant for DON-induced oxidative stress.
Figure 1.
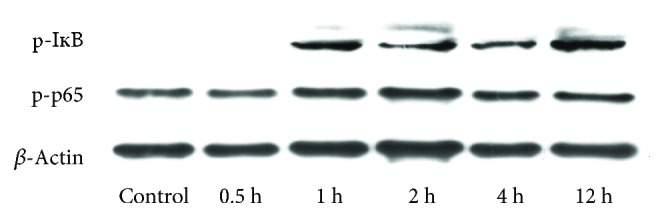
DON-induced IκBα and NF-κB p65 phosphorylation in GH3 cells.
Figure 2.

Nuclear translocation of phosphoryl-NF-κB p65 (p-p65 (Ser536)) induced by DON treatment (1200 ng/mL) and PDTC pretreatment (20 μM, 45 min) followed by DON treatment (1200 ng/mL) in GH3 cells, visualized through indirect immunofluorescence, using Alexa Fluor-conjugated secondary antibody. The nucleus was stained with PI. The panels show PI staining, Alexa Fluor staining, overlay, and the 3D plane of the cells. All photos were captured at 400x magnification. Phosphoryl-NF-κB p65 was upregulated and can be observed in the nucleus.
Figure 3.

PDTC protects cells from DON-induced iNOS expression. (a) Cells were treated with DON, PDTC, and PDTC pretreatment followed by DON to assess iNOS transcription by qRT-PCR. (b). Cells were treated with DON, DON, PDTC, and PDTC pretreatment followed by DON to assess iNOS expression by western blotting. P values < 0.05 are indicated by a single asterisk, ∗. P values < 0.01 are indicated by double asterisks, ∗∗.
In several cell lines, treatment with DON results in a loss of ΔΨm, mitochondrial damage, caspase activation, and apoptosis [9–12, 16, 32]. PDTC relieves oxidative stress and improves mitochondrial structural integrity [33]. Hence, we tested the effects of PDTC pretreatment in DON-treated GH3 cells, with particular focus on mitochondrial ultrastructure and apoptosis. The control cells and PDTC-treated cells exhibited normal mitochondria (Figures 4(a), 4(b), 4(e), and 4(f)), whereas cells treated with DON for 12 hours displayed dose-dependent mitochondrial swelling, serious vacuolar degeneration, disarrayed cristae, and reduced electron density of the matrix (Figures 4(c) and 4(g)). PDTC reduced the DON-induced toxicity, and normal mitochondria were observed despite the reduction in vacuole size observed in the PDTC-pretreated cells (Figures 4(d) and 4(h)). DON treatment led to a significant increase in the number of early and late apoptotic cells. The proportion of apoptotic cells significantly decreased in DON-treated cells that were pretreated with PDTC (Figure 5). All of our findings suggest that PDTC inactivates NF-κB, inhibits iNOS expression, and protects cells from cytotoxicity and mitochondrial toxicity via antioxidant effects (Figure 6). Our results are consistent with the known activity of another antioxidant, lutein, that protects cells from DON-induced mitochondrial structural damage, probably via inhibition of NF-κB [16].
Figure 4.

PDTC protects cells from DON-induced mitochondrial injury in GH3 cells. Cells were treated with DON, PDTC, and PDTC pretreatment, followed by treatment with DON for 12 hours. (a, e) Cell treated with DON showing normal mitochondria. (b, f) Cell treated with PDTC showing normal mitochondria. (c, g) Cells with PDTC pretreatment, followed by treatment with DON for 12 hours showing normal mitochondria and tiny vacuoles.
Figure 5.

PDTC protects cells from DON-induced apoptosis in GH3 cells. Cells in the control group (a), cells treated with PDTC (b) and DON (c), and cells pretreated with PDTC followed by DON treatment (d) were used to assess the apoptosis rate. Data are shown as means for three separate experiments performed in triplicate.
Figure 6.

A proposed mechanism of action for the protective effect of PDTC in DON-mediated mitochondrial dysfunction and apoptosis. DON indirectly activates the NF-κB signal pathway via the classical route of IκB/NF-κB p65 signaling. PDTC inhibits the translocation of NF-κB p65 and the transcription of iNOS, and thereby protects cells from mitochondrial dysfunction and cell apoptosis.
4. Conclusion
In DON-treated GH3 cells, DON caused the translocation of NF-κB and induced iNOS expression. PDTC prevented the DON-induced migration of phosphoryl-NF-κB p65 into the nucleus, inhibited DON-induced iNOS expression, and prevented DON-induced mitochondrial dysfunction and apoptosis.
Acknowledgments
This work was financially supported by the National Key R&D Program of China (2016YFD0501201), National Science Foundation of China (31702127, 31772642, 31572575, 31602114, and 51403072), and by the Fundamental Research Funds for the Central Universities (2662016PY115), as well as the long-term development plan UHK.
Abbreviations
- DON:
Deoxynivalenol
- ERK:
Extracellular signal-regulated kinase
- FBS:
Fetal bovine serum
- FITC:
Fluorescein isothiocyanate
- GSH-Px:
Glutathione peroxidase
- Hck:
Hematopoietic cell kinase
- iNOS:
Inducible nitric oxide synthase
- LDH:
Lactate dehydrogenase
- L-NAME:
L-NG-nitro arginine methyl ester
- MDA:
Malondialdehyde
- NF-κB:
Nuclear factor-kappa B
- PBS:
Phosphate-buffered saline
- PDTC:
Pyrrolidine dithiocarbamate
- PI:
Propidium iodide
- qRT-PCR:
Quantitative real-time PCR
- ROS:
Reactive oxygen species
- SMT:
S-methyl-isothiourea
- SOD:
Superoxide dismutase
- TEM:
Transmission electron microscopy.
Contributor Information
Gang Liu, Email: gangle.liu@gmail.com.
Xu Wang, Email: wangxu@mail.hzau.edu.cn.
Data Availability
The data used to support the findings of this study are available from the corresponding author upon request.
Conflicts of Interest
The authors declare that there is no conflict of interest.
References
- 1.Bondy G. S., Coady L., Curran I., et al. Effects of chronic deoxynivalenol exposure on p 53 heterozygous and p 53 homozygous mice. Food and Chemical Toxicology. 2016;96:24–34. doi: 10.1016/j.fct.2016.07.018. [DOI] [PubMed] [Google Scholar]
- 2.Kosawang C., Karlsson M., Jensen D., Dilokpimol A., Collinge D. B. Transcriptomic profiling to identify genes involved in Fusarium mycotoxin deoxynivalenol and zearalenone tolerance in the mycoparasitic fungus Clonostachys rosea. BMC Genomics. 2014;15(1):p. 55. doi: 10.1186/1471-2164-15-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pinton P., Oswald I. Effect of deoxynivalenol and other type B trichothecenes on the intestine: a review. Toxins. 2014;6(5):1615–1643. doi: 10.3390/toxins6051615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rocha O., Ansari K., Doohan F. M. Effects of trichothecene mycotoxins on eukaryotic cells: a review. Food Additives and Contaminants. 2005;22(4):369–378. doi: 10.1080/02652030500058403. [DOI] [PubMed] [Google Scholar]
- 5.Wu Q., Dohnal V., Huang L., Kuča K., Yuan Z. Metabolic pathways of trichothecenes. Drug Metabolism Reviews. 2010;42(2):250–267. doi: 10.3109/03602530903125807. [DOI] [PubMed] [Google Scholar]
- 6.Pestka J. J. Deoxynivalenol-induced proinflammatory gene expression: mechanisms and pathological sequelae. Toxins. 2010;2(6):1300–1317. doi: 10.3390/toxins2061300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shi Y., Porter K., Parameswaran N., Bae H. K., Pestka J. J. Role of GRP 78/BiP degradation and ER stress in deoxynivalenol-induced interleukin-6 upregulation in the macrophage. Toxicological Sciences. 2009;109(2):247–255. doi: 10.1093/toxsci/kfp060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu Q. H., Wang X., Yang W., et al. Oxidative stress-mediated cytotoxicity and metabolism of T-2 toxin and deoxynivalenol in animals and humans: an update. Archives of Toxicology. 2014;88(7):1309–1326. doi: 10.1007/s00204-014-1280-0. [DOI] [PubMed] [Google Scholar]
- 9.Bensassi F., Gallerne C., Sharaf el Dein O., Lemaire C., Hajlaoui M. R., Bacha H. Involvement of mitochondria-mediated apoptosis in deoxynivalenol cytotoxicity. Food and Chemical Toxicology. 2012;50(5):1680–1689. doi: 10.1016/j.fct.2012.01.015. [DOI] [PubMed] [Google Scholar]
- 10.Ma Y., Zhang A., Shi Z., et al. A mitochondria-mediated apoptotic pathway induced by deoxynivalenol in human colon cancer cells. Toxicology in Vitro. 2012;26(3):414–420. doi: 10.1016/j.tiv.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 11.Wang X., Liu Q., Ihsan A., et al. JAK/STAT pathway plays a critical role in the proinflammatory gene expression and apoptosis of RAW264.7 cells induced by trichothecenes as DON and T-2 toxin. Toxicological Sciences. 2012;127(2):412–424. doi: 10.1093/toxsci/kfs106. [DOI] [PubMed] [Google Scholar]
- 12.Tiemann U., Brüssow K. P., Dannenberger D., et al. The effect of feeding a diet naturally contaminated with deoxynivalenol (DON) and zearalenone (ZON) on the spleen and liver of sow and fetus from day 35 to 70 of gestation. Toxicology Letters. 2008;179(3):113–117. doi: 10.1016/j.toxlet.2008.04.016. [DOI] [PubMed] [Google Scholar]
- 13.Amuzie C. J., Pestka J. J. Suppression of insulin-like growth factor acid-labile subunit expression—a novel mechanism for deoxynivalenol-induced growth retardation. Toxicological Sciences. 2010;113(2):412–421. doi: 10.1093/toxsci/kfp225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Voss K. A. A new perspective on deoxynivalenol and growth suppression. Toxicological Sciences. 2010;113(2):281–283. doi: 10.1093/toxsci/kfp287. [DOI] [PubMed] [Google Scholar]
- 15.Wan D., Wang X., Wu Q., et al. Integrated transcriptional and proteomic analysis of growth hormone suppression mediated by trichothecene T-2 toxin in rat GH 3 cells. Toxicological Sciences. 2015;147(2):326–338. doi: 10.1093/toxsci/kfv131. [DOI] [PubMed] [Google Scholar]
- 16.Krishnaswamy R., Devaraj S. N., Padma V. V. Lutein protects HT-29 cells against deoxynivalenol-induced oxidative stress and apoptosis: prevention of NF-κB nuclear localization and down regulation of NF-κB and cyclo-oxygenase-2 expression. Free Radical Biology and Medicine. 2010;49(1):50–60. doi: 10.1016/j.freeradbiomed.2010.03.016. [DOI] [PubMed] [Google Scholar]
- 17.Van De Walle J., Romier B., Larondelle Y., Schneider Y.-J. Influence of deoxynivalenol on NF-κB activation and IL-8 secretion in human intestinal Caco-2 cells. Toxicology Letters. 2008;177(3):205–214. doi: 10.1016/j.toxlet.2008.01.018. [DOI] [PubMed] [Google Scholar]
- 18.Karin M., Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-κB activity. Annual Review of Immunology. 2000;18(1):621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 19.Steinbrecher K. A., Wilson W., Cogswell P. C., Baldwin A. S. Glycogen synthase kinase 3β functions to specify gene-specific, NF-κB-dependent transcription. Molecular and Cellular Biology. 2005;25(19):8444–8455. doi: 10.1128/MCB.25.19.8444-8455.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morgan M. J., Liu Z. G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Research. 2011;21(1):103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu X., Guo P., Liu A., et al. Nitric oxide (NO)-mediated mitochondrial damage plays a critical role in T-2 toxin-induced apoptosis and growth hormone deficiency in rat anterior pituitary GH3 cells. Food and Chemical Toxicology. 2017;102:11–23. doi: 10.1016/j.fct.2017.01.017. [DOI] [PubMed] [Google Scholar]
- 22.Kowaltowski A. J., Vercesi A. E. Mitochondrial damage induced by conditions of oxidative stress. Free Radical Biology and Medicine. 1999;26(3-4):463–471. doi: 10.1016/S0891-5849(98)00216-0. [DOI] [PubMed] [Google Scholar]
- 23.Lee J., Giordano S., Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. The Biochemical Journal. 2012;441(2):523–540. doi: 10.1042/BJ20111451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang X., Jiang L., Geng C., Cao J., Zhong L. The role of oxidative stress in deoxynivalenol-induced DNA damage in HepG2 cells. Toxicon. 2009;54(4):513–518. doi: 10.1016/j.toxicon.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 25.Chaudhari M., Jayaraj R., Bhaskar A. S. B., Lakshmana Rao P. V. Oxidative stress induction by T-2 toxin causes DNA damage and triggers apoptosis via caspase pathway in human cervical cancer cells. Toxicology. 2009;262(2):153–161. doi: 10.1016/j.tox.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 26.Dvorska J. E., Pappas A. C., Karadas F., Speake B. K., Surai P. F. Protective effect of modified glucomannans and organic selenium against antioxidant depletion in the chicken liver due to T-2 toxin-contaminated feed consumption. Comparative Biochemistry and Physiology Part C: Toxicology & Pharmacology. 2007;145(4):582–587. doi: 10.1016/j.cbpc.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 27.Wu J., Tu D., Yuan L. Y., Yuan H., Wen L. X. T-2 toxin exposure induces apoptosis in rat ovarian granulosa cells through oxidative stress. Environmental Toxicology and Pharmacology. 2013;36(2):493–500. doi: 10.1016/j.etap.2013.03.017. [DOI] [PubMed] [Google Scholar]
- 28.Buss H., Dörrie A., Schmitz M. L., Hoffmann E., Resch K., Kracht M. Constitutive and interleukin-1-inducible phosphorylation of p65 NF-κB at serine 536 is mediated by multiple protein kinases including IκB kinase (IKK)-α, IKKβ, IKKϵ, TRAF family member-associated (TANK)-binding kinase 1 (TBK1), and an unknown kinase and couples p65 to TATA-binding protein-associated factor II31-mediated interleukin-8 transcription. Journal of Biological Chemistry. 2004;279(53):55633–55643. doi: 10.1074/jbc.m409825200. [DOI] [PubMed] [Google Scholar]
- 29.Eberhardt W., Kunz D., Hummel R., Pfeilschifter J. Molecular cloning of the rat inducible nitric oxide synthase gene promoter. Biochemical and Biophysical Research Communications. 1996;223(3):752–756. doi: 10.1006/bbrc.1996.0968. [DOI] [PubMed] [Google Scholar]
- 30.Bensassi F., el Golli-Bennour E., Abid-Essefi S., Bouaziz C., Hajlaoui M. R., Bacha H. Pathway of deoxynivalenol-induced apoptosis in human colon carcinoma cells. Toxicology. 2009;264(1-2):104–109. doi: 10.1016/j.tox.2009.07.020. [DOI] [PubMed] [Google Scholar]
- 31.Ji G. E., Park S. Y., Wong S. S., Pestka J. J. Modulation of nitric oxide, hydrogen peroxide and cytokine production in a clonal macrophage model by the trichothecene vomitoxin (deoxynivalenol) Toxicology. 1998;125(2-3):203–214. doi: 10.1016/s0300-483x(97)00178-9. [DOI] [PubMed] [Google Scholar]
- 32.Bouaziz C., Martel C., Sharaf el dein O., et al. Fusarial toxin-induced toxicity in cultured cells and in isolated mitochondria involves PTPC-dependent activation of the mitochondrial pathway of apoptosis. Toxicological Sciences. 2009;110(2):363–375. doi: 10.1093/toxsci/kfp117. [DOI] [PubMed] [Google Scholar]
- 33.Mariappan N., Elks C. M., Sriramula S., et al. NF-κB-induced oxidative stress contributes to mitochondrial and cardiac dysfunction in type ii diabetes. Cardiovascular Research. 2010;85(3):473–483. doi: 10.1093/cvr/cvp305. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon request.