

Abstract
The feasibility of utilizing the cell surface chemokine receptor CXCR4 for human immunodeficiency virus (HIV) entry inhibition and as an intracellular portal for targeted drug delivery was evaluated. Novel DV3 ligands (1DV3, 2DV3, and 4DV3) were designed, synthesized and conjugated to various probes (fluorescein isothiocyanate (FITC) or biotin) and cargos with sizes ranging from 10 to 50 nm (polyethylene glycol (PEG), streptavidin, and a polymeric nanoparticle). 4DV3 conjugated probes inhibited HIV-1 entry into the CXCR4- expressing reporter cell line TZM-bl (IC50 at 553 nM) whereas 1DV3 and 2DV3 did not. 4DV3 also inhibited binding of anti-CXCR4 antibody 44708 to TZM-bl cells with nanomolar potency, while the small-molecule CXCR4 antagonist AMD3100 did not. Molecular modeling suggested simultaneous binding of a single 4DV3 molecule to four CXCR4 molecules. Differences in CXCR4-binding sites could explain the discrete inhibitory effects observed for 4DV3, the 44708 antibody and AMD3100. In the Sup-T1 cell chemotaxis assay, the 4DV3 ligand functioned as a CXCR4 allosteric enhancer. In addition, 4DV3 ligand-conjugated cargos with sizes ranging from 10 to 50 nm were taken up into CXCR4-expressing Sup-T1 and TZM-bl cells, demonstrating that CXCR4 could serve as a drug delivery portal for nanocarriers. The uptake of 4DV3 functionalized nanocarriers combined with the allosteric interaction with CXCR4 suggests enhanced endocytosis occurs when 4DV3 is the targeting ligand. The current results indicate that 4DV3 might serve as a prototype for a new type of dual function ligand, one that acts as a HIV-1 entry inhibitor and a CXCR4 drug delivery targeting ligand.
Keywords: CXCR4, DV3, allosteric enhancer, HIV-1 entry inhibitor, AMD3100, nanocarrier, targeted drug delivery
Introduction
CXCR4 is a member of the chemokine receptor family, which is a subset of the rhodopsin superfamily of G protein-coupled receptors that bind to small protein ligands called chemokines [1–3]. CXCR4 is critical to numerous endogenous functions like T cell activation and migration, embryogenesis, brain development, hematopoiesis, and vascularization [4–13]. The chemokine molecule CXCL12 (or SDF-1α) is the physiological ligand of CXCR4. The CXCL12-CXCR4 signaling axis plays a principal role in the retention/homing of hematopoietic stem and progenitor cells (HSPCs) in/to the bone marrow and the migration/mobilization of some of these cells into peripheral tissues upon tissue injury or stress. Besides its importance to normal physiology and homeostasis, CXCR4 plays a significant role in several pathologies. CXCR4 is used by some strains of human immunodeficiency virus (HIV) as a co-receptor to infect host cells and its overexpression is associated with metastasis of numerous cancers [13–17].The CXCL12-CXCR4 axis is also associated with oncogenesis as well as the pathogenesis of inflammatory diseases and fibro-proliferative diseases [18, 19].
Disruption of CXCL12-CXCR4 interactions is a strategy that has long been pursued for intervention in these pathological conditions [20, 21]. For example, administration of the small- molecule CXCR4 antagonist AMD-3100 (plerixafor) reduced CXCL12 signaling and retention within the bone marrow [21]. CXCL12-CXCR4 signaling is also affected by CXCR4 homo- and hetero-oligomerization and is subject to allosteric modulation [18, 22, 23]. Finally, chemotaxis is the most important cellular activity resulting from CXCL12-CXCR4 signaling, as with the majority of all chemokine receptors. Recently, a pro-inflammatory macrophage migration inhibitory factor (MIF) was identified as the second physiological ligand of CXCR4 and shown to mediate chemotaxis and arrest of CXCR4-expressing T cells, complicating the understanding of CXCR4-mediated signaling and associated biological processes [22].
CXCL12 is rapidly cleared from the blood circulation, by the combined processes of proteolysis and sequestration through binding to heparin on surrounding CXCL12-secreting cells [22]. Nevertheless, damaged or stressed tissues produce and release large amounts of CXCL12 resulting in elevated blood concentrations. Its fast clearance, however, does create a local CXCL12 concentration gradient around CXCL12-secreting cells, thus enabling chemotaxis/homing of CXCR4-expressing immune or stem/progenitor cells, or the arrest of infiltrating leukocytes at the blood-brain barrier during CNS inflammation [24].
An HIV-1 virion, by its size and shape, somewhat resembles a nanoparticle (NP) with an approximate size of 145 nm [25, 26]. Its structure allows for both protection of its cargo [27, 28] and its interaction with target cells (e.g., CD4+ T cells, macrophages, dendritic and microglial cells) through specific ligands displayed on its surface. This interplay is mediated by viral proteins gp120 and gp41, and host CD4 and chemokine receptors (CXCR4 or CCR5) [29]. Often hitchhiking on infected immune cells, HIV-1 is capable of crossing physiological barriers (e.g., the blood-brain barrier, the blood-testes barrier) and penetrating tissues (e.g., gut-associated lymphoid tissue, mucosa-associated lymphoid tissue and other lymphoid tissues) that are traditionally difficult to access with therapeutic agents [30–32]. The communication among the negatively charged amino acid residues of CXCR4, in the N-terminus and extracellular loops, is critical to the interaction with the positively charged amino acids of gp120 [33–35], as well as CXCL12 [36]. Inhibition of the interface between gp120 and CXCR4 by CXCL12 has been shown to reduce viral entry and replication in vitro [37–39]. Conceptually, the HIV-1 entry process demonstrates that it is feasible for a NP with a properly designed targeting ligand to be transported by means of a cell surface receptor into a cell.
Viral macrophage inflammatory protein-II (vMIP-II) is a virus-encoded chemokine that can bind to the major HIV-1 co-receptors CCR5 and CXCR4 [40, 41]. Synthetic peptides derived from the N-terminus of vMIP-II retain affinity for CXCR4, while also demonstrating anti-HIV activity [42, 43]. The derivatives of vMIP-II are antagonists to CXCL12 and HIV-1, yet do not activate CXCR4 receptors [44]. Preliminary research showed significant antagonistic activity of a positively charged, ten-amino acid-long segment of the vMIP-II, the V3 peptide (Leu-Gly-Ala- Ser-Trp-His-Arg-Pro-Asp-Lys). This peptide binds to CXCR4, albeit with an affinity 1400 times less than CXCL12 [41, 42, 44]. In a monoclonal antibody (mAb) competitive binding assay, D- amino acid isomers of V3 (DV3) improved serum stability as well as CXCR4 binding (by 13.7- fold), as compared to its parent 21-residue L-amino acid V1 peptide [45]. The DV3 peptide is unique in that it enhances affinity upon stereoconversion (inverso) from L- to D-amino acids, without reversing the amino acid sequence [45, 46]. DV3 has been used in anti-cancer applications, as part of fusion proteins containing proapoptotic segments that target cancer cells overexpressing CXCR4, resulting in cell death [47, 48] Recently, in an effort to increase DV3’s potency, DV1-K-(DV3) was identified and evaluated [49]. This peptide consists of DV1 and DV3 linked together through lysine. DV1-K-(DV3) exhibited a significant improvement in CXCR4 binding affinity compared to the monovalent DV3 (IC50 of 4 nM vs. 440 nM) and demonstrated micromolar potency in anti-HIV-1 activity (IC50 of about 1 μΜ).
Nanocarriers with multivalent ligand moieties may be advantageous versus monovalent ligands in disrupting the interaction between CXCR4 and its ligands such as HIV-1 and CXCL12, due to the enhanced avidity for the receptor as a result of interacting with multiple receptors [50–53]. This clustering event could also benefit receptor mediated uptake of NPs for drug delivery. A single copy of ligand bound to the bulky carrier group like a branched polyethylene glycol (PEG) can sterically interfere with ligand interaction with its cellular receptor, leading to its reduced binding avidity relative to free peptide; however, this interference can be overcome by multivalency of ligands [50, 53]. (see Fig. 1)
Figure 1.
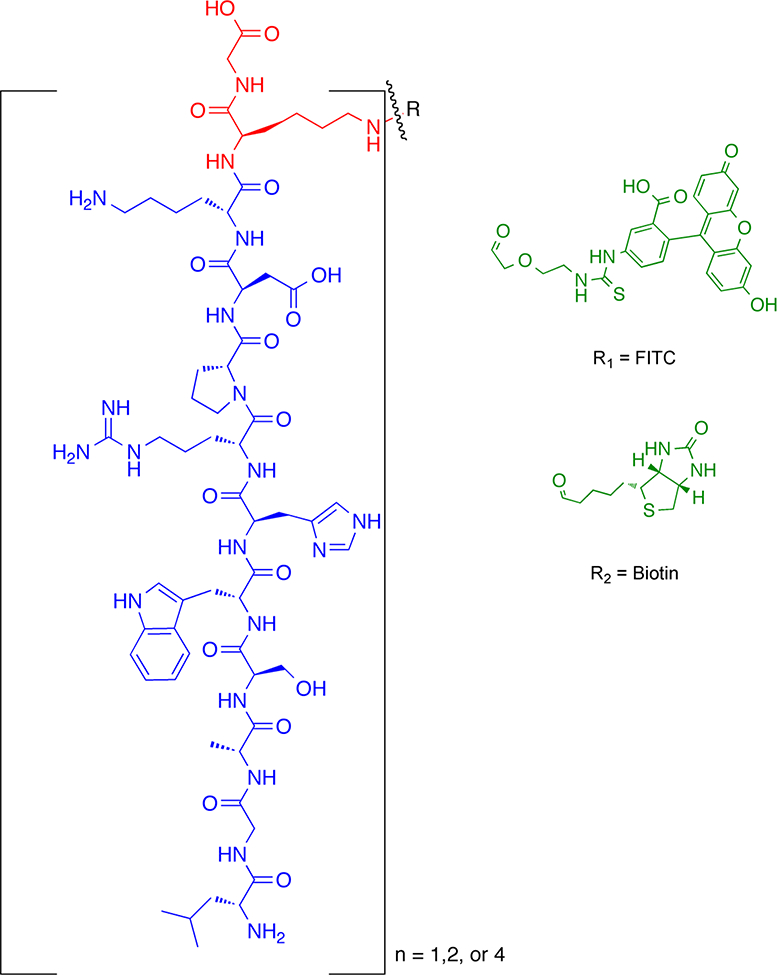
Schematic representation of DV3 (n = 1, 2, 4) conjugates. The DV3 sequence is in blue, the Lys peptidyl core in red, and the tag in green.
In the current investigation, novel DV3 ligands were conjugated to various probes and cargos with sizes ranging from 10 to 50 nm. The constructs were then evaluated for their ability to prevent HIV entry and to utilize CXCR4 as a drug delivery portal. The results of the current study demonstrate that 4DV3 conjugated to various cargos can not only block HIV entry into cells but also enter cells via CXCR4, which serves as a portal for drug delivery. Unexpectedly, 4DV3 demonstrated a novel allosteric enhancement of CXCR4 activity, suggesting that the ligand-receptor interaction altered the configuration of CXCR4 enabling it to be a drug delivery portal. This finding may also prove to be clinically useful, specifically for the mobilization of HSPCs from bone marrow or homing of these cells to tissue repair sites. Given the significance of CXCR4 in HIV/AIDS and cancer, further probing of this mechanism may lead to better utilization of CXCR4 as a drug delivery portal. The results of the current investigation suggest that 4DV3, through a novel allosteric interaction, may play a role in a dual mechanism anti-HIV drug delivery system (i.e., HIV entry inhibition and utilizing CXCR4 as a portal).
Materials and Methods
Materials
Reagents for solid-phase peptide synthesis are listed in the Supplementary Information. The following reagents were used for labeling and conjugation: fluorescein isothiocynate (FITC) (Chem-Impex International, Wood Dale, IL); 10 kDa linear PEG (Nanocs, New York, NY); FITC-labeled streptavidin (FITC-SA) (AnaSpec, Fremont, CA); α-amino ω−hydroxyl terminated polycaprolactone(6000)-b-polyethylene glycol(5000) (PCL-b-PEG) (Polymer Source, Dorval, Canada); and a click chemistry reagent DBCO-PEG5-NHS (Quanta Biodesign, Plain City, OH).
Some biologics and chemicals were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH, and included: TZM-bl cells from Dr. John C. Kappes, Dr. Xiaoyun Wu, and Tranzyme Inc.; HTLV-IIIB/H9 (HIV-1 IIIB) from Dr. Robert Gallo; HIV-1 BaL from Dr. Suzanne Gartner, Dr. Mikulas Popovic and Dr. Robert Gallo; CXCR4 mAb clone 44708 and Sup-T1 cells from Dr. Dharam Ablashi; and bicyclam JM-2987 (the octahydrobromide salt of AMD-3100, referred to as AMD3100 throughout this paper). The following reagents were purchased for in vitro studies: Steady Glo® Luciferase System (Promega, Madison, WI); Rhodamine-dextran 10,000 Da (Rho-DEX), Hank’s Balanced Salt Solutions (HBSS), Dulbecco’s Modified Eagle Medium (DMEM), RPMI 1640 Medium (RPMI), heat- inactivated fetal bovine serum (FBS), penicillin-streptomycin (100x, PenStrep), phosphate buffered saline (PBS), non-essential amino acids (NEAA) and LysoTracker (Life Technologies, Grand Island, NY); SuperBlock Blocking Buffer (Thermo Fisher Scientific, Rockford, IL); 5- carboxyfluorescein-streptavidin (FAM-SA) and 4’,6-diamidino-2-phynylindole dihydrochloride (DAPI) (AnaSpec); goat anti-mouse IgG conjugated with horseradish peroxidase (HRP) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA); 3,3’,5,5’-tetramethylbenzidine (TMB) substrate solution (eBioscience.com, San Diego, CA); and 3-(4,5,-dimethylthiazol-2-yl)-2,5- diphenyltetrazolium bromide (MTT) (Sigma-Aldrich, St. Louis, MO).
Synthesis of DV3 conjugates and nanoparticles
DV3 conjugates were synthesized utilizing Fmoc solid phase peptide synthesis followed by standard cleavage protocol. The compounds were purified by semi-preparative HPLC and the fractions were analyzed by HPLC-MS. The fractions containing the desired compounds were pooled and lyophilized. Synthetic details and structures of the individual compounds appear in the Supplementary Information.
To emulate NPs to which 4DV3 could be conjugated, FITC-SA (MW ~ 60 kDa) and linear PEG (MW ~ 10 kDa) were used as surrogates. 4DV3-Biotin (4DV3-Biot) was mixed with FITC-SA, to allow for the formation of FITC-SA-4DV3-Biot (F-SA-4DV3). 4DV3-PEG12-N3 was reacted with FITC-PEG-DBCO to form FITC-PEG-4DV3 (F-PEG-4DV3), via copper-free click chemistry. Both F-SA-4DV3 and F-PEG-4DV3 were purified and analyzed by HPLC and MALDI-TOF MS and showed at least 90% purity.
4DV3, as a ligand on NP surfaces, was covalently conjugated to PCL-PEG diblock copolymer via copper-free click chemistry. PCL-b-PEG was derivatized by DBCO-PEG5-NHS in the presence of N,N-diisopropylethylamine (DIEA). This was followed by click chemistry reaction with 4DV3-PEG12-N3. The polymer-ligand conjugate was then premixed with unmodified PCL-b-PEG prior to preparation of 4DV3-conjugated NPs, to provide the desired surface coverage of either 1 or 5%.
HIV-1 entry inhibition assay
HIV-1 entry reporter cell line TZM-bl cells were maintained in DMEM containing 10% FBS/100 U/ml of PenStrep/1X NEAA (the medium). Cells were plated to 96-well plates at a density of 2.5 × 103 cells/well in triplicate and allowed to attach overnight. The medium was replaced using serial dilutions: for 4DV3-FITC, from 5 to 0 μΜ; for CXCR4 mAb clone 44708, from 167 to 0 nM; and for AMD3100, from 25 to 0 nM. The plates were then incubated for 1 hour. The set of wells of cells without any treatment (0 nM) were designated as the control. HIV-1 IIIB or HIV-1 BaL, at a multiplicity of infection (MOI) of 0.038 in DMEM, was added to the treated wells, while the medium with no virus was added to the control wells. Cells were incubated for 72 hours at 37 °C with 5% CO2. After lysing, luminescence was measured using Steady Glo® reagent.
Cytotoxicity studies
Cell viability was determined using the MTT assay (56). TZM-bl cells were seeded to 96-well plates at a density of 1.5 × 103 cells/well and allowed to attach to the well surfaces overnight. The medium was replaced with various concentrations of 1DV3-FITC, 2DV3-FITC, or 4DV3-FITC, to designated triplicate wells. This was immediately followed by incubation for 72 hours at 37 °C with 5% CO2. Control cells were left untreated. The contents of the wells were replaced with PBS containing 5 mg/ml of MTT and incubated for at least 3 hours at 37 °C. The formazan crystals that formed were dissolved in DMSO. Absorbance at 570 nm was then measured. The absorbance was normalized (100%) to the untreated controls.
Confocal microscopy
TZM-bl cells were seeded to a chambered cover glass at a density of 1.0 × 104 cells/chamber and allowed to attach overnight. 1DV3-FITC, 2DV3-FITC, 4DV3- FITC, or F-SA-4DV3 were added to individual chambers, at a concentration of 1 μΜ in HBSS containing 1 ng/ml DAPI (a DNA dye) and 250 ng/ml Rho-DEX (a fluid phase endocytosis marker). The cells were incubated for 2 hours (1.5 hours for F-SA-4DV3) at 37 °C. Cells were washed with PBS, fixed with 10% formalin and rewashed with PBS. Cells were then imaged in PBS on a Leica TCS SP5 confocal microscope (Leica Microsystems, Buffalo Grove, IL). The brightness of each color was adjusted to the same extent for all images using Adobe PhotoShop software or Zeiss fluorescent microscopy SPOT software, to ensure good presentation and unbiased comparison. For TZM-bl cells treated with 4DV3 nanoparticles, an Olympus IX81 inverted confocal microscope (Olympus America, Center Valley, PA) was used. Again, color brightness was similarly corrected among all images, to assure their clear display and provide equitable contrasting.
CXCR4 specificity studies
CXCR4 specificity was studied using biotin-labeled 4DV3 (4DV3- Biot) and three known CXCR4-binding compounds: (1) AMD3100, (2) CXCR4 mAb clone 44708, and (3) DV3. Since the DV3 peptide in the 4DV3-FITC conjugate intramolecularly quenched FITC fluorescence, 4DV3-Biot was substituted for 4DV3-FITC.
(A). Inhibition of 4DV3-Biot uptake into TZM-bl cells by AMD3100.
TZM-bl cells were plated to a 24-well plate, in 0.75 ml at a cellular density of 1.0 × 105 cells/ml, to triplicate wells in medium. Cells were allowed to attach overnight. AMD3100 was added to the medium at the final concentration of either 20 μΜ or 200 μM, while control wells were left untreated. The plate was incubated for 30 min at 37 °C and washed with HBSS. Cells were treated for 1 hour at 37 °C with HBSS (control), 1 μM 4DV3-Biot, 1 μM 4DV3-Biot + 20 μM AMD3100, or 1 μM 4DV3- Biot + 200 μM AMD3100, then washed with HBSS. Cells were incubated with 20μg/ml of FAM-SA in HBSS for 30 min at room temperature and washed with HBSS. Cells were lysed with 2N NaOH overnight, neutralized with 2 N HCl, and then the lysate transferred to a black 96-well plate. Fluorescence was measured on a microplate reader at Ex 485 nm/Em 535 nm.
(B). Inhibition of CXCR4 mAb-binding to TZM-bl cells by 4DV3-Biot and AMD3100.
TZM- bl cells were plated to triplicate wells of a 96-well plate at 2.0 × 103 cells/well. Cells were incubated until they reached confluence. Cells were fixed with 10% formalin, washed with PBS, and incubated with serial dilutions of 4DV3-Biot or AMD3100 in 1 μg/ml CXCR4 mAb in SuperBlock buffer for 40 min on ice. Control cells were treated with SuperBlock buffer only. Cells were washed three times with TBST (0.1% (v/v) Tween 20 in tris-buffered saline) and incubated with the goat anti-mouse IgG conjugated with HRP diluted to 1:50000 at room temperature for 1 hour. Cells were washed three times with TBST and incubated with TMB solution. Enzymatic conversion was stopped with 0.1N HCl, and absorbance at 450 nm was measured. The IC50 was calculated after normalizing absorbance to both control cells (0%), and cells treated with CXCR4 mAb alone (100%). Calculations were made using GraphPad Prism (GraphPad Software, La Jolla, CA), after non-linear curve fitting with the software’s dose- response equation.
(C). Inhibition of 4DV3-Biot uptake into TZM-bl cells by free DV3.
TZM-bl cells were seeded to 6 wells of a 24 well plate at 1.0 × 105 cells/well and allowed to attach. One pair of wells was left untreated as control. A second pair of wells was treated with 1 μΜ 4DV3-Biot in HBSS. A third pair of wells was treated with 1 μM 4DV3-Biot/100 μM DV3. The plate was incubated for 1 hour at 37 °C with 5% CO2. Cells were washed with PBS, fixed with formalin, then washed with PBS again. Cell membranes were permeated by using 0.2% Triton-X 100 in PBS. Cells were washed with PBS and incubated with 10μg/ml of FAM-SA in PBS for 1 hour at room temperature. The cells were then lysed overnight with 2N NaOH, neutralized with 2N HCl, and the lysate transferred to an opaque microplate. The fluorescence was measured at Ex 485 nm/Em 535 nm. Relative intracellular fluorescence for the wells treated with 1 μΜ 4DV3- Biot/100 μΜ DV3 was calculated and normalized to cells treated with 4DV3-Biot only.
Molecular modeling of 4DV3-CXCR4 interactions
A 4DV3-bound CXCR4 structure was modeled using Discovery Studio 3.5 software suite (Accelrys, Inc., San Diego, CA). The dimeric X-ray structure of CXCR4 (PDB entry ID: 3ODU, consisting of A and B chains) was used as the starting data set [54] Two identical copies of CXCR4 dimers were positioned to form a tetramer that would bind to one 4DV3 molecule (the ligand). The 4DV3 ligand was manually docked to the CXCR4 tetramer, using a set of H-bond interactions published previously by Choi and his colleagues. Their publications describe the binding of a bivalent DV1 ligand (DV1 is a 21-amino acid parent peptide, encompassing DV3 and derived from vMIP-II) [55]. For energy minimization and molecular dynamics calculations, CHARMM force field was used to provide a conformer for the CXCR4 –4DV3 complex at an energy level of −107566.2 kcal/mol, and with a satisfactory gradient tolerance of 0.0001 kcal/mol Å. The molecular dynamics simulation was performed for 100 ps, at a temperature of 300K.
Chemotaxis assay
Sup-Tl cells were maintained in RPMI 1640 medium, supplemented with 10% FBS and 100 units/ml of PenStrep. Cells were counted using a hematocytometer and washed once by centrifugation with serum-free RPMI medium. The cell pellet was then resuspended in RPMI medium containing 0.5% BSA to a density of 20 × 106 cells/ml. 0.15 ml of cell suspension was added to the top compartment of each Transwell insert (8-micron pore size) that was seated in 24-well plates. Each well was filled with 0.6 ml of RPMI medium/0.5% BSA, with or without different concentrations of a test chemoattractant (CXCL12, FC131 or 4DV3). The plates were incubated in a CO2 incubator for 2 hours, to allow for cell migration through the insert membrane and into the wells. The inserts were discarded and migrated cells from each well were counted for 2–10 times. Two inserts were used for each treatment per experiment.
Cellular uptake of 4DV3-conjugated macromolecules
TZM-bl cells were plated to 12-well plates at 5 × 105 cells/well and allowed to attach overnight. Cells were treated with medium containing 1 ng/ml DAPI, 0.2 μΜ LysoTracker, and 2 μΜ of 4DV3-FITC, F-SA-4DV3 or F- PEG-4DV3, with or without 100 μM AMD3100. After treatment, cells were incubated for 3 hours with 5% CO2. Cells were then washed twice with HBSS, detached by trypsinization, and washed once again with HBSS. The cell pellet was then resuspended in 0.5 ml of 10% formalin, for analysis by flow cytometry on their cell-associated green fluorescence. Analysis was conducted with blanking (gating), using cells not treated with FITC-labeled compounds. The value was expressed as the percentage of cell-associated fluorescence relative to that of the cells incubated with no AMD3100, i.e., the fluorescence level of cells treated with 4DV3-FITC, F- SA-4DV3 or F-PEG-4DV3, but without AMD3100, was set at 100%.
Preparation of 4DV3-conjugated nanoparticles:
Plain and 4DV3-conjugated NPs were prepared using Flash Nanoprecipitation. For plain NPs, vitamin E (0.05 mg), PCL-b-PEG (2.5 mg) and coumarin-6 (2.5 mg) were dissolved in THF (1 mL). For 4DV3 NPs with either 1 or 5% surface coverage, 1 or 5% (by weight) of PCL-b-PEG was replaced by the corresponding amount of 4DV3-conjugated PCL-b-PEG. Equal volumes of the THF stream and ultrapure water were rapidly mixed, by injecting into a dual-inlet mixer. The resulting solution was immediately dispersed into ultrapure water (final THF:water 1:9 by volume) and dialyzed against ultrapure water (1:100 volume of nanoparticle sample to volume of water) overnight. NP sizes and zeta potentials were measured by dynamic light scattering (DLS) using a Zetasizer Nano Series DLS (Malvern, UK).
Cellular uptake of 4DV3-conjugated nanoparticles
TZM-bl cells were seeded on 24-well cell culture plates, at a density of 1 × 105 cells/well. After 24 hours, different concentrations of plain, 1 or 5% 4DV3 NPs were added to the wells and incubated for one hour. Cells were washed twice with HBSS. To prepare the samples for flow cytometry, cells were detached from each well via trypsinization and neutralization, and centrifuged at 300 g for 2 min. After the supernatant was discarded, each cell pellet was resuspended in HBSS (200 μL) and transferred to a 96-well microplate. The fluorescence intensity of the cells was recorded for 5000 cell events for each group, using a CytoFLEX flow cytometer (Beckman Coulter, USA).
Statistical analysis
Statistical analyses were performed using GraphPad Prism, using either One Way ANOVA analysis and Tukey’s post-hoc test, or a two-tailed t test between columns. Statistical significance was determined by obtaining a p value of < 0.05. Nonlinear regression analysis from three separate experiments was also performed, using a downhill dose-response model. Statistical analyses between curves and curve parameters were determined by F test, at a value of p < 0.05.
Results
Synthesis of multivalent DV3 conjugates:
1DV3-FITC, 2DV3-FITC, and 4DV3-FITC conjugates, as well as free DV3 peptide and 4DV3-conjugated macromolecules, were successfully synthesized and thoroughly characterized (See Supplementary Information). The purified conjugates were stored lyophilized. For cellular assays, stock solutions were prepared with sterile PBS and stored in aliquots, to avoid excessive freeze-thaw cycles.
HIV-1 entry inhibition by DV3 conjugates:
1DV3-FITC, 2DV3-FITC, and 4DV3-FITC were investigated as HIV-1 entry inhibitors using the TZM-bl HIV entry reporter cell line. TZM-bl luminescence, as compared to the untreated controls (-HIV-1 IIIB group). 4DV3-FITC inhibited the HIV-1 induced luciferase luminescence, as compared to the +HIV-1 IIIB group (p < 0.001), at the tested concentrations of 0.5, 2.5 and 5.0 μΜ (Fig. 2A). However, 1DV3-FITC and 2DV3- FITC were unable to inhibit luminescence at any of the tested concentrations. At a conjugate concentration equivalent to 10 μΜ of 1DV3 peptide, 2.5 μΜ of 4DV3-FITC significantly inhibited viral entry, but 5 μΜ of 2DV3-FITC did not. Similarly, 0.5 μΜ of 4DV3-FITC (equivalent to 2.0 μΜ of 1DV3) significantly inhibited viral entry, but 2.5 μΜ of 1DV3-FITC (equivalent to 2.5 μΜ of 1DV3) did not. Therefore, at concentrations equal to or less than that of 1DV3-equivalent, the four-copy conjugate exhibited HIV entry inhibitory activity, while 2- and 1-copy conjugates did not. This suggests multivalent enhancement of 4DV3-FITC binding to CXCR4, a hallmark of avidity [56]. The results demonstrate that at least 4 copies of the DV3 peptide per conjugate were necessary to inhibit HIV-1 entry, at the concentrations tested.
Figure 2. 4DV3-FITC inhibits luciferase activity in HIV-1 entry reporter cell line TZM-bl demonstrating HIV entry inhibition.
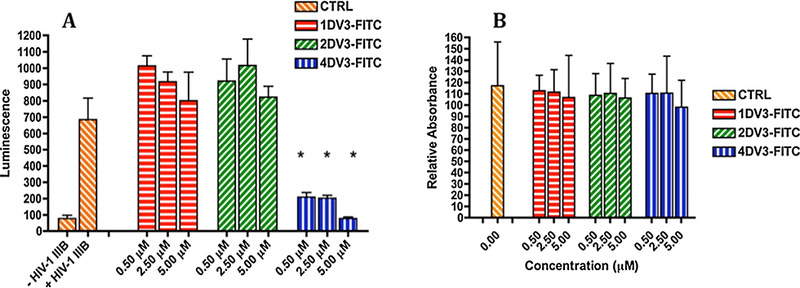
(A) Luciferase activity of 1DV3-FITC, 2DV3-FITC and 4DV3-FITC conjugates. Values are mean ±SD of three independent experiments, with each treatment in an experiment consisting of triplicate wells. Control (CTRL) indicates the wells of cells incubated with no conjugates. The “*” symbol indicates statistical difference (p < 0.001) by One Way ANOVA from “+ HIV-1 MB” in the figure (the CTRL wells exposed to HIV-1 IIIB). Each bar in a group corresponds to a treatment with a DV3 conjugate, at a concentration of 0.5, 2.5, or 5 μΜ. 4DV3-FITC significantly inhibited HIV-1 entry at each of the concentrations investigated. (B) MTT assay activity of 1DV3-FITC, 2DV3-FITC and 4DV3-FITC. Values are mean ± SD of three independent experiments, with each treatment in an experiment consisting of triplicate wells. The values are relative to the CTRL, treated with no conjugates. Each bar in a group corresponds to a treatment with a DV3 conjugate at the concentration of 0.5,2.5, or 5 μM. One Way ANOVA analysis showed no statistical difference between the CTRL and any of the treatment bars.
Concentration-dependent viral entry inhibition by 4DV3-FITC was further investigated in TZM-bl cells, in the absence and presence of known CXCR4 antagonists. The IC50 of 4DV3- FITC for the X4 strain HIV-1 IIIB calculated was 553 nM, with a 95% confidence interval (95% CI) from 246 nM to 1.24 μΜ (Fig. 3A). The IC50 of viral entry of 4DV3-FITC against the R5 strain (HIV-1 BaL) was approximately 10-fold weaker than that of the X4 strain. The calculated IC50 was 4.32 μM (95% CI 2.85 to 6.55 μM) (Fig. 3B). The difference between the X4 and R5 viral strains entry inhibition indicates 4DV3-FITC’s selectivity for CXCR4 over CCR5. Comparison of 4DV3-FITC entry inhibition of HIV-1 IIIB to that of AMD3100 demonstrated that AMD3100 was more potent (1000-fold), with an IC50 equal to 0.415 nM (95% CI = 0.219 –0.785 nM) (Fig. 3C). However, the inhibition (IC50) of HIV-1 entry by the CXCR4 mAb clone 44708 was only about 5-fold better than 4DV3, at 106 nM (95% CI = 60 –187 nM, Fig. 3D). The influence of the FITC tag on the performance of 4DV3 was investigated. 4DV3 (without any tag) was evaluated for anti-HIV-1 activity, measured by both viral replication (p24 production) and entry reporter (luciferase) activity in TZM-bl cells. Neither of these two tested activities was influenced by the presence of FITC (data not shown). The results indicated that 4DV3-FITC inhibited viral entry in the TZM-bl reporter cell system, in a CXCR4-dependent manner.
Figure 3. 4DV3-FITC, as well as known compounds that bind to CXCR4, inhibited HIV-1 entry into TZM-bl reporter cells.
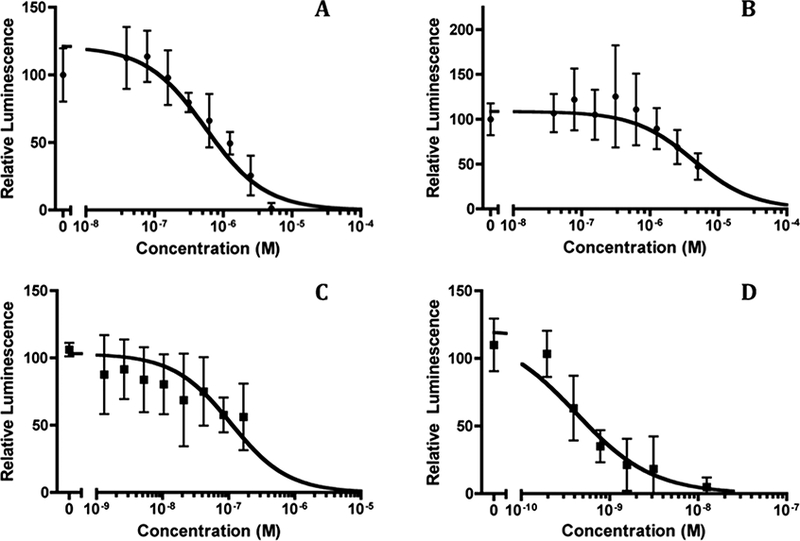
Luminescence values are expressed as values relative to the control wells treated with no compounds. Each panel depicts concentration-dependent inhibition by a compound, compiled from at least three independent experiments (mean ± SD). Non-linear curve fitting was carried out using GraphPad’s Prism software and its dose-response equation, yielding the IC50 values. (A) IC50 of 4DV3-FITC against the X4 strain (HIV-1 MB) is 553nM, with a 95% confidence interval (95% CI) of 246 nM to 1.24 μΜ. (B). IC50 of 4DV3-FITC against the R5 strain (HIV-1 BaL) is 4.32 μM (95% CI = 2.85 –6.55 μM). (C) IC50 of CXCR4 mAb clone 44708 against the HIV-1 IIIB was 106 nM (95% CI = 60 –187 nM). (D) IC50 of AMD3100 against HIV-1 IIIB is 0.415 nM (95% CI = 0.219 –0.785 nM).
Cytotoxicity of DV3 conjugates:
Cell viability was determined by the MTT assay, in the presence of 1DV3-FITC, 2DV3-FITC, or 4DV3-FITC. At all tested concentrations, none of the DV3 conjugates significantly reduced cell viability, compared to the untreated controls (Fig. 2B). The results from the MTT assays demonstrate that the observed anti-HIV activity (i.e., the reduction of luminescence) resulted from viral entry inhibition, and not from a loss in cell viability.
Cellular uptake of DV3 conjugates:
Confocal microscopic images (Fig. 4) depicted that TZM-bl cells treated with 1DV3-FITC had very weak intracellular fluorescence, and cells treated with 2DV3-FITC showed only faint intracellular green fluorescence. By comparison, cells treated with 4DV3-FITC displayed strong intracellular fluorescence. All conjugates exhibited very slight cell surface green FITC fluorescence, indicating that the fluorescence consisted almost exclusively of internalized conjugates. Co-localization of 4DV3-FITC with Rho-Dex (a fluid phase endocytosis marker) was observed in the merged images, suggesting that 4DV3-FITC internalization occurred through endocytosis. This hypothesis is consistent with the known fact that CXCR4 and its bound ligand undergo endocytosis together, following ligand engagement [18].
Figure 4.

4DV3-FITC entered TZM-bl cells by endocytosis and was mainly entrapped in intracellular vesicles. Shown are confocal microscope images of middle sections along the cell Z-axis, acquired in the XYZ mode. TZM-bl cells were incubated with DV3 conjugates, as described in the text. Rows represent 1DV3-FITC, 2DV3-FITC, or 4DV3-FITC, respectively. Columns represent images acquired using the bright field light, fluorescent DAPI (blue), fluorescent FITC (green), fluorescent rhodamine (red), or the blue-green-red merged image (Merged). Red fluorescence originated from the fluid phase endosome marker Rho-Dex. The red fluorescence also shows up in yellow, when co-localized with the green FITC fluorescence in the merged image.
CXCR4 specificity of 4DV3-Biot:
To further assess the specificity of 4DV3 interaction with CXCR4 on TZM-bl cells, competition for CXCR4-interaction between 4DV3-Biot and three known CXCR4 ligands in TZM-bl cells was investigated. Precisely, the three CXCR4 ligands were AMD3100, the CXCR4 mAb clone 44708, and DV3 (equivalent to 1DV3-FITC). 4DV3- Biot was used instead of 4DV3-FITC, because the fluorescence of 4DV3-FITC was weak due to intramolecular fluorescence quenching. The quantity of cell-associated 4DV3-Biot was determined by measuring fluorescence, after non-fluorescent 4DV3-Biot was complexed with fluorescent 5-FAM-streptavidin. The amount of cell-associated CXCR4 mAb clone 44708 was assessed by the horseradish peroxidase (HRP) activity of an HRP-conjugated secondary Ab, one that binds to the mAb. Only at relatively high concentrations did AMD3100 inhibit 4DV3-Biot uptake into TZM-bl cells. At 200 μΜ, AMD3100 inhibited cell-associated 4DV3-Biot by 47%, and at 20 μΜ, by 31% (Fig. 5A). By contrast, 4DV3-Biot was able to inhibit the binding of the CXCR4 mAb to TZM-bl cells at much lower concentrations. Fig. 5B shows that 4DV3-Biot inhibited the mAb’s binding, in a concentration-dependent manner. The IC50 of 4DV3-Biot against 1 μg/ml of CXCR4 mAb 44708 was 398 nM, with a 95% confidence interval of 101nM to 1.57μΜ. In contrast, AMD3100 demonstrated poor ability to inhibit mAb binding to TZM-bl cells (Fig. 5B). Uptake of 4DV3-Biot into TZM-bl cells was inhibited by a 100-fold molar excess of free DV3, which has previously shown a specificity for CXCR4 [45] (Fig. 5C). There was a 65% reduction in cell-associated fluorescence, compared to that which occurred in the absence of the free DV3. The weak competitive inhibition of DV3 to 4DV3-Biot is consistent with the inferior HIV-1 entry inhibition (Fig. 2 and 4) and intracellular fluorescence of 1DV3- FITC, compared to 4DV3-FITC (Fig. 4). Altogether, the data indicate that 4DV3 is specific for CXCR4.
Figure 5. Competition for CXCR4-interaction between 4DV3-Biot and known CXCR4 ligands in TZM-bl cells.

(A) Inhibition of 4DV3-Biot uptake into TZM-bl cells by the small molecule CXCR4 antagonist AMD3100. Values are fluorescence units of FAM-SA, in cell lysates relative to that in the lysate of cells without treatment with AMD3100. Significant inhibition is seen at 20 and 200 μΜ, respectively, by One Way ANOVA (* p< 0.001 and ** p<0.05). (B) Inhibition of CXCR4 mAb clone 44708 binding to TZM-bl cells by 4DV3-Biot or AMD3100. While AMD3100 was ineffective at the tested concentrations, 4DV3-Biot was effective, with an IC50 at 398 nM and a 95% CI = 101nM to 1.57μΜ. Non-linear curve fitting was carried out using GraphPad’s Prism software and its dose-response equation, yielding IC50 values. (C) DV3 at 100 μΜ inhibited 4DV3-Biot uptake (1 μΜ). CTRL: no 4DV3-Biot.
Molecular modeling of 4DV3-CXCR4 interactions:
To gain more insight into the binding site on the CXCR4 receptor, molecular modeling of 4DV3 interaction with CXCR4 was performed. The modeling was based on: (1) the proposed binding site on CXCR4 by another multivalent vMIP-II derivative, and (2) the effect of point mutations of CXCR4 on DV3 binding, respectively [55, 57]. In the 4DV3-(CXCR4)4 bound structure, four individual DV3 chains in 4DV3 contact four individual CXCR4 receptors (Fig. 6A). Interactions between 4DV3 chains 1 and 3 with receptor chains A and C were modeled as follows: Leu1 of 4DV3 formed a hydrogen bond with Asp171 of CXCR4, Ser4 with Arg188, His6 with Asp193, Arg7with Glu32, and Asp9 with Cys28(Fig. 6B). For the interaction of 4DV3 chains 2 and 4 with CXCR4 chains B and D, the binding residues were as follows: Leu1 of 4DV3 formed a hydrogen bond with Glu288 of CXCR4, His6 with Asp193, and Trp5 with Arg188. The simulations demonstrate the ability of 4DV3 to reach into the binding pocket of multiple receptors, simultaneously. Overall, the structural model shows that 4DV3 can bind to 4 CXCR4 receptors at once. This characteristic explains the avidity effect of 4DV3 (4DV3-FITC) in HIV-1 entry inhibition, as compared to free DV3 (1DV3-FITC) at molar equivalent or higher concentrations (Fig. 2).
Figure 6. 4DV3 interacts with multiple binding sites and pockets of CXCR4 clusters.
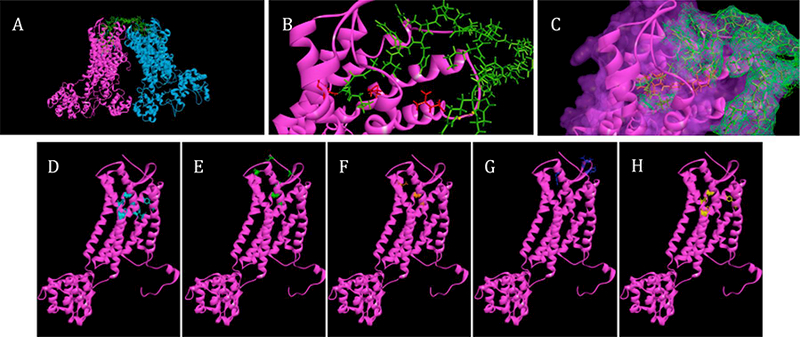
(A) Molecular models of 4DV3 binding to CXCR4 tetramer. CXCR4 receptors are represented as two sets of dimers (PDB 3ODU): one dimer is in magenta and the other is in blue. 4DV3 is represented in green, with each of the DV3 sequences interacting with a receptor binding pocket. (B) A closer view of the interaction between one CXCR4 receptor and one DV3 sequence of 4DV3. (C) Mesh surface representation of a DV3 peptide sequence binding to CXCR4, and displacing AMD3100 (orange). (D) CXCR4 residues identified from alanine scanning, that effect the binding of DV3 peptide (cyan) [55]. (E) CXCR4 residues identified from molecular dynamics, that are important to DV3 binding (green) [55]. (F) CXCR4 binding residues of AMD3100 (orange) [64]. (G) Binding epitope of the CXCR4 mAb clone 44708 (purple) [68]. (H) CXCR4 residues important to the binding of HIV-1 gp120, from the strain IIIB (yellow) [55]. All images generated using Accelrys Discovery Studio software, after 100ps simula t ions at 300K.
Effect of 4DV3 on CXCL12-elicited chemotaxis:
The above results of HIV entry inhibition and CXCR4 specificity were based on the concept of 4DV3 binding to the CXCR4 receptor. To evaluate 4DV3’s CXCR4 signaling property, a chemotaxis assay was performed using Sup-Tl cells and FC131, a known CXCR4 antagonist peptide, was used as control. The physiological agonistic ligand CXCL12 at 50 nM increased chemotaxis by 2.0 to 2.5-fold (Fig. 7). The CXCL12-elicited chemotaxis, however, was reduced to the control level in the presence of 0.5 μΜ FC131 (Fig. 7A), demonstrating that FC131 is an antagonist, as shown previously by others [58] Interestingly, FC131 was neither an agonist nor an allosteric agonist (Fig. 7A). By contrast, 4DV3 boosted CXCL12-elicited chemotaxis of Sup-Tl cells by 1.6-fold, while 4DV3 without CXCL12 did not alter chemotactic activity as compared to the control (Fig. 7B). Therefore, 4DV3 demonstrated a novel allosteric agonistic property towards CXCR4.
Figure 7. 4DV3 boosts CXCL12-elicited chemotaxis, in Sup-T1 T cells.
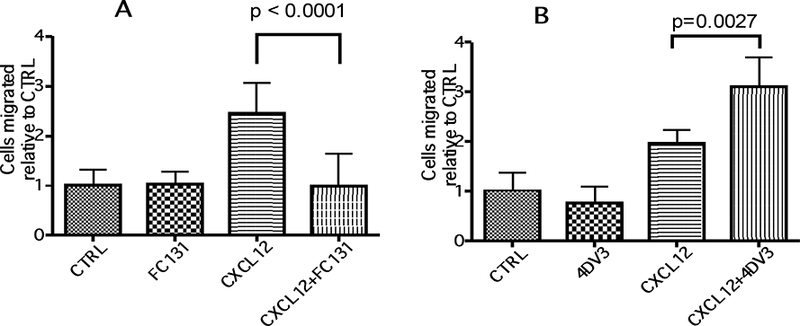
The number of cells migrated in the absence of any test agent was designated as CTRL (expressed as the reference value of 1 in each experiment). The number of cells migrated in the presence of a test agent (FC131 at 0.5 μΜ, CXCL12 at 50 nM, 4DV3 at 0.5 μΜ, a combination of CXCL12/FC131, or a combination of CXCL12/4DV3) are labeled as such (expressed as values relative to that of CTRL, in each experiment). The value of each column represents the mean ± SEM of five independent experiments, each consisting of duplicates of Transwell inserts. (A) Cells treated with FC131 and its control agents. (B) Cells treated with 4DV3 and its control agents.
Cellular uptake of 4DV3-conjugated cargos:
To test whether 4DV3 would be able to deliver a nano-sized cargo into CXCR4-expressing cells, three different cargos with sizes ranging from 10 to 50 nm were studied: (homo-tetrameric) streptavidin, 10-kDa linear PEG and PCL-b-PEG polymeric NP. FITC-SA was complexed with 4DV3-Biot, resulting in F-SA-4DV3 (MW 58.5 kDa). FITC-labeled PEG was conjugated to 4DV3, producing F-PEG-4DV3. Since 4DV3 is highly positively charged, it would be expected that the four copies of 4DV3-Biot in F-SA-4DV3 to be in an extended conformation, due to charge-charge repulsion. As a result, the whole complex is estimated to have a diameter at around 10 nm, compared to the known and comparably sized 80 kDa globular transferrin molecule (10 nm in diameter). Since the hydrodynamic volume of linear PEG is 3 to 10 times greater than that of a globular protein of the same molecular weight [59], F-PEG-4DV3 should have a diameter of about 10 nm, as well. For PCL-b-PEG NPs, the size and zeta potential of NPs in aqueous solution were determined by DLS (Table 1). The number-average diameters of 1% and 5% 4DV3-conjugated NPs were 50.46 and 39.86 nm, respectively, and was not significantly different from that of plain NPs. All the NPs were negatively charged, consistent with other reports on characterization of PCL-PEG NPs [60, 61].
Table 1.
The average diameters, polydispersity indices and zeta potentials of plain and 4DV3- conjugated polymeric NPs (n=3). Numbers are shown as mean ± SD.
| Nanoparticles | Diameter (nm) |
Polydispersity index (PDI) |
Zeta potential (mV) |
|---|---|---|---|
| Plain NP | 41.38 ± 1.78 | 0.246 ±0.010 | −8.74 ±0.48 |
| 1% 4DV3 NP | 50.46 ±2.73 | 0.158 ±0.024 | −12.2 ±0.85 |
| 5% 4DV3 NP | 39.86 ±5.38 | 0.308 ±0.021 | −10.2 ±0.41 |
TZM-bl cells were incubated for 3 hours with 2 μΜ of 4DV3-FITC, F-SA-4DV3, or F- PEG-4DV3, in medium containing DAPI/Lysotracker, in the presence or absence of 100 μΜ of AMD3100. The cells were washed and fixed, followed by flow cytometry and confocal microscopy. All three FITC-labeled conjugates/complex were taken up by TZM-bl cells, but the uptake was essentially abolished by 50x-molar excess of AMD3100 (uptake reduced by 98.2% for 4DV3-FITC, by 96.6% for F-SA-4DV3, and by 97.8% for F-PEG-4DV3). This indicated that all three 4DV3-containing conjugates/complexes were taken up in a CXCR4-dependent manner (Fig. 8). F-SA-4DV3 was localized inside the cytosol with poor cell surface association, demonstrating that the uptake observed in the flow cytometry represented the internalized F-SA- 4DV3 (Fig. 9). Although the intensities of the green fluorescence of 4DV3-FITC and F-PEG- 4DV3 were less than that of F-SA-4DV3, these two conjugates were also localized in the cytosol, with little cell surface binding (images not shown). Therefore, these data show that 4DV3 was indeed able to deliver the two large cargos into the TZM-bl cells.
Figure 8.

AMD3100 abolished cell-associated green fluorescence in TZM-bl cells treated for 3 hours with 2 μM of 4DV3-FITC, F-SA-4DV3, or F-PEG-4DV3, in the absence or presence of AMD3100 (100 μΜ). The treated TZM-bl cells were then washed and analyzed by flow cytometry.
Figure 9. Confocal microscopic images of TZM-bl cells treated with F-SA-4DV3.
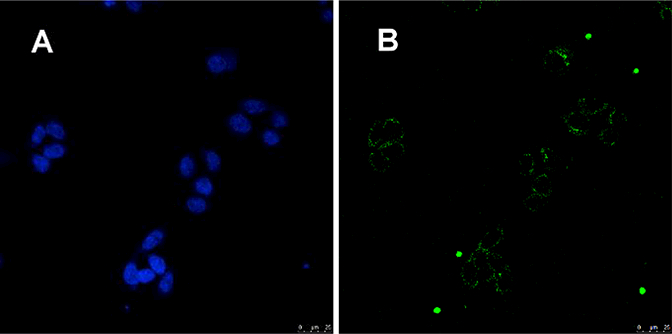
The cells were incubated with 2 μΜ of F-SA-4DV3 for 3 hours, then washed and subjected to confocal microscopic examination. A middle section in Z stacks is shown. (A) Blue fluorescence image showing DAPI-bound cell nuclei; (B) Green fluorescence image showing punctate F-SA-4DV3 within the cytosol, with no noticeable cell surface binding.
To investigate the ability of 4DV3 as a surface ligand to enhance cellular uptake of NPs, NPs were prepared using 4DV3-conjugated di-block copolymer and the fluorescent marker coumarin-6. Using flow cytometry, cell-associated fluorescence was compared with that of plain (surface-unmodified) NPs. The cellular uptake of NPs in TZM-bl cells was improved by surface modification with 4DV3, as both 4DV3-conjugated NP groups showed greater cell-associated fluorescence than plain NPs at different concentrations (Fig. 10).
Figure 10. Cellular uptake of plain and 4DV3-modified NPs.
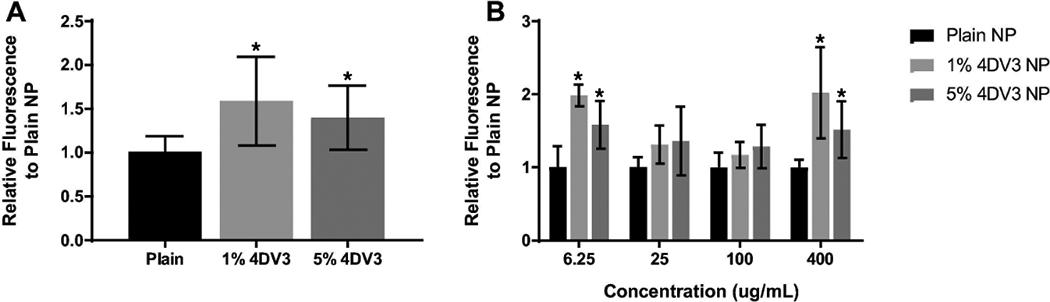
at (A) all concentrations, and (B) various concentrations of NPs. The fluorescence intensity relative to that of plain NPs is presented as mean ± SD of at least five independent experiments. One-way ANOVA was used for graph (A). Two-way ANOVA, followed by Dunnett’s multiple comparisons test, was used for graph (B). An asterisk on a bar indicates a statistical significance (p < 0.05) vs. plain NP.
Discussion
In the current studies, novel multimeric DV3 ligands based on a branched peptide core were synthesized and conjugated to fluorescent probes, biotin or nano-sized cargos in order to investigate their ability to target CXCR4. The DV3 conjugates were designed for use as HIV entry inhibitors and drug delivery targeting ligands. Earlier studies of DV3 demonstrated its ability to inhibit the CXCR4 mAb clone 12G5, but it exhibited weak inhibitory activity of HIV-1 cell entry (V3) [44, 45]. Here, we report its ability to inhibit HIV-1 entry when DV3 is in a multiple copy (i.e., multimeric) format. 4DV3-FITC was shown to have more significant cellular uptake (Fig. 4) and better HIV-1 cell entry inhibition (Fig. 2) than 1DV3-FITC and 2DV3-FITC. The observations made are consistent with trends from previous work done both by our lab and others, supporting the hypothesis that multivalent ligand-receptor interactions improve the cellular association of targeted nanocarriers [51, 53]. The studies also demonstrated that enhanced anti-HIV activity and cellular uptake was dependent on the number of DV3 targeting moieties per conjugate molecule, not the overall concentration of the DV3 targeting moiety, a well-accepted hallmark of avidity [56].
While this study was in progress, a bivalent peptidyl CXCR4 ligand DV1-K-(DV3), which was derived from the same chemokine vMIP-II as was our 4DV3, was published [49] Excluding the core lysine linker, DV1-K-(DV3) is comprised of 31 amino acids, and whereas the tetravalent 4DV3 has 40 amino acids. Similar avidity principles and the use of a lysine core linker were employed in designing both DV1-K-(DV3) and 4DV3 to allow for simultaneous engagement of more than one CXCR4 receptor. A direct comparison of the results of the two peptides is impossible due to the different experimental methods that were used, especially different mAb’s and cell lines used in competitive mAb binding to CXCR4-expressing cells and different cell lines and assays used in anti-HIV-1 activity studies. Nevertheless, the results were similar in that multivalency of a CXCR4 ligand led to greater binding affinity and anti-HIV-1 activity. In terms of the latter, 4DV3 exhibited an IC50 of 553 nM, suggesting 4DV3 is a better anti-HIV-1 peptide than DV1-K-(DV3) (~1 μΜ). Another difference between these two peptidyl CXCR4 ligands is that DV1-K-(DV3) is an antagonist while 4DV3 is an allosteric agonist.
In addition to HIV entry inhibition, 4DV3 was able to deliver three large cargos: the proteinaceous FITC-SA, the polymeric FITC-PEG and the block-co-polymer based PCL-b-PEG NP. Delivery of FITC-SA and FITC-PEG that were conjugated to 4DV3 was made into CXCR4- expressing TZM-bl cells, in a CXCR4-dependent matter (Fig. 8). Confocal images showed that 4DV3-FITC and F-SA-4DV3 had been transported inside the cells, most likely in endosomes (not shown). When the molecular target of a delivered drug is located in the cytosol/nuclear compartment, an endosomal escape mechanism can be engineered into the NP [62] An anti-HIV drug could therefore use 4DV3 to target the viral CXCR4 co-receptor. Moreover, it is conceivable that there could be an additive effect between the entry inhibition property of 4DV3 and the pharmacology of the drug encapsulated within NPs, an effect that could interrupt the HIV life cycle. Our data has shown that different types of nanocarriers could enter cells in a CXCR4-mediated manner. The mechanism of cell entry is likely through endocytosis. Further studies will be needed to find the optimal size of a nanocarrier, which will depend on its physicochemical characteristics as well as the type and mechanism of endocytosis.
4DV3 displayed nanomolar level inhibition toward CXCR4 mAb. AMD3100 was unable to significantly reduce 4DV3-binding to CXCR4, except at high concentrations (200 μΜ). In addition, the difference between the inhibition of 4DV3 binding by mAb and AMD3100 may be attributed to two factors: (1) the size of the molecule (AMD3100 MW = 502 Da, 4DV3-Biot MW = 5.4 kDa and mAb MW = 150 kDa, respectively); and (2) the binding sites of each molecule.
Peptides containing DV3 sequences have been shown to interact with the following CXCR4 residues: Tyr45, Phe87, Asp97, Tyr121, Asp171, Trp252, Tyr255, Glu288 and the N-terminus (Fig. 6D and 6E) [63] The binding of AMD3100 to CXCR4 receptors requires Asp171, Asp262 and Glu288, and overlaps several of the binding residues for 4DV3 (Fig. 6F) [64–66]. Mutations of Asp171, Glu179, Asp181, Tyr184, Tyr190 and Asp193 of CXCR4 reduce the binding capacity of the CXCR4 mAb (Clone 44708) (Fig. 6G) [67, 68]. Insights derived from molecular modeling showed the capacity of 4DV3 to interact with multiple CXCR4 molecules, simultaneously resulting in a capping effect on the receptor tetramer. This binding conformation would explain the competitive inhibition of the mAb, despite a lack of overlap in the binding sites. The extended binding surface of 4DV3 could explain the different inhibitory effects observed for mAb and AMD3100, as well as its anti-viral activity. The steric hindrance from the size and binding proximities of the 4DV3 and the CXCR4 mAb may account for the inhibition of binding observed, based on concentration of 4DV3.
By comparison, the small size of AMD3100 and its lack of binding to the N-terminus weakly inhibited 4DV3-CXCR4 interactions. Yet in contrast to its poor inhibition of the mAb binding, AMD3100 was much more potent than 4DV3 in HIV-1 entry inhibition (Fig. 3). The IC50 of AMD3100 against the X4 strain (HIV-1 IIIB) was 0.4 nM, versus an IC50 of 553 nM of 4DV3-FITC. This may be fully explained by the overlap of AMD3100 binding sites on CXCR4 with that of the X4 HIV-1 strains [65, 67]. Therefore, the analysis suggests that 4DV3 could be an effective CXCR4-targeting moiety on a drug delivery carrier such as a NP, but not a potent HIV-1 entry inhibitor, due to the lack of sufficient overlap of its binding site with the HIV- binding site on CXCR4. This suggestion is supported by the moderate anti-HIV-1 activity that was discovered on a DV1-DV3 dimer, containing two DV3 sequences (IC50 at ~ 1 μM) [49]. The potential synergy between simultaneously blocking HIV-1 and delivering anti-HIV drug cargoes is a novel and potentially promising approach worthy of further study.
4DV3 by itself did not influence chemotaxis of Sup-T1 cells (i.e., not an agonist). Unlike FC131, however, 4DV3 boosted CXCL12-elicited chemotaxis. This behavior conforms to the classification of an allosteric enhancer [69]. To our knowledge, this is the first reported true CXCR4 allosteric enhancer that binds from the extracellular side of the receptor and is not derived from the activation domain of CXCL12 (cf. [70, 71]). These characteristics make it suitable for use as a CXCR4-targeted ligand on a NP. Consequently, this type of allosteric enhancement could have the advantage over standard orthosteric drugs, since allosteric modulators alone are less likely to interfere with the physiological response of normal cells that express the cognate receptor. In theory, this type of allosteric enhancer can be used either alone, or as an adjuvant drug to a CXCR4 antagonist (such as AMD3100) in HSPC mobilization [21, 70]. Although both are based on the same principle of mobilizing CXCR4-expressing HSPCs from bone marrow, the mechanism is different. AMD3100 works by disrupting HSPC retention in the bone marrow. In contrast, this novel type of allosteric enhancement directly mobilizes HSPCs. It could also be used in disease conditions where CXCL12 is beneficial to tissue repair, such as in cardiac infarction [72], and CNS inflammation-mediated tissue damage [24].
Further study of 4DV3’s allosterism may reveal a new step in CXCR4 signaling, where the allosteric agonism occurs. This revelation could help guide the design of new GPCR-targeted drugs, ones that would avoid the pitfalls of orthosteric ligands (i.e., binding to the same site on a GPCR as that of the receptor’s natural ligand) [73]. For example, this discovery could lead to screening for molecules that suppress signaling at this step, which could uncover a new type of antagonist. Such an “allosteric” antagonist could, in turn, have a therapeutic use, such as antagonizing cancer metastasis, which relies on CXCR4 activation.
In conclusion, the uptake of 4DV3 functionalized nanocarriers combined with the allosteric interaction with CXCR4 suggests enhanced endocytosis occurs when 4DV3 is the targeting ligand. The current results indicate that 4DV3 might serve as a prototype for a new type of dual function ligand, one that acts as a HIV-1 entry inhibitor and as a CXCR4 drug delivery targeting ligand.
Supplementary Material
Acknowledgements :
This work was supported by the National Institute of Health (R37 AI051024, R01 AI117776, R01 CA155061); the PhRMA Foundation Pre-doctoral Fellowship in Pharmaceutics; the AFPE Josiah Kirby Lilly, Sr. Memorial Pre-Doctoral Fellowship in Pharmaceutics; and the Parke-Davis Endowed Chair in Pharmaceutics and Drug Delivery. We thank Rutgers University’s Flow Cytometry/Cell Sorting & Confocal Microscopy Core Facility for their assistance. The following reagent was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: TZM-bl from Dr. lohn C. Kappes, Dr. Xiaoyun Wu and Tranzyme, Inc.
Abbreviations:
- MALDI-TOF
matrix-assisted laser desorption ionization - time-of-flight
- HSPC
hematopoietic stem and progenitor cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Federsppiel B, Melhado IG, Duncan AM, Delaney A, Schappert K, Clark-Lewis I, Jirik FR, Molecular cloning of the cDNA and chromosomal localization of the gene for a putative seven-transmembrane segment (7-TMS) receptor isolated from human spleen, Genomics, 16 (1993) 707–712. [DOI] [PubMed] [Google Scholar]
- [2].Baggiolini M, Chemokines and leukocyte traffic, Nature, 392 (1998) 565–568. [DOI] [PubMed] [Google Scholar]
- [3].Baggiolini M, Dewald B, Moser B, Interleukin-8 and related chemotactic cytokines--CXC and CC chemokines, Adv Immunol, 55 (1994) 97–179. [PubMed] [Google Scholar]
- [4].Ma Q, Jones D, Borghesani PR, Segal RA, Nagasawa T, Kishimoto T, Bronson RT, Springer TA, Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice, Proc Natl Acad Sci U S A, 95 (1998) 9448–9453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hernandez-Lopez C, Varas A, Sacedon R, Jimenez E, Munoz JJ, Zapata AG, Vicente A, Stromal cell-derived factor 1/CXCR4 signaling is critical for early human T-cell development, Blood, 99 (2002) 546–554. [DOI] [PubMed] [Google Scholar]
- [6].Segret A, Rucker-Martin C, Pavoine C, Flavigny J, Deroubaix E, Chatel MA, Lombet A, Renaud JF, Structural localization and expression of CXCL12 and CXCR4 in rat heart and isolated cardiac myocytes, J Histochem Cytochem, 55 (2007) 141–150. [DOI] [PubMed] [Google Scholar]
- [7].Lu M, Grove EA, Miller RJ, Abnormal development of the hippocampal dentate gyrus in mice lacking the CXCR4 chemokine receptor, Proc Natl Acad Sci U S A, 99 (2002) 7090–7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bagri A, Gurney T, He X, Zou YR, Littman DR, Tessier-Lavigne M, Pleasure SJ, The chemokine SDF1 regulates migration of dentate granule cells, Development, 129 (2002) 4249–4260. [DOI] [PubMed] [Google Scholar]
- [9].Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR, Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development, Nature, 393 (1998) 595–599. [DOI] [PubMed] [Google Scholar]
- [10].Petit I, Jin D, Rafii S, The SDF-1-CXCR4 signaling pathway: a molecular hub modulating neo-angiogenesis, Trends Immunol, 28 (2007) 299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rosu-Myles M, Gallacher L, Murdoch B, Hess DA, Keeney M, Kelvin D, Dale L, Ferguson SS, Wu D, Fellows F, Bhatia M, The human hematopoietic stem cell compartment is heterogeneous for CXCR4 expression, Proc Natl Acad Sci U S A, 97 (2000) 14626–14631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Murdoch C, CXCR4: chemokine receptor extraordinaire, Immunol Rev, 177 (2000) 175–184. [DOI] [PubMed] [Google Scholar]
- [13].Le Y, Zhou Y, Iribarren P, Wang J, Chemokines and chemokine receptors: their manifold roles in homeostasis and disease, Cell Mol Immunol, 1 (2004) 95–104 [PubMed] [Google Scholar]
- [14].Feng Y, Broder CC, Kennedy PE, Berger EA, HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor, Science, 272 (1996) 872–877. [DOI] [PubMed] [Google Scholar]
- [15].Balkwill F, The significance of cancer cell expression of the chemokine receptor CXCR4, Semin Cancer Biol, 14 (2004) 171–179. [DOI] [PubMed] [Google Scholar]
- [16].Loetscher P, Moser B, Baggiolini M, Chemokines and their receptors in lymphocyte traffic and HIV infection, Adv Immunol, 74 (2000) 127–180. [DOI] [PubMed] [Google Scholar]
- [17].Baggiolini M, Loetscher P, Chemokines in inflammation and immunity, Immunol Today, 21 (2000) 418–420. [DOI] [PubMed] [Google Scholar]
- [18].Pozzobon T, Goldoni G, Viola A, Molon B, CXCR4 signaling in health and disease, Immunol Lett, 177 (2016) 6–15. [DOI] [PubMed] [Google Scholar]
- [19].Zhu Z, Ding J, Tredget EE, The molecular basis of hypertrophic scars, Burns Trauma, 4 (2016) 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Xu C, Zhao H, Chen H, Yao Q, CXCR4 in breast cancer: oncogenic role and therapeutic targeting, Drug Des Devel Ther, 9 (2015) 4953–4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Liu T, Li XB, You S, Bhuyan SS, Dong L, Effectiveness of AMD3100 in treatment of leukemia and solid tumors: from original discovery to use in current clinical practice, Experimental Hematolgy & Oncology, 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Pawig L, Klasen C, Weber C, Bernhagen J, Noels H, Diversity and Inter-Connections in the CXCR4 Chemokine Receptor/Ligand Family: Molecular Perspectives, Front Immunol, 6 (2015) 429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rajasekaran D, Groning S, Schmitz C, Zierow S, Drucker N, Bakou M, Kohl K, Mertens A, Lue H, Weber C, Xiao A, Luker G, Kapurniotu A, Lolis E, Bernhagen J, Macrophage Migration Inhibitory Factor-CXCR4 Receptor Interactions: Evidence for Partial Allosteric Agonism in Comparison with CXCL12 Chemokine, J Biol Chem, 291 (2016) 15881 –15895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Durrant DM, Williams JL, Daniels BP, Klein RS, Chemokines Referee Inflammation within the Central Nervous System during Infection and Disease, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Briggs JA, Wilk T, Welker R, Krausslich HG, Fuller SD, Structural organization of authentic, mature HIV-1 virions and cores, EMBO J, 22 (2003) 1707–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sougrat R, Bartesaghi A, Lifson JD, Bennett AE, Bess JW, Zabransky DJ, Subramaniam S, Electron Tomography of the Contact between T Cells and SIV/HIV-1: Implications for Viral Entry, PLoS Pathog, 3 (2007) e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Perelson AS, Neumann AU, Markowitz M, Leonard JM, Ho DD, HIV-1 Dynamics in Vivo: Virion Clearance Rate, Infected Cell Life-Span, and Viral Generation Time, Science, 271 (1996) 15. [DOI] [PubMed] [Google Scholar]
- [28].Marsh M, Helenius A, Virus entry: open sesame, Cell, 124 (2006) 729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kazmierski WM, Kenakin TP, Gudmundsson KS, Peptide, peptidomimetic and small- molecule drug discovery targeting HIV-1 host-cell attachment and entry through gp120, gp41, CCR5 and CXCR4, Chem Biol Drug Des, 67 (2006) 13–26. [DOI] [PubMed] [Google Scholar]
- [30].Wu D, Clement JG, Pardridge WM, Low blood-brain barrier permeability to azidothymidine (AZT), 3TC, and thymidine in the rat, Brain Res, 791 (1998) 313–316. [DOI] [PubMed] [Google Scholar]
- [31].Pomerantz RJ, Reservoirs of human immunodeficiency virus type 1: the main obstacles to viral eradication, Clin Infect Dis, 34 (2002) 91–97. [DOI] [PubMed] [Google Scholar]
- [32].Park S, Sinko PJ, P-glycoprotein and mutlidrug resistance-associated proteins limit the brain uptake of saquinavir in mice, J Pharmacol Exp Ther, 312 (2005) 1249–1256. [DOI] [PubMed] [Google Scholar]
- [33].Kajumo F, Thompson DA, Guo Y, Dragic T, Entry of R5X4 and X4 human immunodeficiency virus type 1 strains is mediated by negatively charged and tyrosine residues in the amino-terminal domain and the second extracellular loop of CXCR4, Virology, 271 (2000) 240–247. [DOI] [PubMed] [Google Scholar]
- [34].Chabot DJ, Broder CC, Substitutions in a homologous region of extracellular loop 2 of CXCR4 and CCR5 alter coreceptor activities for HIV-1 membrane fusion and virus entry, Journal of Biological Chemistry, 275 (2000) 23774–23782. [DOI] [PubMed] [Google Scholar]
- [35].Tian S, Choi WT, Liu D, Pesavento J, Wang Y, An J, Sodroski JG, Huang Z, Distinct functional sites for human immunodeficiency virus type 1 and stromal cell-derived factor 1alpha on CXCR4 transmembrane helical domains, Journal of Virology, 79 (2005) 12667–12673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Szpakowska M, Fievez V, Arumugan K, van Nuland N, Schmit JC, Chevigne A, Function, diversity and therapeutic potential of the N-terminal domain of human chemokine receptors, Biochem Pharmacol, 84 (2012) 1366–1380. [DOI] [PubMed] [Google Scholar]
- [37].Bleul CC, Farzan M, Choe H, Parolin C, Clark-Lewis I, Sodroski J, Springer TA, The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry, Nature, 382 (1996) 829–833. [DOI] [PubMed] [Google Scholar]
- [38].Oberlin E, Amara A, Bachelerie F, Bessia C, Virelizier JL, Arenzana-Seisdedos F, Schwartz O, Heard JM, Clark-Lewis I, Legler DF, Loetscher M, Baggiolini M, Moser B, The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line- adapted HIV-1, Nature, 382 (1996) 833–835. [DOI] [PubMed] [Google Scholar]
- [39].Amara A, Gall SL, Schwartz O, Salamero J, Montes M, Loetscher P, Baggiolini M, Virelizier JL, Arenzana-Seisdedos F, HIV coreceptor downregulation as antiviral principle: SDF- 1alpha-dependent internalization of the chemokine receptor CXCR4 contributes to inhibition of HIV replication, J Exp Med, 186 (1997) 139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Boshoff C, Endo Y, Collins PD, Takeuchi Y, Reeves JD, Schweickart VL, Siani MA, Sasaki T, Williams TJ, Gray PW, Moore PS, Chang Y, Weiss RA, Angiogenic and HIV- inhibitory functions of KSHV-encoded chemokines, Science, 278 (1997) 290–294. [DOI] [PubMed] [Google Scholar]
- [41].Crump MP, Elisseeva E, Gong J, Clark-Lewis I, Sykes BD, Structure/function of human herpesvirus-8 MIP-II (1–71) and the antagonist N-terminal segment (1–10), FEBS Lett, 489 (2001) 171–175. [DOI] [PubMed] [Google Scholar]
- [42].Luo Z, Fan X, Zhou N, Hiraoka M, Luo J, Kaji H, Huang Z, Structure-function study and anti-HIV activity of synthetic peptide analogues derived from viral chemokine vMIP-II, Biochemistry, 39 (2000) 13545–13550. [DOI] [PubMed] [Google Scholar]
- [43].Kumar S, Choi WT, Dong CZ, Madani N, Tian S, Liu D, Wang Y, Pesavento J, Wang J, Fan X, Yuan J, Fritzsche WR, An J, Sodroski JG, Richman DD, Huang Z, SMM- chemokines: a class of unnatural synthetic molecules as chemical probes of chemokine receptor biology and leads for therapeutic development, Chem Biol, 13 (2006) 69–79. [DOI] [PubMed] [Google Scholar]
- [44].Zhou N, Luo Z, Luo J, Hall JW, Huang Z, A novel peptide antagonist of CXCR4 derived from the N-terminus of viral chemokine vMIP-II, Biochemistry, 39 (2000) 3782–3787. [DOI] [PubMed] [Google Scholar]
- [45].Zhou NM, Luo ZW, Luo JS, Fan XJ, Cayabyab M, Hiraoka M, Liu DX, Han XB, Pesavento J, Dong CZ, Wang YL, An J, Kaji H, Sodroski JG, Huang ZW, Exploring the stereochemistry of CXCR4-peptide recognition and inhibiting HIV-1 entry with D-peptides derived from chemokines, Journal of Biological Chemistry, 277 (2002) 17476–17485. [DOI] [PubMed] [Google Scholar]
- [46].Mori M, Liu D, Kumar S, Huang Z, NMR structures of anti-HIV D-peptides derived from the N-terminus of viral chemokine vMIP-II, Biochem Biophys Res Commun, 335 (2005) 651–658. [DOI] [PubMed] [Google Scholar]
- [47].Snyder EL, Saenz CC, Denicourt C, Meade BR, Cui XS, Kaplan IM, Dowdy SF, Enhanced targeting and killing of tumor cells expressing the CXC chemokine receptor 4 by transducible anticancer peptides, Cancer Res, 65 (2005) 10646–10650. [DOI] [PubMed] [Google Scholar]
- [48].Liu Y, Li Y, Wang H, Yu J, Lin H, Xu D, Wang Y, Liang A, Liang X, Zhang X, BH3-based fusion artificial peptide induces apoptosis and targets human colon cancer, Molecular Therapy, 17 (2009) 1509–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Xu Y, Duggineni S, Espitia S, Richman DD, An J, Huang Z, A synthetic bivalent ligand of CXCR4 inhibits HIV infection, Biochem Biophys Res Commun, 435 (2013) 646–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Gunaseelan S, Gunaseelan K, Deshmukh M, Zhang X, Sinko PJ, Surface modifications of nanocarriers for effective intracellular delivery of anti-HIV drugs, Adv Drug Deliv Rev, 62 (2010) 518–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wan L, Zhang X, Pooyan S, Palombo MS, Leibowitz MJ, Stein S, Sinko PJ, Optimizing size and copy number for PEG-fMLF (N-formyl-methionyl-leucyl-phenylalanine) nanocarrier uptake by macrophages, Bioconjug Chem, 19 (2008) 28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wan L, Pooyan S, Hu P, Leibowitz MJ, Stein S, Sinko PJ, Peritoneal macrophage uptake, pharmacokinetics and biodistribution of macrophage-targeted PEG-fMLF (N-formyl- methionyl-leucyl-phenylalanine) nanocarriers for improving HIV drug delivery, Pharm Res, 24 (2007) 2110–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Pooyan S, Qiu B, Chan MM, Fong D, Sinko PJ, Leibowitz MJ, Stein S, Conjugates bearing multiple formyl-methionyl peptides display enhanced binding to but not activation of phagocytic cells, Bioconjug Chem, 13 (2002) 216–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC, Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists, Science, 330 (2010) 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Choi WT, Kumar S, Madani N, Han X, Tian S, Dong CZ, Liu D, Duggineni S, Yuan J, Sodroski JG, Huang Z, An J, A novel synthetic bivalent ligand to probe chemokine receptor CXCR4 dimerization and inhibit HIV-1 entry, Biochemistry, 51 (2012) 7078–7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Mammen M, Choi SK, Whitesides GM, Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors, Angewandte Chemie-International Edition, 37 (1998) 2755–2794. [DOI] [PubMed] [Google Scholar]
- [57].Choi WT, Tian S, Dong CZ, Kumar S, Liu D, Madani N, An J, Sodroski JG, Huang Z, Unique ligand binding sites on CXCR4 probed by a chemical biology approach: implications for the design of selective human immunodeficiency virus type 1 inhibitors, Journal of Virology, 79 (2005) 15398–15404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Tamamura H, Otaka A, Fujii N, Development of anti-HIV agents targeting dynamic supramolecular mechanism: entry and fusion inhibitors based on CXCR4/CCR5 antagonists and gp41-C34-remodeling peptides, Curr HIV Res, 3 (2005) 289–301. [DOI] [PubMed] [Google Scholar]
- [59].Pasut G, Veronese FM, State of the art in PEGylation: the great versatility achieved after forty years of research, J Control Release, 161 (2012) 461–472. [DOI] [PubMed] [Google Scholar]
- [60].Dubey N, Varshney R, Shukla J, Ganeshpurkar A, Hazari PP, Bandopadhaya GP, Mishra AK, Trivedi P, Synthesis and evaluation of biodegradable PCL/PEG nanoparticles for neuroendocrine tumor targeted delivery of somatostatin analog, Drug Deliv, 19 (2012) 132–142. [DOI] [PubMed] [Google Scholar]
- [61].Li R, Li X, Xie L, Ding D, Hu Y, Qian X, Yu L, Ding Y, Jiang X, Liu B, Preparation and evaluation of PEG-PCL nanoparticles for local tetradrine delivery, Int J Pharm, 379 (2009) 158–166. [DOI] [PubMed] [Google Scholar]
- [62].El-Sayed A, Futaki S, Harashima H, Delivery of Macromolecules Using Arginine-Rich Cell-Penetrating Peptides: Ways to Overcome Endosomal Entrapment, Aaps Journal, 11 (2009) 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Princen K, Hatse S, Vermeire K, Bridger GJ, Skerlj RT, De Clercq E, Schols D, The antiviral activity of the CXCR4 antagonist AMD3100 is independent of the cytokine-induced CXCR4/HIV coreceptor expression level, AIDS Res Hum Retroviruses, 19 (2003) 1135–1139. [DOI] [PubMed] [Google Scholar]
- [64].Gerlach LO, Skerlj RT, Bridger GJ, Schwartz TW, Molecular interactions of cyclam and bicyclam non-peptide antagonists with the CXCR4 chemokine receptor, Journal of Biological Chemistry, 276 (2001) 14153–14160. [DOI] [PubMed] [Google Scholar]
- [65].Hatse S, Princen K, Gerlach LO, Bridger G, Henson G, De Clercq E, Schwartz TW, Schols D, Mutation of Asp(171) and Asp(262) of the chemokine receptor CXCR4 impairs its coreceptor function for human immunodeficiency virus-1 entry and abrogates the antagonistic activity of AMD3100, Mol Pharmacol, 60 (2001) 164–173. [DOI] [PubMed] [Google Scholar]
- [66].Rosenkilde MM, Gerlach L-O, Hatse S, Skerlj RT, Schols D, Bridger GJ, Schwartz TW, Molecular Mechanism of Action of Monocyclam Versus Bicyclam Non-peptide Antagonists in the CXCR4 Chemokine Receptor, Journal of Biological Chemistry, 282 (2007) 27354–27365. [DOI] [PubMed] [Google Scholar]
- [67].Hatse S, Princen K, Vermeire K, Gerlach L-O, Rosenkilde MM, Schwartz TW, Bridger G, De Clercq E, Schols D, Mutations at the CXCR4 interaction sites for AMD3100 influence anti-CXCR4 antibody binding and HIV-1 entry, FEBS Letters, 546 (2003) 300–306. [DOI] [PubMed] [Google Scholar]
- [68].Carnec X, Quan L, Olson WC, Hazan U, Dragic T, Anti-CXCR4 monoclonal antibodies recognizing overlapping epitopes differ significantly in their ability to inhibit entry of human immunodeficiency virus type 1, Journal of Virology, 79 (2005) 1930–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Neubig RR, Spedding M, Kenakin T, Christopoulos A, International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on terms and symbols in quantitative pharmacology, Pharmacol Rev, 55 (2003) 597–606. [DOI] [PubMed] [Google Scholar]
- [70].Sachpatzidis A, Benton BK, Manfredi JP, Wang H, Hamilton A, Dohlman HG, Lolis E, Identification of allosteric peptide agonists of CXCR4, J Biol Chem, 278 (2003) 896–907. [DOI] [PubMed] [Google Scholar]
- [71].Yang Y, Gao M, Zhang Q, Zhang C, Yang X, Huang Z, An J, Design, synthesis, and biological characterization of novel PEG-linked dimeric modulators for CXCR4, Bioorg Med Chem, 24 (2016) 5393–5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Bromage DI, Davidson SM, Yellon DM, Stromal derived factor 1 alpha: A chemokine that delivers a two-pronged defence of the myocardium, Pharmacology & Therapeutics, 143 (2014) 305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Kenakin T, G-protein coupled receptors as allosteric machines, Receptors & Channels, 10 (2004) 51–60. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.