

Abstract
Apart from well-known functions of bile acids in digestion and solubilization of lipophilic nutrients and drugs in the small intestine, the emerging evidence from the past two decades identified the role of bile acids as signaling, endocrine molecules that regulate the glucose, lipid, and energy metabolism through complex and intertwined pathways that are largely mediated by activation of nuclear receptor farnesoid X receptor (FXR) and cell surface G protein-coupled receptor 1, TGR5 (also known as GPBAR1). Interactions of bile acids with the gut microbiota that result in the altered composition of circulating and intestinal bile acids pool, gut microbiota composition and modified signaling pathways, are further extending the complexity of biological functions of these steroid derivatives. Thus, bile acids signaling pathways have become attractive targets for the treatment of various metabolic diseases and metabolic syndrome opening the new potential avenue in their treatment. In addition, there is a significant effort to unveil some specific properties of bile acids relevant to their intrinsic potency and selectivity for particular receptors and to design novel modulators of these receptors with improved pharmacokinetic and pharmacodynamic profiles. This resulted in synthesis of few semi-synthetic bile acids derivatives such as 6α-ethyl-chenodeoxycholic acid (obeticholic acid, OCA), norursodeoxycholic acid (norUDCA), and 12-monoketocholic acid (12-MKC) that are proven to have positive effect in metabolic and hepato-biliary disorders. This review presents an overview of the current knowledge related to bile acids implications in glucose, lipid and energy metabolism, as well as a potential application of bile acids in metabolic syndrome treatment with future perspectives.
Keywords: bile acids, metabolic syndrome, farnesoid X receptor, transmembrane G protein coupled receptor 5, gut microbiota, diabetes, lipoprotein, atherosclerosis
Introduction
Metabolic syndrome is one of the major public health and clinical challenges worldwide. This complex condition constituted of insulin resistance, hyperinsulinemia, glucose intolerance, dyslipidemia, hypertension and obesity, increasing thus the incidence of cardiovascular diseases and mortality (Hanson et al., 2002). The global prevalence of metabolic syndrome largely varies depending on geographic and sociodemographic factors, as well as the diagnostic criteria used (McCracken et al., 2018). Results obtained from National Health and Nutrition Examination Survey estimated that 35% of adults in the United States, and as much as 50% of the people aged over 60, had a diagnosis of metabolic syndrome, with notably higher prevalence in women compared with men (Aguilar et al., 2015). The prevalence of type 2 diabetes mellitus is worldwide increasing health disorder and estimations indicate that is going to affect approximately 8% of the population by 2030 (Lam and LeRoith, 2012).
Both genetic and environmental factors such as high-calorie diet and low level of physical activity are evidently associated with metabolic syndrome (Kaur, 2014). The complexity of the metabolic syndrome etiology in combination with specific metabolic profile and lifestyle of each patient contribute to the lack of adequate efficacy with currently used drugs increasing thus demand for developing novel therapeutic alternatives with new mechanisms of actions.
Apart from classic functions of bile acids in digestion and solubilization of lipophilic nutrients and drugs in the small intestine (Mikov and Fawcett, 2006; Mircioiu et al., 2012), there is emerging body of evidence pointing to the role of bile acids and their derivatives as signaling, endocrine molecules that exert variety of metabolic effects through complex and intertwined pathways, thus becoming the attractive target for metabolic syndrome treatment (Taoka et al., 2016; Chávez-Talavera et al., 2017; de Boer et al., 2018; Molinaro et al., 2018; Shapiro et al., 2018).
This review will provide an overview of the current knowledge related to bile acids implications in glucose, lipid and energy metabolism as well as potential application of bile acids in metabolic syndrome treatment with recommendations for further studies.
Bile Acid Biosynthesis and Enterohepatic Recirculation
Biosynthesis of bile acids represents predominant metabolic pathway of cholesterol catabolism in human organism. Conversion of cholesterol to bile acids is a multiple enzymatic process in which hepatocytes are the only cell type which contains complete set of enzymes necessary for conversion of cholesterol steroid nucleus, side chain removal and conjugation with either glycine (∼75%) or taurine (∼25%). This results in generation of two primary bile acids: cholic acid (3α,7α,12α-trihydroxy-5β-cholan-24-oic acid, CA) and chenodeoxycholic acid (3α,7α-dihydroxy-5β-cholan-24-oic acid, CDCA). Conversion of cholesterol to bile acids involves the processes of hydroxylation, C5-C6 double bond saturation, epimerization of C3 hydroxyl group and oxidative cleavage of three carbon unit of side chain (Gioiello et al., 2014). Abovementioned reactions take place in several subcellular compartments including endoplasmic reticulum, mitochondria, cytoplasm, and peroxisomes. Even though four different biosynthetic pathways have been described, the fact that intermediates in different pathways are substrates for the same enzymes, as well as the transport of bile acids and their precursors between subcellular compartments add complexity to understanding bile acid synthesis (Vaz and Ferdinandusse, 2017).
Two predominant biosynthetic pathways in humans are so called a classical (neutral) pathway and an alternative (acidic) pathway (Figure 1). Through classical pathway, 90% of primary bile acids are synthesized. Cholesterol 7α-hydroxylase (CYP7A1) is a rate-limiting enzyme, which determines quantity of bile acid pool size, catalyzing hydroxylation of cholesterol into 7α-cholesterol. Modifications of steroid nucleus and side chain render CDCA formation. In addition, hydroxylation at C12 position by sterol 12α-hydroxylase (CYP8B1) results in CA synthesis, therefore CYP8B1 is considered as an enzyme that determines hydrophilicity of bile acid pool. In classical pathway CA and CDCA are synthesized in almost equal amounts. In alternative pathway, C27 bile acids and oxysterols produced in different cell types are transported to the liver and further metabolized. Main enzymes that manage side chain shortening and 7α-hydroxylation are mitochondrial sterol 27-hydroxylase (CYP27A1) and microsomal oxysterol 7α-hydroxylase (CYP7B1) with CDCA being the major product (Li et al., 2013; Chiang, 2015). Primary bile acids are not released into biliary tree as free carboxylic acids; instead, these molecules are activated with acetyl-coenzyme A (CoA) by bile acid CoA synthetase (BACS) and formed thioester is then linked by amide bond with amino acids, either glycine or taurine (the reaction is catalyzed by bile acid-CoA: amino acid N-acyltransferase, BAAT). This process of conjugation resulting in bile salt formation reduces cytotoxic and membranolytic potential compared to free bile acids (Chiang, 2009). In addition, whereas unconjugated bile acids are able to diffuse across membranes, bile salts are actively transported.
FIGURE 1.
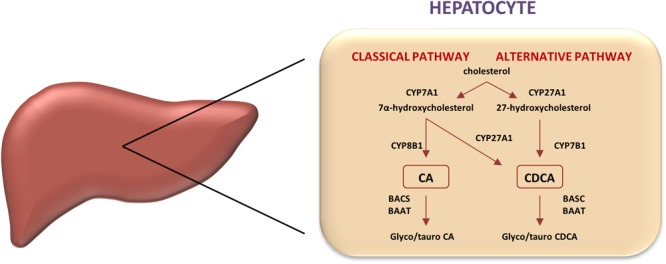
Classic and alternative pathways of bile acids synthesis. In human liver, two primary bile acids, cholic acid (CA), and chenodeoxycholic acid (CDCA), are produced. Key regulatory enzymes in both pathways are shown. Microsomal cholesterol 7α-hydroxylase (CYP7A1) catalyzes the first the rate-limiting step in the classic pathway converting cholesterol into 7-hydroxycholesterol, whereas the mitochondrial sterol 27-hydroxylase (CYP27A1) initiates the alternative pathway by converting cholesterol into 27-hydroxycholesterol, which is then 7-hydroxylated by microsomal oxysterol 7-hydroxylase (CYP7B1). CYP8B1 regulates the cholic acid synthesis in the classic pathway.
Secretion of bile salts from the hepatocyte to the lumen of biliary canaliculi is mediated by two ATP-binding cassette (ABC) proteins – bile salt export pump (BSEP, ABCB11) and multidrug resistance-associated protein-2 (MRP2, ABCC2), which represent the main force for bile secretion (Dawson et al., 2009). Mutations in ABCB11 and ABCC2 gene result in the development of type-two progressive familial intrahepatic cholestasis, and Dubin-Johnson’s syndrome, respectively (Baghdasaryan et al., 2014). Following the meal, bile acids are released into intestinal lumen by the activity of cholecystokinin, and by forming mixed micelles, they facilitate digestion and absorption of lipophilic xenobiotics. Within intestinal lumen, microflora exerts profound effects on bile acids, with the primary aim to reduce bactericidal activity. In the lumen of ileum and colon, non-absorbed bile acids undergo deconjugation and 7α-dehydroxylation by the activity of the enzymes of anaerobic microflora – the process which results in the conversion of primary to the secondary bile acids which affect the host metabolism: deoxycholic acid (3α,12α-dihydroxy-5β-cholan-24-oic acid, DCA) and lithocholic acid (3α-hydroxy-5β-cholan-24-oic acid, LCA) as well as hydrophilic ursodeoxycholic acid (3α,7β-dihydroxy-5β-cholan-24-oic acid, UDCA) which is produced by Clostridia species through 7β-epimerization of CDCA) (Ramirez-Perez et al., 2017).
Deconjugated bile acids are reabsorbed passively whereas approximately 95% of ileal bile acids are efficiently reabsorbed in the distal segments of ileum by the enterocyte apical sodium-dependent bile acid transporter (ASBT, SLC10A2). Trans-enterocyte shuttle of bile acid is facilitated by the gastrotropin, intestinal bile acid-binding protein (IBAP, FABP6), whereas basolateral heterodimeric organic solute transporter (OST) α/β facilitates efflux into the portal circulation. Hepatocytes uptake bile salts through sodium-dependent bile acid transporter (NTCP, SLC10A1) whereas non-conjugated bile acids are reuptaken by hepatocytes through sodium-independent organic anion transporters (OATPs) localized in sinusoidal membrane of hepatocyte (Zwicker and Agellon, 2013). Composition of the bile acid pool, including both the conjugated bile acids/salts following release from gallbladder and deconjugated bile acids metabolized by intestinal microflora, is variable during the day, depending upon dietary habits and composition of intestinal microbiota (Ajouz et al., 2014). Significant cytotoxic propensity of LCA results in fecal excretion of this bile acid species, and small amount of hydrophobic LCA recirculated in the liver undergoes sulfoconjugation in hepatocytes by sulfotransferase 2A1 (SULT2A1) and fecal excretion (Hofmann, 2004).
Bile acid pool is 4–14 times recycled during the day and the fraction lost through feces is compensated by de novo synthesis from cholesterol. Therefore, interruption of enterohepatic recirculation and increased excretion of bile acids represents effective therapeutic strategy in the treatment of hypercholesterolemia (Chiang, 2013). Biosynthesis of bile acids and their enterohepatic circulation with specific transmembrane transport proteins in enterocytes and hepatocytes and are clearly shown and explained in Figure 2.
FIGURE 2.
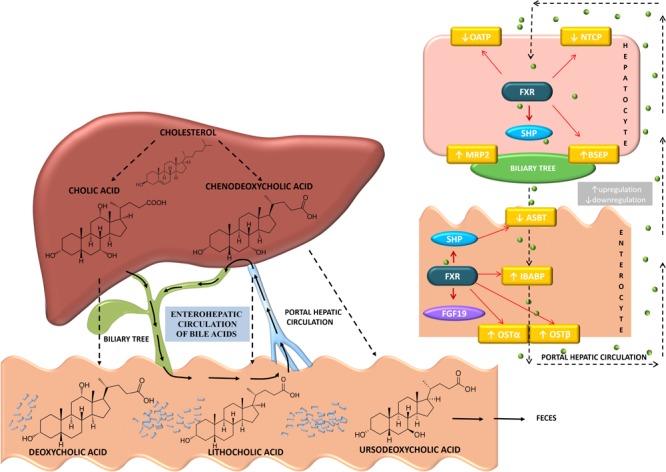
Biosynthesis and enterohepatic circulation of bile acids showing the individual transport proteins expressed in hepatocytes and enterocytes. Primary bile acids synthesized in the liver from cholesterol are cholic acid (CA) and chenodeoxycholic acid (CDCA). The primary bile acids are then conjugated with glycine or taurine in the liver and released into the gallbladder. Gut microbiota present in the intestine, by deconjugation, dehydroxylation, dehydrogenation and epimerization, modify the primary bile acids converting them to the secondary bile acids-deoxycholic acid (DCA), lithocholic acid (LCA), and ursodeoxycholic acid (UDCA). Around 95% of bile acids are reabsorbed into the distal ileum. Transporters involved in enterohepatic circulation of bile acids are apical sodium-dependent bile transporter (ASBT) present in the brush border membrane of the enterocyte, ileal bile acid-binding protein (IBABP) involved in trans-enterocyte shuttle of bile acid, basolateral organic solute transporters, OSTα/OSTβ that facilitate efflux into the portal circulation. Bile acids are actively transported into hepatocytes by sodium sodium-taurocholate cotransporting polypeptide (NTCP) and organic anion transporters (OATP) while bile salt export pump (BSEP) and multidrug resistance-associated protein-2 (MRP2) are involved in the process of exporting bile acids out of hepatocytes. Bile acids activation of FXR signaling pathways regulates the expression of these transporters. In the hepatocyte, bile acid activation of FXR increases SHP expression, decreases the expression of NTCP and OATP, while increases the expression of MRP2 and BSEP. In ileal enterocytes, bile acids signaling leads to upregulation of IBABP and OSTα/OSTβ and downregulation of ASBT. (Bile acid direction is presented by black dashed narrow.)
Bile Acids as Signaling Molecules
Enterohepatic recirculation of bile acids amplifies trans-hepatic flux of bile acids that activate farnesoid X receptor (FXR), the most significant transcription factor involved in the regulation of bile acid biosynthesis and transport. Additionally, trans-intestinal flux of reabsorbed bile acids activates intestinal FXR, regulating enterohepatic recirculation of these amphiphilic molecules. These hepatic and intestinal events prevent hepatocytes overload and accumulation of toxic concentrations of bile acids and prevent hepatocellular injury, by decreasing their own synthesis and uptake, maintaining at the same time sufficient amounts of bile acids in biliary tree and intestinal lumen for emulsification of dietary lipids. Moreover, hepatocellular uptake of bile acids is incomplete and micromolar range of concentrations spills from portal into systemic circulation through hepatic veno-venous anastomoses. These systemic concentrations are sufficient to interact with several currently identified nuclear receptors; FXR, pregnane X receptor (PXR), vitamin D receptor (VDR), and constitutive androstane receptor (CAR), as well as bile acid-coupled membrane receptor Takeda G-protein receptor-5 (TGR5, GPBAR) exerting systemic signaling effects beyond enterohepatic tissues (Gioiello et al., 2014; Comeglio et al., 2017).
Farnesoid X receptor is a metabolic nuclear receptor which primary role is to sense concentration of bile acids and prevent their accumulation to toxic concentration in hepatocyte. Human gene FXRα (NR1H1) which is located at chromosomal locus 12q23.1 encodes for FXR isoforms as a result of difference in transcription initiation site (either exon one or exon three) and alternative splicing between exons five and six (Kemper, 2011). FXR protein has been shown to be widely distributed in the organism and metabolic significance of FXR has been revealed in different cell- and tissue-types particularly by using tissue specific FXR loss-of-function rodent models (Gardmo et al., 2011). Like the other nuclear receptors, FXR contains ligand-binding domain, DNA-binding domain that binds to specific DNA sequences known as the FXR-response elements (FXREs) and co-activator and co-repressor interacting activating domains -1 and -2 (AF1, AF2) bridged by hinge region (Gioiello et al., 2014). FXR regulates the expression of target genes mostly by hetero-dimerization with retinoic acid receptor (RXR), inducing chromatin de-compaction through activating acetylation of histones or by other (epi)genetic mechanisms, resulting in the transactivation of target genes (Pavlovic et al., 2017). FXR also induces gene repression mainly through the small heterodimer partner (SHP), so called an “orphan” nuclear receptor that lacks DNA-binding domain, and binds to regulatory sequences of transcription factors leading to the transcriptional repression of target genes. In the hepatocytes, FXR-SHP axis suppresses bile acid synthesis and uptake transporters, whereas in enterocytes, FXR-SHP axis suppresses uptake and induces portal efflux of already absorbed bile acids (Ferrebee and Dawson, 2015). Ligand-dependent trans-activation of targeted genes by bile acids is induced by the binding of bile acid as the most potent endogenous ligands, with the CDCA being the most potent agonist, whereas hydrophilic UDCA does not activate FXR (Kemper, 2011). The potency of natural bile acids for FXR is summarized in Table 1 (according to Gioiello et al., 2014). In the intestine, FXR activates transcription of enterokine, fibroblast growth factor-19 (FGF-19 or rodent FGF-15 ortholog) through SHP, governing post-prandial bile acid and nutrient metabolism (Mertens et al., 2017).
Table 1.
Potency of natural bile acids for FXR biding (modified according to Gioiello et al., 2014).
| Bile acid species | Free carboxylic acid | Glycine conjugated bile salt | Taurine conjugated bile salt |
|---|---|---|---|
| CDCA (primary) | 4.5 μM | 10 μM | 10 μM |
| CA (primary) | >100 μM | >100 μM | >100 μM |
| LCA (secondary) | 3.8 μM | 4.7 μM | 3.8 μM |
| DCA (secondary) | 100 μM | >100 μM | >100 μM |
| UDCA (secondary) | >100 μM | >100 μM | >100 μM |
Additionally, as endobiotics, bile acids may activate PXR (NR1I2, chromosome 3q13.33), which possesses the large ligand-binding pocket allowing it to interact with a wide range of structurally diverse hydrophobic compounds including drugs, dietary supplements, environmental pollutants. Non-activated PXR most commonly creates inhibitory complexes with silencing mediator for retinoic acid receptor and thyroid hormone receptor (SMRT), SHP and histone deacetylases (Pavlovic et al., 2017). Upon ligand binding, receptor undergoes conformational changes and the release of corepressor factors induces histone acetylation or recruitment of co-activating proteins like steroid receptor coactivator-1 (SRC-1) (Pavek, 2016; Buchman et al., 2018). The main bile acid ligand of PXR is LCA, its oxidized metabolite 3-keto-LCA and acetylated DCA and CA derivatives, while conjugated bile acids do not activate PXR (Carazo et al., 2017). Studies have demonstrated that PXR can alleviate the harmful effects of toxic hydrophobic bile acids such as LCA through activation of cytochromes that hydroxylate bile acids to less toxic, more hydrophilic bile acid species and by induction of conjugation enzymes, sulfotransferase SULT2A1 and uridine 5′-diphospho-glucuronosyltransferases, involved in phase two of metabolism and detoxification of bile acids (Wang et al., 2012). PXR is mostly expressed in the intestine and liver, and the main targets of this nuclear receptor are drug-metabolizing enzymes and transporter proteins. Hence, the influence of bile acids on the PXR activity may exert profound effects on the pharmacokinetics of numerous drugs and therapeutic outcome.
CAR (NR1I3, chromosome 11p15.5) is mainly expressed in the liver and functions predominantly as a xenobiotic sensor and transcriptional regulator of genes influencing drug disposition. Additionally, CAR is unusual among the other nuclear receptors in being constitutively active even in the absence of ligands. Besides, CAR gets its name from two androstane metabolites, which are identified as inverse agonists that repress the constitutive activity of CAR. CAR has been implicated in regulating the expression of multiple genes involved mainly in drug metabolism and disposition (Carazo et al., 2017; Pavlovic et al., 2017). CAR is a receptor for structurally diverse molecules including bile acids as endogenous ligands, with primary bile acid, CA, and keto derivatives 6-keto- and 7-keto-LCA being recognized to exert trans-repressive effect on CAR activity (Stedman et al., 2004; Fiorucci et al., 2010). The accumulation of bile acids during cholestasis and subsequent activation of CAR reduces hepatocellular injury by the reduction of bile acid synthesis as well as by inducing metabolizing enzymes and transporter proteins, which mediates removal of hydrophobic bile acids from hepatocytes (Li and Chiang, 2013; Gabbia et al., 2017). Both CAR and PXR cooperatively diminish bile acids toxicity. Combined loss of CAR and PXR leads to increased sensitivity to bile acid toxicity, since LCA was tolerated by wild-type and PXR-knockout mice, whereas in PXR/CAR double knockout mice, significant accumulation of serum bile acids and liver damage occurred (Uppal et al., 2005). On the other hand, the repression of CAR decreased the bile acid accumulation in cholestasis model of bile duct ligation in mice, indicating the potential of selective CAR antagonists as therapeutic strategy for cholestatic liver disease patients in the future. Even though CAR and PXR display some overlapping functions, and the loss of function of these receptors sensitizes the liver vulnerability to bile acid-mediated toxicity, these receptors do not have capacity to compensate the loss of each other (Stedman et al., 2005).
Bile acids also activate membrane G-protein-coupled receptor TGR5 through which these molecules exert non-genomic effects. TGR5 was first identified as a G-protein-coupled bile acid receptor (GPBAR1, MBAR, GPCR19) in 2002 (Maruyama et al., 2002). Upon bile acids binding, the activation of TGR5 is associated with an intracellular accumulation of cyclic AMP, adenylate cyclase activation and calcium mobilization, followed by downstream modulation of their target signaling pathways. TGR5 is ubiquitously expressed in organism, being even identified in the spinal cord and astrocytes, indicating that bile acids exert numerous previously unanticipated functions. In accordance with wide tissue distribution, bile acid-activated TGR5 receptor is involved in regulation of diverse processes in the organism, ranging from macrophage activation to glucose metabolism regulation. Being expressed in immune cells, bile acids via this receptor reduce production of inflammatory cytokines, whereas in the skeletal muscle and brown adipose tissue bile acids increase energy expenditure through the activity of iodothyronine deiodinase D2 – an enzyme which converts thyroxine to more potent form triiodothyronine, thus preventing obesity (Deutschmann et al., 2018). By activating TGR5 in intestinal entero-endocrine L cells bile acids stimulate the release of insulinotropic glucagon-like peptide-1 (GLP-1), regulating postprandial insulin release and blood glucose concentration (Fiorucci et al., 2009). Studies have also shown that activation of TGR5 on pancreatic β-cells stimulates glucose-induced insulin release through cyclic AMP- and calcium-dependent manner, evading stimulation of the glucagon release from α-cells (Kumar et al., 2016).
Therefore, bile acids are becoming increasingly considered as hormones which may affect various aspects of integral metabolism. However, given that bile acids activate different receptors, significant effort has been made to synthetize, characterize and novel bile acid derivatives (as well as non-bile acid synthetic ligands) with high specificity toward certain receptor, ensuing dissection of signaling pathways and uncovering novel therapeutic targets. Out of numerous bile acid derivatives, only obeticholic acid (OCA, 6-ethyl-chenodeoxycholic acid, 6-ECDCA INT-747) has currently been clinically useful and approved as a potent and specific FXR agonist with 100-fold higher affinity compared to the CDCA – endogenous FXR agonist with the highest potency (Pellicciari et al., 2002). In addition, 6α-ethyl-23(S)-methylcholic acid (INT-777) has been identified as a specific TGR5 agonist with promising effects in treatment of glucose homeostasis disorders, vascular inflammation and atherosclerosis, neuro-inflammation and hepato-gastro-intestinal and metabolic disorders (Pellicciari et al., 2009; Guo et al., 2016).
UDCA is hydrophilic bile acid which β conformation of C7 hydroxyl group interrupts hydrophobic integrity of steroid core’s hydrophobic surface, increasing hydrophilicity but significantly affecting interaction with ligand-binding pocket (LBP) of FXR, making impossible formation of hydrogen bond with tyrosine-366 of LBP and activation of FXR (Katona et al., 2007). Even though UDCA is not potent ligand for other bile acid receptors, this bile acid species exerts various therapeutic effects by different mechanism including antioxidative and anti-apoptotic functions, the role of molecular chaperone regulating endoplasmic reticulum homeostasis, protein folding and post-translational modification of proteins (Perez and Briz, 2009; Cybulsky, 2017; Mueller and Castro, 2018). UDCA modulates apoptosis by tumor suppressor p53, through complexation of p53 with an oncoprotein Mdm 2 (mouse double minute 2), preventing therefore p53 nuclear transportation and activation (Amaral et al., 2010). In addition to regulation of endoplasmic reticulum homeostasis, UDCA stabilizes mitochondrial membrane reducing pro-apoptotic potential of cell, which has profound implications in pathologies with excessive apoptosis (Chun and Low, 2012). This is particularly relevant for tauro-UDCA which has proven beneficial therapeutic effect in central nervous system in models of Parkinson’s disease, neuroinflammation, retinal degenerative diseases, spinal cord injury (Lobysheva et al., 2018; Miao et al., 2018; Rosa et al., 2018). In addition to being a “neuroprotective conjugate,” tauro-UDCA has therapeutic potential, endothelial dysfunction, protecting bile acid composition during inflammatory conditions, reducing the progression of high-fat diet-induced non-alcoholic fatty liver disease (NAFLD), ameliorating intestinal inflammation, improving function of intestinal barrier, and modulating the composition of intestinal microbiota (Walsh et al., 2016; Hou et al., 2017; Wang et al., 2018).
24-Norursodeoxycholic (norUDCA) is specific non-amidated side chain-shortened C23 homolog of UDCA. NorUDCA derivative has a property of targeting biliary canalicular lumen, being shuttled through hepatocyte and excreted in biliary tree as an anion, protonated at biliary pH. NorUDCA induces bicarbonate-rich choleresis with cholangio-protective effects. In addition, norUDCA has direct anti-inflammatory, anti-lipotoxic, anti-fibrotic and anti-proliferative properties having effects on different liver cell populations, representing one of the most promising novel therapeutic approaches targeting both the liver and the system of biliary tree at multi-cell-type and multi-factorial levels (Trauner et al., 2015). NorUDCA protects cholangiocytes from luminal injury as hydrophilic bile acid. This bile acid derivative does not undergo enterohepatic recirculation like conjugated bile salts. Namely, protonated norUDCA is a non-polar molecule that can passively diffuse through cholangiocytes into the circulation of peribiliary plexus to be re-uptaken by the liver cells and re-excreted into the bile in a process known as ‘cholehepatic shunting.’ Since for each norUDCA molecule that passes through cholangiocyte a bicarbonate anion remains in the bile, this process increases biliary bicarbonate concentration, which is known to have protective propensity in cholangiopathies as ‘bicarbonate umbrella,’ favorably changing composition of bile (Halilbasic et al., 2015). Indeed, norUDCA ameliorated both the liver and ductal injury in Mdr2 knockout mice (Fickert et al., 2006). Opposite to UDCA, which increases intra-biliary pressure in obstructive cholestasis due to induced choleresis, norUDCA has been demonstrated not to aggravate hepatocyte’s injury in a rodent model with obstructive cholestasis, which may be explained by the induction of alternative transporter proteins (Fickert et al., 2017).
Overall, the role of bile acids has evolved to the regulatory function which mode of action includes not only receptor-mediated, but also receptor-independent cellular signaling cascades and regulation of epigenetic machinery, making these initially described intestinal emulsifiers as the novel promising therapeutic agents in a plethora of metabolic, inflammatory and even malignant diseases. The discovery that bile acids activate cell receptors, having hence intrinsic property of signaling function rather than exclusively emulsifying function, initiated the renaissance in research in the field of bile acid pharmacology and novel discoveries have been directed toward identification of receptor specific analogs. The mode of action of bile acids is complex and could be determined by several premises: namely, it is structure-dependent since small modifications in the structure significantly alter physiological/pharmacological activity; the activity of these molecules is tissue- and cell-type dependent and displays variable effects in normal and malignant tissues, depending on the context. Deciphering complex network underlying currently described effects of bile acids and their derivatives following administration in various preclinical and clinical settings, as well as the effects of administration of synthetic non-steroid bile-acid receptor modulators is beyond the scope of this article. Therefore, this article is going to provide up to date information regarding the role of bile acids and bile acid derivatives in the treatment of the components of metabolic syndrome.
Structural and Physico-Chemical Properties of Bile Acids Related to Pharmacological Activity
Bile acids are characterized by specific chemical structure derived from a hydrophobic hydrocarbon perhydrocyclopentanophenanthrene with flexible acidic side chain and polar hydroxyl groups. Steroid nucleus with angular methyl groups at position C-18 and C-19 represents a convex hydrophobic β-face, while hydrophilic concave α-face, contains polar hydroxyl groups varying in their number and position (Monte et al., 2009). Therefore, bile acids are amphiphilic molecules that exhibit a great surface activity and tend to self-associate in aqueous solutions forming micelles as long as their concentrations are above a certain concentration, commonly called the critical micellar concentration (CMC) (Hofmann and Hagey, 2008). The key factor that determines the self-association of bile acids is their hydrophobicity that depends on chemical structure, particularly the number and orientation of hydroxyl groups. Bile acids with fewer hydroxyl groups are usually more hydrophobic and vice versa (Ashby et al., 2018). The most hydrophobic bile acid, based on chromatographic behavior is LCA, followed by DCA, CDCA, CA and UDCA (Jia et al., 2017). CMC values of selected bile acids reported by Roda et al. (1989) are given in Table 2. In addition to the concentration, bile acid hydrophobicity is the most important determinant of their toxicity that is associated with cell membrane damage, promotion of the generation of ROS, and eventually necrosis and apoptosis. The more hydrophobic bile acids exhibit more cytotoxic properties compared to hydrophilic bile acids (Perez and Briz, 2009). Since the bile acids have been continuously investigated for therapeutic applications, it is of high importance to take in consideration inter-species variation in bile acid metabolism and composition between animals and humans that might contribute to the discordant toxicity.
Table 2.
Critical micellar concentrations (CMCs) of selected bile acids and their conjugates (reported by Roda et al., 1989).
| Trivial name | Symbol | Substituents | CMC (0.15 M Na+) (mM) | |
|---|---|---|---|---|
| Dihydroxy bile acids | ||||
| Chenodeoxycholic acid | CDCA | 3α,7α | 4 | |
| Taurochenodeoxycholic acid | TCDCA | 6 | ||
| Glycoursodeoxycholic acid | GCDCA | 10 | ||
| Ursodeoxycholic acid | UDCA | 3α,7β | 7 | |
| Tauroursodeoxycholic acid | TUDCA | 2.2 | ||
| Glycoursodeoxycholic acid | GUDCA | 4 | ||
| Deoxycholic acid | DCA | 3α,12α | 3 | |
| Taurodeoxycholic acid | TDCA | 2.4 | ||
| Glycodeoxycholic acid | GDCA | 2 | ||
| Trihydroxy bile acids | ||||
| Cholic acid | CA | 3α,7α,12α | 11 | |
| Taurocholic acid | TCA | 6 | ||
| Glycocholic acid | FCA | 10 | ||
| Ursocholic acid | UCA | 3α,7β,12α | 39 | |
| Tauroursocholic acid | TUCA | 40 | ||
| ;Glycoursocholic acid | GUCA | 30 | ||
As weak acids, bile acids exist in their ionized form, i.e., as bile salts, at physiological conditions. Conjugation of these compounds with either glycine or taurine improves their physico-chemical characteristics by decreasing hydrophobic characteristics and increasing the water-solubility (Hofmann and Hagey, 2008). In the intestinal tract, bile acids are mainly in the form of mixed micelles composed mostly of bile salts, lecithin, and cholesterol, reducing thus bile acid toxicity and representing significant mechanism of cholesterol transport from the liver into the intestine and its excretion from the organism (Roda et al., 1983).
Specific structure of bile acids relating to amphiphilic properties with both hydrophilic and hydrophobic regions is responsible for specific functions of these compounds (Monte et al., 2009). The most important and well-known physiological functions of bile acids are solubilization of fatty acids, cholesterol and liposoluble vitamins in the intestinal tract, thus facilitating their digestion and transport. However, there is a growing body of evidence that the list of their physiological roles is far longer and still not complete (Li and Chiang, 2015; Chávez-Talavera et al., 2017). Discovery of novel functions of bile acids as signaling molecules involved in a plethora of metabolic pathways and signaling cascades represents a driving force to design and synthesize novel selective and potent modulators of these receptors with improved pharmacokinetic and pharmacodynamic profiles. The meaningful challenge in designing agonists of these receptors is to unveil some specific properties that are relevant to their potency and selectivity for particular receptors and to distinguish the desired therapeutic effects from the undesired side effects (Pellicciari et al., 2009).
Introduction of α ethyl moiety in the C6 position of CDCA was found to dramatically increase the agonist activity and selectivity for FXR, thus resulting in discovery of OCA (Pellicciari et al., 2002; Sato et al., 2008). Furthermore, Pellicciari et al. (2009) reported that methylation at the C23-(S) position in the side chain of bile acids such as CDCA and OCA, is a key feature to improve potency and selectivity for TGR5.
Furthermore, the unique and specific structure of bile acids make them attractive compounds in the drug development process, as pharmaceutical tools and potential drug carrier systems that could improve, control and localize drug delivery (Stojančević et al., 2013). Since mid-1990s, much attention has been paid to the investigations of keto derivatives of bile acids, primarily 12-monoketocholic acid (12-MKC), as absorption enhancers through different biological membranes (Al-Salami et al., 2008, 2012; Kuhajda et al., 2009; Yang et al., 2011; Chen et al., 2012; Danic et al., 2016). Replacing hydroxyl with keto group in CA at C12 position produces less lipophilic bile acid with higher CMC and consequently with diminished cytotoxic property, but with preserved absorption-promoting activity (Chen et al., 2012).
Traditional and Current Therapeutic Use of Bile Acids
Beneficial effects on human health and therapeutic use of bile acids have been recognized since ancient times (Mikov et al., 2006). Bear bile has been used in Traditional Chinese Medicine clinical practice for thousands of years. Bear bile was used for detoxification, reduction of inflammation, swelling, fever and pain, several liver diseases, including fibrosis, biliary cirrhosis, and even liver cancer. Bile acids were recognized as the main compounds in bear bile responsible for pharmacodynamic activity (Li et al., 2016).
Clinical studies have confirmed the beneficial effects of bile acids, particularly UDCA in a broad spectrum of cholestatic liver diseases, including primary biliary cirrhosis, pediatric cholestatic disorders, primary sclerosing cholangitis (PSC), and drug-induced cholestasis (Paumgartner and Beuers, 2002). Western medicine recognized therapeutic potential of bile acids during the 20th century. Only UDCA and CDCA were used for the treatment of cholelithiasis for several decades, however, the discovery at the end of 20th century that bile acids activate FXR brought a renaissance in the research of bile acids. The search for selective agonist of bile acid receptor resulted in the synthesis and pre-clinical studies of numerous bile acid derivatives. In the year of 2016, US Food and Drug Administration (FDA) approved the use of CA in inborn disorders of bile acid synthesis and as additional remedy in peroxisomal disorders including Zellweger cerebro-hepato-renal syndrome, as well as a local injection use of DCA for submental fat tissue contouring (Mullard, 2016). During the same year, FDA gave approval for the use of semi-synthetic potent and selective FXR agonist, OCA, for primary biliary cirrhosis therapy in patients with unsatisfactory response to UDCA administration as a monotherapy (Hirschfield et al., 2015). This is the first approval of medicine for primary biliary cirrhosis indication after more than two decades (Gerken and Nitschmann, 2017). In addition, FXR agonist OCA has been extensively investigated in clinical settings. Treatment with OCA has been shown to improve biochemical and histological parameters in patients with NAFLD (Neuschwander-Tetri et al., 2015). During phase II study in a group of patients with NAFLD and type 2 diabetes, OCA improved central and peripheral insulin sensitivity and reduced parameters of hepatic inflammation and fibrosis yielding promising results in larger studies (Mudaliar et al., 2013). NorUDCA, as a novel bile acid derivative, has been successfully implemented during phase II clinical trial in patients with PSC. The safety profile of norUDCA was similar to placebo, whereas serum parameters of cholangiocyte injury were reduced, which gives promising expectations (Fickert et al., 2017).
Many bile acids derivatives have been synthesized and characterized during past decade, and this is continuously growing and promising field of biomedical research worldwide. Today therapeutic role of bile acids appears to expand rapidly attracting the great attention in the field of metabolic syndrome treatment (Taoka et al., 2016).
Bile Acids in Regulation of Glucose Metabolism
Over the past decade, a growing body of evidence has strongly indicated that bile acids regulate glycemic postprandial metabolism displaying also anti-diabetic signaling capacities by acting through bile acid-activating receptors, as well as by improving protein folding and function, in addition to antiapoptotic effects (Hylemon et al., 2009).
There are, however, some unsolved discrepancies in literature between obtained results related to implication of bile acids in glucose metabolism. Initial studies identifying the implication of FXR in glucose metabolism showed that bile acids or synthetic FXR-specific agonists induced the expression of the rate controlling enzyme of gluconeogenesis, phosphoenolpyruvate carboxykinase (PEPCK), increasing the total glucose output in human and rat hepatocyte, as well as in mice in vivo (Stayrook et al., 2005). On the contrary, Yamagata et al. (2004) have shown that bile acids suppress the expression of gluconeogenesis genes PEPCK, glucose-6-phosphatase (G6Pase) and fructose 1,6-bis phosphatase (FBP1) via the interaction between SHP with hepatocyte nuclear factor 4 (HNF-4) or forehead transcription factor Foxo1, in both in vivo and in vitro conditions. Decrease of PEPCK expression appears to be an attractive possibility for type 2 diabetes treatment, given that type 2 diabetes is characterized by an increased hepatic glucose output and hyperglycemia (Cariou et al., 2005). In addition, the activation of hepatic FXR leads to increased glycogen synthase activity by phosphorylation and inactivation of glycogen synthase kinase 3 (GSK3b) (Mencarelli and Fiorucci, 2010).
Zhang et al. (2006) showed that FXR deficiency in mice is associated with glucose intolerance and insulin resistance manifested by hyperglycemia, impaired glucose tolerance, and severely impaired insulin signaling in liver, muscle and adipose tissue. Accordingly, activation of FXR by the synthetic non-steroidal agonist GW4064 in mice (30 mg/kg twice a day) significantly decreased hepatic glucose production, lowered blood glucose levels, increased glycogenesis, improved insulin synthesis and secretion, and improved central (hepatic) and peripheral insulin sensitivity in animals (Zhang et al., 2006).
FXR-induced activation of insulin transcription and secretion in pancreatic β cells are regulated by different mechanisms involving both, genomic as well as non-genomic effects. Genomic effects of FXR activation are based on the induction of the glucose-dependent transcription factor krueppel-like factor 11 (KLF11), which is proven to be an essential factor for insulin gene transcription. Non-genomic effects of FXR activation in βTC6 cells relay on the increase of Akt phosphorylation and translocation of the glucose transporter type 2 (GLUT2), a member of membrane proteins that facilitates transport of glucose along a gradient of concentration at plasma membrane, increasing the glucose uptake by pancreatic β cells (Renga et al., 2010). Furthermore, FXR activation induces the expression of GLUT4 glucose transporter in the liver, which expression is found to be reduced, both in type 1 and 2 diabetic subjects (Garvey et al., 1991, 1992). Activation of FXR was able to upregulate GLUT4 expression through FXRE in the GLUT4 gene promoter. Shen et al. (2008) reported that activation of FXR by CDCA in concentration 10 μM could induce GLUT4 transcription in 3T3-L1 and HepG2 cell lines and increase the GLUT 4 protein expression in C57BL/6J mice that were treated with CDCA (20 mg/kg/day). Overall, these changes resulted in decrease in plasma glucose level, decreased hepatic gluconeogenesis, and increased hepatic glycogen synthesis (Mencarelli and Fiorucci, 2010).
Bile acids may also affect glucose homeostasis in an FXR-independent manner (Stanimirov et al., 2012). In addition to the role in energy expenditure (discussed in further text), the activation of membrane receptor TGR5 by bile acids has been shown to increase intestinal GLP-1 secretion from entero-endocrine L cells, both in vitro and in vivo, stimulating the release of insulin from the pancreatic β-cells without the release of glucagon from α-cells, and reducing postprandial glycaemia (Katsuma et al., 2005; Kumar et al., 2012; Duboc et al., 2014). Furthermore, Maruyama et al. (2006) showed that TGR5-null mice have a 25% decrease in bile acid pool size, while female TGR5-null mice showed significant fat accumulation with body weight gain compared with that of the wild-type mice when fed a high fat diet. Figure 3 clearly shows intertwined pathways of bile acids implication in glucose metabolism mediated by FXR and TGR5 signaling.
FIGURE 3.
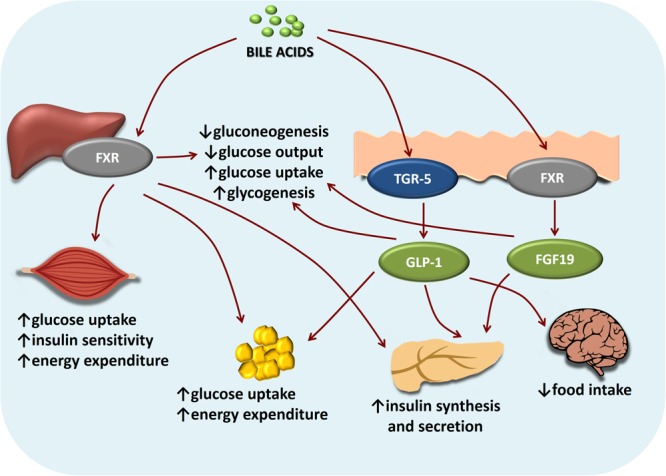
Intertwined bile acids-mediated signaling pathways in glucose metabolism. Bile acids activation of FXR and TGR-5 signaling pathways inhibits gluconeogenesis and promotes glycogen synthesis in the liver, promotes glucose-stimulated insulin release in pancreas, increases energy expenditure especially in skeletal muscles and brown adipose tissue. In the brain, bile acids-TGR5 signaling mediates satiety.
In vitro and in vivo genetic evidence indicate a strong and causal link between the functional capacity of the endoplasmic reticulum and insulin effects. Therefore, the modulation of endoplasmic reticulum function might provide a new approach for diabetes treatment (Ozcan et al., 2006). Ozcan et al. (2006) showed that tauro-UDCA improves insulin resistance by attenuating endoplasmic reticulum stress in type 2 diabetes animals resulting in normalization of hyperglycemia, improvement of systemic insulin sensitivity and insulin action in various tissues.
The efficacy of synthetic FXR agonists as potential therapy for type 2 diabetes mellitus has been proven in an already mentioned phase II clinical trial conducted by Mudaliar et al. (2013) showing that administration of OCA to patients with both type 2 diabetes mellitus and NAFLD for 6 weeks was well-tolerated, improving insulin sensitivity, and reducing markers of liver inflammation and fibrosis.
Bile Acid-Mediated Effects on Diabetes After Bariatric Surgical Procedures
Recently, the search for the mechanisms underlying rapid glycemic improvements following bariatric surgical procedures [Roux-en-Y gastric bypass (RYGB) and vertical sleeve gastrectomy] in patients with morbid obesity and type 2 diabetes confirmed the significant physiological effects of bile acids in glucose homeostasis (Bradley et al., 2012; Pournaras et al., 2012). These procedures caused an increase in free bile acids both in serum and in the lower intestine (Madsbad et al., 2014; Raghow, 2015). The increased concentration of free bile acids in the intestinal lumen had direct incretin effect through TGR5 – GLP-1 axis, enhancing insulin secretion, as well as by activating intestinal FXR and its direct downstream target FGF-15/19 – a postprandial hormone which improves glucose tolerance, the protein which expression is, otherwise, reduced in diabetic patients (Jansen et al., 2011; Schaap, 2012; Madsbad and Holst, 2014). Beneficial effects mediated by the bile acid receptors FXR and TGR5 following bariatric surgery were further confirmed since in the absence of FXR signaling in FXR-null mice or antagonizing GLP-1 signaling by exendin-(9-39), the beneficial effects of bariatric surgery on body weight and glucose metabolism were abolished (Salehi et al., 2011; Ryan et al., 2014). The increase in free bile acid concentrations in the lower parts of intestinal tract after RYGB creates an environment suitable for growth of bile-tolerant bacteria such as phylum Proteobacter taxonomic group (Osto et al., 2013). The overgrowth of Proteobacter results in a decrease of secondary, more toxic bile acid species, consequent increase of primary bile acid levels in plasma and pronounced incretin effect (Vrieze et al., 2014).
Effects of Bile Acids–Gut Microbiota Interactions in Glucose Metabolism
Human intestinal tract is colonized by a diverse collection of microbes, with bacteria as the most numerous members. The composition of gut microbiota is specific to the individual but remains relatively constant over time (Stojančević et al., 2014). Alterations of the human gut microbiome can play a role in disease development (DeGruttola et al., 2016).
Intestinal microbiota regulates host metabolism by producing numerous metabolites that signal through their cognate receptors, thus affecting body weight, bile acid metabolism, pro-inflammatory activity and insulin resistance, and modulation of gut hormones (Han and Lin, 2014; Wahlstrom et al., 2016). One important class of metabolites produced by microbes are bile acids. Bacterial bile salt hydrolase (BSH), which is present in all major bacterial species in the human gut, carry out bile salt deconjugation thus increasing the resistance to bile toxicity, while 7α-dehydroxylases convert the primary bile acids CA and CDCA to the secondary bile acids DCA and LCA, respectively (Wahlstrom et al., 2016). Other bile salt conversions performed by the intestinal microbiota are oxidation and epimerization of hydroxyl groups, 7-dehydroxylation, esterification and desulfation, thus contributing to the great chemical diversity and more hydrophobic bile acid pool (Ridlon et al., 2016). Besides, bile acids can modulate gut microbiota composition both, directly, by destroying the structures of bacterial membranes, and indirectly, via FXR activation by promoting the transcription of antimicrobial agents such as iNOS and IL-18 in the small intestine and thereby inhibiting bacterial growth (Inagaki et al., 2006; Wahlstrom et al., 2016).
Considering the complex relationship of bile acids with intestinal microbiota, it is not surprising that in addition to the changes in the composition of bile acids, diabetic patients also have different composition and activity of intestinal microbiota comparing to healthy individuals (Quercia et al., 2014). Both type 1 and type 2 diabetes mellitus are associated with a decrease in overall microbial diversity, characterized with reduction of Firmicutes and butyrate-producing bacteria, as well as a disturbed intestine barrier function and increased gut permeability (DeGruttola et al., 2016; Knip and Siljander, 2016). Supplementation with probiotic bacteria resulted in beneficial modulation of host’s metabolism in terms of stimulated GLP-1 secretion by specific bacterial metabolites such as short chain fatty acids (SCFAs) through GPR41/43-dependent mechanism (Everard and Cani, 2014). Additionally, administration of probiotic bacteria reduced the food intake and protected from body weight gain and insulin resistance in animal models of obesity and diabetes (Yadav et al., 2013). Studies performed by our group have demonstrated that a multi-therapeutic approach using combination of probiotics and bile acids as adjunct therapy in a rat model of diabetes mellitus exerted even better glycemic regulation and resulted in the alleviation of complications compared to each treatment alone (Al-Salami et al., 2008, 2012). Synergistic effects of bile acids, probiotics and current antidiabetic therapy are reviewed in detail by Mikov et al. (2017) pointing to the potential application of this combination in metabolic disorders with special emphasis on diabetes mellitus.
Thus, the therapeutic manipulation of the intestinal microbiota by probiotic supplementation with secondary effects on bile acid pool composition represents the attractive and promising strategies for such conditions. Although further studies are highly recommended to unveil the exact mechanisms responsible for beneficial effects of bile acids-probiotic co-administration, activation of complex FXR and TGR5 signaling pathways is proposed as one of possible explanations.
Bile Acids and Lipid Metabolism
As a main product of cholesterol catabolism, bile acids exert profound effects, not only on cholesterol metabolism, but also on the metabolism of triacylglycerols, regulating therefore metabolism of various lipoprotein species. Increased synthesis of bile acids increases the utilization of cholesterol as a substrate. By activating FXR bile acids inhibit CYP7A1 – the rate limiting enzyme of bile acid synthesis and cholesterol catabolism in the hepatocytes. In accordance, long term supplementation of either 750 mg or 375 mg/day of CDCA in patients with gallstone disease results in a modest increase in low density lipoprotein cholesterol (LDL) level (Schoenfield and Lachin, 1981). The increase in LDL occurred in 85.2% of patients receiving 750 mg/day, and, in 82.8% of patients receiving 375 mg/day, however, the increase of 67.0% was recorded in a group of patients receiving placebo, possibly as a consequence of the main disease. On the other hand, in vitro studies demonstrate that FXR agonist CDCA (250 μM) stabilizes mRNA of LDL receptor and increases LDL receptor activity in human cultured hepatic cell line increasing thus the uptake and clearance of LDL particles (Nakahara et al., 2002). Changes in circulating cholesterol level through the FXR activation in vivo are distinct among rodent models and humans. Namely, CYP7a1 expression in rodents, is opposite to the humans regulated by two nuclear receptors, the liver X receptor-α (LXR-α) and FXR, both of which abundantly expressed in the liver. LXR-α may be activated by cholesterol derivatives including 24(S),25-epoxycholesterol and 24(S)-hydroxycholesterol, and following its activation LXR interacts with a response element within the CYP7a1 promoter, stimulating thereby gene expression. The translation of effects observed in rodents has been puzzling since in these species circulating cholesterol is predominantly packed in the form of HDL lipoproteins, opposing to LDL species dominant in human. In chimeric mice whose livers mostly contain human hepatocytes and a “humanized” lipoprotein profile, treatment with potent specific FXR agonist, semi-synthetic bile acid derivative OCA (10 mg/kg/day), results in the increase of circulating LDL and HDL reduction, similarly to the FXR activation in humans. The increase in LDL correlated with decreased sterol regulatory element-binding protein-2 (SREBP-2) activity and its target gene expression, including a significant downregulation in the expression of LDL receptor protein (Papazyan et al., 2018). The administration of OCA, 25 or 50 mg/day for 2 weeks, during clinical trials resulted in similar effects (Mudaliar et al., 2013; Walters et al., 2015).
The reduction in HDL level by FXR activation [using chow supplemented with 0.5% w/w taurocholic acid (TLCA) for a 6 days] may be explained by the repression of the apolipoprotein A-I gene, as well as the hetero-exchange of cholesteryl esters and triacylglycerols between plasma HDL and ApoB-containing lipoproteins by inducing cholesteryl ester-transfer protein expression (Lambert et al., 2003; Gautier et al., 2013). On the other hand, FXR targeting may also reinforce the reverse transport of cholesterol, a process in the cholesterol transport from peripheral tissues and cells to the hepatocytes and biliary system, in order to eliminate cholesterol through intestinal route. Northern blot analysis of liver specimens of C57BL/6 male mice, fed by 1% CA supplemented diet for a month, revealed that abovementioned effects are mediated via phospholipid-transfer protein, a protein which mediates the delivery of phospholipids and cholesterol from LDL to HDL lipoprotein particles. Observed effects are also a consequence of the changes in expression of the scavenger receptor-B1 (SR-B1), which is involved in the recognition of HDL particles and their uptake by the hepatocytes (Urizar et al., 2000). Also, the activation of direct FXR target, enterokine FGF15/19 has been shown to stimulate robust secretion of cholesterol into the intestinal lumen via the sterol-exporting heterodimer adenosine triphosphate (ATP)-binding cassette subfamily G member 5/8 (ABCG5/G8) in mice (de Boer et al., 2017). This finding has potential implication in developing strategies aimed at reduction of intestinal cholesterol resorption. The administration of 10 or 25 mg of OCA per day in healthy volunteers induced sustained elevation of serum LDL concentration and reduction of HDL, with a slight elevation of total cholesterol level independently of dose (Pencek et al., 2016). Similar changes were noted in patients with type 2 diabetes mellitus as well as in patients with biopsy-confirmed non-alcoholic steatohepatitis (Mudaliar et al., 2013; Neuschwander-Tetri et al., 2015). However, in patients with primary biliary cholangitis treated with 5–10 mg of OCA per day for 1 year, the rise in LDL and reduction in HDL level was smaller in amplitude and transient (Nevens et al., 2016). Effects of FXR activation by repeated administration of 5, 10, or 25 mg OCA per day for either 2 weeks or 20 days have been estimated in clinical trial recruiting healthy volunteers (Pencek et al., 2016). The results observed in this study confirmed the results of previous clinical studies in terms of slight disturbances in lipid status; namely, treatment with OCA induced a dose-independent reduction in HDL, as a result of reduction in small and medium HDL particles concentration, and increase in LDL cholesterol level (Pencek et al., 2016). Even though the activation of FXR by bile acids and bile acid derivatives may induce potentially pro-atherogenic phenotype in terms of the shift in lipoprotein fractions, large randomized multicenter clinical studies are highly needed to elucidate clinical impact of such dyslipidemic effects treated with bile acids or bile acid derivatives.
On the other hand, bile acids supplementation with 0.5% (w/w) CA for 8 weeks has been shown to modulate triacylglycerol metabolism through several distinct mechanisms, predominantly via FXR activation. By activating FXR bile acids repress triacylglycerol synthesis de novo through FXR downstream target, SHP-mediated inhibition of transcription of sterol regulatory element binding protein-1c (SREBP-1c), a key factor that controls transcription of several genes regulating fatty acids and triacylglycerol synthesis, including a master lipogenic enzyme in the liver, fatty acid synthase (Watanabe et al., 2004). FXR activation has also been demonstrated to induce the expression of nuclear receptor peroxisome-proliferator-activated receptor-α (PPAR-α) and of pyruvate dehydrogenase kinase isoenzyme-4 promoting fatty acid oxidation, whereas by SHP-mediated inhibition of the microsomal triacylglycerol transfer protein expression to reduce the VLDL assembly (Pan et al., 2010). Moreover, FXR promotes the activity of enzyme anchored to the luminal surface of vascular endothelial cells, a lipoprotein lipase (LPL) catalyzing therefore hydrolysis of triacylglycerols by inducing expression of LPL-activating apoC-II in C57BL/6J mice fed for 3 weeks with diet containing 0.5% sodium cholate. Additionally, FXR activation induced the expression of the VLDL receptor, facilitating the clearance of VLDL particles in HepG2 cells incubated with 50 μM of CDCA for 24 h (Jiao et al., 2015). In addition to reduce symptoms of bile acid diarrhea in patients, oral supplementation of 25 mg of OCA per day for 2 weeks resulted in increase in FGF-19 level and triacylglycerol decrease, which implicates the potential role of FGF-19 on triacylglycerol metabolism (Walters et al., 2015).
The additional complexity of signaling network between bile acids and triacylglycerol network arises upon interactions with intestinal microbiota, which is commonly changed in Western-type diet fed persons as well as in obese. Indeed, even short-term administration of oral non-absorbable antibiotics resulted in intestinal dysbiosis followed by the decrease in amounts of secondary bile acids DCA and LCA liver concentrations, and reduced serum triacylglycerol level, which did not recover even after bile acid supplementation (Kuno et al., 2018). The results of the study suggest that secondary bile acids produced by intestinal bacterial species exert significant regulatory role in maintaining serum triacylglycerol levels and metabolism in the host.
The Role of Bile Acids in Non-Alcoholic Fatty Liver Disease
Non-alcoholic fatty liver disease, characterized by excess triglyceride accumulation within hepatocytes, or steatosis, is the most prevalent chronic liver disease. Simple steatosis is regarded as to as relatively benign and non-progressive while non-alcoholic steatohepatitis (NASH) is considered to be the most serious form of NAFLD, characterized by hepatocellular injury, chronic inflammation, and a higher risk of end stage liver diseases such as cirrhosis and liver cancer (Wree et al., 2013; Benedict and Zhang, 2017). NAFLD is strongly associated with the components of metabolic syndrome, mainly type 2 diabetes mellitus and obesity, being thus the important risk factor for both hepatic and cardiovascular mortality (Demir et al., 2015). NAFLD is the most prevalent chronic liver disease, which affects up to 40% of the population, and nearly 30% of patients with NAFLD progress NASH (Sanyal, 2011). In addition, both NAFLD and type 2 diabetes mellitus are dominant health disorders associated with the issue of global overweight and obesity epidemic (Feneberg and Malfertheiner, 2012).
Non-alcoholic fatty liver disease is therefore categorized as a hepatic manifestation of metabolic syndrome. NASH represents advanced grade of NAFLD characterized by hepatocellular injury, inflammatory infiltrate with deposition of collagen. Insulin resistance is known as fundamental feature in the pathogenesis of type 2 diabetes mellitus as well as in NAFLD, being also considered a principal disorder in the initiation and development of NASH (Kitade et al., 2017). In addition to insulin resistance, oxidative stress, hypoadiponectinemia, and visceral adiposity contribute to the progression to NASH (Gruben et al., 2014). The accumulation of triacylglycerols in hepatocytes is a result of lipolysis due to the insulin resistance, increased fatty acid tissue from mal-functional adipose tissue, impaired mitochondrial fatty acid oxidation or impaired VLDL particles assembly and export, as well as by activating inflammatory pathways (Bellanti et al., 2018).
The positive effects of bile acids-mediated FXR and TGR5 activation in a wide range of metabolic processes, including glucose metabolism and insulin signaling, triglyceride and cholesterol metabolism as well as the inflammation, put the focus of research interest on bile acids to develop effective therapeutic strategies for this liver-associated metabolic disease. Due to its characterization and pre-clinical evaluation, OCA has become one of the most commonly ligands used for decoding FXR signaling network both in vivo and in vitro. During preclinical studies OCA administration had significant beneficial effects in numerous disorders of enterohepatic system including improvement of estrogen-induced cholestasis, liver fibrosis, NASH, insulin signaling, portal hypertension, reduction of intestinal inflammation and improvement of ileal barrier function during cholestasis, improved bile acid-induced chronic diarrhea (Fiorucci et al., 2004, 2005; Adorini et al., 2012; Verbeke et al., 2014; Walters et al., 2015). Per os administration of 25 mg of OCA for 72 weeks has been demonstrated to improve histological features of the livers of patients with NASH during phase two of FLINT clinical trial (Neuschwander-Tetri et al., 2015). Beneficial effects of FXR activation in patient with NAFLD include improved central insulin sensitivity in the liver and consequently increased glycogen synthesis, decrease in de novo lipogenesis in liver, improved insulin sensitivity adipose tissue (decreasing lipotoxic effects) and improved function of peroxisome proliferator-activated receptor-gamma (PPAR-γ) and PPAR-α, regulating fatty acid and glucose metabolism both in adipose tissue and liver (Fiorucci et al., 2007). The diet supplementation with OCA (10 mg/kg/day) to fast food diet animal model of NASH, mimicking the metabolic syndrome features ablated micro RNA-21 (miR21) and activated PPAR-α that resulted in significant steatosis reduction, inflammation and lipo-apoptosis, unraveling restoration of miR21/PPAR-α axis in liver and muscle tissue by FXR and OCA (Rodrigues et al., 2017). The activation of FXR by OCA (10 mg/kg/day, administered by oral gavage for 7 weeks) and of TGR5 by 8-week-long diet intervention containing 30 mg/kg/day of INT-777, improved liver steatosis and insulin resistance in rodent model of NAFLD and obesity (Thomas et al., 2009; Cipriani et al., 2010). The administration of OCA (25, 50 mg/day per os during 6 weeks) in patients with type two diabetes mellitus and NAFLD significantly reduced body weight, improved insulin sensitivity and reduced serum level of gamma-glutamyltransferase, whereas increase in serum FGF-19 intestinal enterokine confirmed the activation of FXR in the patients (Mudaliar et al., 2013). Additionally, OCA (10 mg/kg) prevented hepatic inflammation by preventing detrimental nuclear factor κB-mediated immunomodulation and inflammation. Also, through inhibition of hepatic stellate cells activation, incubation with 10 μM of OCA prevented progression toward liver fibrosis and development of cirrhosis (Goto et al., 2018). Treatment with this FXR agonist resulted in improved biochemical and liver histological features in patients with NASH. These functions indicate that FXR is an attractive therapeutic target for liver diseases (Massafra and van Mil, 2018). Further large randomized clinical studies are highly desirable to confirm effects of “knocking on the FXR door” as a potential therapeutic approach in which change in bile acid pool size and composition may be exploited for the treatment of metabolic disorders.
Bile Acids and Atherosclerosis
In addition to the systemic effects on serum lipid profile, bile acids may exert anti-atherosclerosis effects via FXR directly act at the level of the arterial wall. Vascular wall is not typically involved in bile acids metabolism, however, FXR has been found to be expressed in the endothelial cells and vascular smooth muscle cells (Mencarelli and Fiorucci, 2010). Activation of FXR by circulating bile acids may have a beneficial effect on vascular tone by suppressing the potent vasoconstrictive peptide, endothelin-1, and by inducing the production of the vasodilating agent, nitric oxide (NO). In vitro incubation of rat pulmonary artery endothelial cells with CDCA (6.25–50 μM) activated FXR in these cells and reduced expression of endothelin-1 in a concentration-dependent manner. Similarly, the incubation of rat pulmonary microvasculature endothelial cells and bovine aortic endothelial cells with CDCA (12.5 and 50 μM) resulted in increase in endothelial nitric oxide synthase (eNOS) level through transcriptional activation of the eNOS gene promoter by FXR (He et al., 2006; Li et al., 2008). In addition, FXR activation increases the production of physiological vaso-relaxing molecule, hydrogen sulfide (H2S) by activating cystathionase (CSE), the main enzyme in the trans-sulfuration pathway. CSE has been shown to be FXR target gene both in vitro (following incubation of HepG2 cells with 10 μM for 18 h) and in vivo (5 mg/kg of OCA administered intraperitoneally to C57BL/6j mice), reducing the portal pressure and attenuating endothelial dysfunction in the model of isolated and perfused cirrhotic rat livers (Renga et al., 2009).
The incubation of rat aortic smooth muscle cells in medium containing 30 μM of OCA and subsequent FXR activation has been shown to inhibit inflammatory responses as well as migration of vascular smooth muscle cells by inhibiting interleukin 1β-induced expression of inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) genes, by reducing NF-κB activation. The same authors reported that NF-κB activation by IL-1β is downregulated by FXR and SHP. FXR ligands also were shown to reduce platelet-derived growth factor-β (PDGF-β) migration of rodent and human aortic vascular smooth muscle cells (Li et al., 2007). However, in vitro incubation of HUVEC cells with CDCA (100 μM), DCA (100 μM), and LCA (33 μM) induced the expression of adhesion molecules, such as intracellular adhesion molecule-1 (ICAM-1), vascular cellular adhesion molecule-1 (VCAM-1) and E-selectin, promoting adhesion of monocytes to endothelial cells – the pivotal step in initiating the process of atherosclerosis (Qin et al., 2006).
Pharmacological activation of TGR5 by dietary supplementation with 6α-ethyl-23(S)-methyl-cholic acid (INT-777 30 mg/kg/day) for 12 weeks in LDL receptor-knockout mice resulted in inhibition of vascular lesion formation by reducing macrophage inflammation and lipid uptake (Pols et al., 2011). TGR5, which is abundantly expressed in macrophages has been responsive to INT-777 treatment (30 μM) that inhibited cytokine production through cAMP-NF-κB signaling pathway (Pols et al., 2011). Therefore, macrophage inflammation – a central event in the majority of aspects contributing to development of the atherosclerosis, from the initiation of atherosclerosis up to plaque formation and rupture, which results in the initiation of the formation of thrombus, could be prevented by circulatory bile acids and bile acid derivatives as the agents that could affect atherosclerosis development on pleiotropic fashion.
Semi-synthetic bile acid derivative INT-767 (α-ethyl-24-nor-5β-cholane-3α,7α,23-triol-23 sulfate sodium salt) is a dual FXR/TGR5 agonist. Treatment of ApoE- and LDL receptor- knockout mice fed a Western-type diet supplemented with 30 mg/kg of INT-767 reduced atherosclerotic plaque formation downregulating the expression of pro-inflammatory cytokines and chemokines in aortic tissue (Miyazaki-Anzai et al., 2014). Treatment of ApoE knockout mice with INT-767 has also resulted in decreased levels of interleukins IL-1β, IL-6, IL-8, and IL-12 and reduced aortic expression of inflammatory mediators such as TNF-α, IL-6, IL-1β, and MCP-1. The observed anti-inflammatory effects of INT-767 rely on the activation of TGR5 but not of FXR (Miyazaki-Anzai et al., 2014).
In addition, the incubation of TGR5 expressed endothelium of the bovine aorta with TLCA (10–100 μM) resulted in dose-dependent nitric oxide synthesis (Kida et al., 2013). Among other bile acids, LCA (100 μM) also significantly increased nitric oxide production, however, incubation with DCA (100 μM) and CDCA (100 μM) only marginally increased NO production. These results are in agreement with the potency of different bile salts as TGR5 ligands to increase cAMP production in the following order: TLCA > LCA > DCA > CDCA. The established efficiency and good safety profile of UDCA in treatment of hepato-biliary disorders inspired research on the potential benefit of UDCA administration in treating pathological conditions affecting other systems of organs, e.g., cardiac disorders such as atherosclerosis and myocardial infarction. Six weeks long UDCA supplementation (13–19 mg/kg, administered twice a day) in patients with coronary artery disease improved endothelium-dependent nitric oxide-independent vasodilatation (Sinisalo et al., 1999). In addition, UDCA exerts in vitro anti-atherogenic effects in a model of diabetic atherosclerosis by inhibiting non-enzymatic glycation and oxidation of proteins and lipids in diabetes, which is confirmed by incubation of Raw 264.7 and HUVEC cells with 100 μM of UDCA (Chung et al., 2016). UDCA ameliorated endoplasmic reticulum stress and the downstream signaling pathway thereof in endothelial cells and aortic tissues of diabetic ApoE knockout mice, and inhibited reactive oxygen species production through induction Nrf2 as a main transcription factor stimulating transcription and synthesis of antioxidative enzymes, and inhibited NF-κB and Janus N-terminal kinase (JNK)-mediated vascular endothelial inflammation. Also, UDCA suppressed foam cell formation through upregulation of ABCA1 and ABCG1 expression, reduction of hyperglycemia-induced receptor for advanced glycation end-product (RAGE) expression, and suppression of macrophage inflammatory responses, significantly ameliorating atherosclerosis (Chung et al., 2016). These findings indicate that UDCA as a commonly used chemical chaperone could be beneficial therapeutic agent for prevention or treatment of diabetic atherosclerosis. Furthermore, UDCA (100 μM) expressed anti-atherogenic effects by blocking the endoplasmic reticulum stress and inflammatory response in endothelial cells induced by disturbed flow. In vivo, UDCA supplementation (∼400–600 mg/kg/day) abolished development of atherosclerotic lesions in a ApoE KO murine model with disturbed flow-induced atherosclerosis (by partial ligation of the left coronary artery) fed on high-fat-diet, and observed effects are at least partially mediated by inhibiting the activation of endoplasmic stress downstream signaling through XBP-1 and CHOP, and down-regulating expression of adhesion molecules (Chung et al., 2015).
On the other hand, elevated circulating levels of secondary bile acid, DCA (even at in vitro concentration of 5 μM), induced by high-fat diet have been shown to induce vascular smooth muscle cell proliferation and migration by upregulating JNK and platelet-derived growth factor β-receptor (Shimizu et al., 2014). Observed dysfunction of endothelial cells in Western-type diet subjects may the result of long-term change in intestinal microbiota composition and increased production of DCA by enterobacteria, which indicates that disturbances in intestinal microbiota may play role in the pathogenesis of cardiometabolic disorders by changing bile acid pool composition. At the same time this offers potential therapeutic/preventive strategy by manipulating intestinal microflora composition and therefore the pool size and composition of bile acids.
Overall, the influence of bile acids has promising therapeutic effects in different models of atherosclerosis and dyslipidemia, the features of metabolic syndrome. The observed effects are both FXR- and TGR5- dependent and independent (summarized in Figure 4) forcing the need for developing novel bile acid derivatives as potential therapeutic agents. Given the differences between bile acid pool and composition in rodents and humans, tissue- and cell-specific activity of bile acids and context dependency on the mode of their action, the translation of obtained knowledge in preclinical settings could not be straightforwardly implemented in clinical settings. Therefore, significant effort should be carried out to identify and characterize novel bile acid derivatives as useful pharmacodynamic agents and therapeutics in the future.
FIGURE 4.
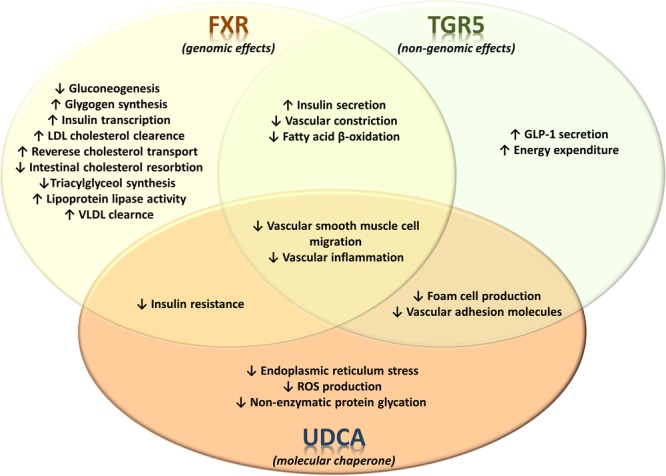
Receptor-dependent and receptor-independent regulation of metabolic and signaling pathways by bile acids in metabolic syndrome. The effects of bile acid-activated receptors FXR and TGR5 on glucose, lipid and energy metabolism as well as on vascular events associated with atherosclerosis. FXR and TGR5 have significant number of currently identified overlapping effects. On the other hand, the effects of UDCA are not associated with activation of these bile acid receptors, instead UDCA exerts various physiological/pharmacological effects which are associated with its specific structural properties.
Bile Acids, Obesity, and Cardiometabolic Diseases
Obesity, a central facet of metabolic syndrome, represents a major health issue in both developed and developing countries, being associated with several comorbidities as independent risk factors for cardiovascular disease. In addition to adipose tissue excess, obese patients often display dysfunctional adipocytes. Obesity is additionally associated with dyslipidemia and elevated circulating levels of cholesterol, triacylglycerols and free fatty acids, and disturbances in lipid metabolism result in ectopic lipid deposits leading to hepatic steatosis and consequently to NAFLD. Subsequent “spill over” of free fatty acids from insulin resistant, dysfunctional adipose tissue initiates a phenomena of lipotoxicity caused by the accumulation of triacylglycerol-derived toxic metabolites in non-adipose tissues, inducing therefore inflammatory pathways, cellular dysfunction including insulin resistance, followed by impaired glucose control and type 2 diabetes (Mendez-Sanchez et al., 2018). Obesity is also associated with hemodynamic alterations and arterial hypertension which contribute to cardiac morphology changes predisposing to impairment of ventricular function and heart failure (Alpert et al., 2018).
Increased circulating bile acid level in obese individuals has been found to positively correlate with body mass index (Prinz et al., 2015). Also, insulin resistance is associated with Foxo-1-mediated downregulation of CYP8B1 resulting in depletion of 12α-hydroxylated bile acid pool, which may be explained by hyperglycemia-induced Foxo degradation, and therefore a lack to activate CYP8B1, whereas in type 2 diabetic patients concentration of DCA has been found to be elevated (Brufau et al., 2010; Haeusler et al., 2013). Thus, interventions manipulating bile acid pool composition could represent novel therapeutic strategies in insulin resistance. Changes in bile acid pool size and composition following bariatric surgery in obese individuals are reflected in improved metabolic homeostasis (Penney et al., 2015). It is therefore, reasonable to assume that supplementation with bile acids or bile acid derivatives, changing bile acid signaling, could be considered as potential cardioprotective intervention improving metabolism and decreasing inflammation level. Indeed, the activation of TGR5 in macrophages and endothelial cells by micromolar levels of circulating bile acids both during fasting and in postprandial state when bile circulating bile acids reach concentration peak, exert anti-atherogenic effects and inhibit atherosclerosis and coronary artery disease (Steiner et al., 2011). TGR5 receptor, expressed in adipocytes regulates energy expenditure and body weight (van Nierop et al., 2017). Bile acid-induced GLP1 also exerts beneficial effects on endothelial function, blood pressure, myocardial metabolism, left ventricular ejection fraction, atherosclerosis and response to oxidative injury induced by ischemia-reperfusion (Kang and Jung, 2016). RYGBP procedure in obese patients results in increase in GLP1 level having therefore cardioprotective effects. In addition, increased intestinal flux of bile acids following RYGBP leads to the activation of intestinal FXR and its downstream target, an enterokine FGF15/19, which has been shown to repress apo(a) gene expression by ERK1/2 phosphorylation-mediated cascade, reducing circulating level of highly atherogenic lipoprotein(a) (Chennamsetty et al., 2012). The administration of recombinant human FGF19 or FGF19 overexpression in leptin deficient diabetic mice was shown to reduce weight gain, reverse diabetes, increase lipid oxidation rate decreasing also hepatic triacylglycerol content (Fu et al., 2004). Bariatric surgery procedures also result in increase in circulating levels of bile acid species, both in fasted and postprandial state, as well as qualitative changes in bile acid pool composition, due to increased hepatic synthesis and reabsorption and reduced intestinal elimination, or by changes in microbiota composition (De Giorgi et al., 2015; Dutia et al., 2016). In accordance, the increase in concentration of bile acids both in intestinal lumen and systemic circulation, as potent ligands for FXR and TGR5, mediates the improvement of metabolic rate, glucose and lipid metabolism, increases thermogenesis resulting in reduction of body weight, ameliorates body-wide inflammation, and even promotes brown adipose tissue browning (Fang et al., 2015). These metabolic improvements implicate that change in bile acid composition or pool size by pharmacological approach or metabolic surgery affects systemic metabolism with favorable outcome, suggesting novel therapeutic approach in treating obesity and components of metabolic syndrome. However, given that bile acids activate multiple nuclear receptors and possibly more than one GPCR, careful dissection and in depth evaluation of bile acid-mediated signaling pathways on tissue specific manner should provide useful information in future development of novel specific and selective bile acid derivatives as novel therapeutic agents in treatment of metabolic syndrome.
Conclusion
Traditional approaches, such as diet and increased physical activity, have shown to be insufficient in decreasing the prevalence of metabolic diseases that are becoming more and more common in overall population. The emerging evidence in the past decade has pointed to the role of bile acids as signaling, endocrine molecules that regulate the glucose, lipid, and energy metabolism through complex and interrelated pathways that mainly include FXR and TGR5 signaling cascade. Thus, the modulation of these signaling pathways by using bile acids and their derivatives and by co-administration with probiotic bacteria with secondary effects on bile acid pool composition has become an attractive area in modern research offering a new approach for metabolic syndrome treatment. However, many of the obtained results derived from studies carried out in animal models that should be taken into consideration during interpretation of results due to major differences in bile acid metabolism and gut microbiota composition between animals and humans. Additionally, interindividual differences in gut microbiota composition, i.e., specific bacterial fingerprint in certain individuals contributes to highly person-specific bile acids profiles as well, which differentially influence pathogenesis of disease, and conceivably the response to bile acid-related preventive and therapeutic interventions, requiring further studies and elucidation. From a therapeutic point of view, a more in depth insight metabolic effects of each of the natural and synthetic bile acid species in vivo is highly desirable. Probiotics, as potential strategy to modulate the composition of gut microbiota, should be further studied to help the restoration of bile acid metabolism and potentially aid in the treatment of metabolic disorders. Future research should be relied on metabolomic, proteomic and lipidomic approaches in both healthy and diseased populations in order to identify bile acid-related biomarkers that may be valuable for prediction of bile acid related therapy of different metabolic disorders.
To conclude, targeting the interactions between bile acids, microbiota, and bile acid receptors signaling seems to derive a promising approach for the treatment of metabolic diseases, but additional detailed pre-clinical research and translation of obtained knowledge in clinical studies are highly recommended in order to confirm the efficacy of bile acids and bile acid derivatives in such conditions.
Author Contributions
MÐ and BS reviewed the literature and drafted the manuscript. MM and HA-S conceived the idea. All authors edited, revised, and approved the final version of this review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This work was supported by Ministry of Education, Science and Technological Development, Republic of Serbia, Project No. 41012, Project for Scientific and Technological Development of Vojvodina No. 114-451-2072-/2016, and HORIZON2020 MEDLEM Project No. 690876.
References
- Adorini L., Pruzanski M., Shapiro D. (2012). Farnesoid X receptor targeting to treat nonalcoholic steatohepatitis. Drug Discov. Today 17 988–997. 10.1016/j.drudis.2012.05.012 [DOI] [PubMed] [Google Scholar]
- Aguilar M., Bhuket T., Torres S., Liu B., Wong R. J. (2015). Prevalence of the metabolic syndrome in the United States, 2003-2012. JAMA 313 1973–1974. 10.1001/jama.2015.4260 [DOI] [PubMed] [Google Scholar]
- Ajouz H., Mukherji D., Shamseddine A. (2014). Secondary bile acids: an underrecognized cause of colon cancer. World J. Surg. Oncol. 12 164–164. 10.1186/1477-7819-12-164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alpert M. A., Karthikeyan K., Abdullah O., Ghadban R. (2018). Obesity and cardiac remodeling in adults: mechanisms and clinical implications. Prog. Cardiovasc. Dis. 61 114–123. 10.1016/j.pcad.2018.07.012 [DOI] [PubMed] [Google Scholar]
- Al-Salami H., Butt G., Tucker I., Golocorbin-Kon S., Mikov M. (2012). Probiotics decreased the bioavailability of the bile acid analog, monoketocholic acid, when coadministered with gliclazide, in healthy but not diabetic rats. Eur. J. Drug Metab. Pharmacokinet. 37 99–108. 10.1007/s13318-011-0060-y [DOI] [PubMed] [Google Scholar]
- Al-Salami H., Butt G., Tucker I., Mikov M. (2008). Influence of the semisynthetic bile acid MKC on the ileal permeation of gliclazide in vitro in healthy and diabetic rats treated with probiotics. Methods Find. Exp. Clin. Pharmacol. 30 107–113. 10.1358/mf.2008.30.2.1159652 [DOI] [PubMed] [Google Scholar]
- Amaral J. D., Xavier J. M., Steer C. J., Rodrigues C. M. (2010). The role of p53 in apoptosis. Discov. Med. 9 145–152. [PubMed] [Google Scholar]
- Ashby K., Navarro Almario E. E., Tong W., Borlak J., Mehta R., Chen M. (2018). Review article: therapeutic bile acids and the risks for hepatotoxicity. Aliment. Pharmacol. Ther. 47 1623–1638. 10.1111/apt.14678 [DOI] [PubMed] [Google Scholar]
- Baghdasaryan A., Chiba P., Trauner M. (2014). Clinical application of transcriptional activators of bile salt transporters. Mol. Aspects Med. 37 57–76. 10.1016/j.mam.2013.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellanti F., Villani R., Tamborra R., Blonda M., Iannelli G., di Bello G., et al. (2018). Synergistic interaction of fatty acids and oxysterols impairs mitochondrial function and limits liver adaptation during nafld progression. Redox Biol. 15 86–96. 10.1016/j.redox.2017.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedict M., Zhang X. (2017). Non-alcoholic fatty liver disease: an expanded review. World J. Hepatol. 9 715–732. 10.4254/wjh.v9.i16.715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley D., Conte C., Mittendorfer B., Eagon J. C., Varela J. E., Fabbrini E., et al. (2012). Gastric bypass and banding equally improve insulin sensitivity and beta cell function. J. Clin. Invest. 122 4667–4674. 10.1172/jci64895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brufau G., Stellaard F., Prado K., Bloks V. W., Jonkers E., Boverhof R., et al. (2010). Improved glycemic control with colesevelam treatment in patients with type 2 diabetes is not directly associated with changes in bile acid metabolism. Hepatology 52 1455–1464. 10.1002/hep.23831 [DOI] [PubMed] [Google Scholar]
- Buchman C. D., Chai S. C., Chen T. (2018). A current structural perspective on PXR and CAR in drug metabolism. Expert Opin. Drug Metab. Toxicol. 14 635–647. 10.1080/17425255.2018.1476488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carazo A., Hyrsova L., Dusek J., Chodounska H., Horvatova A., Berka K., et al. (2017). Acetylated deoxycholic (DCA) and cholic (CA) acids are potent ligands of pregnane X (PXR) receptor. Toxicol. Lett. 265 86–96. 10.1016/j.toxlet.2016.11.013 [DOI] [PubMed] [Google Scholar]
- Cariou B., Duran-Sandoval D., Kuipers F., Staels B. (2005). Farnesoid X receptor: a new player in glucose metabolism? Endocrinology 146 981–983. 10.1210/en.2004-1595 [DOI] [PubMed] [Google Scholar]
- Chávez-Talavera O., Tailleux A., Lefebvre P., Staels B. (2017). Bile acid control of metabolism and inflammation in obesity, type 2 diabetes, dyslipidemia, and nonalcoholic fatty liver disease. Gastroenterology 152 1679–1694.e3. 10.1053/j.gastro.2017.01.055 [DOI] [PubMed] [Google Scholar]
- Chen G., Yang L., Zhang H., Tucker I. G., Fawcett J. P. (2012). Effect of ketocholate derivatives on methotrexate uptake in Caco-2 cell monolayers. Int. J. Pharm. 433 89–93. 10.1016/j.ijpharm.2012.04.077 [DOI] [PubMed] [Google Scholar]
- Chennamsetty I., Claudel T., Kostner K. M., Trauner M., Kostner G. M. (2012). FGF19 signaling cascade suppresses APOA gene expression. Arterioscler. Thromb. Vasc. Biol. 32 1220–1227. 10.1161/atvbaha.111.243055 [DOI] [PubMed] [Google Scholar]
- Chiang J. Y. (2009). Bile acids: regulation of synthesis. J. Lipid Res. 50 1955–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang J. Y. (2013). Bile acid metabolism and signaling. Compr. Physiol. 3 1191–1212. 10.1002/cphy.c120023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang J. Y. (2015). Negative feedback regulation of bile acid metabolism: impact on liver metabolism and diseases. Hepatology 62 1315–1317. 10.1002/hep.27964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun H. S., Low W. C. (2012). Ursodeoxycholic acid suppresses mitochondria-dependent programmed cell death induced by sodium nitroprusside in SH-SY5Y cells. Toxicology 292 105–112. 10.1016/j.tox.2011.11.020 [DOI] [PubMed] [Google Scholar]
- Chung J., An S. H., Kang S. W., Kwon K. (2016). Ursodeoxycholic acid (UDCA) exerts anti-atherogenic effects by inhibiting RAGE signaling in diabetic atherosclerosis. PLoS One 11:e0147839. 10.1371/journal.pone.0147839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung J., Kim K. H., Lee S. C., An S. H., Kwon K. (2015). Ursodeoxycholic acid (UDCA) exerts anti-atherogenic effects by inhibiting endoplasmic reticulum (ER) stress induced by disturbed flow. Mol. Cells 38 851–858. 10.14348/molcells.2015.0094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipriani S., Mencarelli A., Palladino G., Fiorucci S. (2010). FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. J. Lipid Res. 51 771–784. 10.1194/jlr.M001602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeglio P., Morelli A., Adorini L., Maggi M., Vignozzi L. (2017). Beneficial effects of bile acid receptor agonists in pulmonary disease models. Expert Opin. Investig. Drugs 26 1215–1228. 10.1080/13543784.2017.1385760 [DOI] [PubMed] [Google Scholar]
- Cybulsky A. V. (2017). Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat. Rev. Nephrol. 13 681–696. 10.1038/nrneph.2017.129 [DOI] [PubMed] [Google Scholar]
- Danic M., Pavlovic N., Stanimirov B., Vukmirovic S., Nikolic K., Agbaba D., et al. (2016). The influence of bile salts on the distribution of simvastatin in the octanol/buffer system. Drug Dev. Ind. Pharm. 42 661–667. 10.3109/03639045.2015.1067626 [DOI] [PubMed] [Google Scholar]
- Dawson P. A., Lan T., Rao A. (2009). Bile acid transporters. J. Lipid Res. 50 2340–2357. 10.1194/jlr.R900012-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer J. F., Schonewille M., Boesjes M., Wolters H., Bloks V. W., Bos T., et al. (2017). Intestinal farnesoid X receptor controls transintestinal cholesterol excretion in mice. Gastroenterology 152 1126–1138.e6. 10.1053/j.gastro.2016.12.037 [DOI] [PubMed] [Google Scholar]
- de Boer J. F., Bloks V. W., Heiner-Fokkema M. R., Kuipers F. (2018). New insights in the multiple roles of bile acids and their signaling pathways in metabolic control. Curr. Opin. Lipidol. 29 194–202. 10.1097/MOL.0000000000000508 [DOI] [PubMed] [Google Scholar]
- De Giorgi S., Campos V., Egli L., Toepel U., Carrel G., Cariou B., et al. (2015). Long-term effects of Roux-en-Y gastric bypass on postprandial plasma lipid and bile acids kinetics in female non diabetic subjects: a cross-sectional pilot study. Clin. Nutr. 34 911–917. 10.1016/j.clnu.2014.09.018 [DOI] [PubMed] [Google Scholar]
- DeGruttola A. K., Low D., Mizoguchi A., Mizoguchi E. (2016). Current understanding of dysbiosis in disease in human and animal models. Inflamm. Bowel Dis. 22 1137–1150. 10.1097/MIB.0000000000000750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demir M., Lang S., Steffen H. M. (2015). Nonalcoholic fatty liver disease - current status and future directions. J. Dig. Dis. 16 541–557. 10.1111/1751-2980.12291 [DOI] [PubMed] [Google Scholar]
- Deutschmann K., Reich M., Klindt C., Droge C., Spomer L., Haussinger D., et al. (2018). Bile acid receptors in the biliary tree: TGR5 in physiology and disease. Biochim. Biophys. Acta 1864 1319–1325. 10.1016/j.bbadis.2017.08.021 [DOI] [PubMed] [Google Scholar]
- Duboc H., Tache Y., Hofmann A. F. (2014). The bile acid TGR5 membrane receptor: from basic research to clinical application. Dig. Liver Dis. 46 302–312. 10.1016/j.dld.2013.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutia R., Embrey M., O’Brien S., Haeusler R. A., Agenor K. K., Homel P., et al. (2016). Temporal changes in bile acid levels and 12alpha-hydroxylation after Roux-en-Y gastric bypass surgery in type 2 diabetes. Int. J. Obes. 40:554. 10.1038/ijo.2015.250 [DOI] [PubMed] [Google Scholar]
- Everard A., Cani P. D. (2014). Gut microbiota and GLP-1. Rev. Endocr. Metab. Disord. 15 189–196. 10.1007/s11154-014-9288-6 [DOI] [PubMed] [Google Scholar]
- Fang S., Suh J. M., Reilly S. M., Yu E., Osborn O., Lackey D., et al. (2015). Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat. Med. 21 159–165. 10.1038/nm.3760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feneberg A., Malfertheiner P. (2012). Epidemic trends of obesity with impact on metabolism and digestive diseases. Dig. Dis. 30 143–147. 10.1159/000336660 [DOI] [PubMed] [Google Scholar]
- Ferrebee C. B., Dawson P. A. (2015). Metabolic effects of intestinal absorption and enterohepatic cycling of bile acids. Acta Pharm. Sin. B 5 129–134. 10.1016/j.apsb.2015.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fickert P., Hirschfield G. M., Denk G., Marschall H. U., Altorjay I., Farkkila M., et al. (2017). NorUrsodeoxycholic acid improves cholestasis in primary sclerosing cholangitis. J. Hepatol. 67 549–558. 10.1016/j.jhep.2017.05.009 [DOI] [PubMed] [Google Scholar]
- Fickert P., Wagner M., Marschall H. U., Fuchsbichler A., Zollner G., Tsybrovskyy O., et al. (2006). 24-NorUrsodeoxycholic acid is superior to ursodeoxycholic acid in the treatment of sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology 130 465–481. 10.1053/j.gastro.2005.10.018 [DOI] [PubMed] [Google Scholar]
- Fiorucci S., Antonelli E., Rizzo G., Renga B., Mencarelli A., Riccardi L., et al. (2004). The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis. Gastroenterology 127 1497–1512. [DOI] [PubMed] [Google Scholar]
- Fiorucci S., Cipriani S., Baldelli F., Mencarelli A. (2010). Bile acid-activated receptors in the treatment of dyslipidemia and related disorders. Prog. Lipid Res. 49 171–185. 10.1016/j.plipres.2009.11.001 [DOI] [PubMed] [Google Scholar]
- Fiorucci S., Clerici C., Antonelli E., Orlandi S., Goodwin B., Sadeghpour B. M., et al. (2005). Protective effects of 6-ethyl chenodeoxycholic acid, a farnesoid X receptor ligand, in estrogen-induced cholestasis. J. Pharmacol. Exp. Ther. 313 604–612. 10.1124/jpet.104.079665 [DOI] [PubMed] [Google Scholar]
- Fiorucci S., Mencarelli A., Palladino G., Cipriani S. (2009). Bile-acid-activated receptors: targeting TGR5 and farnesoid-X-receptor in lipid and glucose disorders. Trends Pharmacol. Sci. 30 570–580. 10.1016/j.tips.2009.08.001 [DOI] [PubMed] [Google Scholar]
- Fiorucci S., Rizzo G., Donini A., Distrutti E., Santucci L. (2007). Targeting farnesoid X receptor for liver and metabolic disorders. Trends Mol. Med. 13 298–309. 10.1016/j.molmed.2007.06.001 [DOI] [PubMed] [Google Scholar]
- Fu L., John L. M., Adams S. H., Yu X. X., Tomlinson E., Renz M., et al. (2004). Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology 145 2594–2603. 10.1210/en.2003-1671 [DOI] [PubMed] [Google Scholar]
- Gabbia D., Pozza A. D., Albertoni L., Lazzari R., Zigiotto G., Carrara M., et al. (2017). Pregnane X receptor and constitutive androstane receptor modulate differently CYP3A-mediated metabolism in early- and late-stage cholestasis. World J. Gastroenterol. 23 7519–7530. 10.3748/wjg.v23.i42.7519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardmo C., Tamburro A., Modica S., Moschetta A. (2011). Proteomics for the discovery of nuclear bile acid receptor FXR targets. Biochim. Biophys. Acta 1812 836–841. 10.1016/j.bbadis.2011.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garvey W. T., Maianu L., Hancock J. A., Golichowski A. M., Baron A. (1992). Gene expression of GLUT4 in skeletal muscle from insulin-resistant patients with obesity, IGT, GDM, and NIDDM. Diabetes Metab. Res. Rev. 41 465–475. [DOI] [PubMed] [Google Scholar]
- Garvey W. T., Maianu L., Huecksteadt T. P., Birnbaum M. J., Molina J. M., Ciaraldi T. P. (1991). Pretranslational suppression of a glucose transporter protein causes insulin resistance in adipocytes from patients with non-insulin-dependent diabetes mellitus and obesity. J. Clin. Invest. 87 1072–1081. 10.1172/jci115068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier T., de Haan W., Grober J., Ye D., Bahr M. J., Claudel T., et al. (2013). Farnesoid X receptor activation increases cholesteryl ester transfer protein expression in humans and transgenic mice. J. Lipid Res. 54 2195–2205. 10.1194/jlr.M038141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerken G., Nitschmann S. (2017). [Obeticholic acid in primary biliary cholangitis : PBC OCA international study of efficacy (POISE)]. Internist 58 202–204. 10.1007/s00108-016-0185-9 [DOI] [PubMed] [Google Scholar]
- Gioiello A., Cerra B., Mostarda S., Guercini C., Pellicciari R., Macchiarulo A. (2014). Bile acid derivatives as ligands of the farnesoid x receptor: molecular determinants for bile acid binding and receptor modulation. Curr. Top. Med. Chem. 14 2159–2174. [DOI] [PubMed] [Google Scholar]
- Goto T., Itoh M., Suganami T., Kanai S., Shirakawa I., Sakai T., et al. (2018). Obeticholic acid protects against hepatocyte death and liver fibrosis in a murine model of nonalcoholic steatohepatitis. Sci. Rep. 8:8157. 10.1038/s41598-018-26383-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruben N., Shiri-Sverdlov R., Koonen D. P., Hofker M. H. (2014). Nonalcoholic fatty liver disease: a main driver of insulin resistance or a dangerous liaison? Biochim. Biophys. Acta 1842 2329–2343. 10.1016/j.bbadis.2014.08.004 [DOI] [PubMed] [Google Scholar]
- Guo C., Chen W. D., Wang Y. D. (2016). TGR5, not only a metabolic regulator. Front. Physiol. 7:646 10.3389/fphys.2016.00646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler R. A., Astiarraga B., Camastra S., Accili D., Ferrannini E. (2013). Human insulin resistance is associated with increased plasma levels of 12alpha-hydroxylated bile acids. Diabetes Metab. Res. Rev. 62 4184–4191. 10.2337/db13-0639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halilbasic E., Fuchs C., Hofer H., Paumgartner G., Trauner M. (2015). Therapy of primary sclerosing cholangitis–today and tomorrow. Dig. Dis. 33(Suppl. 2) 149–163. 10.1159/000440827 [DOI] [PubMed] [Google Scholar]
- Han J.-L., Lin H.-L. (2014). Intestinal microbiota and type 2 diabetes: from mechanism insights to therapeutic perspective. World J. Gastroenterol. 20 17737–17745. 10.3748/wjg.v20.i47.17737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson R. L., Imperatore G., Bennett P. H., Knowler W. C. (2002). Components of the “metabolic syndrome” and incidence of type 2 diabetes. Diabetes Metab. Res. Rev. 51 3120–3127. [DOI] [PubMed] [Google Scholar]
- He F., Li J., Mu Y., Kuruba R., Ma Z., Wilson A., et al. (2006). Downregulation of endothelin-1 by farnesoid X receptor in vascular endothelial cells. Circ. Res. 98 192–199. 10.1161/01.res.0000200400.55539.85 [DOI] [PubMed] [Google Scholar]
- Hirschfield G. M., Mason A., Luketic V., Lindor K., Gordon S. C., Mayo M., et al. (2015). Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid. Gastroenterology 148 751–761.e8. 10.1053/j.gastro.2014.12.005 [DOI] [PubMed] [Google Scholar]
- Hofmann A. F. (2004). Detoxification of lithocholic acid, a toxic bile acid: relevance to drug hepatotoxicity. Drug Metab. Rev. 36 703–722. 10.1081/dmr-200033475 [DOI] [PubMed] [Google Scholar]
- Hofmann A. F., Hagey L. R. (2008). Bile acids: chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell. Mol. Life Sci. 65 2461–2483. 10.1007/s00018-008-7568-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y., Yang H., Cui Z., Tai X., Chu Y., Guo X. (2017). Tauroursodeoxycholic acid attenuates endoplasmic reticulum stress and protects the liver from chronic intermittent hypoxia induced injury. Exp. Ther. Med. 14 2461–2468. 10.3892/etm.2017.4804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hylemon P. B., Zhou H., Pandak W. M., Ren S., Gil G., Dent P. (2009). Bile acids as regulatory molecules. J. Lipid Res. 50 1509–1520. 10.1194/jlr.R900007-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki T., Moschetta A., Lee Y. K., Peng L., Zhao G., Downes M., et al. (2006). Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc. Natl. Acad. Sci. U.S.A. 103 3920–3925. 10.1073/pnas.0509592103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen P. L., van Werven J., Aarts E., Berends F., Janssen I., Stoker J., et al. (2011). Alterations of hormonally active fibroblast growth factors after Roux-en-Y gastric bypass surgery. Dig. Dis. 29 48–51. 10.1159/000324128 [DOI] [PubMed] [Google Scholar]
- Jia W., Xie G., Jia W. (2017). Bile acid–microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 15 111–128. 10.1038/nrgastro.2017.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y., Lu Y., Li X. Y. (2015). Farnesoid X receptor: a master regulator of hepatic triglyceride and glucose homeostasis. Acta Pharmacol. Sin. 36 44–50. 10.1038/aps.2014.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y. M., Jung C. H. (2016). Cardiovascular effects of glucagon-like peptide-1 receptor agonists. Endocrinol. Metab. 31 258–274. 10.3803/EnM.2016.31.2.258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona B. W., Cummins C. L., Ferguson A. D., Li T., Schmidt D. R., Mangelsdorf D. J., et al. (2007). Synthesis, characterization, and receptor interaction profiles of enantiomeric bile acids. J. Med. Chem. 50 6048–6058. 10.1021/jm0707931 [DOI] [PubMed] [Google Scholar]
- Katsuma S., Hirasawa A., Tsujimoto G. (2005). Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem. Biophys. Res. Commun. 329 386–390. [DOI] [PubMed] [Google Scholar]
- Kaur J. (2014). A comprehensive review on metabolic syndrome. Cardiol. Res. Pract. 2014:943162. 10.1155/2014/943162 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kemper J. K. (2011). Regulation of FXR transcriptional activity in health and disease: emerging roles of FXR cofactors and post-translational modifications. Biochim. Biophys. Acta 1812 842–850. 10.1016/j.bbadis.2010.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kida T., Tsubosaka Y., Hori M., Ozaki H., Murata T. (2013). Bile acid receptor TGR5 agonism induces NO production and reduces monocyte adhesion in vascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 33 1663–1669. 10.1161/atvbaha.113.301565 [DOI] [PubMed] [Google Scholar]
- Kitade H., Chen G., Ni Y., Ota T. (2017). Nonalcoholic fatty liver disease and insulin resistance: new insights and potential new treatments. Nutrients 9:E387. 10.3390/nu9040387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knip M., Siljander H. (2016). The role of the intestinal microbiota in type 1 diabetes mellitus. Nat. Rev. Endocrinol. 12 154–167. 10.1038/nrendo.2015.218 [DOI] [PubMed] [Google Scholar]
- Kuhajda I., Poša M., Jakovljeviæ V., Ivetiæ V., Mikov M. (2009). Effect of 12-monoketocholic acid on modulation of analgesic action of morphine and tramadol. Eur. J. Drug Metab. Pharmacokinet. 34 73–78. [DOI] [PubMed] [Google Scholar]
- Kumar D. P., Asgharpour A., Mirshahi F., Park S. H., Liu S., Imai Y., et al. (2016). Activation of transmembrane bile acid receptor TGR5 modulates pancreatic islet alpha cells to promote glucose homeostasis. J. Biol. Chem. 291 6626–6640. 10.1074/jbc.M115.699504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D. P., Rajagopal S., Mahavadi S., Mirshahi F., Grider J. R., Murthy K. S., et al. (2012). Activation of transmembrane bile acid receptor TGR5 stimulates insulin secretion in pancreatic beta cells. Biochem. Biophys. Res. Commun. 427 600–605. 10.1016/j.bbrc.2012.09.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuno T., Hirayama-Kurogi M., Ito S., Ohtsuki S. (2018). Reduction in hepatic secondary bile acids caused by short-term antibiotic-induced dysbiosis decreases mouse serum glucose and triglyceride levels. Sci. Rep. 8:1253. 10.1038/s41598-018-19545-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam D. W., LeRoith D. (2012). The worldwide diabetes epidemic. Curr. Opin. Endocrinol. Diabetes Obes. 19 93–96. 10.1097/MED.0b013e328350583a [DOI] [PubMed] [Google Scholar]
- Lambert G., Amar M. J., Guo G., Brewer H. B., Jr., Gonzalez F. J., Sinal C. J. (2003). The farnesoid X-receptor is an essential regulator of cholesterol homeostasis. J. Biol. Chem. 278 2563–2570. 10.1074/jbc.M209525200 [DOI] [PubMed] [Google Scholar]
- Li J., Wilson A., Kuruba R., Zhang Q., Gao X., He F., et al. (2008). FXR-mediated regulation of eNOS expression in vascular endothelial cells. Cardiovasc. Res. 77 169–177. 10.1093/cvr/cvm016 [DOI] [PubMed] [Google Scholar]
- Li S., Tan H. Y., Wang N., Hong M., Li L., Cheung F., et al. (2016). Substitutes for bear bile for the treatment of liver diseases: research progress and future perspective. Evid. Based Complement. Alternat. Med. 2016:4305074. 10.1155/2016/4305074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T., Chiang J. Y. (2013). Nuclear receptors in bile acid metabolism. Drug Metab. Rev. 45 145–155. 10.3109/03602532.2012.740048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T., Chiang J. Y. L. (2015). Bile acids as metabolic regulators. Curr. Opin. Gastroenterol. 31 159–165. 10.1097/MOG.0000000000000156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T., Francl J. M., Boehme S., Chiang J. Y. (2013). Regulation of cholesterol and bile acid homeostasis by the cholesterol 7alpha-hydroxylase/steroid response element-binding protein 2/microRNA-33a axis in mice. Hepatology 58 1111–1121. 10.1002/hep.26427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. T., Swales K. E., Thomas G. J., Warner T. D., Bishop-Bailey D. (2007). Farnesoid x receptor ligands inhibit vascular smooth muscle cell inflammation and migration. Arterioscler. Thromb. Vasc. Biol. 27 2606–2611. 10.1161/atvbaha.107.152694 [DOI] [PubMed] [Google Scholar]
- Lobysheva E., Taylor C. M., Marshall G. R., Kisselev O. G. (2018). Tauroursodeoxycholic acid binds to the G-protein site on light activated rhodopsin. Exp. Eye Res. 170 51–57. 10.1016/j.exer.2018.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsbad S., Dirksen C., Holst J. J. (2014). Mechanisms of changes in glucose metabolism and bodyweight after bariatric surgery. Lancet Diabetes Endocrinol. 2 152–164. 10.1016/s2213-8587(13)70218-3 [DOI] [PubMed] [Google Scholar]
- Madsbad S., Holst J. J. (2014). GLP-1 as a mediator in the remission of type 2 diabetes after gastric bypass and sleeve gastrectomy surgery. Diabetes Metab. Res. Rev. 63 3172–3174. 10.2337/db14-0935 [DOI] [PubMed] [Google Scholar]
- Maruyama T., Miyamoto Y., Nakamura T., Tamai Y., Okada H., Sugiyama E., et al. (2002). Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Commun. 298 714–719. [DOI] [PubMed] [Google Scholar]
- Maruyama T., Tanaka K., Suzuki J., Miyoshi H., Harada N., Nakamura T., et al. (2006). Targeted disruption of G protein-coupled bile acid receptor 1 (Gpbar1/M-Bar) in mice. J. Endocrinol. 191 197–205. 10.1677/joe.1.06546 [DOI] [PubMed] [Google Scholar]
- Massafra V., van Mil S. W. C. (2018). Farnesoid X receptor: a “homeostat” for hepatic nutrient metabolism. Biochim. Biophys. Acta 1864 45–59. 10.1016/j.bbadis.2017.10.003 [DOI] [PubMed] [Google Scholar]
- McCracken E., Monaghan M., Sreenivasan S. (2018). Pathophysiology of the metabolic syndrome. Clin. Dermatol. 36 14–20. 10.1016/j.clindermatol.2017.09.004 [DOI] [PubMed] [Google Scholar]
- Mencarelli A., Fiorucci S. (2010). FXR an emerging therapeutic target for the treatment of atherosclerosis. J. Cell. Mol. Med. 14 79–92. 10.1111/j.1582-4934.2009.00997.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez-Sanchez N., Cruz-Ramon V. C., Ramirez-Perez O. L., Hwang J. P., Barranco-Fragoso B., Cordova-Gallardo J. (2018). New aspects of lipotoxicity in nonalcoholic steatohepatitis. Int. J. Mol. Sci. 19:E2034. 10.3390/ijms19072034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens K. L., Kalsbeek A., Soeters M. R., Eggink H. M. (2017). Bile acid signaling pathways from the enterohepatic circulation to the central nervous system. Front. Neurosci. 11:617. 10.3389/fnins.2017.00617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao L., Dong Y., Zhou F. B., Chang Y. L., Suo Z. G., Ding H. Q. (2018). Protective effect of tauroursodeoxycholic acid on the autophagy of nerve cells in rats with acute spinal cord injury. Eur. Rev. Med. Pharmacol. Sci. 22 1133–1141. 10.26355/eurrev_201802_14402 [DOI] [PubMed] [Google Scholar]
- Mikov M., Ðanić M., Pavlović N., Stanimirov B., Goloèorbin-Kon S., Stankov K., et al. (2017). Potential applications of gliclazide in treating type 1 diabetes mellitus: formulation with bile acids and probiotics. Eur. J. Drug Metab. Pharmacokinet. 43 269–280. 10.1007/s13318-017-0441-y [DOI] [PubMed] [Google Scholar]
- Mikov M., Fawcett J. P. (2006). Chemistry, biosynthesis, analysis, chemical & metabolic transformations and pharmacology. Eur. J. Drug Metab. Pharmacokinet. 31 133–134. [DOI] [PubMed] [Google Scholar]
- Mikov M., Fawcett J. P., Kuhajda K., Kevresan S. (2006). Pharmacology of bile acids and their derivatives: absorption promoters and therapeutic agents. Eur. J. Drug Metab. Pharmacokinet. 31 237–251. [DOI] [PubMed] [Google Scholar]
- Mircioiu I., Anuta V., Ibrahim N., Mircioiu C. (2012). Dissolution of tamoxifen in biorelevant media. A two phase release model. Farmacia 6 315–324. [Google Scholar]
- Miyazaki-Anzai S., Masuda M., Levi M., Keenan A. L., Miyazaki M. (2014). Dual activation of the bile acid nuclear receptor FXR and G-protein-coupled receptor TGR5 protects mice against atherosclerosis. PLoS One 9:e108270. 10.1371/journal.pone.0108270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinaro A., Wahlstrom A., Marschall H. U. (2018). Role of bile acids in metabolic control. Trends Endocrinol. Metab. 29 31–41. 10.1016/j.tem.2017.11.002 [DOI] [PubMed] [Google Scholar]
- Monte M. J., Marin J. J. G., Antelo A., Vazquez-Tato J. (2009). Bile acids: chemistry, physiology, and pathophysiology. World J. Gastroenterol. 15 804–816. 10.3748/wjg.15.804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudaliar S., Henry R. R., Sanyal A. J., Morrow L., Marschall H. U., Kipnes M., et al. (2013). Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 145 574–582.e1. 10.1053/j.gastro.2013.05.042 [DOI] [PubMed] [Google Scholar]
- Mueller M., Castro R. E. (2018). Ursodeoxycholic acid: effects on hepatic unfolded protein response, apoptosis and oxidative stress in morbidly obese patients. Liver Int. 38 523–531. 10.1111/liv.13562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullard A. (2016). 2015 FDA drug approvals. Nat. Rev. Drug Discov. 15 73–76. 10.1038/nrd.2016.15 [DOI] [PubMed] [Google Scholar]
- Nakahara M., Fujii H., Maloney P. R., Shimizu M., Sato R. (2002). Bile acids enhance low density lipoprotein receptor gene expression via a MAPK cascade-mediated stabilization of mRNA. J. Biol. Chem. 277 37229–37234. 10.1074/jbc.M206749200 [DOI] [PubMed] [Google Scholar]
- Neuschwander-Tetri B. A., Loomba R., Sanyal A. J., Lavine J. E., Van Natta M. L., Abdelmalek M. F., et al. (2015). Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet 385 956–965. 10.1016/s0140-6736(14)61933-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevens F., Andreone P., Mazzella G., Strasser S. I., Bowlus C., Invernizzi P., et al. (2016). A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N. Engl. J. Med. 375 631–643. 10.1056/NEJMoa1509840 [DOI] [PubMed] [Google Scholar]
- Osto M., Abegg K., Bueter M., le Roux C. W., Cani P. D., Lutz T. A. (2013). Roux-en-Y gastric bypass surgery in rats alters gut microbiota profile along the intestine. Physiol. Behav. 119 92–96. 10.1016/j.physbeh.2013.06.008 [DOI] [PubMed] [Google Scholar]
- Ozcan U., Yilmaz E., Ozcan L., Furuhashi M., Vaillancourt E., Smith R. O., et al. (2006). Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313 1137–1140. 10.1126/science.1128294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X., Zhang Y., Wang L., Hussain M. M. (2010). Diurnal regulation of MTP and plasma triglyceride by CLOCK is mediated by SHP. Cell Metab. 12 174–186. 10.1016/j.cmet.2010.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papazyan R., Liu X., Liu J., Dong B., Plummer E. M., Lewis R. D., II, et al. (2018). FXR activation by obeticholic acid or non-steroidal agonists induces a human-like lipoprotein cholesterol change in mice with humanized chimeric liver. J. Lipid Res. 59 982–993. 10.1194/jlr.M081935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paumgartner G., Beuers U. (2002). Ursodeoxycholic acid in cholestatic liver disease: mechanisms of action and therapeutic use revisited. Hepatology 36 525–531. 10.1053/jhep.2002.36088 [DOI] [PubMed] [Google Scholar]
- Pavek P. (2016). Pregnane X receptor (PXR)-mediated gene repression and cross-talk of PXR with other nuclear receptors via coactivator interactions. Front. Pharmacol. 7:456. 10.3389/fphar.2016.00456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlovic N., Stanimirov B., Mikov M. (2017). Bile acids as novel pharmacological agents: the interplay between gene polymorphisms, epigenetic factors and drug response. Curr. Pharm. Des. 23 187–215. 10.2174/1381612822666161006161409 [DOI] [PubMed] [Google Scholar]
- Pellicciari R., Fiorucci S., Camaioni E., Clerici C., Costantino G., Maloney P. R., et al. (2002). 6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J. Med. Chem. 45 3569–3572. [DOI] [PubMed] [Google Scholar]
- Pellicciari R., Gioiello A., Macchiarulo A., Thomas C., Rosatelli E., Natalini B., et al. (2009). Discovery of 6alpha-ethyl-23(S)-methylcholic acid (S-EMCA, INT-777) as a potent and selective agonist for the TGR5 receptor, a novel target for diabesity. J. Med. Chem. 52 7958–7961. 10.1021/jm901390p [DOI] [PubMed] [Google Scholar]
- Pencek R., Marmon T., Roth J. D., Liberman A., Hooshmand-Rad R., Young M. A. (2016). Effects of obeticholic acid on lipoprotein metabolism in healthy volunteers. Diabetes Obes. Metab. 18 936–940. 10.1111/dom.12681 [DOI] [PubMed] [Google Scholar]
- Penney N. C., Kinross J., Newton R. C., Purkayastha S. (2015). The role of bile acids in reducing the metabolic complications of obesity after bariatric surgery: a systematic review. Int. J. Obes. 39 1565–1574. 10.1038/ijo.2015.115 [DOI] [PubMed] [Google Scholar]
- Perez M. J., Briz O. (2009). Bile-acid-induced cell injury and protection. World J. Gastroenterol. 15 1677–1689. 10.3748/wjg.15.1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pols T. W., Nomura M., Harach T., Lo Sasso G., Oosterveer M. H., Thomas C., et al. (2011). TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell Metab. 14 747–757. 10.1016/j.cmet.2011.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pournaras D. J., Glicksman C., Vincent R. P., Kuganolipava S., Alaghband-Zadeh J., Mahon D., et al. (2012). The role of bile after roux-en-Y gastric bypass in promoting weight loss and improving glycaemic control. Endocrinology 153 3613–3619. 10.1210/en.2011-2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz P., Hofmann T., Ahnis A., Elbelt U., Goebel-Stengel M., Klapp B. F., et al. (2015). Plasma bile acids show a positive correlation with body mass index and are negatively associated with cognitive restraint of eating in obese patients. Front. Neurosci. 9:199. 10.3389/fnins.2015.00199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin P., Tang X., Elloso M. M., Harnish D. C. (2006). Bile acids induce adhesion molecule expression in endothelial cells through activation of reactive oxygen species, NF-kappaB, and p38. Am. J. Physiol. Heart Circ. Physiol. 291 H741–H747. 10.1152/ajpheart.01182.2005 [DOI] [PubMed] [Google Scholar]
- Quercia I., Dutia R., Kotler D. P., Belsley S., Laferrere B. (2014). Gastrointestinal changes after bariatric surgery. Diabetes Metab. 40 87–94. 10.1016/j.diabet.2013.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghow R. (2015). Menage-a-trois of bariatric surgery, bile acids and the gut microbiome. World J. Diabetes 6 367–370. 10.4239/wjd.v6.i3.367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Perez O., Cruz-Ramon V., Chinchilla-Lopez P., Mendez-Sanchez N. (2017). The role of the gut microbiota in bile acid metabolism. Ann. Hepatol. 16 s15–s20. 10.5604/01.3001.0010.5494 [DOI] [PubMed] [Google Scholar]
- Renga B., Mencarelli A., Migliorati M., Distrutti E., Fiorucci S. (2009). Bile-acid-activated farnesoid X receptor regulates hydrogen sulfide production and hepatic microcirculation. World J. Gastroenterol. 15 2097–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renga B., Mencarelli A., Vavassori P., Brancaleone V., Fiorucci S. (2010). The bile acid sensor FXR regulates insulin transcription and secretion. Biochim. Biophys. Acta 1802 363–372. 10.1016/j.bbadis.2010.01.002 [DOI] [PubMed] [Google Scholar]
- Ridlon J. M., Harris S. C., Bhowmik S., Kang D. J., Hylemon P. B. (2016). Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 7 22–39. 10.1080/19490976.2015.1127483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roda A., Grigolo B., Pellicciari R., Natalini B. (1989). Structure-activity relationship studies on natural and synthetic bile acid analogs. Dig. Dis. Sci. 34 24s–35s. [DOI] [PubMed] [Google Scholar]
- Roda A., Hofmann A. F., Mysels K. J. (1983). The influence of bile salt structure on self-association in aqueous solutions. J. Biol. Chem. 258 6362–6370. [PubMed] [Google Scholar]
- Rodrigues P. M., Afonso M. B., Simão A. L., Carvalho C. C., Trindade A., Duarte A., et al. (2017). miR-21 ablation and obeticholic acid ameliorate nonalcoholic steatohepatitis in mice. Cell Death Dis. 8:e2748. 10.1038/cddis.2017.172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa A. I., Duarte-Silva S., Silva-Fernandes A., Nunes M. J., Carvalho A. N., Rodrigues E., et al. (2018). Tauroursodeoxycholic acid improves motor symptoms in a mouse model of Parkinson’s disease. Mol. Neurobiol. 55 9139–9155. 10.1007/s12035-018-1062-4 [DOI] [PubMed] [Google Scholar]
- Ryan K. K., Tremaroli V., Clemmensen C., Kovatcheva-Datchary P., Myronovych A., Karns R., et al. (2014). FXR is a molecular target for the effects of vertical sleeve gastrectomy. Nature 509 183–188. 10.1038/nature13135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi M., Prigeon R. L., D’Alessio D. A. (2011). Gastric bypass surgery enhances glucagon-like peptide 1-stimulated postprandial insulin secretion in humans. Diabetes Metab. Res. Rev. 60 2308–2314. 10.2337/db11-0203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal A. J. (2011). NASH: a global health problem. Hepatol. Res. 41 670–674. 10.1111/j.1872-034X.2011.00824.x [DOI] [PubMed] [Google Scholar]
- Sato H., Macchiarulo A., Thomas C., Gioiello A., Une M., Hofmann A. F., et al. (2008). Novel potent and selective bile acid derivatives as TGR5 agonists: biological screening, structure-activity relationships, and molecular modeling studies. J. Med. Chem. 51 1831–1841. 10.1021/jm7015864 [DOI] [PubMed] [Google Scholar]
- Schaap F. G. (2012). Role of fibroblast growth factor 19 in the control of glucose homeostasis. Curr. Opin. Clin. Nutr. Metab. Care 15 386–391. 10.1097/MCO.0b013e3283547171 [DOI] [PubMed] [Google Scholar]
- Schoenfield L. J., Lachin J. M. (1981). Chenodiol (chenodeoxycholic acid) for dissolution of gallstones: the National Cooperative Gallstone Study. A controlled trial of efficacy and safety. Ann. Intern. Med. 95 257–282. [DOI] [PubMed] [Google Scholar]
- Shapiro H., Kolodziejczyk A. A., Halstuch D., Elinav E. (2018). Bile acids in glucose metabolism in health and disease. J. Exp. Med. 215 383–396. 10.1084/jem.20171965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H., Zhang Y., Ding H., Wang X., Chen L., Jiang H., et al. (2008). Farnesoid X receptor induces GLUT4 expression through FXR response element in the GLUT4 promoter. Cell. Physiol. Biochem. 22 1–14. 10.1159/000149779 [DOI] [PubMed] [Google Scholar]
- Shimizu H., Hagio M., Iwaya H., Tsuneki I., Lee J. Y., Fukiya S., et al. (2014). Deoxycholic acid is involved in the proliferation and migration of vascular smooth muscle cells. J. Nutr. Sci. Vitaminol. 60 450–454. 10.3177/jnsv.60.450 [DOI] [PubMed] [Google Scholar]
- Sinisalo J., Vanhanen H., Pajunen P., Vapaatalo H., Nieminen M. S. (1999). Ursodeoxycholic acid and endothelial-dependent, nitric oxide-independent vasodilatation of forearm resistance arteries in patients with coronary heart disease. Br. J. Clin. Pharmacol. 47 661–665. 10.1046/j.1365-2125.1999.00940.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanimirov B., Stankov K., Mikov M. (2012). Pleiotropic functions of bile acids mediated by the farnesoid X receptor. Acta Gastroenterol. Belg. 75 389–398. [PubMed] [Google Scholar]
- Stayrook K. R., Bramlett K. S., Savkur R. S., Ficorilli J., Cook T., Christe M. E., et al. (2005). Regulation of carbohydrate metabolism by the farnesoid X receptor. Endocrinology 146 984–991. 10.1210/en.2004-0965 [DOI] [PubMed] [Google Scholar]
- Stedman C., Robertson G., Coulter S., Liddle C. (2004). Feed-forward regulation of bile acid detoxification by CYP3A4: studies in humanized transgenic mice. J. Biol. Chem. 279 11336–11343. 10.1074/jbc.M310258200 [DOI] [PubMed] [Google Scholar]
- Stedman C. A., Liddle C., Coulter S. A., Sonoda J., Alvarez J. G., Moore D. D., et al. (2005). Nuclear receptors constitutive androstane receptor and pregnane X receptor ameliorate cholestatic liver injury. Proc. Natl. Acad. Sci. U.S.A. 102 2063–2068. 10.1073/pnas.0409794102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner C., Othman A., Saely C. H., Rein P., Drexel H., von Eckardstein A., et al. (2011). Bile acid metabolites in serum: intraindividual variation and associations with coronary heart disease, metabolic syndrome and diabetes mellitus. PLoS One 6:e25006. 10.1371/journal.pone.0025006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojančević M., Bojic G., Salami H. A., Mikov M. (2014). The influence of intestinal tract and probiotics on the fate of orally administered drugs. Curr. Issues Mol. Biol. 16 55–68. [PubMed] [Google Scholar]
- Stojančević M., Pavlović N., Goločorbin-Kon S., Mikov M. (2013). Application of bile acids in drug formulation and delivery. Front. Life Sci. 7 112–122. 10.1080/21553769.2013.879925 [DOI] [Google Scholar]
- Taoka H., Yokoyama Y., Morimoto K., Kitamura N., Tanigaki T., Takashina Y., et al. (2016). Role of bile acids in the regulation of the metabolic pathways. World J. Diabetes 7 260–270. 10.4239/wjd.v7.i13.260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C., Gioiello A., Noriega L., Strehle A., Oury J., Rizzo G., et al. (2009). TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 10 167–177. 10.1016/j.cmet.2009.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trauner M., Halilbasic E., Claudel T., Steinacher D., Fuchs C., Moustafa T., et al. (2015). Potential of nor-ursodeoxycholic acid in cholestatic and metabolic disorders. Dig. Dis. 33 433–439. 10.1159/000371904 [DOI] [PubMed] [Google Scholar]
- Uppal H., Toma D., Saini S. P., Ren S., Jones T. J., Xie W. (2005). Combined loss of orphan receptors PXR and CAR heightens sensitivity to toxic bile acids in mice. Hepatology 41 168–176. 10.1002/hep.20512 [DOI] [PubMed] [Google Scholar]
- Urizar N. L., Dowhan D. H., Moore D. D. (2000). The farnesoid X-activated receptor mediates bile acid activation of phospholipid transfer protein gene expression. J. Biol. Chem. 275 39313–39317. 10.1074/jbc.M007998200 [DOI] [PubMed] [Google Scholar]
- van Nierop F. S., Scheltema M. J., Eggink H. M., Pols T. W., Sonne D. P., Knop F. K., et al. (2017). Clinical relevance of the bile acid receptor TGR5 in metabolism. Lancet Diabetes Endocrinol. 5 224–233. 10.1016/s2213-8587(16)30155-3 [DOI] [PubMed] [Google Scholar]
- Vaz F. M., Ferdinandusse S. (2017). Bile acid analysis in human disorders of bile acid biosynthesis. Mol. Aspects Med. 56 10–24. 10.1016/j.mam.2017.03.003 [DOI] [PubMed] [Google Scholar]
- Verbeke L., Farre R., Trebicka J., Komuta M., Roskams T., Klein S., et al. (2014). Obeticholic acid, a farnesoid X receptor agonist, improves portal hypertension by two distinct pathways in cirrhotic rats. Hepatology 59 2286–2298. 10.1002/hep.26939 [DOI] [PubMed] [Google Scholar]
- Vrieze A., Out C., Fuentes S., Jonker L., Reuling I., Kootte R. S., et al. (2014). Impact of oral vancomycin on gut microbiota, bile acid metabolism, and insulin sensitivity. J. Hepatol. 60 824–831. 10.1016/j.jhep.2013.11.034 [DOI] [PubMed] [Google Scholar]
- Wahlstrom A., Sayin S. I., Marschall H. U., Backhed F. (2016). Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. 24 41–50. 10.1016/j.cmet.2016.05.005 [DOI] [PubMed] [Google Scholar]
- Walsh L. K., Restaino R. M., Neuringer M., Manrique C., Padilla J. (2016). Administration of tauroursodeoxycholic acid prevents endothelial dysfunction caused by an oral glucose load. Clin. Sci. 130 1881–1888. 10.1042/cs20160501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters J. R., Johnston I. M., Nolan J. D., Vassie C., Pruzanski M. E., Shapiro D. A. (2015). The response of patients with bile acid diarrhoea to the farnesoid X receptor agonist obeticholic acid. Aliment. Pharmacol. Ther. 41 54–64. 10.1111/apt.12999 [DOI] [PubMed] [Google Scholar]
- Wang W., Zhao J., Gui W., Sun D., Dai H., Xiao L., et al. (2018). Tauroursodeoxycholic acid inhibits intestinal inflammation and barrier disruption in mice with non-alcoholic fatty liver disease. Br. J. Pharmacol. 175 469–484. 10.1111/bph.14095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. G., Zhou J. M., Ma Z. C., Li H., Liang Q. D., Tan H. L., et al. (2012). Pregnane X receptor mediated-transcription regulation of CYP3A by glycyrrhizin: a possible mechanism for its hepatoprotective property against lithocholic acid-induced injury. Chem. Biol. Interact. 200 11–20. 10.1016/j.cbi.2012.08.023 [DOI] [PubMed] [Google Scholar]
- Watanabe M., Houten S. M., Wang L., Moschetta A., Mangelsdorf D. J., Heyman R. A., et al. (2004). Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Invest. 113 1408–1418. 10.1172/JCI200421025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wree A., Broderick L., Canbay A., Hoffman H. M., Feldstein A. E. (2013). From NAFLD to NASH to cirrhosis—new insights into disease mechanisms. Nat. Rev. Gastroenterol. Hepatol. 10 627–636. 10.1038/nrgastro.2013.149 [DOI] [PubMed] [Google Scholar]
- Yadav H., Lee J. H., Lloyd J., Walter P., Rane S. G. (2013). Beneficial metabolic effects of a probiotic via butyrate-induced GLP-1 hormone secretion. J. Biol. Chem. 288 25088–25097. 10.1074/jbc.M113.452516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata K., Daitoku H., Shimamoto Y., Matsuzaki H., Hirota K., Ishida J., et al. (2004). Bile acids regulate gluconeogenic gene expression via small heterodimer partner-mediated repression of hepatocyte nuclear factor 4 and Foxo1. J. Biol. Chem. 279 23158–23165. 10.1074/jbc.M314322200 [DOI] [PubMed] [Google Scholar]
- Yang L., Zhang H., Fawcett J. P., Mikov M., Tucker I. G. (2011). Effect of bile salts on the transport of morphine-6-glucuronide in rat brain endothelial cells. J. Pharm. Sci. 100 1516–1524. 10.1002/jps.22390 [DOI] [PubMed] [Google Scholar]
- Zhang Y., Lee F. Y., Barrera G., Lee H., Vales C., Gonzalez F. J., et al. (2006). Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc. Natl. Acad. Sci. U.S.A. 103 1006–1011. 10.1073/pnas.0506982103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwicker B. L., Agellon L. B. (2013). Transport and biological activities of bile acids. Int. J. Biochem. Cell Biol. 45 1389–1398. 10.1016/j.biocel.2013.04.012 [DOI] [PubMed] [Google Scholar]