

Alpha granules are the most abundant of the platelet secretory granules, and are packaged with over 300 proteins including von Willebrand Factor, platelet factor 4, thrombospondin, and fibrinogen (Blair P, et al., 2009). Release of alpha granule contents is essential for all platelet functions including hemostasis, inflammation, immunity, wound healing, and angiogenesis (Blair P, et al., 2009). Alpha granules are packaged in multivesicular bodies in the megakaryocyte, their cargo consisting of both endocytosed and biosynthesized proteins, and are then trafficked individually along proplatelets to their final destination in the platelet bud (Heijnen HF, et al., 1998). Alpha granule packaging appears to be a highly regulated process, but the exact mechanism of alpha granule biogenesis is not completely understood.
Much of what is known about alpha granule biogenesis comes from studying genetic disorders in which alpha granules are absent or abnormal. Fibrinogen endocytosis and trafficking into platelet alpha granules was first described by tracking infused fibrinogen in a patient with congenital afibrinogenemia (Harrison P, et al., 1989). Additionally, patients with ARC (Arthrogryposis, Renal dysfunction and Cholestasis) syndrome have platelets that are completely void of alpha granules. The genes associated with ARC syndrome, VPS33b and VPS16b, localize to the trans-Golgi network, multivesicular bodies, and alpha granules (Lo B, et al., 2005, Urban D, et al., 2012). VBS16b and VPS33b are thus thought to be associated with granule biogenesis and trafficking of biosynthesized alpha granule proteins. Finally, Gray Platelet Syndrome (GPS), a rare autosomal recessive bleeding disorder, is characterized by improperly formed alpha granules, macrothrombocytopenia, and myelofibrosis (Nurden AT, et al., 2007). The alpha granules in GPS patients are also called ‘ghost granules’ due to their empty vacuole appearance in platelets -- these granules contain membrane proteins such as P- selectin but lack both endocytosed and biosynthesized alpha granule proteins (Maynard DM, et al., 2010). The genetic etiology of GPS was described in 2011 to be deleterious mutations in the gene NBEAL2 (Albers CA, et al., 2011, Gunay-Aygun M, et al., 2011, Kahr WH, et al., 2011). The NBEAL2 (neurobeachin-like protein 2) protein is a member of a family of proteins that contain a BEACH (Beige and Chediak Higashi) domain, a highly conserved domain associated with vesicular trafficking, membrane dynamics, and receptor signaling (Wang X, et al., 2000). The GPS phenotype is consistent with defects in other members of the BEACH domain protein family, such as LYST, which causes Chediak Higashi Syndrome, a syndrome characterized by melanosome, lysosome, and platelet dense granule defects (Karim MA, et al., 1997). Apart from what can be inferred from homology with other proteins, very little is known about the molecular contribution of NBEAL2 to alpha granule biogenesis and platelet production. Nbeal2 knockout mice have impaired megakaryocyte maturation and ploidy as well as increased extracellular secretion of VWF when compared to wild-type (WT) mice (Kahr WH, et al., 2013). This knockout mouse also exhibits impaired platelet function resulting in decreased arterial thrombosis, protection from inflammatory cerebral ischemia, and impaired wound healing (Kahr WH, et al., 2013, Deppermann C, et al., 2013). Although the mouse model of GPS has been well- characterized, highlighting the multifactorial roles of platelet alpha granules, the function of the NBEAL2 protein in the process of alpha granule biogenesis remains elusive.
In this issue of ATVB, Kahr and colleagues dissect the molecular mechanism of this obscure protein, significantly furthering the field of alpha granule biogenesis. Lo et al use the Nbeal2 knockout mouse to show that the resulting defect in alpha granule biogenesis does not occur in early endocytosis of alpha granule proteins, namely fibrinogen, but rather in the retention of these proteins at later stages. Specifically, this paper elegantly describes the trafficking of fibrinogen in both WT and Nbeal2 knockout megakaryocytes, showing that fibrinogen co- localizes with RAB5-associated early endosomes in both WT and knockout megakaryocytes at early stages of endocytosis. However, while WT megakaryocytes traffic fibrinogen from early endosomes to RAB7-associated late endosomes, and finally to P-selectin positive alpha granules, Nbeal2 knockout megakaryocytes fail to retain fibrinogen into RAB7 or P-selectin structures. Interestingly, Nbeal2 knockout megakaryocytes instead traffic fibrinogen to RAB11- associated recycling endosomes at later time points. Labeled fibrinogen was also shown to be more rapidly released into the media of Nbeal2 knockout megakaryocytes in vitro. Similarly, von Willebrand factor significantly associates with RAB11 in Nbeal2 knockout megakaryocytes, indicating that this altered trafficking process involves both biosynthesized and endocytosed alpha granule proteins. These data show that Nbeal2 is responsible for retention of proteins in alpha granules, and that its absence does not disrupt early endocytosis, but results in trafficking of alpha granule proteins to recycling endosomes and secretion into the extracellular space. Furthermore, this paper probes the question of the normal function of NBEAL2 by examining localization with other proteins. Using a novel antibody, Lo et al showed co-localization of NBEAL2 with P-selectin by immunofluorescence, electron microscopy, and co- immunoprecipitation in primary human megakaryocytes. The authors posit that this interaction with P-selectin stabilizes alpha granules and prevents cargo loss to recycling RAB11 endosomes. Thus, the phenotype of Nbeal2 knockout is likely due to loss of this interaction with P-selectin and aberrant secretion of alpha granule proteins through the recycling endosome and exocytosis pathways, or as Lo et al adeptly stated: “In essence, loss of NBEAL2 turns MKs from packaging cells into secreting cells.”
This work has progressed the study of alpha granule biogenesis by demonstrating that alpha granule packaging involves a balance of protein trafficking to alpha granules and retention of proteins to prevent release into secretory pathways. Here, NBEAL2 is shown to be responsible for retention of proteins into the alpha granules, potentially through its association with P- selectin, and loss of NBEAL2 results in trafficking to recycling and secretory endosomes. Additionally, this work contributes to the knowledge of the GPS phenotype. The hallmark of this rare disease is a lack of alpha granules; however, patients and mice lacking NBEAL2 have macrothrombocytopenia, bone marrow fibrosis, and extramedullary hematopoiesis (Nurden AT, et al., 2007). Excessive secretion of alpha granule proteins from megakaryocytes lacking Nbeal2 suggests that the alpha granule proteins, many of which are inflammatory cytokines, are secreted into the bone marrow space in vivo, causing inflammation and the potential for fibrosis. Furthermore, factors contained in platelet alpha granules, such as platelet factor 4 and CCL5, have been previously shown to alter the potential for megakaryocyte proplatelet formation (Lambert MP, et al., 2007, Machlus KR, et al., 2016). One hypothesis for the thrombocytopenia seen in GPS is an auto-inhibitory mechanism of aberrantly secreted alpha granule proteins acting on the megakaryocyte itself. This work demonstrates that alpha granule proteins are indeed secreted through a RAB11-mediated mechanism in the absence of Nbeal2. Future directions in this field would include determining whether excess secretion of alpha granule proteins is inhibitory to proplatelet formation, or whether NBEAL2 and/or proper alpha granule biogenesis are essential for complete platelet production.
Platelets and their alpha granules contain factors involved in inflammatory and antimicrobial responses, angiogenesis, as well as cancer progression. Thus, understanding the function of a gene such as NBEAL2 can contribute to our knowledge of not only rare bleeding diseases such as GPS, but of thrombopoiesis and alpha granule biogenesis at large. Further investigation of alpha granule packaging may lead to discovering ways to selectively load proteins into alpha granules for creation of ‘designer platelets.’ Lo et al provide a significant first step in understanding this process through their work in GPS and NBEAL2.
Figure 1.
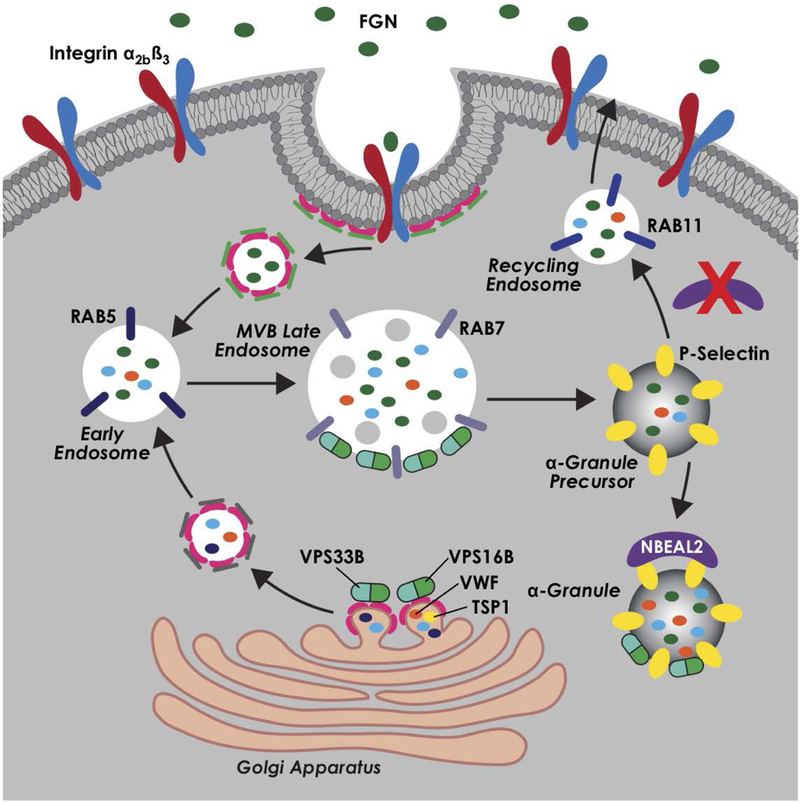
Current model for alpha-granule biogenesis and roles of alpha-granule packaging proteins VPS16B, VPS33B, and NBEAL2. Professional illustration by Kristin Johnson
Footnotes
Disclosures
J.E.I. has financial interest in and is a founder of Platelet BioGenesis, a company that aims to produce donor-independent human platelets from human-induced pluripotent stem cells at scale. J.E.I. is an inventor on this patent. The interests of J.E.I. were reviewed and are managed by the Brigham and Women’s Hospital and Partners HealthCare in accordance with their conflict-of-interest policies. The remaining authors declare no competing financial interests.
References
- Blair P and Flaumenhaft R. Platelet alpha-granules: basic biology and clinical correlates. Blood Rev. 2009; 23:177–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijnen HF, Debili N, Vainchencker W, Breton-Gorius J, Geuze HJ and Sixma JJ. Multivesicular bodies are an intermediate stage in the formation of platelet alpha-granules. Blood. 1998; 91:2313–25. [PubMed] [Google Scholar]
- Harrison P, Wilbourn B, Debili N, Vainchenker W, Breton-Gorius J, Lawrie AS, Masse JM, Savidge GF and Cramer EM. Uptake of plasma fibrinogen into the alpha granules of human megakaryocytes and platelets. J Clin Invest. 1989; 84:1320–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo B, Li L, Gissen P, Christensen H, McKiernan PJ, Ye C, Abdelhaleem M, Hayes JA, Williams MD, Chitayat D and Kahr WH. Requirement of VPS33B, a member of the Sec1/Munc18 protein family, in megakaryocyte and platelet alpha-granule biogenesis. Blood. 2005; 106:4159–66. [DOI] [PubMed] [Google Scholar]
- Urban D, Li L, Christensen H, Pluthero FG, Chen SZ, Puhacz M, Garg PM, Lanka KK, Cummings JJ, Kramer H, Wasmuth JD, Parkinson J and Kahr WH. The VPS33B-binding protein VPS16B is required in megakaryocyte and platelet alpha-granule biogenesis. Blood. 2012; 120:5032–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurden AT and Nurden P. The gray platelet syndrome: clinical spectrum of the disease. Blood Rev. 2007; 21:21–36. [DOI] [PubMed] [Google Scholar]
- Maynard DM, Heijnen HF, Gahl WA and Gunay-Aygun M. The alpha-granule proteome: novel proteins in normal and ghost granules in gray platelet syndrome. J Thromb Haemost. 2010; 8:1786–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albers CA, Cvejic A, Favier R, et al. Exome sequencing identifies NBEAL2 as the causative gene for gray platelet syndrome. Nat Genet. 2011; 43:735–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunay-Aygun M, Falik-Zaccai TC, Vilboux T, et al. NBEAL2 is mutated in gray platelet syndrome and is required for biogenesis of platelet alpha-granules. Nat Genet. 2011; 43:732–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahr WH, Hinckley J, Li L, et al. Mutations in NBEAL2, encoding a BEACH protein, cause gray platelet syndrome. Nat Genet. 2011; 43:738–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Herberg FW, Laue MM, Wullner C, Hu B, Petrasch-Parwez E and Kilimann MW. Neurobeachin: A protein kinase A-anchoring, beige/Chediak-higashi protein homolog implicated in neuronal membrane traffic. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2000; 20:8551–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim MA, Nagle DL, Kandil HH, Burger J, Moore KJ and Spritz RA. Mutations in the Chediak- Higashi syndrome gene (CHS1) indicate requirement for the complete 3801 amino acid CHS protein. Hum Mol Genet. 1997; 6:1087–9. [DOI] [PubMed] [Google Scholar]
- Kahr WH, Lo RW, Li L, Pluthero FG, Christensen H, Ni R, Vaezzadeh N, Hawkins CE, Weyrich AS, Di Paola J, Landolt-Marticorena C and Gross PL. Abnormal megakaryocyte development and platelet function in Nbeal2(−/−) mice. Blood. 2013; 122:3349–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deppermann C, Cherpokova D, Nurden P, et al. Gray platelet syndrome and defective thrombo- inflammation in Nbeal2-deficient mice. J Clin Invest. 2013; 123(8):3331–3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Rauova L, Bailey M, Sola-Visner MC, Kowalska MA and Poncz M. Platelet factor 4 is a negative autocrine in vivo regulator of megakaryopoiesis: clinical and therapeutic implications. Blood. 2007; 110:1153–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machlus KR, Johnson KE, Kulenthirarajan R, Forward JA, Tippy MD, Soussou TS, El-Husayni SH, Wu SK, Wang S, Watnick RS, Italiano JE Jr., and Battinelli EM . CCL5 derived from platelets increases megakaryocyte proplatelet formation. Blood. 2016; 127:921–6. [DOI] [PMC free article] [PubMed] [Google Scholar]